

Abstract
Painful intervertebral disc (IVD) degeneration is a common cause for spinal surgery. There is a clinical need to develop injectable biomaterials capable of promoting IVD regeneration, yet many available biomaterials do not mimic the native extracellular matrix (ECM) or promote matrix production. This study aimed to develop a decellularized injectable bovine ECM material that maintains structural and compositional features of native tissue and promotes nucleus pulposus (NP) cell (NPC) and mesenchymal stem cell (MSC) adaption. Injectable decellularized ECM constructs were created using 3 NP tissue decellularization methods (con.A: sodium deoxycholate, con.B: sodium deoxycholate & sodium dodecyl sulfate, con.C: sodium deoxycholate, sodium dodecyl sulfate & TritonX-100) and evaluated for protein, microstructure, and for cell adaptation in 21 day human NPC and MSC culture experiments. Con.A was most efficient at DNA depletion, preserved best collagen microstructure and content, and maintained the highest glycosaminoglycan (GAG) content. NPCs in decellularized constructs of con.A&B demonstrated newly synthesized GAG production, which was apparent from “halos” of GAG staining surrounding seeded NPCs. Con.A also promoted MSC adaption with high cell viability and ECM production. The injectable decellularized NP biomaterial that used sodium deoxycholate without additional decellularization steps maintained native NP tissue structure and composition closest to natural ECM and promoted cellular adaptation of NP cells and MSCs. This natural decellularized biomaterial warrants further investigation for its potential as an injectable cell seeded supplement to augment NP replacement biomaterials and deliver NPCs or MSCs.
Keywords: intervertebral disc, decellularization, nucleus pulposus replacement, extracellular matrix biomaterial, tissue engineering
Low back pain is a major cause of disability in Western society, with a lifetime prevalence of ~80%.1,2 Low back pain is often related to intervertebral disc (IVD) degeneration and herniation, which are the most common causes for lumbar spinal surgery.3,4 Nuclectomy is a common spine surgery procedure, where the affected degenerated and/or herniated nucleus pulposus (NP) tissue is removed, leaving an enlarged hole in the IVD. While the procedure results in pain relief, it cannot reverse degeneration and may even accelerate degenerative changes resulting in long-term clinical problems.5–8
Biological and tissue engineering strategies for IVD repair are of great current interest and several growth factor and cell-based studies have been performed in vivo and in vitro.7,9,10 However, growth factors have a short half-life, which limits their suitability for long term regeneration.11 Using cell-based therapies nucleus pulposus cells (NPCs) or mesenchymal stem cells (MSCs) can be injected directly into the injury site of herniated or early degenerated IVDs to stimulate the metabolism of host cells and to produce more matrix.12 Thus, in diseased IVD tissues undifferentiated MSCs may have difficulties surviving due to the harsh IVD environment that is typically hypoxic, has a low pH, and a higher osmolarity compared to other tissues.13–15
Several tissue-engineering techniques have recently been developed to replace the NP and are a promising approach to treat accelerated degeneration complications following IVD herniation.16–18 Yet, little is known about whether these techniques provide the appropriate extracellular environment for NPCs and the success of NP replacement strategies are highly dependent on the choice of an appropriate biomaterial.
Decellularized NP from xenogeneic tissue combines the advantages of a native NP structure with a reduced risk of rejection or disease transmission since the decellularization process removes cellular antigens while maintaining many structural proteins of the extracellular matrix (ECM). Recently, native decellularized tissue scaffolds from various animal sources have been developed19,20 to mimic the native 3D matrix for cell survival, to promote glycosaminoglycan (GAG) production, and to restore the mechanical properties of the IVD.21–23 One disadvantage of scaffolds is that during surgery, scaffolds have to be resized according to the injury size. Injectable hydrogels that can gel within minutes have been studied extensively.7,24,25 In gel form they can fill up the entire hole and adhere to the host tissue, minimizing the risk of herniation; thus, these gels cannot represent the native ECM. The addition of an injectable decellularized ECM would provide the advantage of a native 3D environment. The optimal decellularized NP tissue should be processed in a manner that allows it to maintain important native ECM structure and composition characteristics while allowing human NPCs and MSCs to survive, proliferate, and produce new matrix.
The objectives of this study were to: (I) develop a procedure to decellularize healthy xenogeneic NP tissue fragments that best maintain native tissue structure and composition and process it in a form that can be used as an injectable cell-seeded construct, (II) evaluate the capacity of human NPCs to survive, proliferate and adapt to produce extracellular matrix when in a variety of decellularized matrix conditions, and (III) evaluate the capacity of human MSCs to survive, proliferate and adapt to produce extracellular matrix within the optimized decellularized matrix condition.
METHODS
Tissue Harvest
Caudal IVDs from skeletally mature bovines were obtained from a local abattoir and 5–6 IVDs were dissected from each bovine. NP tissue was separated from the annulus fibrosus. NPs from two tails were combined, minced, and then split into four groups: Non-treated (nat.ECM), condition A (con.A), condition B (con.B), and condition C (con.C).
Tissue Preparation
All specimens (n = 7) underwent the first stage of tissue preparation involving 5 freeze-thaw cycles to break up cell membranes, lyophilization (Freeze-zone 1, LABCONCO, MO), and tissue-grinding (Geno/Grinder 2010, SPEX SamplePrep, NJ), which increased the surface area and created small pieces of tissue that could be suspended for easy implantation via pipette or syringe. Control samples (nat. ECM) underwent the same pre-processing steps in order to most closely resemble the native ECM and were stored at −20°C until further usage.
Tissue Decellularization
Decellularization chemicals and timing in this study were selected from 9 initial treatment conditions modified from methods of Vavken et al. 200926 and Stapleton et al. 2008.27 Treatment conditions varied in incubation duration (24, 2 and 1 hours) to determine the minimal necessary exposure times, adding sodium chloride to the decellularization, dissolving the detergents in PBS and performing the washes in PBS or distilled water The 3 protocols that retained maximum GAG were considered most promising and chosen for more in-depth analysis. All conditions, including the control (nat.ECM), underwent 5 freeze thaw cycles and tissue grinding before decellularization. The chosen protocols included three groups with following steps: con.A: treatment with 2% sodium-deoxycholate and DNase; con.B: treatment with 2% sodium dodecyl sulfate (SDS), 2% sodium deoxycholate and DNase; and con.C: treatment with 2% SDS, 0.1% Triton-X 100, 2% sodium deoxycholate and DNase (Table 1). All solutions were prepared under sterile conditions in Millipore water. Each decellularization step was performed for 1 h under agitation at 37°C and washed in sterile Millipore water after each decellularization step. Samples were then lyophilized overnight and stored at −20°C until further usage.
Table 1.
Decellularization Protocols
| Condition | Tissue Preparation | Tissue Decellularization | ||||||
|---|---|---|---|---|---|---|---|---|
| con.A | 5 Freeze thaw cycles | Lyophilize | Tissue grind | 2% Sodium deoxycholate | DNase | Lyophilize | ||
| con.B | 2% SDS | 2% Sodium deoxycholate | DNase | |||||
| con.C | 2% SDS | 0.1% Triton-X 100 | 2% Sodium deoxycholate | DNase | ||||
| nat.ECM | ||||||||
For each treatment the samples were incubated for 1 h followed by 3 washes in dH2O. SDS, Sodium dodecyl sulfate.
Tissue Characterization
Tissue Loss
The amount of tissue loss was calculated from the total tissue dry weight before and after decellularization.
Fragment Size
After lyophilization, the decellularized ECM was carefully re-separated and the fragment size was determined by using brightfield microscopy (5×; Axiovert 200M, Zeiss, Germany) and MATLAB (R2015a) using a custom made macro.
Extra Cellular Matrix Composition
Glycosaminoglycan (GAG) content of papain digested tissue was determined using the dimethylmethylene blue (DMMB) assay (n = 5)28 and collagen type 2 content was determined by ELISA (Type II Collagen Detection kit #6018; Chondrex, Redmond, WA; n = 4) according to manufacturer instructions. Briefly: All samples were digested with pepsin at 4°C. Then, 1–2 mg (dry weight) of each sample was re-hydrated in 0.5 ml cold H2O overnight and centrifuged, the supernatant was discarded and the swollen tissue was incubated overnight in 0.5 ml of 3 M guanidine. After centrifuging, the supernatant was transferred into a 15 ml graduated centrifuge tube containing 0.2 ml normal goad serum that was prepared to collect all following supernatants. The precipitate was then washed in H2O, incubated overnight in acetic acid and centrifuged. The combined supernatants were dialyzed against tris-buffered saline and stored at −20°C until further usage. The precipitate was incubated in 0.5 ml pepsin solution for 24–48 h and the solution was replaced until all tissue fragments were solubilized. After 3 changes of pepsin, the precipitate was incubated in 0.1 mg/ml elastase solution and incubated for 24 h. Supernatants of each step were combined and neutralized with 1/50 1 M Tris base to neutralize the solution and the volume was adjusted to a final volume of 5 ml by adding tris-buffered saline, pH 7.5. All incubation steps were performed at 4°C. COL2 content was determined using the Type II Collagen Detection Kit.
DNA Content
Tissues were papain digested and DNA content was determined by QuantiFluor® dsDNA System (Promega, WI) according to manufacturer instructions.
Microstructure
After lyophilization, the decellularized ECM fragments formed a firm sponge-like matrix (Fig. 1A). To assess the microstructure, lyophilized constructs (n = 3/condition; Fig. 1B), or fresh NP tissue (n = 2) were cut into cubes (~2 mm3) and fixed in 3% Glutaraldehyde in 0.2 M Sodium Cacodylate Buffer pH 7.3 for 48 h. Alternatively, to evaluate the microstructure of constructs for cell culture, the sponge like lyophilized decellularized ECM matrices were loosened up into fine fragments, and rehydrated with media (Fig. 1C). Rehydration resulted in formation of viscose tissue matrices, which were formed to ~5 mm3 constructs and immersed in low melting agarose for 30 s to create a shell to prevent dissociation of the matrix (n = 1/condition; Fig. 1D). The hydrated constructs were then fixed for 48 h (as described above), cut into cubes (~2 mm3) and fixed for an additional 48 h.
Figure 1.

Sample processing of decellularized constructs: (A) Matrix fragments form a firm sponge like matrix after lyophilization (B) To assess the microstructure, the constructs are either cut into a small cubes or are separated into fine fragments (C) For culture and to assess the microstructure (SEM) the fragments are re-hydrated in culture media until they form a viscose matrix (D) Culture-constructs with agarose shell (E + F) To assess potential injectivity of the (C) hydrated matrix is transferred into a (E) dual barrel syringe and injected through a mixing tip and (F) 25 G needle into the injury site of an IVD after discectomy.
After fixation, all specimens were washed in 0.2 M Sodium Cacodylate Buffer pH 7.3, postfixed in 1% Osmium Tetroxide in 0.2 M Sodium Cacodylate Buffer, washed and dehydrated in graded ethyl alcohol steps, infiltrated and embedded (EPON A&B + DMP-30), and polymerized overnight. Next, the specimens were mounted onto metal stubs and coated in an Electron Microscopy Sciences sputter coater 550× (EMS550×, Electron Microscopy Sciences, Hatfield, PA). Observations of the specimen surface were conducted using a S-4300 Hitachi field emission scanning electron microscope (Hitachi High Technologies America, Inc.). Electron micrographs were obtained at 70×, 3,000×, and 50,000×. The approximate collagen fiber thickness was determined by randomly measuring the diameters of the thickest and thinnest fibers using imageJ (http://imagej.nih.gov/ij/).
Matrix Injectivity
To assess matrix injectivity, rehydrated decellularized ECM fragments (Fig. 1C) were transferred into a dual barrel syringe (Fig. 1E) and carefully pressed through a mixing tip, which was attached to a 25 gauche needle to simulate injection into an IVD after discectomy (Fig. 1F).
NPC Isolation and Culture
Human NPCs were harvested from 7 surgical IVD tissues that were obtained from patients undergoing surgery for discectomy following a protocol approved by the Institutional Review Board at the Icahn School of Medicine at Mount Sinai. NPCs were isolated from NP tissue that could be clearly distinguished from annulus fibrosus tissue and did not contain herniated or other unidentifiable tissue as previously described.29 Briefly, NP tissue was diced, rinsed in phosphate buffered saline, and NPCs were enzymatically released using 0.2% protease for 1 h followed by 0.2% collagenase for 4 hours. NPCs were then pressed through a 70 μm cell strainer and expanded in standard DMEM (4.5 g glucose) under normoxia (21% O2) and 5% CO2 at 37°C in a humidified atmosphere. Passages 3–4 were used for all experiments.
MSC Culture
Bone marrow derived MSCs (obtained from Texas A&M University, Texas) were expanded in standard DMEM (4.5g glucose) under normoxia (21% O2) and 5% CO2 at 37°C in a humidified atmosphere. Passage 3 MSCs were used for all experiments.
Cell Seeded Constructs Generation and Culture
To archive a cell density similar to mature IVDs,30 lyophilized ECM constructs were minced into fine fragments (Figs. 1B & 2C) and formed to cell seeded constructs by re-hydrating the samples with a suspension of 100μl low glucose media (standard DMEM, 1 g glucose) containing human NPCs in a final concentration of 1 × 106 cells/100 mg tissue (dry weight). The relative low media-volume to cell ratio was chosen to ensure homogenous cell distribution within the matrix, while avoiding oversaturation of the matrix, which would impede formation of the cell seeded constructs. Additional media was immediately added until all tissue fragments were hydrated forming a viscous matrix. The matrices were then formed to 10 cell seeded constructs for each lyophilized condition and immersed in low melting agarose for 30 s to create a shell to prevent swelling and dissociation of the matrix (Fig. 1D). The resulting cell seeded constructs were cultured for up to 21 days under hypoxia (5% O2) and 5% CO2 at 37°C in a humidified atmosphere, and culture media was changed every 3–4 days. To evaluate the potential of the optimized decellularized matrix as a human MSC delivery supplement, the cell culture study was repeated with human MSCs for con.A (n = 5, 2 donors).
Figure 2.

Matrix fragment size after decellularization. (A + B) Decellularized fragments of con.A highly varied in size with most fragments being less than 200 μm in length and 5000 μm2 in area (C) Fragments were randomly shaped with a rough surface. Note that y-axis is log scale (***p < 0.001).
Cell Viability
Cell viability at day 1, 7, and 21 was determined by calceinAM and ethidium-homodimer. Cell seeded constructs were incubated in serum free media supplemented with 5 μmol/l calceinAM and 1 μmol/l ethidium-homodimer for 1 h at 37°C. Constructs were then washed in PBS and visualized on a confocal laser scanning microscope (Leica SP5 DMI; human NPC: con.A+B n = 7, con.C n = 5, agarose n = 4, nat. ECM n = 3; human MSC: con.A n = 5, ECM n = 4, agarose n = 4; Life Technologies, NY). Images of 175 μm deep scans were reconstructed to a z-projection after excluding images of scans through agarose to exclude potential cells that grew on the construct surface or within the agarose shell. Cells were quantified using ImageJ (http://imagej.nih.gov/ij/) and Cell Profiler (www.cellprofiler.org/).
Histology
Cultured cell seeded constructs (day 1and 21) were embedded either in methacrylate31 (human NPC, n = 3) or in paraffin (human MSC, n = 3) and 5 μm thick sections were used for histology. Sections were de-plasticized with a combination of xylene and 2-ethoxyethanol, or de-paraffinized with a combination of petroleum ether and 2-ethoxyethanol. For ECM content assessment GAGs were stained by alcian blue and toluidine blue, and collagen was stained by picosirius red. All samples were analyzed using brightfield microscopy (Axio Imager Z1, Zeiss, Germany). Cell-construct morphology was determined by using toluidine blue and observed by Differential Interference Contrast (DIC) microscopy.
Histological GAG and Collagen Content Quantification
Due to the interference of the remaining agarose after culture, semi-quantitative GAG or collagen intensities were assessed histologically rather than with biochemical measurements. Analysis was performed on histology slides of methacrylate embedded sections by light microscopy analysis (n = 3). Three images/section were randomly chosen and staining intensities were evaluated independently by 5 researchers, ranking the blinded samples by intensity from 5 (most intense stain) to 1 (weakest stain). Only sections of the same staining and imaging procedure were compared.
Statistics
For statistical analysis of tissue characteristics, One-way ANOVA repeated measures tests were performed with subsequent post hoc testing (Newman–Keuls Multiple Comparison Test). Fragment sizes, culture effects of cell viability and cell number were analyzed by Mann–Whitney U-Test. Effects of culture conditions were assessed with Kruskal–Wallis test with subsequent post hoc testing (Dunn’s Multiple Comparison Test). All statistical analyses were performed with GraphPad Prism5; for all analyses, p < 0.05 was considered significant, and descriptive statistics are presented as mean±SD.
RESULTS
Tissue Loss
Tissue loss from decellularization occurred in all groups and was highest in con.C (con.A: 58.70% ± 0.27; con.B: 64.3% ± 0.09; con.C: 67.44 ± 0.53; n = 7).
Fragment Size and Injectivity
The fragment size after decellularization con.A ranged from 20–1,000 μm (length) and 100–74,000 μm2 (area) with the majority of fragments being under 200 μm (87%) and 5,000 μm2 (84%; Fig. 2). Hydrated fragments were successfully injected through a 25 g needle (inner ø 260 μm) into the injury site after a simulated discectomy in a bovine IVD (Fig. 1E & F).
Collagen Type 2 Content
The decellularization process led to significantly increased collagen type 2 (COL2) content in all groups compared to nat.ECM, while no differences were observed between the different decellularization conditions (p < 0.0002; Fig. 3A).
Figure 3.

Matrix composition after decellularization. (A) After decellularization, a higher COL2 content was observed in all conditions. (n = 4). (B) Decellularization caused significant GAG loss in all conditions. GAG loss was significantly higher in con. B&C compared to con.A (n = 5). (C) DNA content was significantly decreased in all conditions (n = 5; *p < 0.05, **p < 0.0002).
GAG Content
All decellularization processing conditions resulted in significant GAG loss compared to nat.ECM (p < 0.0001). GAG content was significantly higher in con.A compared to con.B&C (p < 0.05; Fig. 3B).
DNA Content
After decellularization DNA content significantly decreased in all conditions and was lowest in con.A (p < 0.0001; Fig. 3C).
Microstructure (SEM)
With additional decellularization steps the matrices became more porous with larger spaces between fragments (Fig. 4B–D). At a higher magnification, the collagen fibers of con.B&C appeared less loose, while the structures of con.A were loosely connected to each other (Fig. 4F–H). Examining the collagen fibers in more detail revealed that the collagen fibers in nat. ECM were randomly and loosely organized, and varied in diameter (ø 73–18 nm; Fig. 4I); small particles were attached to the surface of some fibers giving them a rough appearance (Fig. 4I). Fiber orientation in con.A was similar to nat.ECM while the portion of thin fibers was decreased (ø 71–25 nm; Fig. 4J). With increasing decellularization steps the collagen fibers became organized in a tight network, often aligned to each other, and appeared shortened and homogenous in diameter (con.B: ø 81–33; con.C: ø 125–39 nm; Fig. 4K + L).
Figure 4.

Scanning electron micrographs of decellularized constructs. (A–D): With increasing decellularization steps the matrices became more porous while (E–H) the collagen fibers of con.B&C appear less lose. (I–L): (I) nat.ECM and (J) con.A contain collagen fibers of various diameters while collagen fibers in (K) con.B and (L) con.C appear shorter and more homogenous with few thin fibers.
NPC Viability and Cell Number
To assess cytotoxicity and determine the effect of the decellularized matrix conditions on cell proliferation, native NPCs were seeded in 3D constructs and cultured for up to 21 days. During the 21 day culture, cell viability increased in all conditions, which was significant for con.A and nat.ECM (Fig. 5A + Fig. 6). In con. A&B constructs, the number of live cells increased during the 21 days culture (con.A: d1 = 22.1±24.7/mm2, d21 = 37.01±26.4/mm2; con.B: d1 = 20.0±15.7/mm2, d21 = 43.9±27.7/mm2) while cell number in con.C, nat. ECM, and agarose decreased from d1 to d21 (con.C: d1 = 20.0±18.4/mm2, d21 = 18.3±10.1/mm2; nat.ECM: d1 = 31.6±15.4/mm2, d21 = 17.5±2.9/mm2; agarose: d1 = 29.1±16.8/mm2, d21 = 12.8±4.8/mm2; Fig. 3B+ Fig. 4), however, these changes were not significant (p > 0.1). No significant differences were observed between conditions at d1 or d21, for neither cell viability nor cell number (p > 0.1).
Figure 5.
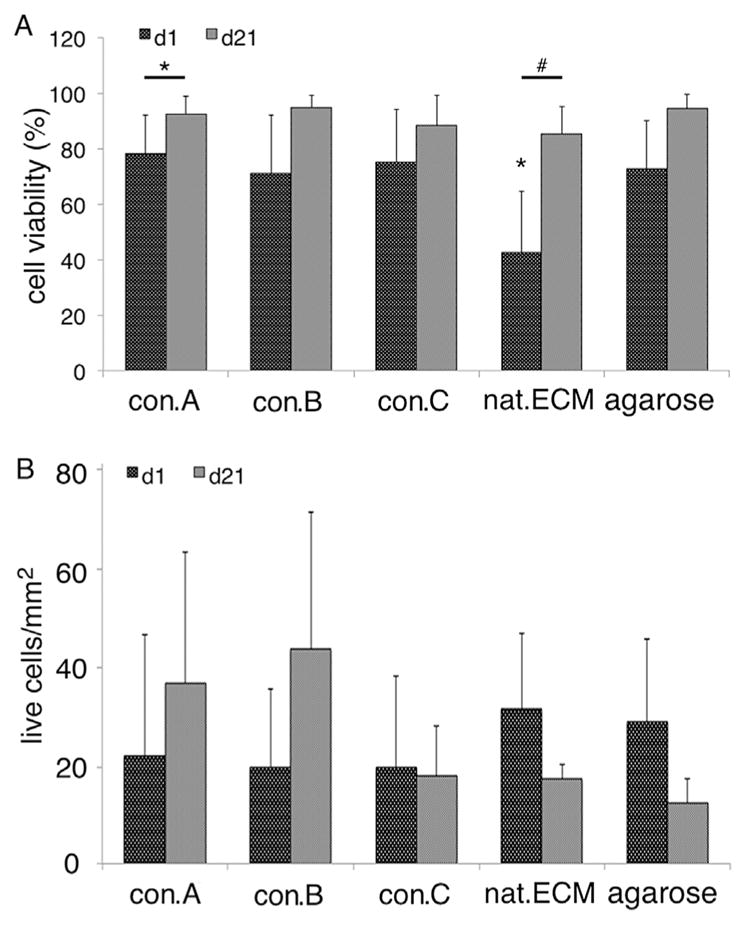
Cell analysis in constructs after 21 days culture. (A) Cell viability and (B) cell number in decellularized constructs after 1 and 21 day cultures (d1 vs. d21: *=p < 0.05).
Figure 6.

Confocal laser scanning micrographs of NPCs seeded in decellularized constructs. Images of con.A, B, C, nat.ECM, and agarose constructs at d1 and d21 using calcein AM and ethidium homodymer to identify live (green) and dead (red) cells, respectively, as visualized with confocal microscopy.
GAG and Collagen Intensity on Histological Sections
Histological quantification of GAG and collagen contents exhibited similar observations as for the ECM composition measurements. All decellularization conditions suggested a loss of GAG content compared to nat.ECM, with con.A retaining more GAG than con. B&C. While not as drastic, at d1 the GAG content was highest in nat.ECM and decreased from con.A to C (Fig. 5 upper row, Table 2). After culture, GAG loss was observed in all conditions (Fig. 5, second row, Table 2). In line with the ECM composition measurements for COL2, histological collagen content was higher in the decellularized constructs than in the nat. ECM (Fig. 5 middle rows, Table 2); however the trends in these histological evaluations were not significantly different, perhaps due to the small sample size.
Table 2.
GAG and Collagen Content
| con.A | con.B | con.C | nat.ECM | agarose | |
|---|---|---|---|---|---|
| GAG | |||||
| d1 | 3.8 ± 0.4 | 3.5 ± 0.7 | 2.7 ± 0.7 | 4.2 ± 0.7 | 1 ± 0 |
| d21 | 3.3 ± 0.9 | 3.2 ± 0.7 | 2.9 ± 0.5 | 4.3 ± 0.5 | 1 ± 0 |
| COL | |||||
| d1 | 4.4 ± 0.6 | 3.7 ± 0.4 | 3.8 ± 0.5 | 2.8 ± 0.9 | 1 ± 0 |
| d21 | 3.8 ± 0.4 | 4.1 ± 0.6 | 3.4 ± 0.6 | 2.9 ± 1.3 | 1 ± 0 |
Semi-quantitative GAG and collagen staining intensity of histological sections after 1 and 21 day culture of human NPC seeded constructs (ranking 1 = least staining intensity, 5 = highest staining intensity).
Matrix Production and NPC Morphology (Histology)
Collagen and GAGs were visualized with a light toluidine blue stain. In constructs of nat.ECM, con.A and con.B, a blue-purple matrix stain surrounded the NPCs like halos, indicative of newly synthesized matrix production. When cultured in con.C constructs, newly synthesized matrix was rarely observed near cells, and after culture in agarose no staining was observed around any cells (Fig. 7, lower rows).
Figure 7.

Representative images of matrix composition of decellularized constructs after 1 and 21 day culture. (A) GAG and (B) collagen content staining intensity and (C) NP cell morphology. Arrows mark blue/purple halos surrounding the cells.
MSC Viability and Cell Number
Con.A was the most promising condition since it was the decellularization condition that most effectively retained composition and structure of nat.ECM, therefore, it was chosen to evaluate the potential of decellularized ECM as biomaterial supplement for MSC survival. During culture MSC cell numbers remained stable in con.A constructs while they slightly decreased in both, nat.ECM and agarose, however, these changes were not significant. Similarly, at d1 more live cells were observed in nat.ECM; yet, the cell viability of con.A was significantly higher compared to nat.ECM (p < 0.01); after 21 days, culture high cell viability was observed in all groups (Fig. 8).
Figure 8.

MSC viability and proliferation. (A) Confocal laser scanning micrographs of MSC seeded constructs, (B) cell proliferation, and (C) cell viability after 1 and 21 day culture (*p < 0.5; **p < 0.01).
Matrix Production and MSC Morphology
Toluidine blue staining was performed to assess cell morphology and GAG production of MSCs. At day 1, MSCs in tissue of con.A and in the nat.ECM were surrounded by blue-purple stain. After 21 days culture, the matrix of con.A and nat.ECM appeared less stained while the halos around the MSCs were clearly visible; no staining was observed around MSCs cultured in agarose (Fig. 9).
Figure 9.

MSC morphology. A light toluidine blue stain was performed to visualize newly synthesized GAG. Arrows mark blue/purple halos of newly synthesized GAG surrounding the cells.
DISCUSSION
An ideal decellularized NP matrix would maintain the important tissue structure and composition characteristics of native ECM while being depleted of cells and membrane associated antigens, and this study further prioritized processing of decellularized NP matrix for injectable delivery. Here we compared three decellularization conditions to assess their efficacy to decellularize bovine NP tissue while maintaining the native NP matrix structure. Decellularization conditions included the anionic detergents sodium deoxycholate and SDS and the non-ionic detergent Triton X-100. Results indicated that NPCs could successfully survive and proliferate in tissues of all decellularization conditions, yet con.A best retained structure and composition. Furthermore, culture of MSCs in the optimized con.A demonstrated that MSCs could survive and adapt to decellularized matrix conditions.
Decellularization of xenogeneic tissue has been used for tissue engineering applications in lung, heart, liver, cartilage, and also in the IVD.21,22,26,32–37 Decellularization of lung is quite effective because of the high surface area to volume ratio and is advancing rapidly with feasibility as demonstrated in large animals and human tissue models.38,39 The relatively low surface area to volume ratio of IVD and the dense tissue of healthy IVDs make decellularization more challenging. Tissue-grinding was therefore used to fragment the tough IVD tissue to better decellularize this material and to also make it amenable to be used in an injectable form. Sodium deoxycolate and SDS detergents as anionic hydrophilic detergents of varying strength are very effective in removing cellular remnants and were used to decellularize the NP tissue by solubilizing cytoplasmic and nuclear cellular membranes.20 Triton-x100 is a non-ionic (neutrally charged) detergent capable of disrupting lipid-protein and lipid-lipid interactions, while leaving protein-protein interactions intact and maintaining a functional protein confirmation after decellularization.20 DNAse was used in all conditions to promote maximum decellularization. These detergents have been used in various combinations in the literature and are known to break protein bonds and loosen the collagen network, allowing penetration to lyse cell membranes and denature membrane-bound antigens.20,23,37,40 The best decellularization technique, which used the fewest detergents for the least amount of time, showed acceptable decellularization and best maintained NP composition and structure.
Decellularization of whole IVD tissue is challenging since the decellularization solutions have to diffuse long distances through the dense IVD tissue matrix and solubilized materials have to be transported out of the IVD. Yet, decellularization of whole IVD tissues has been attempted with some success. Chan et al. reported whole IVD decellularization of up to 70% of the endogenous cells, while retaining GAG content and collagen architecture.22 Decellularization was performed with a combination of freeze-thaw cycles and incubation in SDS. Reintroduction of NPCs into the scaffold indicated a high survival rate but showed only minor cell penetration after 7 days culture.22 The dense structure of the IVD likely inhibited cell and fluid transport throughout the IVD and therefore impeded the complete depletion of native cells. Additionally, remnants of the negatively charged SDS might have prevented deeper cell penetration of the reintroduced cells. Nevertheless, Chan et al. demonstrated the potential for decellularization of whole IVDs as scaffolds for bioengineering. In the context of the current study, decellularizing whole IVDs retained native ECM structure and compositions better than grinded decellularized NP tissue, while grinded and decellularized NP provided an injectable biomaterial with shorter diffusion distances that served as a better carrier for cell delivery.
Collagen and GAGs are the main ECM protein components of the NP, which are important for determining cellular behavior such as proliferation, differentiation, migration and maturation.41,42 Decellularization of the ECM resulted in a loose structure of the matrix within the small tissue fragments (Fig. 4A–D), which likely provided the necessary space for cell migration into these tissue fragments. Some GAG remnant within con.A matrix might have facilitated the observed maintenance of the loose collagen structure that was only observed in con.A (Fig. 4F). While collagen type 2 content was maintained after decellularization, the GAG content decreased with prolonged decellularization. Loss of GAG has been reported in several studies on decellularized ECM20,21,43 and is likely caused by the decellularization detergents, which disrupt collagen fibers and matrix architecture so that ECM proteins are prone to dissociate and to be washed out of the matrix.21
Successful decellularized matrices for tissue regeneration have to be cytocompatible, and provide a structure that promotes cell ingrowth and proliferation of the host cells. All decellularized constructs in this study were successfully depleted of DNA and were cytocompatible as indicated by the high cell viability after the 21 day culture. Con.A, with the shortest procedure and the least decellularization steps resulted in sufficient collagen matrix loosening while maintaining the highest amount of GAG of the 3 decellularization processes. GAG halos surrounding the cells of con.A&B after culture further suggested newly synthesized GAG; all indicating that a loosened ECM structure and quality are important for cell maintenance and viability. Moreover, the spindle like morphology of some cells within the cell seeded constructs of con.A&B (Fig. 6) that was observed after 3D reconstruction, resembled NPC morphology that can be observed in fresh, healthy bovine and sheep NP tissue (personal observation). Unfortunately, due to the processing steps for generating decellularized matrix, it was not possible to include cultured, untreated NP tissue (tissue pieces) of the same bovine as additional controls.
Con.A was chosen as the most promising decellularization process and was used to evaluate the potential of maintaining MSCs in decellularized matrix constructs. MSCs are more readily available than NPCs and in vitro co-culture of NPCs with MSCs has been shown to enhance NPC proliferation, activate DNA and GAG synthesis, and have anti-apoptotic effects on inflammatory factor-stimulated NP.44–46 A recent clinical study by Mochida et al. demonstrated in a 3 year follow up that transplantation of activated NPC had the potential to slow the further degeneration of human IVDs.47
One limitation of this study is that the agarose shell that was chosen to prevent dissociation of the decellularized ECM could not be removed completely after culture. Remaining agarose made it impossible to determine tissue weights for biochemical analysis after culture, and also interfered with gene expression analysis. Yet, others demonstrated that cultures of MSCs under IVD like conditions with oxygen and nutrition deprivation have the potential to induce MSC differentiation towards a chondrogenic phenotype13,48 which supports our hypothesis that the observed round morphology with the newly synthesized GAG halos that are surrounding the cells might have induced a potential pathway of MSCs differentiation towards a more chondrogenic phenotype.
MSCs seeded in decellularized NP constructs maintained high viability suggesting that they adapted well to the 3D decellularized environment. The decrease of MSCs and also NPCs in nat.ECM constructs was likely due to the dense structure of this tissue (Fig. 4E), which potentially prevented cell migration into the tissue resulting in an initially higher detectable cell number at the beginning of the culture since cells were more clustered on the nat.ECM surface. The more pronounced decrease of cell number during culture suggested an effect of the native DNA and cellular antigens, which were present in greater abundance in the nat.ECM. The observed increase in cell viability and decrease in cell number after a 21 day culture suggested that the cells surviving during the culture period in nat.ECM remained in a steady state with high cell viability but no proliferation.
Cell viability and cell morphology of NPCs and MSCs within decellularized matrix constructs remained rounded and suggestive of a chondrocytic phenotype. Results therefore demonstrated cytocompatibility with the matrix, and strongly suggested that the decellularization detergents were transported completely out of the ECM following our processing. Potential remaining DNA fragments did not have a detrimental effect on cell viability of either cell type. Yet, in vitro studies can only give limited information about the effects of DNA residues on exogenous cells. To assess potential immune responses after long term exposure to the decellularized ECM further in vivo studies will be necessary.
CONCLUSION
The optimized decellularized matrix has the potential to be used as an injectable supplement for NP replacement biomaterials and could be used to pre-condition MSCs before injection. One future application could be to administer the decellularized matrix into the herniated IVD, where the MSCs within the decellularized matrix could potentially activate native NP cells of the host IVD. The structure of the con.A decellularized matrix allowed NP migration and proliferation and supported generation of native ECM. The decellularized ECM is not expected to act as a framework for native cells, but to populate and produce matrix in order to promote NP regeneration of moderately degenerated IVDs. Future studies will evaluate the combination of decellularized ECM with additional biomaterials.
Acknowledgments
Grant sponsor: National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health; Grant number: R01 AR057397; Grant sponsor: AO Exploratory Research Board of AO Foundation.
The Research reported in this publication was supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (R01 AR057397), a consortium grant from AO Exploratory Research Board of AO Foundation, and internal funding of the Icahn School of Medicine at Mount Sinai. Confocal laser-scanning microscopy was performed at the microscope CORE at the Icahn School of Medicine at Mount Sinai. The authors thank Dr. Steven B. Nicoll for his valuable intellectual contribution. This work would not have been possible without the help of Heather Bell from the Mount Sinai Electron Microscopy Facility, Xue Chen, Young Lu, and Olivia Torre.
Footnotes
Conflict of interest: None.
This manuscript has not been previously published and is not simultaneously being submitted elsewhere. Relevant disclosures for Dr. Hecht include royalties, consultant and Scientific Advisory Board of Zimmer Spine. Disclosures for Dr. Qureshi include royalties of Zimmer Spine, consultant for Stryker Spine, Medtronic, and Orthofix, and Speaking and/or Teaching Arrangements with Globus. Dr. Qureshi is on the Board of Directors of Musculoskeletal Transplant Foundation, and is on the Scientific Advisory Board of Zimmer Spine and Orthofix Spine.
AUTHORS’ CONTRIBUTIONS
SIJ made substantial contributions to research design, acquisition, analysis, interpretation of data and drafting the paper. DDS made substantial contributions to acquisition, analysis of data and revising the manuscript critically. DML made substantial contributions to acquisition of data and revising the manuscript critically. ACH made substantial contributions to research design and revising the manuscript critically. SAQ made substantial contributions to research design and revising the manuscript critically. JCI made substantial contributions to research design, interpretation of data and revising the manuscript critically. All authors have read and approved the final submitted manuscript.
References
- 1.The Burden of Musculoskeletal Diseases in the United States: Prevalence, Societal and Economic Cost. Amer Acad Orthop Surg. 2008 http://www.boneandjointburden.org/
- 2.Vos T, Flaxman AD, Naghavi M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2163–2196. doi: 10.1016/S0140-6736(12)61729-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim CH, Chung CK, Park CS, et al. Reoperation rate after surgery for lumbar herniated intervertebral disc disease: nationwide cohort study. Spine (PhilaPa 1976) 2013;38:581–590. doi: 10.1097/BRS.0b013e318274f9a7. [DOI] [PubMed] [Google Scholar]
- 4.Martin BI, Mirza SK, Comstock BA, et al. Reoperation rates following lumbar spine surgery and the influence of spinal fusion procedures. Spine (PhilaPa 1976) 2007;32:382–387. doi: 10.1097/01.brs.0000254104.55716.46. [DOI] [PubMed] [Google Scholar]
- 5.Hanley EN, Jr, Shapiro DE. The development of low-back pain after excision of a lumbar disc. J Bone Joint Surg Am. 1989;71:719–721. [PubMed] [Google Scholar]
- 6.Loupasis GA, Stamos K, Katonis PG, et al. Seven- to 20-year outcome of lumbar discectomy. Spine (PhilaPa 1976) 1999;24:2313–2317. doi: 10.1097/00007632-199911150-00005. [DOI] [PubMed] [Google Scholar]
- 7.Iatridis JC, Nicoll SB, Michalek AJ, et al. Role of biomechanics in intervertebral disc degeneration and regenerative therapies: what needs repairing in the disc and what are promising biomaterials for its repair? Spine J. 2013;13:243–262. doi: 10.1016/j.spinee.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fakouri B, Shetty NR, White TC. Is sequestrectomy a viable alternative to microdiscectomy? A systematic review of the literature. Clin Orthop Relat Res. 2014 doi: 10.1007/s11999-014-3904-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakai D, Andersson GB. Stem cell therapy for intervertebral disc regeneration: obstacles and solutions. Nat Rev Rheumatol. 2015;11:243–256. doi: 10.1038/nrrheum.2015.13. [DOI] [PubMed] [Google Scholar]
- 10.Hudson KD, Alimi M, Grunert P, et al. Recent advances in biological therapies for disc degeneration: tissue engineering of the annulus fibrosus, nucleus pulposus and whole intervertebral discs. Curr Opin Biotechnol. 2013;24:872–879. doi: 10.1016/j.copbio.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 11.Lee K, Silva EA, Mooney DJ. Growth factor delivery-based tissue engineering: general approaches and a review of recent developments. J R Soc Interface. 2011;8:153–170. doi: 10.1098/rsif.2010.0223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Z, Perez-Terzic CM, Smith J, et al. Efficacy of intervertebral disc regeneration with stem cells—a systematic review and meta-analysis of animal controlled trials. Gene. 2015;564:1–8. doi: 10.1016/j.gene.2015.03.022. [DOI] [PubMed] [Google Scholar]
- 13.Wuertz K, Godburn K, Iatridis JC. MSC response to pH levels found in degenerating intervertebral discs. Biochem Biophys Res Commun. 2009;379:824–829. doi: 10.1016/j.bbrc.2008.12.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan SC, Burki A, Bonel HM, et al. Papain-induced in vitro disc degeneration model for the study of injectable nucleus pulposus therapy. Spine J. 2013;13:273–283. doi: 10.1016/j.spinee.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 15.Huang YC, Urban JP, Luk KD. Intervertebral disc regeneration: do nutrients lead the way? Nat Rev Rheumatol. 2014;10:561–566. doi: 10.1038/nrrheum.2014.91. [DOI] [PubMed] [Google Scholar]
- 16.Feng G, Zhang Z, Jin X, et al. Regenerating nucleus pulposus of the intervertebral disc using biodegradable nanofibrous polymer scaffolds. Tissue Eng Part A. 2012;18:2231–2238. doi: 10.1089/ten.tea.2011.0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kirzner Y, Marcolongo M, Bhatia SK. Advances in biomaterials for the treatment of intervertebral disc degeneration. J Long Term Eff Med Implants. 2012;22:73–84. doi: 10.1615/jlongtermeffmedimplants.v22.i1.80. [DOI] [PubMed] [Google Scholar]
- 18.Mern DS, Beierfuss A, Thome C, et al. Enhancing human nucleus pulposus cells for biological treatment approaches of degenerative intervertebral disc diseases: a systematic review. J Tissue Eng Regen Med. 2014;8:925–936. doi: 10.1002/term.1583. [DOI] [PubMed] [Google Scholar]
- 19.Badylak SF. The extracellular matrix as a biologic scaffold material. Biomaterials. 2007;28:3587–3593. doi: 10.1016/j.biomaterials.2007.04.043. [DOI] [PubMed] [Google Scholar]
- 20.Gilbert TW, Sellaro TL, Badylak SF. Decellularization of tissues and organs. Biomaterials. 2006;27:3675–3683. doi: 10.1016/j.biomaterials.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 21.Xu H, Xu B, Yang Q, et al. Comparison of decellularization protocols for preparing a decellularized porcine annulus fibrosus scaffold. PLoS ONE. 2014;9:e86723. doi: 10.1371/journal.pone.0086723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan LK, Leung VY, Tam V, et al. Decellularized bovine intervertebral disc as a natural scaffold for xenogenic cell studies. Acta Biomater. 2013;9:5262–5272. doi: 10.1016/j.actbio.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 23.Fitzpatrick JC, Clark PM, Capaldi FM. Effect of decellularization protocol on the mechanical behavior of porcine descending aorta. Int J Biomater. 2010;2010:620503. doi: 10.1155/2010/620503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang X, Li X. Nucleus pulposus tissue engineering: a brief review. Eur Spine J. 2009;18:1564–1572. doi: 10.1007/s00586-009-1092-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomas JD, Fussell G, Sarkar S, et al. Synthesis and recovery characteristics of branched and grafted PNIPAAm-PEG hydrogels for the development of an injectable load-bearing nucleus pulposus replacement. Acta Biomater. 2010;6:1319–1328. doi: 10.1016/j.actbio.2009.10.024. [DOI] [PubMed] [Google Scholar]
- 26.Vavken P, Joshi S, Murray MM. TRITON-X is most effective among three decellularization agents for ACL tissue engineering. J Orthop Res. 2009;27:1612–1618. doi: 10.1002/jor.20932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stapleton TW, Ingram J, Katta J, et al. Development and characterization of an acellular porcine medial meniscus for use in tissue engineering. Tissue Eng Part A. 2008;14:505–518. doi: 10.1089/tea.2007.0233. [DOI] [PubMed] [Google Scholar]
- 28.Farndale RW, Buttle DJ, Barrett AJ. Improved quantitation and discrimination of sulphated glycosaminoglycans by use of dimethylmethylene blue. Biochim Biophys Acta. 1986;883:173–177. doi: 10.1016/0304-4165(86)90306-5. [DOI] [PubMed] [Google Scholar]
- 29.Abbott RD, Purmessur D, Monsey RD, et al. Regenerative potential of TGFbeta3 + Dex and notochordal cell conditioned media on degenerated human intervertebral disc cells. J Orthop Res. 2012;30:482–488. doi: 10.1002/jor.21534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boubriak OA, Watson N, Sivan SS, et al. Factors regulating viable cell density in the intervertebral disc: blood supply in relation to disc height. J Anat. 2013;222:341–348. doi: 10.1111/joa.12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laudier D, Schaffler MB, Flatow EL, et al. Novel procedure for high-fidelity tendon histology. J Orthop Res. 2007;25:390–395. doi: 10.1002/jor.20304. [DOI] [PubMed] [Google Scholar]
- 32.Schwarz S, Koerber L, Elsaesser AF, et al. Decellularized cartilage matrix as a novel biomatrix for cartilage tissue-engineering applications. Tissue Eng Part A. 2012;18:2195–2209. doi: 10.1089/ten.TEA.2011.0705. [DOI] [PubMed] [Google Scholar]
- 33.Song JJ, Guyette JP, Gilpin SE, et al. Regeneration and experimental orthotopic transplantation of a bioengineered kidney. Nat Med. 2013;19:646–651. doi: 10.1038/nm.3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scarritt ME, Pashos NC, Bunnell BA. A review of cellularization strategies for tissue engineering of whole organs. Front Bioeng Biotechnol. 2015;3:43. doi: 10.3389/fbioe.2015.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanchez PL, Fernandez-Santos ME, Costanza S, et al. Acellular human heart matrix: a critical step toward whole heart grafts. Biomaterials. 2015;61:279–289. doi: 10.1016/j.biomaterials.2015.04.056. [DOI] [PubMed] [Google Scholar]
- 36.Jiang WC, Cheng YH, Yen MH, et al. Cryo-chemical decellularization of the whole liver for mesenchymal stem cells-based functional hepatic tissue engineering. Biomaterials. 2014;35:3607–3617. doi: 10.1016/j.biomaterials.2014.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teebken OE, Puschmann C, Breitenbach I, et al. Preclinical development of tissue-engineered vein valves and venous substitutes using re-endothelialised human vein matrix. Eur J Vasc Endovasc Surg. 2009;37:92–102. doi: 10.1016/j.ejvs.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 38.Price AP, Godin LM, Domek A, et al. Automated decellularization of intact, human-sized lungs for tissue engineering. Tissue Eng Part C Methods. 2015;21:94–103. doi: 10.1089/ten.tec.2013.0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weymann A, Patil NP, Sabashnikov A, et al. Perfusion-decellularization of porcine lung and trachea for respiratory bioengineering. Artif Organs. 2015 doi: 10.1111/aor.12481. Epub 2015/04/22. [DOI] [PubMed] [Google Scholar]
- 40.Courtman DW, Pereira CA, Kashef V, et al. Development of a pericardial acellular matrix biomaterial: biochemical and mechanical effects of cell extraction. J Biomed Mater Res. 1994;28:655–666. doi: 10.1002/jbm.820280602. [DOI] [PubMed] [Google Scholar]
- 41.Guillaume O, Naqvi SM, Lennon K, et al. Enhancing cell migration in shape-memory alginate-collagen composite scaffolds: in vitro and ex vivo assessment for intervertebral disc repair. J Biomater Appl. 2015;29:1230–1246. doi: 10.1177/0885328214557905. [DOI] [PubMed] [Google Scholar]
- 42.Bosnakovski D, Mizuno M, Kim G, et al. Chondrogenic differentiation of bovine bone marrow mesenchymal stem cells (MSCs) in different hydrogels: influence of collagen type II extracellular matrix on MSC chondrogenesis. Biotechnol Bioeng. 2006;93:1152–1163. doi: 10.1002/bit.20828. [DOI] [PubMed] [Google Scholar]
- 43.Reing JE, Brown BN, Daly KA, et al. The effects of processing methods upon mechanical and biologic properties of porcine dermal extracellular matrix scaffolds. Biomaterials. 2010;31:8626–8633. doi: 10.1016/j.biomaterials.2010.07.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu J, Deng G, Tian Y, et al. An in vitro investigation into the role of bone marrowderived mesenchymal stem cells in the control of disc degeneration. Mol Med Rep. 2015;12:5701–5708. doi: 10.3892/mmr.2015.4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watanabe T, Sakai D, Yamamoto Y, et al. Human nucleus pulposus cells significantly enhanced biological properties in a coculture system with direct cell-to-cell contact with autologous mesenchymal stem cells. J Orthop Res. 2010;28:623–630. doi: 10.1002/jor.21036. [DOI] [PubMed] [Google Scholar]
- 46.Yang SH, Wu CC, Shih TT, et al. In vitro study on interaction between human nucleus pulposus cells and mesenchymal stem cells through paracrine stimulation. Spine (Phila Pa 1976) 2008;33:1951–1957. doi: 10.1097/BRS.0b013e31817e6974. [DOI] [PubMed] [Google Scholar]
- 47.Mochida J, Sakai D, Nakamura Y, et al. Intervertebral disc repair with activated nucleus pulposus cell transplantation: a three-year, prospective clinical study of its safety. Eur Cell Mater. 2015;29:202–212. doi: 10.22203/ecm.v029a15. discussion 212. [DOI] [PubMed] [Google Scholar]
- 48.Li H, Tao Y, Liang C, et al. Influence of hypoxia in the intervertebral disc on the biological behaviors of rat adipose—and nucleus pulposus-derived mesenchymal stem cells. Cells Tissues Organs. 2013;198:266–277. doi: 10.1159/000356505. [DOI] [PubMed] [Google Scholar]