

Abstract
Objective
Since 2006, our institution has cared for 70 patients with pulmonary arterial hypertension (PAH) and severe right ventricular (RV) hypertension; 44 with congenital heart disease (CHD) had Eisenmenger syndrome (ES) and 26 had PAH without CHD. We sought to determine and compare ‘real-world’ survival outcomes and cause of death in these two distinct groups.
Methods
An observational study of consecutive adult patients from our CHD or PAH clinics with RV systolic pressure ≥80 mm Hg (on echocardiography or right-heart catheter). Detailed patient demographics, investigations and outcomes at baseline and follow-up were reviewed.
Results
Patients with ES were younger than non-CHD group (39±9 vs 64±14 years, p<0.001) with a lower proportion of women (61% vs 85%; p=0.04), and higher RV systolic pressure. Estimated 1-year, 3-year and 7-year survival were not significantly different between the groups (98%, 95% and 74% ES; 100%, 92% and 63% non-CHD, p=0.52). In patients with non-CHD, poorer survival was associated with a diagnosis of connective tissue disease (HR 6.90, 95% CI 1.21 to 39.3) and number of PAH therapies (HR 2.8, 95% CI 1.03 to 7.59). Mortality was directly attributed to PAH in 75% of non-CHD group compared with 31% in patients with ES (p=0.049), many of whom died from infection or bleeding.
Conclusions
We report favourable 7-year survival in adults with PAH and systemic RV pressures—equivalent in ES and non-CHD groups. Those with non-CHD predominately died of cardiac complications of PAH, as distinct from those with ES, many of whom died from complications of chronic cyanosis.
Keywords: CONGENITAL HEART DISEASE
Introduction
Pulmonary hypertension (PH) complicates a number of diverse medical conditions. PH is characterised by progressive vascular remodelling and proliferation of the pulmonary arterioles, with subsequent elevation in pulmonary vascular resistance (PVR) and an increase in right ventricular (RV) afterload.1–3 Significant elevation in RV afterload leads variably to RV dilatation, hypertrophy, impaired systolic function and failure. Furthermore, RV dysfunction is independently associated with poor prognosis in patients with PH, regardless of the underlying aetiology.4–6
Previously published work (from registries, including patients with PH of any severity) suggests that patients with PH in the context of congenital heart disease (CHD) (such as patients with Eisenmenger syndrome, ES) have a better prognosis, compared with those with pulmonary arterial hypertension (PAH) associated with a structurally normal heart.7 Data from the French registries (untreated patients, enrolled 2002–2003) and the Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL, 2006 onwards), since the advent of pulmonary vasodilator therapies, demonstrate 3-year survival rates in patients with non-congenital PAH of 58%–74%, as compared with 3-year survival of greater than 80% in patients with ES.1 8 Possible explanations suggested include that the RV in ES may be better adapted to high afterload (as the RV pressure may have never fallen postnatally), or a beneficial effect of a ‘blow off’ for the hypertensive right heart, through a septal defect.
Less examined, however, are the outcomes of patients with systemic or near-systemic RV pressures, especially in a non-registry setting where there might be less (or different) ‘selection bias’ than for voluntary multicentre databases. Accordingly, we sought to determine and compare ‘real-world’ survival rates in our institution for patients with PH with ES versus those with non-congenital causes for PAH and systemic or near-systemic pulmonary pressures, on optimal medical therapy. Further, we sought to characterise the demographic factors which might be associated with survival, in these groups. Finally, we aimed to evaluate and compare causes of death in these two different patient groups.
Methods
Inclusion criteria
Adults (age≥18 years) seen in our institution (PHT or CHD clinics) at least once since January 2006 with a diagnosis of severe PAH or ES were included in the study. We chose this entry date as January 2006 is approximately when specific oral pulmonary vasodilator therapy became available in Australia. None of the included patients were enrolled in a clinical trial. Severe RV hypertension in the patients with non-CHD was defined as a systolic RV pressure of 80 mm Hg or greater based on either transthoracic echocardiogram estimates and/or right-heart catheterisation measurements. ES was diagnosed based on clinical, transthoracic and/or haemodynamic evidence of right-to-left blood flow and subpulmonary systolic pressures of 80 mm Hg or greater.
The primary diagnosis in each patient was determined by their treating physician, according to current guidelines (mean PAP≥25 mm Hg, pulmonary artery wedge pressure <15 mm Hg and cardiac output normal or reduced on right-heart catheter), with 100% of patients with non-CHD undertaking right-heart catheterisation.2 Patients with ‘non-congenital PAH’ were separated into idiopathic PAH (Type 1.1) and PAH associated with connective tissue disease (CTD-PAH, Type 1.4).2 ES was categorised based on their structural abnormality. Those with a solitary atrial septal defect (ASD), ventricular septal defect (VSD) or persistent ductus arteriosus (PDA) were ‘simple’ and all others were defined as ‘complex’.
A review of each case included patient's medical files, transthoracic echocardiograms and right-heart catheters. Local ethics committee approval was obtained for this retrospective data analysis with patient consent obtained at the time of their inclusion in either registry. Demographic data including age and gender were recorded. Further information, including survival and cause of death, was then obtained from the local CHD database and National PH database. Cause of death as recorded in the databases was obtained directly from death certificates. In the non-CHD group, a documented primary cause of death of ‘Pulmonary Hypertension’ or ‘Heart Failure’ was recorded as a death related to PH, in this study. In the CHD cohort, a documented primary cause of death of ‘Congenital heart disease’, ‘Heart Failure’ or ‘Pulmonary Hypertension’ was recorded as a death related to PH. Comprehensive data were available as patients on pulmonary vasodilator therapy have mandatory 6-monthly reviews in order to qualify for continued supply of medication.
Investigations
Each patient's 6-monthly transthoracic echocardiograms were reviewed when available; however, the majority of patients’ had baseline studies performed outside our referral centre. Given the limited availability of high-quality baseline data, we did not attempt any detailed analysis of echocardiography predictors of outcome.
Six-minute walk tests were recorded when they had been performed in patients on pulmonary vasodilator therapy. A stable 6 min walk test was defined as a less than 10% decline in distance post-therapy versus pretherapy.
Statistical analysis
Data were analysed using SPSS V.22.0 (IBM, New York, USA). Descriptive data were expressed as percentages with 95% CI, medians with IQR or means with SD depending on data distribution. Comparisons between groups were undertaken using χ2 for categorical data and Mann–Whitney U test for continuous data. A two-tailed p value of <0.05 was considered statistically significant.
Survival analysis for mortality was estimated using Kaplan–Meier survival curves and analysed using the log rank test, comparing congenital and non-congenital cohorts. The Kaplan–Meier survival curves were terminated when the number at risk in either group was five or fewer subjects. The Cox proportional hazards model was used to identify factors associated with survival. This used a backwards, stepwise approach including in the first model all the factors associated with the outcomes using bivariate analysis with a p value <0.1. Factors with a p value ≥0.05 were progressively removed from the models starting with those variables with a regression coefficient closet to 0 or an OR closest to 1. Final models were limited to predictive factors with significant coefficients (p<0.05).
Results
Demographics, haemodynamics and therapies
Between 2006 and 2015, 114 patients with PH and structurally normal hearts were seen in our multidisciplinary PH clinic, of whom 26 had systolic pulmonary pressures of greater than or equal to 80 mm Hg and were included in the study. Over the same time period, 2633 adults with CHD were seen in our institution. Of these, all those with severe PH (as defined) had ES (n=44) and were included. The diagnoses of these individuals can be seen in figure 1.
Figure 1.

Flowchart of included patients. ASD, atrial septal defect; VSD, ventricular septal defect; complex, complex congenital heart disease; IPAH, idiopathic pulmonary arterial hypertension; CTD-PHT, connective tissue related pulmonary hypertension; CTEPH, chronic thromboembolic pulmonary hypertension; RVSP, right ventricular systolic pressure.
The mean age was 39 years (8.7 SD) for those with ES and significantly older in the patients with non-congenital PAH (64 years, 14.2 SD; p<0.001), with ES having slightly but significantly higher estimated systolic pulmonary artery pressures (116.8 mm Hg vs 109.7 mm Hg, p=0.01). Both groups were predominantly female (39% men in ES, 15% men in non-CHD; p=0.04). Diuretics were used significantly less in the patients with ES (24% vs 50%; p=0.038); however, there was no significant difference in oral anticoagulant use (26% in ES, 27% in non-CHD; p=0.90). In the non-CHD group, all were on at least one PAH-specific drug (50% one agent, 42% two agents, 8% three agents), whereas only 61% of the patients with ES were on PAH-specific therapy (41% one agent, 20% two agents). The agents used differed between the two groups. In the non-CHD group, 69% were on sildenafil, 46% bosentan, 19% inhaled iloprost, 19% ambrisentan and 4% macitentan. In the treated ES, 44% were on sildenafil, 70% bosentan, 15% macitentan and 4% sitaxentan. The 39% of patients with ES who were untreated at the time of their last review, were not functionally limited by their cardiorespiratory system (WHO 1 or 2), with all 61% patients with treated ES being in WHO 3 or 4 at the time that treatment was commenced. Of those not treated with PAH-specific therapy, eight had Down's syndrome. In those who were treated, 58% patients with CHD had stable 6MWT post-therapy versus pretherapy and 71% patients with non-CHD; p=0.370. (tables 1 and 2)
Table 1.
Cohort demographics and therapies
| Congenital (n=44) | Non-congenital (n=26) | p Value | |
|---|---|---|---|
| Age (years) | |||
| Mean (SD) | 39 (8.7) | 64 (14.2) | <0.001 |
| Median (IQR) | 39 (32.0–44.8) | 65 (56.5–74.0) | |
| Sex (% men) | 39 | 15 | 0.040 |
| Mortality (% dead) | 30 | 31 | 0.914 |
| Diuretics (%) | 24 | 50 | 0.038 |
| Anticoagulants (%) | 26 | 27 | 0.90 |
| Treated (%) | 61 | 100 | <0.001 |
| Heart–lung transplant (%) | 7 | 4 | 0.605 |
| Peak SPAP (mm Hg) | |||
| Mean (SD) | 117 (14.2) | 110 (25.2) | 0.010 |
| Median (IQR) | 115 (110–124) | 103 (93–120) | |
| 6-min walk distance (m) | |||
| Mean (SD) | 425 (105) | 370 (114) | 0.50 |
| Median (IQR) | 446 (351–485) | 369 (293–437) | |
| WHO functional class (%) | |||
| I | 9 | 0 | <0.001 |
| II | 54 | 12 | |
| III | 35 | 83 | |
| IV | 2 | 0 | |
WHO functional class measured at time of initial referral to centre; 6-min walk distance represents average distance on most recent test. Data represented as percentages (%), mean with SD and/or median with IQR.
Table 2.
Targeted PAH therapy
| Number of PAH- specific therapies (%) | Congenital (n=44) | Non-congenital (n=26) | p Value |
|---|---|---|---|
| 0 | 39 | 0 | 0.001 |
| 1 | 41 | 50 | |
| 2 | 20 | 42 | |
| 3 | 0 | 8 |
Data represented as percentages (%).
PAH, pulmonary arterial hypertension.
Survival
Two survival analyses were performed, one for death and the other for a composite end point of death/transplant. Kaplan–Meier survival curves confirmed no significant difference in death (log rank p=0.52) or death/transplant (log rank p=0.75). In the non-CHD group 1-year, 3-year, 5-year and 7-year survival was 100%, 92%, 82% and 63%, respectively; and in the CHD group, it was 98%, 95%, 80% and 74%, respectively. (figures 2 and 3).
Figure 2.
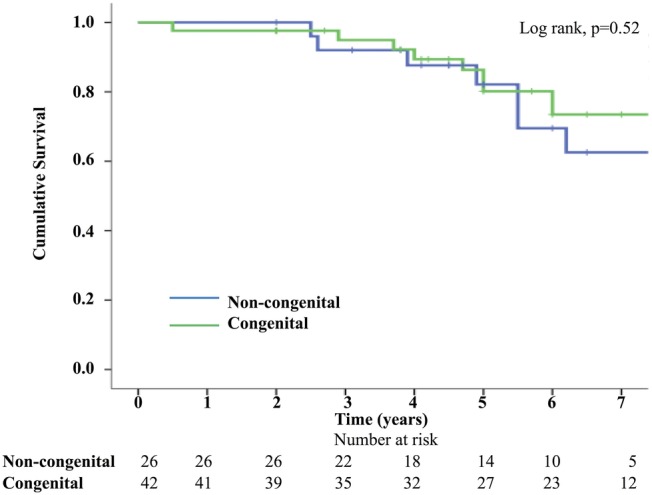
Kaplan–Meier survival curve—death.
Figure 3.
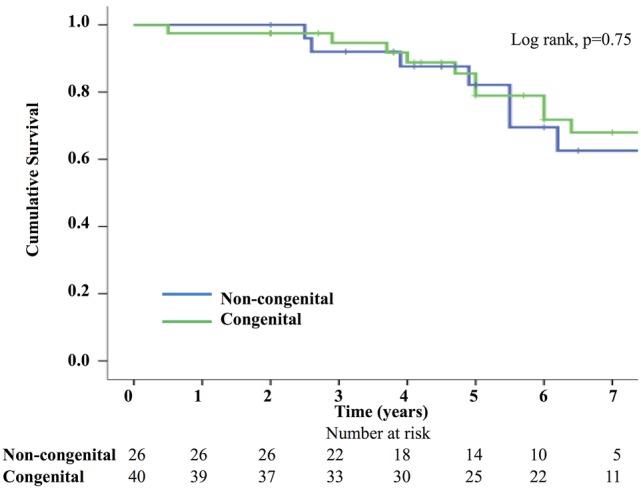
Kaplan–Meier survival curve—composite of death or transplant.
Cox regression analysis was performed in both groups for age, sex, diuretic therapy, number of vasodilators, peak pulmonary pressure and underlying diagnosis. It demonstrated that in the non-CHD group, the two factors associated with survival were the number of PAH agents received (mortality HR 2.8 for each additional vasodilator, 95% CI 1.0 to 7.6; p=0.043), and a diagnosis of PAH associated with CTD (mortality HR 6.9, 95% CI 1.2 to 39.3; p=0.029). None of the tested factors were significant in the CHD group.
Cause of death was reviewed in both groups. In the non-CHD cohort, 75% deaths were directly attributable to their PH and associated heart failure, as compared with only 31% in the CHD group (p=0.049). In the non-CHD group, the other causes of death (25%) were transplant rejection and multiorgan failure. In the CHD cohort, the non-cardiac causes of death (69%) were gastrointestinal haemorrhage, pulmonary haemorrhage, septicaemia, appendicitis, endocarditis, influenza, pneumonia and thyroid neoplasm.
Discussion
The most salient finding from this study is the lack of any significant difference in 7-year survival between ES and severe non-congenital PAH with systemic or near-systemic PA pressure, potentially driven by recent improvements in outcomes in both groups, related to specific medical therapies. Furthermore, patients with non-CHD mostly died from direct complications of their PH, whereas patients with CHD had a variety of ‘non-cardiac’ causes of death such as haemorrhage and infection, complications that are more likely sequelae of chronic cyanosis.
Since the advent of targeted PAH therapy, median survival in PH, based on registry data, has improved significantly. PAH had a median survival of 2.8 years from diagnosis in the 1980s National Institutes of Health (NIH)-supported US registry, as compared with >7 years in the REVEAL study (2006–2009).1 8 9 When looking specifically at PAH associated with systemic sclerosis, a prospective 4-year study of 794 patients demonstrated a 3-year survival of 56%.10 Indeed, PAH associated with CTD, specifically systemic sclerosis, has previously been shown to have a poorer prognosis than other subtypes, with the best prognosis being documented in patients with CHD.8–11
In our study of severe PAH with systolic PAP ≥80 mm Hg 7-year survival for non-CHD patients was 63%. Notably, however, this is in a cohort in which PAH-specific therapy was used in 100% of cases (with 50% on more than one agent). In both our ES cohort and the non-CHD cohorts, 7-year survival was greater than recently published registry data (74% in ES, 63% non-CHD; as compared with 67% in ES, 57% in idiopathic pulmonary arterial hypertension (IPAH) and 36% in CTD associated-PAH in the REVEAL study).1 A recent subanalysis of the REVEAL study compared 7-year survival in IPAH and ES and found slightly lower survival than our cohort but no significant difference between the two groups (55% IPAH vs 64% ES; p=0.25). However, it is difficult to make direct comparisons between this study and ours given that their data were derived in the registry setting, across 55 US sites with the potential for selection bias. Furthermore, their IPAH cohort represented all disease severities (mean PAP 53 mm Hg) as distinct from this current ‘severe’ cohort with right ventricular systolic pressure (RVSP) ≥80 mm Hg.12
Factors predictive of survival are difficult to elucidate in this complex condition, especially with relatively small patient numbers. Previous studies have shown prognosis to be dependent on WHO functional class, RV function, underlying cause, mean PAP and rate of deterioration.9–11 13 14 Other factors associated with improved survival in PAH include female sex, greater 6MWT, higher cardiac output and normal serum albumin. Further, in a study of 84 patients, from 1994 to 2002, reduced survival was associated with African–American or Asian race.8 15 Specifically in systemic-sclerosis-associated PAH, a raised mean right atrial pressure was the strongest haemodynamic predictor of mortality. In the majority of cases of PAH, RV failure is the direct cause of death.3 10
When looking specifically at the comparison of survival between patients with congenital and non-congenital heart disease with severe PAH, Hopkins et al found in a study of 100 adults that there was a trend to higher pulmonary pressures in ES (mean SPAP 107 mm Hg in ES, 97 mm Hg non-congenital PAH, p=0.06) however with higher cardiac indexes and lower mean right atrial pressures than non-congenital PAH. ES had better 1-year, 2-year and 3-year survival than non-congenital PAH.16 This contrasts with our study, where in spite of a significantly higher PAP in ES, there was no significant difference in 7-year survival rates between the two cohorts in both the end point of death and the composite end point of death/transplant. Specific factors independently associated with survival in our non-CHD cohort were number of pulmonary vasodilators, with the addition of each vasodilator increasing the risk of mortality (HR 2.8 for each additional vasodilator, 95% CI 1.0 to 7.6; p=0.043); and diagnosis, with poorer prognosis in those with connective-tissue-associated disease (HR 6.9, 95% CI 1.2 to 39.3; p=0.029). It is important to note, however, that only single agent therapy is supported by government funding in Australia and thus combination therapy is reserved for the sickest patients (self-funded or by compassionate access schemes). This makes the association between number of pulmonary vasodilators and survival difficult to interpret. No independent risk factors were identified in the CHD group.
Survival analysis from registry data are often confounded by enrolment of exclusively incident cases or a combination of incident and prevalent cases. Previous studies have demonstrated that incident cases of PAH have worse survival compared with prevalent cases from time of diagnosis, due to the effect of survivor bias.17 Our non-CHD group included only incident cases of PAH referred to our clinic for initial diagnostic assessment and management. Thus, the favourable long-term survival in the non-CHD group is unlikely to be confounded by the effect of survivor bias.
Notably, the majority of deaths in the non-CHD cohort were related to PH-RV failure (75%) as opposed to only 31% in the CHD cohort (p=0.049). Those with ES predominantly died of complications of their CHD and chronic cyanosis, such as infection and catastrophic haemorrhage; rather than directly from PH.
Study limitations
There are several limitations to this study, including the relatively small numbers studied, and that they were all obtained from a single centre. There is also the risk of selection bias, as these represent patients from a tertiary referral centre who are motivated to attend regular review and investigation; and are given intensive, evidence-based therapies and management. Further, due to our limited access to baseline echocardiographic data, combined with relatively small subject numbers, there was limited value to an analysis of echocardiographic predictors of outcome and thus it was not performed. Finally, we defined severe disease based on an RVSP≥80 mm Hg. The inherent issue with this definition is that pulmonary pressures can fall as RV function declines and thus some individuals with severe disease may have been unintentionally excluded from the study. Future studies that have access to more comprehensive transthoracic echocardiography data, such as RV fractional area change, RV S prime and tricuspid annular plane systolic excursion, will help to address this limitation.
Conclusion
We report favourable 7-year survival data from a small cohort of patients with PH and systemic or near-systemic RV pressures—equivalent in ES and non-CHD groups. This may represent a beneficial effect of the more widespread use of targeted pulmonary vasodilatory therapy in severe PH and/or care in a tertiary referral centre; however, larger studies are needed to confirm this. Further, those with non-CHD were more likely to die of severe PH, as distinct from those with ES whom died of complications of their chronic cyanosis.
Key messages.
What is already known about this subject?
Previously published data (largely registry based) state that those with pulmonary hypertension (PH) associated with congenital heart disease have a better prognosis than those with PH in the context of a structurally normal heart. There is, however, very limited ‘real-world’ data, particularly in the context of current optimal medical therapy. Further, there is also a paucity of data on outcome comparisons in those with severe PH, factors associated with survival and the differing causes of death in the two groups.
What does this study add?
This study provides ‘real-world’ data on survival outcomes, factors associated with survival and cause of death in those with severe PH with and without a structurally normal heart. It is one of the first papers to demonstrate a comparable survival rate between the two groups in the context of current optimal medical therapy.
How might this impact on clinical practice?
The relatively favourable survival rates seen in both cohorts are important in terms of patient counselling and management by clinicians. The data on cause of death are also important as it highlights that both cohorts are distinctly different in terms of their risk profile and this should be instructive in their management.
Footnotes
Contributors: All authors are responsible for the reported research and have participated in the concept and design, analysis and interpretation of data, drafting or revising of the manuscript, and have approved the manuscript for submission. No authors have any conflicting interests.
Competing interests: None declared.
Ethics approval: Sydney Local Health District Ethics Committee.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: No unpublished data from this study is available to any outside parties. Professor David Celermajer has all data stored securely in his research laboratory that cannot be accessed by external individuals.
References
- 1.Benza RL, Miller DP, Barst RJ et al. . An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 2012;142:448–56. doi:10.1378/chest.11-1460 [DOI] [PubMed] [Google Scholar]
- 2.Galiè N, Hoeper MM, Humbert M et al. . Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009;30:2493–537. doi:10.1093/eurheartj/ehp297 [DOI] [PubMed] [Google Scholar]
- 3.Vonk Noordegraaf A, Galiè N. The role of the right ventricle in pulmonary arterial hypertension. Eur Respir Rev 2011;20:243–53. doi:10.1183/09059180.00006511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Forfia PR, Fisher MR, Mathai SC et al. . Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med 2006;174:1034–41. doi:10.1164/rccm.200604-547OC [DOI] [PubMed] [Google Scholar]
- 5.Voelkel NF, Natarajan R, Drake JI et al. . Right ventricle in pulmonary hypertension. Compr Physiol 2011;1:525–40. doi:10.1002/cphy.c090008 [DOI] [PubMed] [Google Scholar]
- 6.Badano LP, Ginghina C, Easaw J et al. . Right ventricle in pulmonary arterial hypertension: haemodynamics, structural changes, imaging, and proposal of a study protocol aimed to assess remodelling and treatment effects. Eur J Echocardiogr 2010;11:27–37. doi:10.1093/ejechocard/jep152 [DOI] [PubMed] [Google Scholar]
- 7.Beghetti M, Galiè N. Eisenmenger syndrome a clinical perspective in a new therapeutic era of pulmonary arterial hypertension. J Am Coll Cardiol 2009;53:733–40. doi:10.1016/j.jacc.2008.11.025 [DOI] [PubMed] [Google Scholar]
- 8.Humbert M, Sitbon O, Chaouat A et al. . Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010;122:156–63. doi:10.1161/CIRCULATIONAHA.109.911818 [DOI] [PubMed] [Google Scholar]
- 9.McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation 2002;106:1477–82. doi:10.1161/01.CIR.0000029100.82385.58 [DOI] [PubMed] [Google Scholar]
- 10.Mukerjee D, St George D, Coleiro B et al. . Prevalence and outcome in systemic sclerosis associated pulmonary arterial hypertension: application of a registry approach. Ann Rheum Dis 2003;62:1088–93. doi:10.1136/ard.62.11.1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McLaughlin VV, Presberg KW, Doyle RL et al. . Prognosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest 2004;126(1 Suppl):78S–92S. doi:10.1378/chest.126.1_suppl.78S [DOI] [PubMed] [Google Scholar]
- 12.Barst RJ, Ivy DD, Foreman AJ et al. . Four- and seven-year outcomes of patients with congenital heart disease-associated pulmonary arterial hypertension (from the REVEAL Registry). Am J Cardiol 2014;113:147–55. doi:10.1016/j.amjcard.2013.09.032 [DOI] [PubMed] [Google Scholar]
- 13.Chung L, Liu J, Parsons L et al. . Characterization of connective tissue disease-associated pulmonary arterial hypertension from REVEAL: identifying systemic sclerosis as a unique phenotype. Chest 2010;138:1383–94. doi:10.1378/chest.10-0260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Wolferen SA, Marcus JT, Boonstra A et al. . Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur Heart J 2007;28:1250–7. doi:10.1093/eurheartj/ehl477 [DOI] [PubMed] [Google Scholar]
- 15.Kawut SM, Horn EM, Berekashvili KK et al. . New predictors of outcome in idiopathic pulmonary arterial hypertension. Am J Cardiol 2005;95: 199–203. doi:10.1016/j.amjcard.2004.09.006 [DOI] [PubMed] [Google Scholar]
- 16.Hopkins WE, Ochoa LL, Richardson GW et al. . Comparison of the hemodynamics and survival of adults with severe primary pulmonary hypertension or Eisenmenger syndrome. J Heart Lung Transplant 1996;15(1 Pt 1):100–5. [PubMed] [Google Scholar]
- 17.Humbert M, Sitbon O, Yaïci A et al. . Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J 2010;36:549–55. doi:10.1183/09031936.00057010 [DOI] [PubMed] [Google Scholar]