

Abstract
Heart failure (HF) is a growing public health concern as a consequence of the ageing of the population and the improved survival of patients with HF. HF is defined as impaired organ perfusion and/or high filling pressure. It is a systemic and chronic disease and as such involves many organs, not least the liver and kidney. The complex vascular system of the liver and its high metabolic activity render it vulnerable to circulation disturbances and trigger many molecular and haemodynamic changes in patients. There are many studies describing the impact of liver disease on patient outcomes. Hepatic dysfunction is commonly seen in HF patients and is closely correlated with a poor outcome. Knowledge about the mechanisms and impacts of liver disease in HF helps us to know the stage of the disease and treat it properly. Moreover, many drugs and toxins that are metabolised in the liver and contribute to drug interactions should also be taken into account when prescribing medication for HF patients. In light of the above-mentioned points, the authors have compiled this review on congestive hepatopathy with the aim of providing physicians and cardiologists with a succinct and useful guide on the role of the liver in HF.
Keywords: Ventricular tachycardia, congenital heart disease, fallots tetralogy, coronary artery disease, risk stratification, systolic heart failure, heart failure treatment, heart failure with normal ejection fraction
Introduction
Heart failure (HF) is defined as impaired organ perfusion, also known as forward failure, and/or high filling pressure and venous congestion, also known as backward failure. HF is a systemic and chronic disease and as such involves many organs, not least the liver and kidney. The complex vascular system of the liver and its high metabolic activity render it vulnerable to circulation disturbances and trigger many molecular and haemodynamic changes in patients.
There are many studies in the existing literature describing the impact of liver disease on patient outcomes. Hepatic dysfunction is commonly seen in HF patients and is closely correlated with haemodynamic parameters.1 It appears that hepatic dysfunction and HF are inter-related. Moreover, many drugs and toxins that are metabolised in the liver and contribute to drug interactions should be taken into account when prescribing medications for HF patients (table 1).2
Table 1.
Different cardiovascular drugs and their potential effects on liver
| Potential liver injury effects | |
|---|---|
| Cardiovascular drugs | |
| Amiodaron | Centrilobular necrosis |
| Quinidine | Hypersensitivity reaction |
| Procainamide | Delayed drug hypersensitivity |
| Propafenone | Hepatocellular and cholestatic liver injury |
| Flecainide | Hepatocellular injury |
| Dysopiramide | Hepatocellular damage, intrahepatic cholestasis |
| AntiPlt | Mostly hepatocellular damage |
| Statins | Hepatocellular necrosis |
| Fibrate | Hepatocellular injury |
| Niacin | Hepatocellular and cholestatic injury |
| Ezetimibe | Hepatotoxicity in combination with statins |
| Ca channel blockers | Hepatocellular injury |
| Warfarin | Rare but cholestatic type |
| Heparin | Mild elevation in aminotransferase levels |
| Enoxaparin | Mostly hepatocellular |
| HF therapies | |
| ACE inhibitors | Cholestatic injury |
| β-Blockers | Hepatocellular injury |
| ARB | Cholestatic injury and/or hepatocellular injury |
| Loop diuretics | Cholestatic injury |
| Thiazides | Mixed hepatocellular and cholestatic injury |
| Hydralazin | Hepatocellular injury |
| Aldostrone antagonists | Elevated aminotransferase |
| Antidiabetics | |
| Insulin | Elevation in γ-glutamyl transpeptidase |
| Glibenclamide | Mixed pattern of liver injury |
| Glithazones | Mostly hepatocellular injury |
ARB, agniotensin receptor blocker; HF, heart failure.
It is very important for physicians and cardiologists to know about the functioning of the liver in patients with HF and its impact on the whole body and they should also have the ability to identify clinical clues of hepatic dysfunction in HF based on haemodynamic parameters. In this paper, we provide an overview of liver function in HF based on reviews of previous texts and what we have learnt from our own studies on the liver and uric acid.3
Anatomy
The liver can be divided into hemi-livers: right and left, each having its own blood supply. The liver receives 25% of the cardiac output, even though it makes up only 2–3% of the total body weight. It has a dual circulatory supply: the portal veins provide approximately two-thirds of the blood supply, while the hepatic artery provides the rest. The blood flowing through the liver is drained via the right, left and middle hepatic veins into the inferior vena cava and then into the right side of the heart.4 There is a difference in the composition of the portal venous and hepatic arterial blood. The portal venous blood is rich in basic nutrients such as glucose, amino acids and triglycerides, yet it is relatively deficient in oxygen. By contrast, the hepatic artery supplies oxygen-rich blood, accounting for more than 50% of the oxygen delivered to the liver and 100% of the oxygen delivered to the major bile ducts; however, it contains fewer basic nutrients. The right hemi-liver comprises 50–70% of the liver mass.5 The liver can be further subdivided into eight segments based on the vascular or bile duct distribution. The liver parenchyma consists of acinuses. The acinus is a cluster of hepatocytes (approximately 2 mm in diameter) that are grouped around the terminal branches of a hepatic arteriole and portal venule. Blood enters the acinus via a branch of the portal vein and the hepatic artery in the portal and periportal regions and flows through the sinusoids before it drains into a terminal hepatic venule. The centre of the acinus (periportal) is known as zone 1, the periphery (perivenular) as zone 3 and the region in between as zone 2. Zone 1 receives blood with the highest levels of oxygen and nutrients compared with zone 3. Thus, zone 3 is particularly vulnerable to a circulatory insult. Zone 1 hepatocytes have high oxidative activities and contain many large mitochondria. Their dominant processes include gluconeogenesis, β oxidation of fatty acids, amino acid catabolism, ureagenesis, cholesterol synthesis and bile acid secretion. By contrast, zone 3 hepatocytes are more involved in glycolysis and lipogenesis. The hepatic circulation is regulated by smooth muscle microcirculation activity that surrounds the hepatic arteriole via adenosine. The portal flow does not have an autoregulatory system. Hepatocytes are more vulnerable than biliary, Kupffer and endothelial cells.6
Pathology
The severity and characteristics of liver injury depend on the vessels involved and correspond to hepatic congestion and reduced perfusion. The first description of the nutmeg liver was made by Kiernan and Mallory, who showed central congestion and focal necrosis and association with impaired liver circulation.4 They also reported high capillary pressure and fluid collection between the cordal cells and capillaries. In their study, the hepatic blood supply was lower by two SDs than that of the normal control groups, which was proportionate to a lower cardiac output. Also, in their study, not only did the total blood volume increase significantly in HF patients but the visceral blood volume was also higher in HF patients, which correlated with venous congestion. The reduction in the renal blood supply was much greater than that in the cardiac output.7
The nutmeg liver includes sinusoidal congestion, bleeding in atrophic regions, haemorrhagic necrosis in zone 3, fatty change and cholestasis (figure 1). Fibrosis involves the liver tissue and terminal hepatic venules (phlebosclerosis).8 The portal vein pressure and hepatic venous wedge pressure are nearly equal in as much as their mean is 8–8.5 mm Hg.9 There is no significant correlation between the hepatic circulation and wedge pressure. In HF patients, an increased right atrial pressure is parallel to the central and hepatic vein wedge pressure and is approximately 14.5 mm Hg. There is no gradient between the central and wedge pressures. An increased sinusoidal pressure is associated with a disruption in the sinusoid endothelium, and this pressure is transmitted to the hepatocytes and the junctions in between.
Figure 1.

Central necrosis with passive congestion.
Zonula occludens are the tight junctions between hepatocytes that separate the extravascular spaces from the biliary canaliculi. A disruption in these junctions leads to an opening of these canaliculi to the sinusoids and the formation of fistulas due to increased sinusoidal pressure. Histopathological characteristics are hyperaemia, centrilobular congestion, collagen deposition and septal fibrosis10 (figure 2). A significant reduction in the functional liver mass is seen in acute decompensated HF, which correlates significantly with the ejection fraction, left ventricular (LV) end diastolic pressure and left atrial diameter, but not with right ventricular end diastolic pressure. The main targets in congestive hepatopathy are the hepatocytes and biliary epithelia. Most patients have congestion, pericentral necrosis and fibrosis, and dilatation of the sinusoids.11 The liver is spotty in appearance or resembles brown, grated nutmeg-like kernels. Black spots correspond to congestive hepatic venules and small distal liver veins. Pallor regions are uninvolved areas and contribute to necrosis and fibrosis in severe and chronic cases, also known as cardiac cirrhosis. Pericentral hepatocytes, which surround the periportal veins, are deoxygenated. Zone 3 is more sensitive to necrosis. There is no significant correlation between congestion and necrosis in zone 3.
Figure 2.
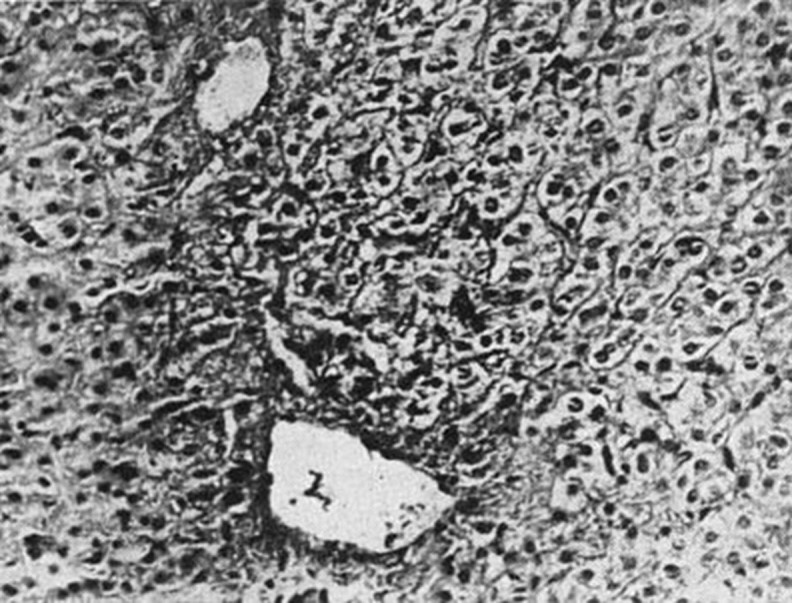
Pericentral vein fibrosis.
Cardiac cirrhosis is a continuum of liver diseases that are seen in right-sided HF; it is an unknown and subclinical disease and its incidence has decreased during the last decade due to a decreasing incidence of rheumatic heart disease or constrictive pericarditis. Cardiac cirrhosis is more common in men due to the higher prevalence of HF in this sex. There is no evidence about age as a factor. The fibrosis pattern in the liver is variable with focal sinusoidal fibrosis and venous occlusion. A prominent cardiac cirrhosis pattern is reverse lobulation, which is bridging fibrosis between the central vein and portal tracts. Histopathological studies and subcutaneous transjugular biopsy are the gold standard for the diagnosis of cardiac cirrhosis. There is no significant correlation between the pattern of fibrosis, age, sex, length of symptoms, severity of right ventricular and LV dysfunction, and filling pressure and the pattern of liver enzymes and the type of cardiomyopathy. The grading of fibrosis is as follows:
0—no fibrosis
1—sinusoidal or pericentral and portal fibrosis (restrict)
2—bridging fibrosis
3—nodular formation.
A mixed or obstructive test disorder or moderate to severe tricuspid regurgitation significantly correlates with hepatic fibrosis in liver biopsy. In the cirrhotic liver, fibrosis distorts the structure and vessels and brings about a reduction in venular and sinusoidal anastomosis. Renal dysfunction is more common in these patients. The severity of liver damage is very difficult to estimate in HF patients as liver fibrosis must be established before abnormal liver function tests or typical portal congestion symptoms manifest.
Ischaemic hepatitis consists of massive hepatocellular necrosis, which can be accompanied by cardiogenic shock or haemodynamic collapse. It seems that low blood pressure and low cardiac output are more important than hypoxia in this regard. This abnormality mostly occurs in left-sided HF, whereas cardiac cirrhosis is predominantly seen in right-sided HF. Additionally, passive congestion is more prominent; however, it has been observed that patients with portal hypertension are more susceptible to a low cardiac output and cardiogenic shock. Zone 3 necrosis can be detected in left-sided HF in the absence of right-sided HF, hence the significance of the cardiac output.12
Pathophysiology
The prevalence of liver injury in HF is 15–65%. Different liver injuries include veno-occlusive disease, congestion, infarction and ischaemia. Congestive hepatopathy mostly occurs in right-sided HF in constrictive pericarditis, mitral stenosis, tricuspid regurgitation and cor pulmonale. Tricuspid regurgitation in particular coincides with hepatic congestion. Necrosis in the central zone of the hepatic lobules is comprised of compression and congestion.13 In conjunction with systemic venous hypertension, advanced HF is in tandem with inflammatory cytokines like interleukin 13 and tumour necrosis factor and in circulation and oxidative stress, all of which are important factors in liver fibrosis. Biventricular failure, not least right ventricular failure, gives rise to a transmission of the central vein hypertension to the liver via the inferior vena cava and hepatic veins and engorged and dammed veins that are more prevalent in chronic HF. On a cellular level, a rise in venous pressure causes sinusoidal blood drainage into the distal venules and thus leads to stasis. Additionally, venous pooling gives rise to deoxygenating and parenchymal atrophy, necrosis, collagen deposition and ultimately fibrosis.
A reduction in the liver metabolic activity can also be seen and is evaluated by the C peptide Methacin test in congestive HF patients.14 A different theory suggests that cardiac cirrhosis is not a simple response to chronic pressure and stasis but needs high-grade vessel occlusion due to intrahepatic thrombosis. This theory posits that sinusoidal thrombosis spreads to moderate-sized hepatic venules and portal branches and results in parenchymal involvement and fibrosis. Furthermore, pericentral hepatocytes, which surround the periportal veins, are deoxygenated. These lesions are observed in pulmonary diseases like interstitial pneumonitis, pleural effusion and intrathoracic tumours. There are two distinct patterns: hepatocellular and cholestatic.
Hepatic involvement in HF is reduced liver circulation, hepatic congestion and decreased oxygen saturation. Centrilobular congestion per se can result in reduced oxygen delivery. Also, we should consider some drugs routinely used in HF that contribute to liver injury like lisinopril (table 1), which can cause a chronic inflammatory and necrotic reaction such as autoimmune chronic hepatitis, and many comorbidities in HF, which influence the liver such as sepsis and pulmonary disease.
Another factor of significance is poor nutrition in HF patients in that it contributes to fatty liver and fibrosis.
We have implied in our study, which is currently under revision, that the cardiac output is a more important factor than is right-sided pressure in congestive hepatopathy. Twenty-seven pulmonary arterial hypertension (PAH) patients were studied and there were no correlations between liver enzymes and pressure data, but an inverse correlation was observed between uric acid levels and carbon monoxide (CO) in this group. PAH puts a significant after-load pressure on the right ventricle. The mean cardiac index levels in the PAH group in our study is significantly higher than in the HF group and close to normal values. This finding might suggest that liver function and uric acid levels are deteriorated more by low cardiac output than systemic congestion since the right-sided filling pressures are much higher in PAH patients but cardiac output remains nearly normal before advanced stages.15
Clinical signs and symptoms
Patients are often asymptomatic and present with elevated liver enzymes. Sometimes, jaundice is also diagnosed and hepatomegaly and hepatojugular reflux are also positive in patients. HF symptoms are prominent and often mask gastrointestinal symptoms. The disease progresses insidiously or it can progress suddenly to constrictive pericarditis or right ventricular failure. Fulminant hepatitis is also reported. A history of coronary artery disease, myocardial infarction, hypertension, diabetes mellitus, valvular heart disease, chronic obstructive pulmonary disease, core pulmonale, constrictive pericarditis, PAH and alcoholism has been observed alongside HF in patients. Patients are liable to complain of discomfort in the right upper quadrant and report jaundice, weight gain, oedema, ascites, nocturia, progressive fatigue, anorexia, and nausea and vomiting. Oedema typically occurs in the lower extremities and dependent regions and may progress to anasarc oedema in advanced and untreated patients. Chronic oedema may be associated with lower extremity pigmentation, induration and cellulites. In clinical examination, abnormal sternal or left parasternal lift as well as S3 and S4 sounds at right side, holosystolic murmur, high-pitched, blowing murmur of tricuspid regurgitation, splitting of S2 and loud P2 can be detected. Hepatomegaly usually presents as a hard, firm liver, and there is also a possibility of splenomegaly. Fewer than 10% of all patients exhibit jaundice. Hepatic encephalopathy is rare, and so is haematemesis. Ascites, albeit not very common, cannot be entirely ruled out and is characterised by increased albumin (Alb) gradient and protein concentration in the ascites fluid. Spider angioma and varices and variceal haemorrhage secondary to HF do not often occur.4
Laboratory data
The most common laboratory abnormality is a mild increase in serum bilirubin levels in up to 70% of patients. Total bilirubin is usually >3 mg/dl. Causes can be hepatocellular disease, haemolysis, pulmonary infarction, canalicular obstruction, drugs and sepsis. Bilirubin levels are correlated with right atrial pressure but not with the cardiac output. Alkaline phosphatase (ALP) is normal or mildly elevated, and increased aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels can be seen in a third of patients not more than two to three times the upper normal limit. There is also a very small possibility of AST and ALT rising even further possibly due to a reduced cardiac output. Elevated aminotransferase levels correlate with zone 3 necrosis in biopsy. Alb reduces in 30–50% of patients but it is not usually less than 2.5 g/dl. Furthermore, Alb does not correlate with liver injury but it is linked with malnutrition and loss of proteins.16 Prothrombin time is slightly elevated, the increase being secondary to impaired coagulation factor synthesis. Disseminated intravascular coagulation itself brings about a slight elevation in serum ammonia. The protein content of ascites is usually more than 25 g/dl, and Alb serum gradient to Alb ascites levels is more than 1.1, which is a sign of portal hypertension. Some studies have reported a significant correlation between the central venous pressure, cardiac index and total bilirubin, AST and ALT levels.17 The central venous pressure interacts with all liver function tests, but the cardiac index is associated significantly with AST, ALT and total bilirubin. Increased liver enzyme levels correlate with hepatocellular injury due to reduced perfusion, but increased direct bilirubin and ALP and low ALT/ALP levels are linked to cholestatic injury and increased central venous pressure levels. Increased bilirubin levels are reported to correlate with high inotrope doses, intensive diuresis and weight loss in HF patients; bilirubin is associated with acute decompensate HF, high central venous pressure, pulmonary capillary wedge pressure, low cardiac output, poor prognosis in early admission, rehospitalisation and long-term outcome of HF patients.18
γ-Glutamine transferase (GGT) is an important factor, and an increase in GGT levels is not only associated with HF severity, high New York Heart Association class, lower ejection fraction and increased B-Type natriuretic peptide levels but is also an independent predictor of death and heart transplantation.19
Mild HF tends to exert an additive prognostic impact on patients, too. Total serum Alb, sodium levels, total cholesterol and body mass index are significantly lower in this group of patients. ALP, GGT and lactate dehydrogenase are independent of both congestion and reduced perfusion. Direct bilirubin and GGT are correlated with the central venous pressure and not with the cardiac index. Studies have demonstrated that elevated bilirubin levels are associated with acute decompensated HF, increased central venous pressure and pulmonary capillary wedge pressure, lower cardiac index and poor prognosis in HF patients. Additionally, liver function tests are correlated significantly with total iron-binding capacity. Elevated seruloplasmin activity can be seen in congestive HF, which is an antioxidant enzyme and a marker of oxidative stress. ALP and GGT are produced in the biliary epithelium and when the biliary canaliculi are injured, they are released in the circulation.
Cholestatic profile and reduced Alb levels are more common than are increased ALT levels, and this pattern is parallel to tricuspid regurgitation severity. Also, it correlates with PAH severity, high right atrial pressure and severe LV dysfunction according to another study.20 These three parameters are linked to bilirubin significantly, but tricuspid regurgitation severity independently correlates with ALP. Decrease in functional liver mass in acute decompensated HF sometimes coincides with normal liver function tests, and this decrease correlates significantly with the ejection fraction, LV end diastolic diameter and left atrial diameter;21 nevertheless, its relation to right ventricular end diastolic diameter is insignificant.22 As mentioned before, we showed a significant relation between total bilirubin and direct bilirubin and cardiac output and right ventricular pressure, pulmonary arterial pressure and especially pulmonary capillary wedge pressure in HF patients. Nonetheless, in PAH patients, although right-sided pressure is very high, it is only total bilirubin that correlates with the cardiac output. Therefore, cardiac output is considered an extremely important factor in congestive hepatopathy.15
Also, in one of our previous studies on uric acid, we demonstrated a significant correlation between uric acid and right ventricular pressure, cardiac output and, again interestingly, pulmonary capillary wedge pressure, which can be used as a marker in advanced HF.3
Prothrombin time is prolonged in >75% of patients and is resistant to vitamin K and it shows cellular injury. It is noteworthy that liver tests cannot present liver function accurately: ALT is dependent on the body mass index, normal Alb levels present acute pressure and low Alb levels show chronic disease. A bilirubin excretion level in urine of >0.05 mg/dl or >0.9 mmol/l is a primary sign of liver disease. ALP is higher in younger patients. Abnormal thyroid function tests can lead to abnormal liver function tests. History and physical examination, toxin exposure, duration of abnormality, history of cholestasis and blood transfusion, intravenous drugs, alcohol consumption, jugular venous pressure and ascites should be taken into account. In alcohol abuse, AST/ALT levels are >2 and GGT levels can increase up to twofold but AST is not more than eightfold and ALT not more than fivefold levels. Commonly used drugs that cause elevated liver enzymes should be considered: NSAIDs, antibiotics, antiseizure drugs, anti-TB drugs, statins, herbal and illicit drugs or even acetaminophen.
GGT has been recently referred to as a marker of atherosclerosis and instability of plaques. It is also effective in the prediction of cardiovascular and cerebrovascular events like myocardial infarction, stroke and cardiac death independent of liver disease. GGT is also associated with most risk factors like diabetes mellitus, hypertension and metabolic syndrome. GGT is also linked to elevated uric acid, NT pro-brain natriuretic peptide, CRP levels and hepatobiliary enzymes such as AST, ALT and ALP. Increased GGT levels are secondary to hepatic congestion or local injury to biliary canalliculation due to increased sinusoidal pressure, ischaemia and freedom of inflammatory cytokines in blood. Studies show that there is no strong correlation between GGT and right atrial and pulmonary artery pressures, so other different factors come into play in this regard; this is indeed true for the markers that we mentioned above. GGT bound to the membrane catalyses the first stage of antioxidant glutathione degradation, which produces amino acids, cysteine and glycine. These glycine thiols can produce anion radicals and hydrogen peroxide in reaction with Fe. GGT reactions catalyse lipoprotein oxidation and can be effective in plaque rupture. GGT is a rescue enzyme for cellular glutation synthesis and is very important in the defensive antioxidant system and its increased activation can be a marker of oxidative stress. There is a correlation between GGT, inflammation and oxidative stress, and endothelial dysfunction and HF progression. There is also a correlation between GGT and vascular stiffness as a marker of endothelial dysfunction; it strongly correlates with ALP but has a modest relation to AST. Furthermore, GGT is more valuable in mild HF patients and in patients younger than 70 years of age.
Uric acid is the end point of the metabolism of purine compounds produced in the liver. Uric acid is eliminated in the gut (33%) and kidneys (67%).
Uric acid itself may have a role in the cardiovascular and renal metabolism. Uric acid can be protective itself and by promoting superoxide dismutase activity; however, it can promote vascular smooth muscle cell proliferation. Both uric acid and xanthine oxidase may play a role in nitric oxide signalling, and may have a pathophysiological role in HF patients.23
Elevated uric acid levels are a marker of inflammation, metabolic disturbances, oxidative stress (free oxygen radicals), vascular and endothelial dysfunction, and perhaps myocardial injury. Hyperuricemia can be in consequence of renal hypoperfusion and reduced urate excretion (shown to be related to high renin expression and high endothelin and aldostrone levels). Uric acid serum levels show xanthine oxidase activation that is upregulated in HF. There is an association among increased xanthine oxidase activity and insulin resistance, tissue hypoxia and inflammatory cytokine activation.24
High uric acid levels correlate well directly with increasing pulmonary artery pressure (PAP), right ventricular pressure(RVP), pulmonary capillary wedge pressure (PCWP) and clinical congestive signs. Therefore, measuring serum uric acid level in patients with systolic HF may be due to high LV filling pressures, low CO, low renal perfusion and tissue hypoxia, high RA and RV pressures and hepatic and renal congestion also.25
Imaging
Chest radiography may show cardiomegaly, pulmonary venous hypertension, interstitial or pulmonary oedema, or pleural effusion. Pleural effusions typically are larger on the right.
Transthoracic echocardiogram with Doppler may diagnose the underlying cause of cardiac cirrhosis. Evaluation of biventricular size, mass, function, wall motion and valves are indicated. Because restrictive cardiomyopathy and pericardial constriction can lead to cardiac cirrhosis, specific attention should be paid to diastolic function parameters such as mitral inflow, pulmonary vein flow, mitral annular flow and their responses to respiration. Tissue Doppler imaging and 2D speckle tracking echocardiography have been introduced as quantitative and more objective methods to quantify regional and global LV systolic and diastolic functions recently.
Abdominal Doppler ultrasonography is useful in the setting of ascites, jaundice or abnormal serum LFTs that are refractory to effective treatment of underlying HF. The test is performed to search for an alternative diagnosis, such as Budd–Chiari syndrome.
CT scan and MRI can diagnose restrictive and constrictive pericardial disease. These studies also may identify enlarged chamber size, ventricular hypertrophy, diffuse cardiomyopathy, valvular disease and other structural abnormalities such as arrhythmogenic dysplasia of the right ventricle. Body imaging may reveal evidence of cardiac cirrhosis, including hepatomegaly, hepatic congestion, inferior vena cava enlargement and splenomegaly. Non-homogenous enhancement in the liver, increased thickness in the gall bladder and, rarely, splenomegaly can also be seen.
Cardiac catheterisation may be indicated in patients with cardiac cirrhosis and HF to diagnose pulmonary hypertension.
In less than 1% of patients with chronic liver failure, pulmonary hypertension occurs in the absence of underlying pulmonary or cardiac disease.
Needle liver biopsy is not indicated routinely. It is indicated in heart transplant candidates with ascites to rule out cirrhosis.
Endomyocardial biopsy may be indicated in patients with cardiac cirrhosis with deteriorating clinical condition and a strong clinical suspicion for myocarditis or in the presence of a systemic disease with possible cardiac involvement, such as haemochromatosis or sarcoid.
There are a few diagnostic findings in the imaging studies of the liver that could indicate or determine the severity of the disease and HF; imaging is only for the purposes of differentiating between the various causes of liver involvement and cardiac causes.26
Cirrhotic cardiomyopathy
In cirrhosis, cardiac contractile function has been extensively documented to be abnormal. This phenomenon has been termed ‘cirrhotic cardiomyopathy.‘
The initial studies in the early 1950s documented the existence of hyperdynamic circulation in cirrhosis, manifested by increased cardiac output and reduced systemic vascular resistance.24
Kowalski was the first to report that patients with cirrhosis had abnormal cardiovascular function and a prolonged QT interval.27
Early histological investigations demonstrated myocardial hypertrophy and ultra structural changes including cardiomyocyte oedema, fibrosis, exudation, nuclear vacuolation and unusual pigmentation.
The size of the left atrium and ventricle in patients with cirrhosis is normal to increased. The change in the LV dimensions in cirrhosis is related to haemodynamic dysfunction.
Many of the patients present with dyspnoea, fluid retention and limited exercise capacity.
Physical exercise, pharmacological stress and therapeutic procedures may affect cardiac pressures. Thus, the LV end diastolic pressure increases but the cardiac stroke index and LV ejection fraction fall during exercise, which indicates an abnormal ventricular response to an increase in ventricular filling pressure. Aerobic exercise capacity and maximal heart rate are lower in most patients with cirrhosis.28
Diastolic dysfunction
Abnormal LV diastolic function, caused by decreased LV compliance and relaxation, implies an abnormal filling pattern of the ventricles. The transmitral blood flow is changed, with an increased atrial contribution to the late ventricular filling.
The pathophysiological background of the diastolic dysfunction in cirrhosis is an increased stiffness of the myocardial wall, most likely because of a combination of mild myocardial hypertrophy, fibrosis and subendothelial oedema.29
Prolongation of the QT interval
The main electrocardiographic change in cirrhosis is a prolongation of the QT interval adjusted for heart rate. It has been known for a long time that it is prolonged in up to 50% of patients with cirrhosis. In alcoholic patients, prolonged QT interval is associated with an increased risk of sudden cardiac death. In patients with cirrhosis, the prolonged QT interval is unrelated to the aetiology of the liver disease, and it is seen in both alcoholic and non-alcoholic liver diseases.
However, in cirrhosis, the duration of the QT interval is associated with indicators of autonomic dysfunction and is partly reversible after liver transplantation.30
Cardiac autonomic dysfunction
Reduced baroreflex sensitivity has been shown to occur in cirrhosis as part of a general cardiovascular autonomic dysfunction. A reduced baroreflex sensitivity was seen, which was significantly related to central haemodynamic and biochemical characteristics. These results suggest that a reduced bar receptor sensitivity owing to the severity of the liver disease is associated with the cardiac dysfunction in cirrhosis.
This cardiac dysfunction may affect the prognosis of the patients and aggravate the course during invasive procedures such as surgery, insertion of a transjugular intrahepatic portosystemic shunts and liver transplantation. On the other hand, liver transplantation has also been shown to ameliorate the cardiac and circulatory disturbances.28
Mechanisms underlying cirrhotic cardiomyopathy
Alcohol: In alcoholic heart muscle disease, impairment of contractile protein synthesis and formation of cardiac protein–acetaldehyde adducts are the major causes leading to cardiac impairment.
Hyperdynamic circulation: In the hyperdynamic circulation, the heart is overloaded by the persistent increase in cardiac output, associated with the expansion of the circulating blood volume. It is well known that prolonged haemodynamic overload of the heart can lead to impairment of cardiac contractility.
Other potential mechanisms of impaired cardiac function in cirrhosis are production of other cardio depressant substances, such as endotoxins, endothelins, cytokines and bile acids; also, downregulation of β-receptors along with an impaired β-adrenergic signalling, changed plasma membrane fluidity with altered potassium, calcium channels and electrophysiological abnormalities, and activation of the cannabinoid can be mentioned, Nitric oxide may also play a part in cardiac contractility.31
Treatment
At present, no specific treatment can be recommended and evident ventricular failure in patients with cirrhotic cardiomyopathy should be treated as non-cirrhotic causes with sodium restriction, diuretics and after-load reduction.
Cardiac glycosides are not be expected to be of significant value in cirrhotic cardiomyopathy as these failed to improve cardiac contractility after angiotensin infusion.
Special caution should be taken during and after stressful procedures such as surgery, shunt implantation and liver transplantation.
QT interval prolongation and mortality are subjects for future research. Newer treatments with anticytokines may also prove to be of interest in patients with cirrhotic cardiomyopathy.30
Acute ischaemic hepatitis (liver shock)
Acute ischaemic hepatitis or liver shock is generally defined on the basis of histological evidence of centrolobular hepatocytic necrosis. It usually results from severe, although sometimes transient, circulatory failure. The pathophysiology of acute hypoxic hepatitis remains unclear. Controversy remains about the respective roles of acute low hepatic blood flow (‘forward failure’) and venous congestion (‘backward failure’) in its pathophysiology. It occurs during the course of myocardial infarction complicated by cardiogenic shock or pulmonary oedema.
The diagnosis of acute hypoxic hepatitis is usually based on a combination of severe cardiac clinical signs (signs of congestion and low blood flow) and major hepatic disorders.32
Clinical manifestations
Gastrointestinal manifestations generally predominated, with nausea, vomiting and right hypochondrial pain and jaundice. Acute diarrhoea and altered general status can be the presentation in some cases. Ascites, lower-limb oedema and encephalopathy can be the signs. All the patients have relatively low blood pressure (mean 100/60 mm Hg). Mean heart rate usually is 100 bpm.
Laboratory data
In acute hypoxic hepatitis, transaminases can increase to more than 100 times the normal values in relation with the importance of centro-lobular hepatic necrosis, peaking at 12–24 h and normalising within 10 days with treatment. Alkaline phosphates and bilirubin increase less. Decreased prothrombin time has an important prognostic value. Other coagulation factors may decrease with the importance of hepatic failure. Thrombopenia, when present, occurs simultaneously with the decrease of the prothrombin time.
Importantly, renal failure is found in nearly every case of acute hypoxic hepatitis whereas it occurs in only approximately 30% of the patients with fulminant hepatitis, in association with encephalopathy.32
Treatment
Treatment involves identifying and removing the precipitating cause by:
using low-dose intravenous dopamine to augment splanchnic perfusion
focusing on the low CO state.
The ischaemic liver injury is usually self-limiting when it affects the normal liver, but more serious changes may occur when the liver has been previously damaged.32
Clinical implication
Hepatic dysfunction is commonly seen in HF patients and is closely correlated with haemodynamic parameters. As mentioned above, there are many studies describing the impact of liver disease on patient outcomes. Van Deursen et al 1 and Amin et al 3 reported the correlation between liver enzymes and serum uric acid levels and the haemodynamic profile and posited that AST, ALT and lactate dehydrogenase were significant predictors of all-cause mortality; nevertheless, these parameters were not independent of the cardiac index and central venous pressure in their study and were markers of a poor haemodynamic status in HF patients. Shinagava et al 32 also suggested that abnormal liver function tests correlated with increased mortality and cardiovascular events.33 The CHARM trial not only showed an independent prognostic relation between total bilirubin and mortality due to HF34 but also cited studies maintaining that congestive hepathopathy rather than influencing the prognosis independently was related to the severity of the main disease. Moreover, many drugs and toxins that are metabolised in the liver and contribute to drug interactions should also be taken into account when prescribing medication to HF patients.
Treatment
Medical care
No prospective studies have been performed to evaluate the medical treatment of cardiac cirrhosis. Because no data suggest that the presence of cardiac cirrhosis worsens mortality or morbidity rates, direct treatment must focus on the underlying source of elevated right-sided heart pressure and hepatic venous congestion.
In most cases, diuresis is the cornerstone of initial medical therapy for symptomatic relief. As cardiac cirrhosis is a direct complication of elevated central venous pressures, effective diuresis should improve hepatic derangements. Initial treatment of cardiac cirrhosis usually requires a loop diuretic (eg, furosemide). Spironolactone may provide an additional source for diuresis through its aldosterone antagonism effects. Also, it is better to consider adherence to a low-sodium diet.
Once the patient is euvolaemic, β-blockers and ACE inhibitors should be added if the underlying cause is LV dysfunction.
Beyond diuretics, medical therapy should be directed at treating the underlying HF and correcting the source of elevated right-sided heart pressures.
Failure to resolve elevated pressure levels despite HF resolution should prompt evaluation of non-cardiac sources of liver disease.35
Surgical care
Definitive treatment of cardiac cirrhosis sometimes requires surgical intervention, particularly when the underlying structural or anatomic lesion remains symptomatic despite maximal medical therapy. Some surgical intervention should be considered as follows:
coronary artery bypass surgery for ischaemic cardiomyopathy
tricuspid valve repair or replacement for tricuspid regurgitation or tricuspid stenosis
pericardiectomy (cardiac decortication) for constrictive pericarditis
transjugular intrahepatic portosystemic shunt is generally contraindicated because of the risk of acute right-sided decompensation from increased venous return
cardiac transplantation can be considered for end-stage cardiomyopathy; the presence of cardiac cirrhosis with significant liver fibrosis is considered a contraindication to transplantation.34
Conclusion
The liver is an important and complex organ and its high metabolic activity is associated with many molecular and haemodynamic changes in patients. Hepatic dysfunction is commonly seen in HF patients and is closely correlated with haemodynamic parameters. The liver receives 25% of the cardiac output. It has a dual circulatory supply, which is regulated by smooth muscle microcirculation activity. Zone 3 is more susceptible to tissue hypoxia. The characteristics of liver injury depend on the involved vessels and correspond to hepatic congestion and reduced perfusion. The main targets in congestive hepatopathy are the hepatocytes and biliary epithelia. Most patients have congestion, pericentral necrosis and fibrosis, and dilatation of the sinusoids. Cardiac cirrhosis is a continuum of liver diseases that are seen due to right-sided HF. There is no significant correlation between the pattern of fibrosis, filling pressure and the type of cardiomyopathy. Ischaemic hepatitis consists of massive hepatocellular necrosis, which can be accompanied by cardiogenic shock or haemodynamic collapse. Also, there would be significant LV dysfunction in cirrhosis, which is known as cirrhotic cardiomyopathy. In this article, we discussed the clinical and laboratory manifestations, imaging clues and some treatment options for liver diseases in HF patients and made some new observations on liver markers and serum uric acid levels in relation to CO in the hope it will serve as a guide for future research.
Acknowledgments
We would like to thank Dr Pedram Amouzade and all colleagues in the Heart Failure and Transplantation Department.
Footnotes
Competing interests: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Van Deursen VM, Damman K, Hillieg HL, et al. Abnormal liver function in relation to hemodynamic profile in heart failure patients. J Card Fail 2010;16:84–90. [DOI] [PubMed] [Google Scholar]
- 2.Bhardwaj SS, Chalasani NP. Hepatotoxicity of cardiovascular and antidiabetic medications. In: Drug-Induced Liver Disease. 2nd edn New York, London: Informa Health Care, 2007:593–631. [Google Scholar]
- 3.Amin A, Vakilian F, Maleki M. Serum uric acid levels correlate with filling pressures in systolic heart failure. Congest Heart Fail 2011;17:80–4. [DOI] [PubMed] [Google Scholar]
- 4.Friedman LS. Congestive Hepathopathy. 2010. Version 17.2. http://www.uptodate.com/css/amages/ui-icones (accessed Jul 2010).
- 5.Giallourakis CC, Rosenberg PM, Friedman LS. The liver in heart failure. Clin Liver Dis 2002;6:947–67. [DOI] [PubMed] [Google Scholar]
- 6.White TJ, Wallace RB, Gnassi AM. Hepatic abnormalities in congestive heart failure: needle biopsy studies. Circulation 1951;3:501–7. [DOI] [PubMed] [Google Scholar]
- 7.Rapaport E, Weisbart MH, Levine M. The splanchnic blood volume in congestive heart failure. Circulation 1958;18:581–7. [DOI] [PubMed] [Google Scholar]
- 8.Gelow JM, Desai AS, Hochberg CP, et al. Clinical predictors of hepatic fibrosis in chronic advanced heart failure. Circ Heart Fail 2010;3:59–64. [DOI] [PubMed] [Google Scholar]
- 9.Myers JD, Taylor WJ. Liver, cirrhosis of the liver and no cirrhotic portal hypertension occlusive hepatic venous catheterization in the study of the normal. Circulation 1956;13:368–80. [DOI] [PubMed] [Google Scholar]
- 10.Ren X, Andrews AH, Holtzmuller KC. Cardiac Cirrhosis. 2010. (accessed Nov 2010). Available from emedicine specialties>cardiology>myocardial disease and cardiomyopathy. [Google Scholar]
- 11.Cogger VC, Fraser R, Le Couteur DG, et al. Liver dysfunction and heart failure. Am J Cardiol 2003;91:1399. [DOI] [PubMed] [Google Scholar]
- 12.Friedman LS, Chapora S, Bonis PA. Pathogenesis of Liver Injury in Circulatory Failure. 2010. Version 18.1. http://www.uptodate.com/css/amages/ui-icones (accessed Nov 2010).
- 13.Lau GT, Tan HC, Kritharides L. Type of liver dysfunction in heart failure and its relation to the severity of tricuspid regurgitation. Am J Cardiol 2002;90:1405–8. [DOI] [PubMed] [Google Scholar]
- 14.Hendrichová M, Málek F, Kopřivová H, et al. Correlation of NT-proBNP with metabolic liver function as assessed with 13C-methacetin breath test in patients with acute decompensate heart failure. Int J Cardiol 2009;144:321–2. [DOI] [PubMed] [Google Scholar]
- 15.Amin A, Vakilian F, Maleki M. Liver function tests and uric acid serum levels in relation to hemodynamic profile a comparison between heart failure and PAH patients. Iranian Heart Journal 2011;6:148–52. [Google Scholar]
- 16.Felder L, Mund A, Parker JG. Liver function tests in chronic congestive heart failure. Circulation 1950;2:286–97. [DOI] [PubMed] [Google Scholar]
- 17.Anand IS, Ferrari R, Kalra GS, et al. Edema of cardiac origin. Studies of body water and sodium, renal function hemodynamic indexes, and plasma hormones in untreated congestive cardiac Failure. Circulation 1989;80:299–305. [DOI] [PubMed] [Google Scholar]
- 18.Shinagava H, Inomata T, Koitabashi T, et al. Increased serum bilirubin levels coincident with heart failure decompensation indicate the need for intravenous inotropic agents. Int Heart J 2007;48:195–204. [DOI] [PubMed] [Google Scholar]
- 19.Poelzl G, Eberl C, Achrainer H, et al. Prevalence and prognostic significance of elevated-glutamyltransferase in chronic heart failure. Circ Heart Fail 2009;2:294–302. [DOI] [PubMed] [Google Scholar]
- 20.Davidson CJ, Bonow RO. Cardiac catheterization. In: Braunwald's Heart Disease: A Text Book of Cardiovascular Medicine. 8th edn New York, NY: Saunders, 2008:439–62. [Google Scholar]
- 21.Málek F, Hendrichová M, Krátká K, et al. Correlation of the functional liver mass with left ventricular ejection fraction and left atrial diameter in patients with congestive heart failure. Int J Cardiol 2008;127:271–3. [DOI] [PubMed] [Google Scholar]
- 22.Rudski LG, Lai WW, Afilalo J, et al. New guidelines for the echocardiography assessment of the right heart in adults: a report from the American Society of echocardiography. J Am Soc Echocardiography 2010;23:685–713. [DOI] [PubMed] [Google Scholar]
- 23.Becker MA. Uric Acid Balance. 2010. Version 18.1 http://www.uptodate.com/css/images/ui-icons-000000-256*240.png (accessed Jul 2010).
- 24.Alimonda AL, Nunez J, Nunez E, et al. Hyperuricemia in acute heart failure. More than a simple spectator? Eur J Intern Med 2009;20:74–9. [DOI] [PubMed] [Google Scholar]
- 25.Juan P, Canella C, Perna RE, et al. Elevated serum uric acid levels in outpatients with chronic heart failure: side effect or marker of myocardial damage? J Cardiac Fail 2003;9:S91. [Google Scholar]
- 26.Vasconcelos LA, de Almeida EA, Bachur LF. Clinical evaluation and hepatic laboratory assessment in individuals with congestive heart failure. Arq Bras Cardiol 2007;88:524–9. [DOI] [PubMed] [Google Scholar]
- 27.Kowalski HJ, Abelmann WH. The cardiac output at rest in Laennnecs cirrhosis. J Clin Invest 1953;32:1025–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma Z, Lee SS. Cirrhotic cardiomyopathy: getting to the heart of the matter. Hepatology 1996;24:451–9. [DOI] [PubMed] [Google Scholar]
- 29.Møller S, Henriksen JH. Cirrhotic cardiomyopathy: a pathophysiological review of circulatory dysfunction in liver disease. Heart J 2002;87:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al-Hamoudi W, Lee SS. Cirrhotic cardiomyopathy. Ann Hepatol 2006;5:132–9. [PubMed] [Google Scholar]
- 31.Møller S, Henriksen JH. Cirrhotic cardiomyopathy. J Hepatol 2010;53:179–90. [DOI] [PubMed] [Google Scholar]
- 32.Denis C, De Kerguennec C, Bernuau J, et al. Acute hypoxic hepatitis (‘liver shock’): still a frequently overlooked cardiological diagnosis. Eur J Heart Fail 2004;6:561–5. [DOI] [PubMed] [Google Scholar]
- 33.Ferriera S, Azevodo A, Frioes F, et al. Uric acid and prognosis in HF patients. Eur J Intern Med 2003;14:151–9. [Google Scholar]
- 34.Pfeffer M, Yusuf S. CHARM: Candesartan Benefits in Heart Failure. http://www.theheart.org/
- 35.Ren X, Oei HH, Pearl man JD. Cardiac Cirrhosis and Congestive Hepatopathy Treatment and Management. http://emedicine.medscape.com/article/151792-overview (accessed 20 Aug 2010).