

Summary
Phospholipid biosynthesis is critical for the development, differentiation and pathogenesis of several eukaryotic pathogens. Genetic studies have validated the pathway for phosphatidylethanolamine synthesis from phosphatidylserine catalyzed by phosphatidylserine decarboxylase enzymes (PSD) as a suitable target for development of antimicrobials; however no inhibitors of this class of enzymes have been discovered. We show that the Plasmodium falciparum PSD can restore the essential function of the yeast gene in strains requiring PSD for growth. Genetic, biochemical and metabolic analyses demonstrate that amino acids between positions 40 and 70 of the parasite enzyme are critical for proenzyme processing and decarboxylase activity. We used the essential role of Plasmodium PSD in yeast as a tool for screening a library of anti-malarials. One of these compounds is 7-chloro-N-(4-ethoxyphenyl)-4-quinolinamine, an inhibitor with potent activity against P. falciparum, and low toxicity toward mammalian cells. We synthesized an analog of this compound and showed that it inhibits PfPSD activity and eliminates Plasmodium yoelii infection in mice. These results highlight the importance of 4-quinolinamines as a novel class of drugs targeting membrane biogenesis via inhibition of PSD activity
Introduction
Malaria caused by Plasmodium parasites remains an important global health problem and a major obstacle to economic development in many parts of the world. The World Malaria Report 2014 concluded that in the African continent alone, malaria is responsible for about 430,000 early childhood deaths every year. Equally concerning, approximately 15 million pregnant women do not have access to preventive treatment for malaria (WHO, 2010). The widespread emergence of resistance to currently approved anti-malarials and insecticides, and the impact outbreaks such as Ebola have on the control of malaria, emphasize the urgent need to develop new, effective and safe strategies to prevent and treat malaria.
Transmission of Plasmodium parasites from Anopheles mosquitoes to humans is accompanied by a rapid multiplication of the parasite first in hepatocytes and subsequently in erythrocytes. The growth and multiplication of the parasite relies heavily on its ability to scavenge host factors, including precursors for phospholipid biosynthesis (Vial and Ben Mamoun, 2005; Pessi and Ben Mamoun, 2006). Metabolic labeling studies and mass spectrometry analyses have shown that phosphatidylcholine (PC) and phosphatidylethanolamine (PE) are the major phospholipids in Plasmodium membranes during all phases of the parasite life cycle. The distribution, structural diversity and role in development, differentiation and pathogenesis of these two phospholipids as well as others such as phosphatidylserine (PS) and phosphatidylinositol (PI) have only started to be elucidated. In fungi PS decarboxylases (PSDs), which catalyze the synthesis of PE from PS have been shown to play a critical role in cell survival, division and virulence (Chen et al., 2010). PE is a phospholipid with a small polar head group and a conical molecular structure. Genetic studies demonstrated a crucial role for this phospholipid in several important biological functions including signaling, autophagy, cytokinesis and viral replication (Emoto and Umeda, 2000; Emoto et al., 2005; Xie and Klionsky, 2007; Paulick and Bertozzi, 2008; Vance and Tasseva, 2013; Xu and Nagy, 2015). The presence of PE in membrane bilayers increases membrane curvature, a physical process known to play a critical role in membrane budding, fusion and fission (Pecheur et al., 2002; Marsh, 2007; Joshi et al., 2012). To date, no inhibitors of bacterial or eukaryotic PSD enzymes have been developed as antimicrobials.
In Plasmodium falciparum, metabolic reconstitution and expression profiling of genes encoding enzymes involved in phospholipid biosynthesis indicated that PE synthesis in this parasite can be achieved either from PS by decarboxylation catalyzed by a single PSD enzyme, or via the cytidine diphosphate (CDP)-ethanolamine pathway, which requires cellular import of ethanolamine (Fig. 1) (Vial and Ben Mamoun, 2005). Previously, no genetic studies to determine whether the two pathways for PE synthesis are essential for parasite development and survival have been reported. The activity of a reconstituted P. falciparum PfPSD was previously reported and immunofluorescence analyses indicated that the native enzyme is localized to the endoplasmic reticulum (ER) of the parasite (Baunaure et al., 2004). However, the role PfPSD plays in parasite development and survival was not determined. Previous studies using yeast as a model system identified the Plasmodium knowlesi PkPSD gene as a functional homolog of the yeast PSD enzymes (Choi et al., 2012). Unlike mammalian, yeast and bacterial PSDs, which are membrane bound, PkPSD was found in both soluble and membrane-bound forms (Choi et al., 2012; 2015). This property not only demonstrates the uniqueness of the Plasmodium PSD enzyme compared with its human counterparts, but also provides a unique opportunity to investigate its structure. In this study, we have determined several catalytic and physical properties of PfPSD expressed in yeast, tested yeast as a biological platform for screening for PfPSD inhibitors, and report the identification of an inhibitor of PfPSD from the Malaria Box (Spangenberg et al., 2013), a library of chemicals with known activity against P. falciparum in vitro.
Fig. 1.
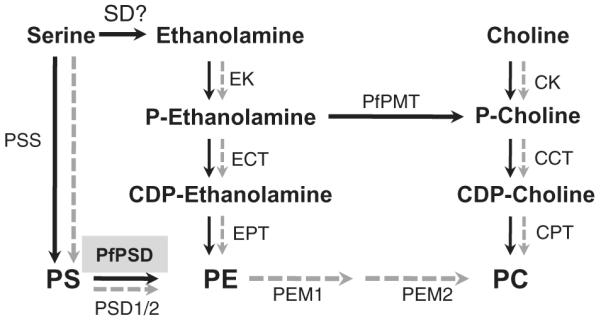
Schematic outline of phospholipid biosynthesis pathways in Plasmodium falciparum and yeast. P. falciparum pathways are depicted with black arrows and yeast pathways are depicted in gray. The gene encoding this activity has not been identified in Plasmodium and no homologs of plant SD enzymes exist in Plasmodium parasites. EK, ethanolamine kinase; ECT, ethanolamine phosphate cytidylyltransferase; EPT, ethanolamine phosphotransferase; CK, choline kinase; CCT, choline phosphate cytidylyltransferase; CPT, choline phosphotransferase; PSD, phosphatidylserine decarboxylase; PSS, phosphatidylserine synthase; PEM1/2, phophatidylethanolamine methyltransferases (1 and 2); PfPMT, phosphoethanolamine methyltransferase; PS, phosphatidylserine; PE, phosphatidylethanolamine; PC, phosphatidylcholine; PSD, PS decarboxylase; SD, serine decarboxylase.
Results
Plasmodium falciparum PfPSD complements the loss of PSD activity in yeast
To establish a functional assay to characterize an active PfPSD in vivo, we utilized a yeast mutant devoid of PSD activity because of deletion of the genes encoding mitochondrial PSD1 and non-mitochondrial PSD2. The yeast mutant we chose also lacks the DPL1 gene encoding the sphingosine-1-P lyase that generates phosphoethanolamine from sphingolipid degradation (Choi et al., 2012). The psd1Δpsd2Δdpl1Δ mutant is not viable in minimal medium lacking ethanolamine, but grows to wild-type levels in minimal medium supplemented with ethanolamine (Choi et al., 2012). Because of the high A+T content of PfPSD (71.3%), the gene was first codon-optimized by upgrading the distribution of codon usage frequency along the length of the gene sequence from an index of 0.70 to 0.83 and changing the G+C content from 28.7% found in the original gene to 35.03%. The codon-optimized PfPSD was then cloned into the pBEVY-U yeast expression vector containing the URA3 selectable marker and the resulting vector was used to transform the psd1Δpsd2Δdpl1Δ mutant. Transformants were first selected on solid medium lacking uracil but containing 2 mM ethanolamine and after sub-cloning in the same medium, tested for their ability to grow in the absence of ethanolamine.As shown in Fig. 2A, expression of PfPSD in the psd1Δpsd2Δdpl1Δ mutant restores growth of this auxotrophic mutant in the absence of exogenous ethanolamine. As a positive control, expression of the P. knowlesi PkPSD complements ethanolamine auxotrophy of the mutant as previously described (Choi et al., 2012), whereas a mutant expressing an empty vector failed to grow in the absence of ethanolamine (Fig. 2A). Previous studies have suggested that serine can be converted into ethanolamine via direct serine decarboxylation in Plasmodium falciparum-infected erythrocytes (Elabbadi et al., 1997). Ethanolamine is then subsequently incorporated into PE and PC biosynthesis via the CDP-ethanolamine and PMT pathways (Bobenchik et al., 2011) (Fig. 1). However no serine decarboxylase genes have been identified in the Plasmodium databases. To critically test whether the PfPSD enzyme has any serine decarboxylase activity, the enzyme was expressed in the yeast pss1 mutant strain lacking PS synthase activity. Although the pss1 mutant cannot synthesize PS from serine, it is rescued by ethanolamine supplementation (Atkinson et al., 1980a,b), which provides the precursor to synthesize an essential pool of PE. We reasoned that any significant serine decarboxylase activity exhibited by PfPSD should provide adequate ethanolamine to rescue the pss1 yeast mutant. As shown in Fig. 2B, expression of PfPSD in the pss1 mutant failed to rescue the growth defect of the mutant strain, indicating that the enzyme cannot execute direct decarboxylation of serine to ethanolamine.
Fig. 2.

Genetic evidence for PfPSD-mediated phosphatidylserine decarboxylation, but not serine decarboxylation activity in vivo.
A. PfPSD is a functional PSD enzyme. WT and psd1Δpsd2Δdpl1Δ mutants harboring an empty vector (V), or vectors containing either the full-length PfPSD or its ortholog from P. knowlesi, PkPSD, were grown overnight in minimal medium containing ethanolamine. The strains were harvested by centrifugation in water, to remove medium, and spotted on SD solid media lacking, or containing 2 mM ethanolamine, at a cell density ranging between 10 and 104 cells and incubated at 30°C for up to 9 days.
B. PfPSD does not catalyze serine decarboxylase activity. The yeast pss mutant strains harboring an empty vector (pBEVY) or pBEVY-PfPSD were spotted using serial fivefold dilutions starting with 3 × 104 cells, on SD media lacking, or supplemented with 2 mM ethanolamine and incubated at 30°C for 2 days.
The steady state phospholipid composition of the psd1Δpsd2Δdpl1Δ strain expressing PfPSD was determined and compared with that of a wild-type strain and a psd2Δdpl1Δ strain that harbors wild-type yeast PSD1. Lipids were separated by two-dimensional thin layer chromatography (TLC), visualized by iodine staining and quantified by phosphate analysis (Fig. 3A and B). Expression of PfPSD in a psd1Δpsd2Δdpl1Δ strain resulted in ethanolamine prototrophy, and a phospholipid composition that showed a 34% increase in the steady state levels of PE, a doubling in phosphatidyldimethylethanolamine (PDME) content and 42% reduction in PS, when compared with a yeast strain expressing only PSD1. In yeast, PE can be sequentially methylated to form phosphatidylmonomethylethanolamine (PMME), PDME and PC. Therefore, an accurate index of the efficiency of PfPSD-mediated decarboxylation of PS is the ratio of downstream products (PE+PDME+PC) to PS. As shown in Fig. 3C this ratio is approximately double that of the psd1Δpsd2Δdpl1Δ strains expressing the yeast PSD1 and approximately double that of wild-type yeast. The PE : PC ratio in the wild-type strain is 0.46, and the ratio in the PSD1psd2Δdpl1Δ strain is 0.74, and the ratio is 1.07 in the PfPSD psd1Δpsd2Δdpl1Δ strain.
Fig. 3.

Phospholipid composition of wild-type yeast and mutant strains expressing only yeast or parasite PfPSD. The WT strain (BY4741-pVUT-102U)), and a Δpsd2Δdpl1Δ strain harboring yeast PSD1 (ScPSD1), and the psd1Δpsd2Δdpl1Δ mutant strain harboring pBEVY-PfPSD (PfPSD were grown to log phase in synthetic uracil dropout medium, and lipids were extracted and separated by two-dimensional TLC.
A. The lipids were visualized by iodine staining.
B. The isolated lipids were quantified by phosphorus analysis and the results are expressed as the percentage of total lipid phosphorus. Data are expressed as mean ± range of two experiments.
C. The ratio of product, PE + PDME + PC, over precursor, PS, is indicative of PSD function in vivo. Abbreviations: PC, phosphatidylcholine; PE, phosphatidylethanolamine; PS, phosphatidylserine; PI, phosphatidylinositol; CL, cardiolipin; PA, phosphatidic acid, PDME, phosphatidyldimethylethanolamine.
Soluble and membrane-bound forms of PfPSD catalyze PS decarboxylation
The PfPSD expressed in psd1Δpsd2ΔdplΔ yeast strain was analyzed to characterize the activity and processing of the protein. Western blot analyses using anti-PfPSD antibodies on whole-cell extracts, soluble fractions and membrane fractions, showed the presence of both the unprocessed PfPSD proenzyme and the mature β subunit (Fig. 4A). The majority of PfPSD was in the form of mature β subunit, which indicates that the newly synthesized PfPSD was efficiently processed through an auto-endoproteolytic cleavage mechanism (Choi et al., 2015). Unlike mammalian and other eukaryotic PSDs, which are tightly associated with membranes, PfPSD was detected in both soluble and membrane fractions (Fig. 4B). Quantification of the PfPSD enzyme activity revealed that both fractions were catalytically active with a membrane/soluble distribution of 3/1. In contrast, the distribution of the yeast PSD1 between membrane and soluble fractions was 9.5/1.0 (Fig. 4B). The membrane association of the PfPSD was examined further by expressing an MBP-PfPSD fusion protein in Escherichia coli and testing binding of the protein to multilamellar liposomes (Figs 4C and 5D). In the absence of phospholipid, both the proenzyme and the β subunit partitioned mostly (80%) to the soluble fraction and this distribution was unaffected by the presence of PC liposomes. In contrast, both the proenzyme and β-subunit showed high affinity (75–82% binding) to PS and PG liposomes, but minimal binding to PC liposomes. This latter finding is interesting because both PG and PS have been implicated in regulating processing of PSD (Choi et al., 2012).
Fig. 4.

Biochemical properties of heterologously expressed PfPSD.
A. Western blot analysis was performed with rabbit anti-PfPSD antibody and HRP conjugated goat anti-rabbit antibody to detect PfPSD in the psd1Δpsd2Δdpl1Δ mutant strain expressing PfPSD. Total cell free extracts (T), soluble fractions (S), and membrane fractions (M) were analyzed using anti-PfPSD. The upper band and the lower band are the PfPSD proenzyme and the mature β-subunit, respectively.
B. PSD enzyme assays were performed with soluble fractions and membrane fractions extracted from yeast psd1Δpsd2Δdpl1Δ mutant strains expressing PfPSD. Measurement of PSD catalytic activity utilized Ptd[1′-14C]Ser as the substrate, and the reaction product was trapped as 14CO2 on 2 M KOH impregnated filter paper. Data are means ± standard deviation for two experiments each performed in duplicate.
C. An MBP-PfPSD fusion protein was expressed in Escherichia coli and affinity purified using amylose resin. The binding of MBP-PfPSD to phospholipids was measured after incubation at 37°C for 40 min, using co-sedimentation of the enzyme with multilamellar liposomes (200 μg ml−1) centrifuged at 10,000 × g × 5 min. Aliquots of the supernatant (S) were taken for gel electrophoresis and Western blotting. The resultant pellets were collected and diluted to the same volume as the supernatants, and analyzed by electrophoresis and Western blotting. Mock, indicates no incubation of extracts with liposomes and PS, PC and PG indicate the lipid classes used in separate incubations.
D. Quantification of the enzyme distribution data from panel C. The protein band intensities of the proenzymes and processed β-subunits of the supernatant and the liposome pellet on the Western blot were quantified using Image J software. Data are means ± range for two experiments.
Fig. 5.

Functional analysis of PfPSD deletion mutants.
A. Growth of psd1Δpsd2Δdpl1Δ mutant strain harboring an empty pBEVY-U vector, or the same vector encoding full length, or truncated versions of PfPSD, in the absence, or presence of 2 mM ethanolamine. The three conserved sequence motifs harboring the D-H-S residues required for endoproteolytic processing and activity are highlighted.
B. Growth of the same strains in liquid medium lacking or supplemented with 2 mM ethanolamine.
Role of the N-terminal domain of PfPSD in processing and decarboxylase activity
Sequence analysis of PfPSD identified the D-H-S catalytic triad, previously shown to be critical for auto-endoproteolytic cleavage of the PkPSD proenzyme into mature α- and β-subunits (Choi et al., 2015). Further analysis also revealed an unusual hydrophobicity profile in the N-terminal domain, consisting of the first 70 amino acids in the polypeptide upstream of the aspartate residue of the catalytic triad. To examine the importance of this N-terminal domain, a step-wise deletion approach was undertaken to progressively remove 10 amino acid segments between positions 1 and 70 of PfPSD. The consequences of these deletions were examined using the functional complementation assay in the psd1Δpsd2Δdpl1Δ mutant background. As shown in Fig. 5, deletions up to residue 50 did not reduce ability of the enzyme to complement the ethanolamine auxotrophy of the psd1Δpsd2Δdpl1Δ strain. In contrast, deletions of the first 60 or 70 residues resulted in loss of complementation in both solid (Fig. 5A) and liquid media (Fig. 5B). Metabolic analyses using [3H]-serine as a precursor to monitor synthesis of PS, PE and PC provided good correlation between the growth profile of the strains and their lipid metabolism; with the complemented strains producing radiolabeled PE at levels similar to those found in the mutant expressing the full-length enzyme (Fig. 6A). The increase in PE labeling in complemented strains is paralleled by a decrease in PS labeling, indicating increased turnover of the latter. In strains producing either full-length PfPSD or enzymes with deletions up to the first 50 amino acids, approximately 20% of the radiolabel appears in PS and 60% of the label appears in PE. Most of the label appearing in PC is due to the 3H-serine entering the 1-carbon pathway (Fig. 6A). The (PE + PC)/PS ratio in these cells ranged between 2..0 and 3.8, consistent with the turnover of PS to form PE (Fig. 6B). The failure of the Δ60 and Δ70 PfPSD constructs to complement the mutant yeast strain was accompanied by defects in turnover of PS to PE, that were nearly identical to the defects found in strains lacking any PSD (Figs 6A and 7B).
Fig. 6.

The N-terminal hydrophobic domain encompassing amino acids 1–50 of PfPSD is not essential for PSD enzyme activity in vivo.
A. Incorporation of radioactive serine into phospholipids in the psd1Δpsd2Δdpl1Δ mutant strain harboring full-length PfPSD (F.L.), or truncated versions lacking the first 10, 20, 30, 40, 50, 60 and 70 amino acid residues. Labeled phospholipids were extracted from cells grown for 3 h in synthetic uracil dropout medium containing 10 μCi ml−1 L-[3H(G)] serine. The lipid classes were resolved by thin layer chromatography, and radioactivity was quantified by liquid scintillation spectrometry. Data are the mean ± standard deviation from two experiments performed in duplicate. Results are the percentage of total radiolabel incorporated into each phospholipid.
B. The ratio of products (PE + PC) over precursor (PS) is indicative of the in vivo efficiency of PSD activity.
Fig. 7.

Effect of the N-terminal deletions on the processing of PfPSD proenzymes and the PSD activities.
A. Western blot analysis was performed using anti-PfPSD to detect unprocessed and processed PfPSDs in the psd1Δpsd2Δdpl1Δ mutant strain, expressing full-length and truncated versions lacking the first 10, 20, 30, 40, 50, 60 and 70 amino acid residues of the PfPSD. Sample loading was normalized to the amount of protein present in host cell extracts. The top panel shows the PfPSD proenzyme (marked by asterisk) and the mature β-subunit (lower band), respectively. The lower panel shows the mature α-subunit.
B. The protein band intensities of the proenzymes and processed β-subunits on the Western blot were quantified using Image J software. Data are means ± range for two experiments.
C. PSD enzyme assays were performed as described in Fig. 5B. The PSD enzyme activities were normalized to the quantified β-subunits. Data are means ± standard deviation for two experiments each performed in duplicate.
To examine the effect of the deletions on the processing of PfPSD, anti-PfPSD antibodies were used to detect proenzyme (41.5 kDa) and mature α (5.4 kDa) and β (36 kDa) subunits in extracts from cells expressing full-length and truncated version of PfPSD. As shown in Fig. 7A, the rate of PfPSD processing in cells expressing full length through Δ40 constructs of the enzyme, was similar. However, processing of PfPSD constructs with larger deletions was significantly reduced for Δ50 and Δ60 variants, and absent for the Δ70 variant, with a concomitant increase in the levels of the proenzyme (Fig. 7B). Immunoblotting analyses showed the presence of the α subunit in cells expressing full length through Δ60 constructs of the enzyme and its absence in cells expressing Δ70 (Fig. 7A). The detection of the α-subunit was more variable than the precursor and β-subunits because of its low molecular mass. The lack of both mature α and β subunit in the Δ70 construct suggests an essential role of amino acids between residues 60 and 70 in processing. The catalytic activity of the truncated enzymes ranging from Δ10 to Δ40 was comparable with that of the full-length enzyme, when normalized to the amount of β-subunit formed (Fig. 7C). However, the Δ50 PfPSD showed ~ 40% of the activity of the full-length enzyme, and the Δ60 PfPSD showed only ~ 5% of the activity of the full-length enzyme. This defect is not due to interference in the decarboxylase reaction from unprocessed pro-enzyme, as the activity of the mature full-length PfPSD was not affected by the presence of the unprocessed Δ70 form (Fig. 8A). Thus, in addition to a reduced rate of processing, the Δ50 and Δ60 variants also show a loss in intrinsic decarboxylase activity. In additional experiments we demonstrated that neither increasing the incubation time, nor admixture of the processing competent Δ50 enzyme with the Δ70 enzyme resulted in processing of the Δ70 form (Fig. 8B). These data identify structural elements between positions 60 and 70 of PfPSD as necessary for the proteolytic cleavage of the proenzyme into mature α-and β-subunits. These findings are also consistent with PfPSD processing occurring primarily in cis (Fig. 8B) (Choi et al., 2015).
Fig. 8.

Unprocessed forms of PfPSD proenzyme do not interfere with processing or catalysis reactions of other variants.
A. Extracts were prepared from yeast psd1Δpsd2Δdpl1Δ strains expressing Mock, full-length PfPSD or PfPSDΔ70. The extracts as indicated in the figure were mixed and pre-incubated for 10 min prior start of the enzyme assay for decarboxylase activity. Data are means ± standard deviation for two experiments each performed in duplicate.
B. Cell free extracts of the psd1Δpsd2Δdpl1Δ mutant strain expressing Mock, PfPSDΔ50 or PfPSDΔ70 were mixed for 0 or 2 h as indicated. Western blot analysis was performed to detect the proenzyme and the mature β-subunit of PfPSDΔ70 and PfPSDΔ50 using anti-PfPSD.
PfPSD inhibitors with anti-malarial activity
The finding that PfPSD can complement the lethal phenotype of yeast cells lacking PSD activity made it possible to use yeast as a model system to search for inhibitors of the enzyme. Our initial screening assay is described in Fig. 9 and aimed to identify compounds that can inhibit the growth of psd1Δpsd2Δdpl1Δ cells harboring PkPSD, but can also be rescued by ethanolamine supplementation. We initially limited the screening to compounds already known to have anti-malarial activity by focusing on the Malaria Box, provided by Medicines for Malaria Venture (MMV), which contains 200 drug-like and 200 probe-like compounds (Spangenberg et al., 2013). Because of the generally reduced activity of drugs in yeast, due to active drug efflux and detoxification, the initial screen was performed at 100 μM for each of the compounds in the Malaria Box in a 96-well format assay (Fig. 10). Eighteen compounds showed differential inhibitory activity in the absence vs. presence of ethanolamine (Fig. 10). Available data show that these compounds inhibit growth of P. falciparum 3D7 parasites with EC50 values ranging between 0.2 (MMV011099) and 15 μM (MMV001239) (Table 1) (Spangenberg et al., 2013). Several of these compounds display excellent selectivity indices including MMV011099, MMV019266 with indices of > 132 and > 52, respectively (Table 1). We have initially focused upon MMV007285 because its structure suggested a relatively easy path for synthesis of the molecule and structural analogs.
Fig. 9.

Schematic outline of the genetic assay used to screen the Malaria Box. The screening assay to identify specific inhibitors of PfPSD based on the activity of the compounds to inhibit the growth of psd1Δpsd2Δdpl1Δ-pBEVY-PkPSD strain in the absence, but not the presence of 2 mM ethanolamine. Compounds that inhibit equally well under both conditions are considered non-specific and included compounds found to have a general antifungal activity. The abbreviations used are SP Inhib, specific inhibitor; Non-SP Inhib, non-specific inhibitor.
Fig. 10.

The anti-malarial compound MMV007285 inhibits growth of yeast strains dependent upon PfPSD enzyme activity.
A. Heat-map representation of the results of the chemical screen using yeast as a model system.
B. Enlarged section of (A) with the list of 18 compounds that show enhanced PfPSD inhibitory activity in the absence, but not in the presence of ethanolamine.
C. Percentage growth of yeast cells harboring PkPSD in the absence (black bars) or presence of 100 μM (gray bars) of the compounds.
Table 1.
Activity of Plasmodium PSD inhibitors against Plasmodium falciparum 3D7 and human foreskin fibroblast (HFF).
| Compound ID | MW | ALogP |
P. falciparum 3D7 (EC50 in μM) |
HFF (IC50 in μM) |
SI: HFF/3D7 |
|---|---|---|---|---|---|
| MMV665927 | 282.55 | 2.87 | 0.555 | 16.2 | 29 |
| MMV666106 | 436.57 | 5.13 | 1.07 | >32 | >30 |
| MMV019555 | 478.67 | 8.06 | 0.376 | 4.06 | 11 |
| MMV666026 | 288.30 | 3.66 | 3.845 | >32 | >8 |
| MMV019918 | 383.71 | 3.93 | 0.8 | 4.028 | 5 |
| MMV665949 | 281.13 | 4.12 | 2.47 | >32 | >13 |
| MMV666025 | 500.19 | 5.97 | 1.26 | >32 | >25 |
| MMV396594 | 490.66 | 5.52 | 1.06 | >32 | >30 |
| MMV665971 | 484.95 | 4.86 | 1.14 | 16.594 | >15 |
| MMV019064 | 493.60 | 4.62 | 1.3 | >32 | 25 |
| MMV001239 | 400.45 | 3.74 | 15 | >32 | >2 |
| MMV007764 | 256.39 | 4.93 | 0.716 | 19.329 | >27 |
| MMV019266 | 312.41 | 4.46 | 0.615 | >32 | >52 |
| MMV000356 | 379.26 | 4.16 | 1.03 | >32 | >31 |
| MMV007285 | 298.77 | 4.56 | 4 | >32 | >8 |
| MMV011099 | 275.31 | 3.52 | 0.242 | >32 | >132 |
| MMV665797 | 364.44 | 5.02 | 0.584 | 14.254 | 24 |
| MMV020243 | 362.43 | 2.54 | 1.15 | 23.776 | 21 |
The selectivity index (SI) for each compound was determined as a ratio of HFF IC50/3D7 EC50. Data were obtained from MMV and Chembl2028072.
The MMV007285 analog 7-chloro-N-(4-propylphenyl)-4-quinolinamine (7CPQA) inhibits PfPSD activity and blocks Plasmodium blood stage development in the malaria murine model
MMV007285 – a 7-chloro-N-(4-ethoxyphenyl)-4-quinolinamine, has an EC50 against P. falciparum strain 3D7 of 4 μM and an IC50 against human foreskin fibroblasts of > 32 μM (Table 1) (Spangenberg et al., 2013). The compound shares the 7-chloroquinoline core and the phenyl group at the 4-position found in amodiaquine, a potent anti-malarial drug. However, instead of a hydroxyl group found in amodiaquine, MMV007285 harbors an ethoxy group at the para position of the phenyl group at the 4-position. Furthermore the compound lacks a side chain quaternary amine, suggesting a possible mode of action outside the digestive vacuole. We synthesized an analog of MMV007285, 7CPQA, and assessed its activity as an inhibitor of PfPSD and parasite development using the Plasmodium yoelii murine malaria model. Addition of 7CPQA to purified PfPSD-Δ40 resulted in a concentration dependent inhibition of decarboxylase activity, with 10 μM of the compound inhibiting catalysis by 29 % and 100 μM of the compound inhibiting catalysis by 76 % (Fig. 11B). In vitro efficacy studies showed that the compound was equally effective against the P. falciparum drug-sensitive NF54 and chloroquine-resistant W2 strains with an IC50 ~ 1 μM (Fig. 11C). To determine whether 7CPQA inhibits growth of the parasite at specific stages during of the intraerythrocytic life cycle, cultures of NF54 were synchronized and treated with either vehicle alone or 7CPQA at times 0, 24 h and 36 h and parasite development from the ring to the trophozoite and then to the schizont stage was monitored by light microscopy. 7CPQA inhibited both parasite maturation as well as division, blocking the formation of daughter merozoites (Fig. 11D). This inhibition profile is reminiscent of that of other inhibitors of lipid metabolism in malaria parasites (Pessi et al., 2004; Roggero et al., 2004; Vial and Ben Mamoun, 2005; Le Roch et al., 2008; Bobenchik et al., 2013). In vivo efficacy studies showed that mice infected with 103 P. yoelii reached peak parasitemia of 10–14% by day 16, before parasite clearance. In contrast, infected mice given an oral dose of 30 mg kg−1 of 7CPQA once a day for 3 consecutive days cleared the infection by day 6, and no parasites could be detected for the entire 25-day period of the study (Fig. 11E). Together, these studies demonstrate potent anti-malarial activity of 7CPQA in vivo and in vitro, and indicate that the parasite PSD enzyme is at least one target of drug action.
Fig. 11.

PSD enzyme inhibition and in vivo efficacy of 7CPQA.
A. Chemical structures of MMV007285 and 7CPQA.
B. In vitro inhibition of PfPSD by 7CPQA. MBP-PfPSDΔ40 was expressed in Escherichia coli BL21 codon plus strain and purified by amylose column affinity chromatography. PSD enzyme analysis was performed with the purified enzyme in the presence of DMSO alone, 10 μM or 100 μM of 7CPQA for 45 min at 30°C as described in Experimental Procedures. PSD activity of 7CPQA-treated MBP-PfPSDΔ40 was normalized to 100% of MBP-PfPSDΔ40 treated with DMSO control. Data are means ± standard deviation for three experiments each performed in duplicate.
C. Inhibition of NF54 (closed circles) and W2 (closed squares) parasite clones as a function of CPQA concentrations. Values are means of triplicates.
D. Stage specific inhibition of Plasmodium falciparum (NF54) parasite by 7CPQA. Highly synchronized cultures of the parasites were grown in the absence or presence of 10 μM of 7CPQA, stained by Giemsa stain, and analyzed by light microscopy.
E. In vivo efficacy of 7CPQA against Plasmodium yoelii XNL strain. Two groups of 6-week-old female Swiss Webster mice were injected with 103 infected red blood cells. On days 4, 5 and 6 after infection, three mice were administered a single daily dose of vehicle alone (PEG-400) and four mice were administered vehicle containing 30 mg kg−1 of 7CPQA by oral gavage. Parasitemia was monitored by collecting blood from animals and determining the number of infected red blood cells by Giemsa staining (counting at least 5000 red blood cells).
Discussion
In this study we have used yeast as a reporter system to characterize the activity of the P. falciparum PfPSD gene, determine its post-translational processing and lipid binding properties, and developed an in vivo screen in yeast to identify new lead inhibitors.
In P. falciparum, most ethanolamine available to the parasite comes from serine, which can be imported from human serum or obtained from degradation of host cytoplasm, primarily hemoglobin (Elabbadi et al., 1997; Pessi et al., 2004). Studies in P. knowlesi suggested the presence of a unique serine decarboxylase reaction to form ethanolamine from serine (Elabbadi et al., 1997; Pessi et al., 2004). However, the putative enzyme could not be purified and no serine decarboxylase gene has been identified in Plasmodium species. In addition, our data in Fig. 2B clearly demonstrate that the PfPSD enzyme does not catalyze an SD reaction in vivo. An alternative interpretation of previous data implicating a parasite SD is that serine is first converted into PS, which is subsequently decarboxylated to produce PE by PSD. Further metabolism of PE by parasite phospholipases could produce phosphoethanolamine and possibly ethanolamine. Human malaria parasites use a unique phosphoethanolamine methyltransferase PMT for synthesis of phosphocholine from phosphoethanolamine (Pessi et al., 2004; Pessi and Ben Mamoun, 2006; Bobenchik et al., 2010; 2011; 2013; Bezsonova et al., 2013). The phosphocholine generated from the latter pathway can subsequently be metabolized to CDP-choline and PC. However, metabolic labeling studies using 14C-ethanolamine showed that the PMT enzyme uses only a third of newly synthesized phosphoethanolamine (Witola et al., 2008). Thus PS decarboxylation in malaria parasites may produce unique molecular species of PE, different from those produced via the CDP-ethanolamine pathway, or that PS decarboxylation and de novo synthesis of PE from ethanolamine occur in different cellular compartments.
PSD enzymes contain a pyruvoyl prosthetic group that is generated autocatalytically via a recently identified canonical serine protease mechanism (Li and Dowhan, 1988; 1990; Choi et al., 2015), which cleaves the pro-enzyme into a small C-terminal α-subunit and a large N-terminal β-subunit. The pyruvoyl moiety is produced at the N-terminus of the α-subunit in a concerted reaction that accompanies formation of both subunits. Characterization of the soluble P. knowlesi PkPSD demonstrated D139, H198 and S308 form the protease active site (Li and Dowhan, 1988; 1990; Choi et al., 2015). The yeast PSD1 and PSD2 proenzymes follow this same general pathway (Kitamura et al., 2002; Schumacher et al., 2002; Gulshan et al., 2010; Horvath et al., 2012; Onguka et al., 2015) and all contain the positionally conserved D-H-S catalytic triad. However, the processing of the parasite enzyme shows distinct phospholipid regulation by PS and PG (Choi et al., 2012) that is apparently not required for yeast PSD1 processing (Onguka et al., 2015). The role of the N-terminal portion of the parasite PSD upstream of the aspartic residue involved in auto-proteolytic processing has not previously been investigated in detail. Our analysis using step-wise deletions of the first 70 amino acid residues revealed important information about the link between PfPSD function, processing and catalysis. The N-terminal domain of PfPSD is predicted to bind phospholipids and our lipid binding assays indicated a preference of the enzyme for PS and PG over PC.
To date, only one report has described a chemical screen to identify inhibitors of a human PSD enzyme; however, no structures were provided and no follow-up analyses were reported (Forbes et al., 2007). The present study describes our initial attempt to identify inhibitors of PSD enzymes as a strategy to develop drugs against eukaryotic pathogens. We have used yeast as a model system to screen for inhibitors of this class of enzymes in a library of drug-like and probe-like compounds with potent anti-malarial activity and excellent safety profile (Spangenberg et al., 2013). The screen searched for compounds that inhibited yeast growth in a parasite PSD-dependent manner, which was also bypassed by ethanolamine supplementation. Out of 400 compounds, our analysis identified 18 candidates for PSD inhibitors based on their differential growth inhibitory activity of a yeast mutant dependent upon parasite PSD. Of these compounds, 7-chloro-N-(4-ethoxyphenyl)-4-quinolinamine (MMV007285) was chosen for further analysis because of the ready availability of precursors and ease of synthesis. MMV007285 has been shown to be equally effective against P. falciparum drug-sensitive and drug-resistant parasites (chloroquine- and pyrimethamine-resistant parasites) with IC50 values ranging between 0.5 and 1 μM (MMV, unpubl. data) and has a selectivity index of > 8 in vitro. Our studies using 7CPQA, an analog of MMV007285, showed the compound has potent activity in mice infected with P. yoelii, eliminating malaria infection at 30 mg kg−1 following a daily oral administration for 3 consecutive days. The 7CPQA also inhibits all stages of parasite intraerythrocytic development. Most importantly, 7CPQA inhibits the catalytic activity of purified PfPSD in enzyme reactions. Despite the fact that the compound appears to be a weak enzyme inhibitor, with an IC50 between 10 and 100 uM; the results demonstrate that the screen is useful for identifying such compounds, which can serve as novel leads for developing more potent inhibitors through chemical modification. As with any inhibitor, we cannot rule out off target effects, but these effects at the cellular level would need to be dependent upon the presence of parasite PSD and also be reversible by ethanolamine supplementation.
MMV007285 and 7CPQA share some structural similarity with the 4-aminoquinolines, chloroquine and amodiaquine, known to act in the digestive vacuole of the parasite. However, the presence of an ethoxy group in MMV007285, and a propyl group in 7CPQA, at the para position of the phenyl group at the 4-position in MMV007285, and the lack of a side chain quaternary amine, suggest a mode of action that is mechanistically different from those previously reported for amodiaquine action within the digestive vacuole, as well as outside the vacuole against parasite methyltransferases (Bobenchik et al., 2010; 2013).
While MMV007285 has IC50 values between 0.5 and 1 μM against P. falciparum, its efficacy against yeast cells expressing Plasmodium PSD requires higher concentrations of the compound. This is likely due to major differences in drug uptake and efflux, and drug concentration and detoxification in specialized organelles. Several anti-malarials with IC50s in the nM or low μM range (e.g. miltefosine, G25, T16, amodiaquine, fenpropimoprph) against parasites grown in red blood cells, show activities in the mM range when tested in yeast (Pessi et al., 2004; 2005; Roggero et al., 2004; Le Roch et al., 2008; Bobenchik et al., 2010; 2013; Augagneur et al., 2013; Garg et al., 2015).
Baunaure and colleagues have previously reported that in P. falciparum only 6% of the PSD enzyme is in a soluble form (Baunaure et al., 2004). Thus, it is possible that the membrane association of the enzyme may influence its susceptibility to inhibition by MMV007285 and 7-CPQA. Efforts are now underway to solve the structure of malarial PSDs and such information will help us better understand the mechanism of inhibition and mode of interaction of the inhibitors with soluble and membrane associated PSD enzymes. These efforts may also prove useful for structural optimization of the inhibitors.
The finding of new inhibitors of PSD with potent anti-malarial activity and excellent pharmacological properties will help diversify the pool of drugs that could be advanced for future evaluation as new anti-malarial drugs including those that inhibit membrane biogenesis in the parasite.
Experimental procedures
Cell culture and materials
Parasites were cultured by the method of Trager and Jensen (1976) using a gas mixture of 3% O2, 3% CO2 and 94% N2. Complete medium used for propagation of P. falciparum cultures consists of RPMI medium 1640 supplemented with 30 mg l−1 hypoxanthine (Sigma), 25 mM Hepes (Sigma), 0.225% NaHCO3 (Sigma), 0.5% Albumax I (Life Technologies, Grand Island, NY, USA), and 10 μg ml−1 gentamycin (Life Technologies). All chemicals for E. coli and Saccharomyces cerevisiae cell cultures were purchased from Sigma, Fisher Scientific and Difco. Phospholipids were purchased from Avanti Polar Lipids. Reagents for protein determination were from Bio-Rad. Pre-cast SDS-polyacrylamide gels were purchased from Invitrogen. Assays to determine the inhibitory activity of 7CPQA against NF54 and W2 and stage specificity of inhibition were performed as previously described (Reynolds et al., 2007; Bobenchik et al., 2013).
Vectors for PfPSD expression in E. coli and yeast
To characterize the activity and function of full length and truncated forms of PfPSD, PCR reactions were performed to clone codon-optimized versions of the cDNA, lacking either none, or the first 10, 20, 30, 40, 50, 60 and 70 amino acid residues. The resultant cDNAs were ligated into the pMAL-c2x E. coli expression vector and the pBEVY S. cerevisiae expression vector. Cloning into pMAL-c2x results in addition of an N-terminal maltose binding protein, MBP. The constructs were introduced into E. coli strain BL21 (DE3) and following induction with IPTG, recombinant proteins were purified. Purified full-length and Δ40 proteins were used to generate polyclonal antibodies in rabbits, which was performed by Cocalico Biologicals, Inc. For expression of MBP-PfPSD and MBP-PfPSDΔ40 in E. coli, BL21 codon plus strains harboring these constructs were grown to saturation overnight on LB-0.2% glucose, ampicillin (100 μg ml−1) and chloramphenicol (34 μg ml−1), then diluted 100-fold, and grown to A600 ~ 0.5 at 37°C. MBP-PfPSD expression was induced by addition of IPTG (0.3 mM) for 2 hours. The cells were harvested by centrifugation (4,000 × g, 20 min, 4°C) and washed by resus-pension in water and re-centrifugation. The cells were resus-pended in column buffer (20 mM Tris-HCl, pH 7.4, 200 mM NaCl, 1 mM Ethylenediaminetetraacetic acid (EDTA), and 10 mM β mercaptoethanol), frozen in a dry ice-ethanol bath, stored overnight at −20°C and subsequently thawed on ice water. Cell free extracts were obtained by sonication (15 second burst at 25% amplitude, 8 times with 30 second cooling intervals) followed by centrifugation at 20,000 × g for 20 min.
Yeast growth assays
The psd1Δpsd2Δdpl1Δ strains carrying the empty vector pBEVY-U, or the same vector harboring full-length or truncated versions of PfPSD were pre-grown overnight in uracil dropout, synthetic glucose (2%) (SD-U) medium supplemented with ethanolamine. Cells were washed and diluted to 105 cells ml−1 and used to start liquid cultures or perform limiting dilutions on agar plates, consisting of SD-U either without, or with 2 mM ethanolamine. Cells were incubated at 30°C and cell number in liquid cultures was determined at A660 on samples collected at different times points.
Liposome binding assays
For lipid binding assays, MBP-PfPSD proteins were partially purified from cell free extracts. A 250 μl aliquot of cell free extracts was incubated with 250 μl of the amylose resin slurry for 30 min at 4C. MBP fusion proteins bound to amylose resin were collected by centrifugation at 6,000 × g for 1 min. Nonspecific binding proteins were removed by washing the pellet with 1 ml of column buffer. MBP-PfPSD proteins were eluted from the resin after re-suspension in the column buffer containing 10 mM maltose for 5 min followed by centrifugation at 6,000 × g for 1 min. The elution step was repeated and the resultant purified PfPSD proteins were used for the liposome binding assay. A 30 ul aliquot of each purified MBP-PfPSD variant was diluted in 170 ul of 10 mM Tris, pH 7.4 and 0.1 M KCl buffer and 50 ul of multilamellar liposomes (1 mg ml−1 of DOPS, DOPC, DOPG, or Mock in 10 mM Tris, pH 7.4 and 0.1 M KCl buffer) was added. The mixture was incubated for 40 min at 37C with gentle shaking at 150 rpm and then centrifuged at 10,000 × g for 5 min to recover liposomes (P). Supernatants were transferred to a fresh tube and the liposome pellets were re-suspended in 250 ul of column buffer. The supernatant and the liposome pellet were analyzed by western blotting.
PSD activity and processing
Yeast extracts from psd1Δpsd2Δdpl1Δ strains harboring empty vector, full-length PfPSD, or truncated PfPSD forms were prepared as described previously, and used for PSD enzyme assay (Choi et al., 2012). The assay for PSD activity utilized [1′-14C Serine]-PS as the substrate, and the reaction product was trapped as 14CO2 on 2 M KOH-impregnated filter paper, as described previously (Trotter and Voelker, 1995). PSD activities are presented as enzyme specific activity (nmol/mg-protein/45 min, or nmol/relative pixel intensity of β-subunit/45 min). All enzyme reactions were performed at substrate and protein concentrations that produced a linear response with the amount of enzyme added to the reaction. To test if enzyme activity of full-length PfPSD can be inhibited by catalytically inactive PfPSD-Δ70, cell free extracts containing full-length PfPSD and cell free extracts containing PfPSD-Δ70 were pre-incubated at 30°C for 10 min ahead of PSD enzyme assay. For in vitro processing, cell free extracts containing PfPSD-Δ70 (1.2 μg μl−1) were incubated with cell free extracts containing full-length PfPSD (1.2 μg μl−1) and PS liposomes (0.1 mg ml−1) at 30°C for 2 h. The reaction was stopped by the addition of SDS gel loading buffer. The processed β forms and unprocessed forms of PfPSDs were analyzed by Western blot analysis using Rabbit-Anti-PfPSDΔ40 antibodies and horse-radish peroxidase-conjugated goat anti-rabbit secondary antibody, which were diluted 1.5 × 104-fold and 1 × 104-fold, respectively. For the analysis of the processed α forms of PfPSDs, the primary and the secondary antibodies were diluted 1.5 × 103-fold and 5 × 103-fold, respectively.
In vitro inhibition of PfPSD activity by 7CPQA
Escherichia coli cell free extracts containing MBP-PfPSD-Δ40 were prepared as described earlier. MBP-PfPSD-Δ40 proteins were purified from the cell free extracts by amylose column affinity chromatography. 7CPQA dissolved in DMSO was pre-incubated with the 80 μl buffer (20 mM Tris-HCl, pH 7.4, 200 mM NaCl, 1 mM EDTA, 10 mM β mercaptoethanol, and 10 mM maltose) containing 0.4 μg of purified MBP-PkPSDΔ40 proteins for 10 min at 30°C. The PSD enzyme analysis was started by adding 320 μl of buffer (31 mM potassium phosphate, pH 6.8, 0.125% Triton X-100, 5.6 mM β mercaptoethanol, 0.56 mM EDTA, 0.15 M Sucrose, 0.28 mM PMSF, and 28 mM Tris-HCl, pH 8.0) containing the substrate, 0.2 mM Ptd[1′-14C]Ser (400 cpm nmol−1) to the 7CPQA treated MBP-PkPSD-Δ40 protein. The enzyme activities were measured as described earlier.
Phospholipid analyses
The yeast mutant psd2Δdpl1Δ harboring the yeast PSD1 gene and psd1Δpsd2Δdpl1Δ strains harboring the empty pBEVY-U vector, or this vector encoding codon optimized full-length PfPSD, or Δ10, Δ20, Δ30, Δ40, Δ50,Δ60, or Δ70 truncated PfPSD forms, were pre-grown to mid-log phase at 30°C in SC-URA medium supplemented with ethanolamine (2 mM). The cells were harvested by centrifugation, washed twice with H2O, and diluted to A600 = 0.02 in 100 ml of SC-URA medium, and then grown to A600 = 1. The lipids were extracted, and phospholipids were separated by two-dimensional TLC on Silica 60 plates using chloroform/methanol/ammonium hydroxide (65/35/5 v/v/v) followed by chloroform/acetic acid/methanol/water (75/25/5/2.2 v/v/v). Lipids were visualized by staining with iodine vapor and quantified by measuring phosphate (Rouser et al., 1966). In some experiments, for comparative purposes, we also included lipids from the yeast strain BY4741, which has wild-type alleles for PSD1, PSD2 and DPL1 genes.
Chemical screening in yeast
For screening of the Malaria Box, the psd1Δpsd2Δdpl1Δ strain harboring the PkPSD construct was pre-cultured overnight in SD medium without ethanolamine. Cells were then washed in water, diluted and dispensed into 96-well plates at 2 × 104 cells ml−1 in SD medium lacking or supplemented with ethanolamine in the absence or presence of 10 μM of each compound diluted in 1% DMSO. Controls included wells containing wild-type yeast cells, psd1Δpsd2Δdpl1Δ harboring an empty vector in SD medium, either lacking or supplemented with ethanolamine, and containing 1% DMSO. The plates were incubated at 30°C and read at A660, 48 h following inoculation.
In vivo efficacy of 7CPQA in mice
To examine the efficacy of 7CQA against P. yoelii XNL strain, two groups of 6-week-old female Swiss Webster mice were injected with 103 infected red blood cells. On days 4, 5 and 6 after infection, three mice were administered a single daily dose of vehicle alone (PEG-400 (Sigma-Aldrich #202398) and four mice were administered vehicle containing 30 mg kg−1 of 7CQA by oral gavage. Parasitemia was monitored by collecting blood from animals and determining the number of infected red blood cells by Giemsa staining (counting at least 5000 red blood cells). All animal work was approved by the Yale AICUC Committee and was performed at a Yale-approved animal care facility.
Statistical analyses
Statistical analyses were performed using an unpaired Student’s t-test. Differences were considered statistically significant when P < 0.05.
Acknowledgements
We are grateful to Medicines for Malaria Venture (MMV) for providing the chemicals, and biological and pharmacological data on the compounds in the Malaria Box. We thank Dr. Michael Van Zandt for the synthesis of 7CPQA. We also thank Dr. Michael Riscoe and Dr. Wesley Van Voorhis for their suggestions and comments on the structures and properties of the compounds. This work was supported by National Institutes of Health (AI097218, AI09486 and AI077603) and the Bill and Melinda Gates Foundation (OPP1086229, OPP1069779 and 1021571) grants to CBM; and by NIH grant GM104485 to DRV.
References
- Atkinson K, Fogel S, Henry SA. Yeast mutant defective in phosphatidylserine synthesis. J Biol Chem. 1980a;255:6653–6661. [PubMed] [Google Scholar]
- Atkinson KD, Jensen B, Kolat AI, Storm EM, Henry SA, Fogel S. Yeast mutants auxotrophic for choline or ethanolamine. J Bacteriol. 1980b;141:558–564. doi: 10.1128/jb.141.2.558-564.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augagneur Y, Jaubert L, Schiavoni M, Pachikara N, Garg A, Usmani-Brown S, et al. Identification and functional analysis of the primary pantothenate transporter, PfPAT, of the human malaria parasite Plasmodium falciparum. J Biol Chem. 2013;288:20558–20567. doi: 10.1074/jbc.M113.482992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baunaure F, Eldin P, Cathiard AM, Vial H. Characterization of a non-mitochondrial type I phosphatidylserine decarboxylase in Plasmodium falciparum. Mol Microbiol. 2004;51:33–46. doi: 10.1046/j.1365-2958.2003.03822.x. [DOI] [PubMed] [Google Scholar]
- Bezsonova I, Rujan I, Bobenchik AM, Gorbatyuk V, Maciejewski MW, Gorbatyuk O, et al. (1)H, (13)C, and (15)N chemical shift assignments for PfPMT, a phosphoethanolamine methyltransferase from Plasmodium falciparum. Biomol NMR Assign. 2013;7:17–20. doi: 10.1007/s12104-012-9372-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobenchik AM, Choi JY, Mishra A, Rujan IN, Hao B, Voelker DR, et al. Identification of inhibitors of Plasmodium falciparum phosphoethanolamine methyltransferase using an enzyme-coupled transmethylation assay. BMC Biochem. 2010;11:4. doi: 10.1186/1471-2091-11-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobenchik AM, Augagneur Y, Hao B, Hoch JC, Ben Mamoun C. Phosphoethanolamine methyltransferases in phosphocholine biosynthesis: functions and potential for antiparasite therapy. FEMS Microbiol Rev. 2011;35:609–619. doi: 10.1111/j.1574-6976.2011.00267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobenchik AM, Witola WH, Augagneur Y, Nic Lochlainn L, Garg A, Pachikara N, et al. Plasmodium falciparum phosphoethanolamine methyltransferase is essential for malaria transmission. Proc Natl Acad Sci USA. 2013;110:18262–18267. doi: 10.1073/pnas.1313965110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YL, Montedonico AE, Kauffman S, Dunlap JR, Menn FM, Reynolds TB. Phosphatidylserine synthase and phosphatidylserine decarboxylase are essential for cell wall integrity and virulence in Candida albicans. Mol Microbiol. 2010;75:1112–1132. doi: 10.1111/j.1365-2958.2009.07018.x. [DOI] [PubMed] [Google Scholar]
- Choi JY, Augagneur Y, Ben Mamoun C, Voelker DR. Identification of gene encoding Plasmodium knowlesi phosphatidylserine decarboxylase by genetic complementation in yeast and characterization of in vitro maturation of encoded enzyme. J Biol Chem. 2012;287:222–232. doi: 10.1074/jbc.M111.313676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JY, Duraisingh MT, Marti M, Ben Mamoun C, Voelker DR. From protease to decarboxylase: the molecular metamorphosis of phosphatidylserine decarboxylase. J Biol Chem. 2015;290:10972–10980. doi: 10.1074/jbc.M115.642413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elabbadi N, Ancelin ML, Vial HJ. Phospholipid metabolism of serine in Plasmodium-infected erythrocytes involves phosphatidylserine and direct serine decarboxylation. Biochem J. 1997;324:435–445. doi: 10.1042/bj3240435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emoto K, Umeda M. An essential role for a membrane lipid in cytokinesis. Regulation of contractile ring disassembly by redistribution of phosphatidylethanolamine. J Cell Biol. 2000;149:1215–1224. doi: 10.1083/jcb.149.6.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emoto K, Inadome H, Kanaho Y, Narumiya S, Umeda M. Local change in phospholipid composition at the cleavage furrow is essential for completion of cytokinesis. J Biol Chem. 2005;280:37901–37907. doi: 10.1074/jbc.M504282200. [DOI] [PubMed] [Google Scholar]
- Forbes CD, Toth JG, Ozbal CC, Lamarr WA, Pendleton JA, Rocks S, et al. High-throughput mass spectrometry screening for inhibitors of phosphatidylserine decarboxylase. J Biomol Screen. 2007;12:628–634. doi: 10.1177/1087057107301320. [DOI] [PubMed] [Google Scholar]
- Garg A, Lukk T, Kumar V, Choi JY, Augagneur Y, Voelker DR, et al. Structure, function and inhibition of the phosphoethanolamine methyltransferases of the human malaria parasites Plasmodium vivax and Plasmodium knowlesi. Sci Rep. 2015;5:9064. doi: 10.1038/srep09064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulshan K, Shahi P, Moye-Rowley WS. Compartment-specific synthesis of phosphatidylethanolamine is required for normal heavy metal resistance. Mol Biol Cell. 2010;21:443–455. doi: 10.1091/mbc.E09-06-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath SE, Bottinger L, Vogtle FN, Wiedemann N, Meisinger C, Becker T, Daum G. Processing and topology of the yeast mitochondrial phosphatidylserine decarboxylase 1. J Biol Chem. 2012;287:36744–36755. doi: 10.1074/jbc.M112.398107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi AS, Thompson MN, Fei N, Huttemann M, Greenberg ML. Cardiolipin and mitochondrial phosphatidylethanolamine have overlapping functions in mitochondrial fusion in Saccharomyces cerevisiae. J Biol Chem. 2012;287:17589–17597. doi: 10.1074/jbc.M111.330167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura H, Wu WI, Voelker DR. The C2 domain of phosphatidylserine decarboxylase 2 is not required for catalysis but is essential for in vivo function. J Biol Chem. 2002;277:33720–33726. doi: 10.1074/jbc.M205672200. [DOI] [PubMed] [Google Scholar]
- Le Roch KG, Johnson JR, Ahiboh H, Chung DW, Prudhomme J, Plouffe D, et al. A systematic approach to understand the mechanism of action of the bisthiazolium compound T4 on the human malaria parasite, Plasmodium falciparum. BMC Genomics. 2008;9:513. doi: 10.1186/1471-2164-9-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QX, Dowhan W. Structural characterization of Escherichia coli phosphatidylserine decarboxylase. J Biol Chem. 1988;263:11516–11522. [PubMed] [Google Scholar]
- Li QX, Dowhan W. Studies on the mechanism of formation of the pyruvate prosthetic group of phosphatidylserine decarboxylase from Escherichia coli. J Biol Chem. 1990;265:4111–4115. [PubMed] [Google Scholar]
- Marsh D. Lateral pressure profile, spontaneous curvature frustration, and the incorporation and conformation of proteins in membranes. Biophys J. 2007;93:3884–3899. doi: 10.1529/biophysj.107.107938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onguka O, Calzada E, Ogunbona OB, Claypool SM. Phosphatidylserine decarboxylase 1 autocatalysis and function does not require a mitochondrialspecific factor. J Biol Chem. 2015;290:12744–12752. doi: 10.1074/jbc.M115.641118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulick MG, Bertozzi CR. The glycosylphosphatidylinositol anchor: a complex membrane-anchoring structure for proteins. Biochemistry. 2008;47:6991–7000. doi: 10.1021/bi8006324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecheur EI, Martin I, Maier O, Bakowsky U, Ruysschaert JM, Hoekstra D. Phospholipid species act as modulators in p97/p47-mediated fusion of Golgi membranes. Biochemistry. 2002;41:9813–9823. doi: 10.1021/bi0259195. [DOI] [PubMed] [Google Scholar]
- Pessi G, Ben Mamoun C. Pathways for phosphatidylcholine biosynthesis: targets and strategies for antimalarial drugs. Future Medicine, Future Lipidology. 2006:173–180. [Google Scholar]
- Pessi G, Kociubinski G, Mamoun CB. A pathway for phosphatidylcholine biosynthesis in Plasmodium falciparum involving phosphoethanolamine methylation. Proc Natl Acad Sci USA. 2004;101:6206–6211. doi: 10.1073/pnas.0307742101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessi G, Choi JY, Reynolds JM, Voelker DR, Mamoun CB. In vivo evidence for the specificity of Plasmodium falciparum phosphoethanolamine methyltransferase and its coupling to the Kennedy pathway. J Biol Chem. 2005;280:12461–12466. doi: 10.1074/jbc.M414626200. [DOI] [PubMed] [Google Scholar]
- Reynolds JM, El Bissati K, Brandenburg J, Gunzl A, Mamoun CB. Antimalarial activity of the anticancer and proteasome inhibitor bortezomib and its analog ZL3B. BMC Clin Pharmacol. 2007;7:13. doi: 10.1186/1472-6904-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roggero R, Zufferey R, Minca M, Richier E, Calas M, Vial H, Ben Mamoun C. Unraveling the mode of action of the antimalarial choline analog G25 in Plasmodium falciparum and Saccharomyces cerevisiae. Antimicrob Agents Chemother. 2004;48:2816–2824. doi: 10.1128/AAC.48.8.2816-2824.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouser G, Siakotos AN, Fleischer S. Quantitative analysis of phospholipids by thin-layer chromatography and phosphorus analysis of spots. Lipids. 1966;1:85–86. doi: 10.1007/BF02668129. [DOI] [PubMed] [Google Scholar]
- Schumacher MM, Choi JY, Voelker DR. Phosphatidylserine transport to the mitochondria is regulated by ubiquitination. J Biol Chem. 277:51033–51042. doi: 10.1074/jbc.M205301200. [DOI] [PubMed] [Google Scholar]
- Spangenberg T, Burrows JN, Kowalczyk P, McDonald S, Wells TN, Willis P. The open access malaria box: a drug discovery catalyst for neglected diseases. PLoS ONE. 2013;8:e62906. doi: 10.1371/journal.pone.0062906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- Trotter PJ, Voelker DR. Identification of a nonmitochondrial phosphatidylserine decarboxylase activity (PSD2) in the yeast Saccharomyces cerevisiae. J Biol Chem. 1995;270:6062–6070. doi: 10.1074/jbc.270.11.6062. [DOI] [PubMed] [Google Scholar]
- Vance JE, Tasseva G. Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim Biophys Acta. 2013;1831:543–554. doi: 10.1016/j.bbalip.2012.08.016. [DOI] [PubMed] [Google Scholar]
- Vial HJ, Ben Mamoun C. Plasmodium lipids: metabolism and function. In: Sherman IW, editor. Molecular Approaches to Malaria. ASM Press; Washington D.C.: 2005. pp. 327–352. [Google Scholar]
- WHO World Health Organization Malaria Fact Sheet NO. 94. 2010 URL http://www.who.int/mediacentre/factsheets/fs94/en/print.html.
- Witola WH, El Bissati K, Pessi G, Xie C, Roepe PD, Mamoun CB. Disruption of the Plasmodium falciparum PfPMT gene results in a complete loss of phosphatidylcholine biosynthesis via the serine-decarboxylasephosphoethanolamine-methyltransferase pathway and severe growth and survival defects. J Biol Chem. 2008;283:27636–27643. doi: 10.1074/jbc.M804360200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–1109. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- Xu K, Nagy PD. RNA virus replication depends on enrichment of phosphatidylethanolamine at replication sites in subcellular membranes. Proc Natl Acad Sci USA. 2015;112:E1782–E1791. doi: 10.1073/pnas.1418971112. [DOI] [PMC free article] [PubMed] [Google Scholar]