

Abstract
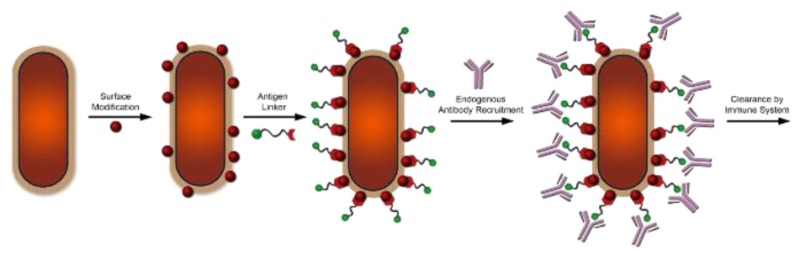
The number of antibiotic-resistant bacterial infections has increased dramatically over the past decade. To combat these pathogens, novel antimicrobial strategies must be explored and developed. We previously reported a strategy based on hapten-modified cell wall analogues to induce recruitment of endogenous antibodies to bacterial cell surfaces. Cell surface remodeling using unnatural single d-amino acid cell wall analogues led to modification at the C-terminus of the peptidoglycan stem peptide. During peptidoglycan processing, installed hapten-displaying amino acids can be subsequently removed by cell wall enzymes. Herein, we disclose a two-step dipeptide peptidoglycan remodeling strategy aimed at introducing haptens at an alternative site within the stem peptide to improve retention and diminish removal by cell wall enzymes. Through this redesigned strategy, we determined size constraints of peptidoglycan remodeling and applied these constraints to attain hapten–linker conjugates that produced high levels of antibody recruitment to bacterial cell surfaces.
Keywords: antibiotics, bacterial, surface, remodeling, d-amino acids
The introduction of antibiotics into clinical settings signaled a revolution in modern medicine.1 Despite the almost immediate detection of drug resistance nearly seven decades ago, antibiotics continued to have tremendous utility due to the pace of their development.2 However, the emergence of extensive resistance among bacterial pathogens has rendered several antimicrobial agents ineffective.3,4 A major health crisis currently looms as the number of antibiotic-resistant pathogens has drastically increased over the past few decades.5,6 Concomitantly, we have recently witnessed historically low levels of U.S. FDA antibiotic approvals.7 To reverse this disconcerting trend, it is essential to explore innovative antibiotic strategies that target pathogenic bacteria through unique and previously unexplored mechanisms of action.
Treatment of bacterial infections traditionally impairs vital cellular processes such as cell wall biosynthesis, DNA replication, or protein synthesis.8 In particular, targeting of peptidoglycan biosynthesis has been highly successful. Peptidoglycan, a major component of bacterial cell walls, is composed of repeating units of N-acetylglucosomine (GlcNAc) and N-acetylmuramic acid (MurNAc) that constitute a mesh-like network that resides to the exterior of cytoplasmic membranes. Stem peptides are attached to the MurNac sugar units that include both l- and d-amino acids.9−11 Cross-linking of neighboring stem peptides is carried out through the enzymatic activity of penicillin-binding protein (PBPs) transpeptidase domains to endow bacterial cell walls with the ability to resist osmotic shock and turgor pressure.12 Due to the critical role of these proteins, chemical inactivators that target PBPs are the basis of several potent classes of antibiotics, such as β-lactams.13
Recently, it has been established that several of the enzymes in the peptidoglycan biosynthetic pathway display broad substrate promiscuity.14−16 This feature opens the door to the metabolic labeling of peptidoglycan using unnatural cell wall analogues. In particular, both Gram-positive and -negative bacteria readily incorporate d-amino acids displaying unnatural side chains into the stem peptide of the peptidoglycan.17−23 This cell surface remodeling process occurs during the cross-linking step mediated by transpeptidase domains, which swap the terminal d-alanine residue of the oligopeptide with an exogenous d-amino acid from the media (Figure S1).24 Together, synthetic cell wall probes are providing unprecedented insight into cell wall biosynthesis and host–cell interactions.25
The use of immunological agents to trigger the selective clearance of cancer cells is an emerging strategy with an impressive track record.26−31 In fact, a significant number of current anticancer agents under clinical evaluations rely on some variation of immune modulation. We propose that immune-modulatory therapeutics may provide an alternative to traditional bactericidal agents. Our research group previously demonstrated the successful remodeling of bacterial cell surfaces with the ultimate goal of inducing a selective immune response (Figure 1A).32,33 Treatment of bacteria with d-amino acids conjugated to dinitrophenol (DNP) led to the surface tagging of bacterial surfaces followed by the recruitment of the endogenous anti-DNP antibodies. In this paper, we describe an alternative mode of metabolic labeling that hijacks the intracellular peptidoglycan biosynthetic pathway. By targeting an earlier point in the biosynthetic pathway, unnatural dipeptides installed haptens with improved retention at the bacterial cell surface.
Figure 1.

(A) Labeling of bacterial cell surfaces with DNP containing d-amino acids results in anti-DNP IgG antibody recruitment and host clearance of infection. (B) Transpeptidase “swapping” results in the incorporation of DNP-d-amino acids into the C-terminal position of the stem peptide. Further transpeptidase and carboxypeptidase activity may result in removal of DNP hapten-containing amino acids. (C) Intracellular dipeptide ligation via MurF results in installation of DNP on the fourth position of the stem peptide and permits greater hapten retention.
Results and Discussion
A prominent feature of bacterial cell walls is the continuous remodeling that occurs during cell growth and division.34 Chemical modifications to the polymeric peptidoglycan chains by endogenous enzymes are essential for maintaining cellular viability. Two of the processes mediated by cell wall-linked enzymes (carboxypeptidase and transpeptidase activities) can potentially lead to the removal of the C-terminal position on the peptidoglycan stem peptide (Figure 1B). d,d-Transpeptidase peptidoglycan cross-linking involves the hydrolysis of the terminal residue to activate the acyl-donor chain. Additionally, d,d-carboxypeptidases catalyze the hydrolysis of the terminal residue to generate truncated tetrapeptides (Figure S1).35 Both of these two reactions may hydrolyze hapten-conjugated amino acids installed onto the terminal position, thus reducing the hapten valency at the cell surface. To achieve greater cell surface retention, we considered metabolic labeling strategies that deliver haptens at alternative positions within the stem peptide (Figure 1C). In addition to transpeptidase substrate promiscuity, several additional enzymes operating in the peptidoglycan biosynthesis pathway demonstrate extensive substrate flexibility. Recently, the intracellular MurF, which is the enzyme responsible for the ligation of d-Ala-d-Ala dipeptide onto the fourth and fifth positions of the growing peptidoglycan precursor molecule, was found to tolerate dipeptides displaying unnatural side chains.22,36 By utilizing a dipeptide-based metabolic labeling strategy, we hypothesized that it would be possible to achieve greater hapten retention on bacterial cell surfaces.
At first, we set out to explore the possibility that epitopes could be conjugated directly to the side chain of d-amino acid-based dipeptides. Treatment of bacterial cells with d-amino acid-based dipeptides with unnatural side chains handles on the N-terminus of the dipeptide unit should lead to epitope installment at the fourth position within the stem peptide (Figure S2). Therefore, by conjugating epitopes on the N-terminal amino acid of the dipeptide, greater protection from d,d-carboxypeptide- and -transpeptidase-mediated hapten removal was expected. We previously found in our single amino acid strategy that d-Lys was the most effective unnatural side chain in the recruitment of anti-DNP antibodies. To evaluate labeling levels, we synthesized an analogous fluorescent amino acid d-Lys(NBD) (1), in which the ε-amine of the lysine side chain was modified with a nitrobenzoxadiazole (NBD) handle. NBD served as an excellent surrogate for the similarly structured DNP hapten, and it enabled quantification of relative metabolic labeling levels.
Labeling levels were assessed with Bacillus subtilis, which was chosen due to its extensive similarity to pathogenic bacteria.37 Consistent with our prior studies, treatment of B. subtilis cells with 1 led to a 20-fold increase in fluorescence relative to untreated cells (Figure 2). To assess the possibility that d,d-carboxypeptidase may reduce the valency of the unnatural cell surface handle, 1 was incubated with a genetically modified strain of B. subtilis devoid of dacA (d,d-carboxypeptidase). Cellular fluorescence in B. subtilis ΔdacA variants increased ∼7.5-fold relative to wild type cells, thus indicating epitopes installed on the terminal position via unnatural single amino acids are susceptible to removal by endogenous enzymes. Removal by d,d-transpeptidase could not be evaluated because the complete knockout of these genes is lethal to bacteria.
Figure 2.

(A) Chemical structures of NBD conjugates. (B) Flow cytometry analysis of B. subtilis cells when incubated overnight with 1 mM of NBD conjugates. Data are represented as the mean ± SD (n = 3).
In an effort to minimize enzymatic removal of unnatural modifications, we sought a strategy that installed epitopes onto the fourth position within the stem peptide via the MurF dipeptide pathway (Figure S2). Dipeptide 2 was synthesized to mimic the lysine side chain in the single amino acid metabolic labeling strategy (Figure 2A). Treatment of B. subtilis cells with dipeptide 2 led to an insignificant increase over untreated cells. From these results, it became apparent that side-chain size may have contributed to drastically reduced labeling levels. We proceeded to synthesize a similar dipeptide using d-2,3-diaminopropionic acid (3) with the aim of minimizing the overall size of the amino acid side chain. However, flow cytometry analysis of B. subtilis bacteria treated with dipeptides 2 and 3 revealed minimal increases in labeling. (Figure 2B and Figure S3). Interestingly, it is apparent from these results that various levels of tolerance for unnatural cell wall analogues exists depending on the entry point into the peptidoglycan biosynthesis pathway. We recently demonstrated that side-chain structural features control incorporation efficiency of single d-amino acids.38 Finally, we synthesized a dipeptide conjugated with a DNP hapten analogous to dipeptides 2 and 3 and observed no discernible anti-DNP antibody recruitment to the cell surface (data not shown). With these early results in hand, we set out to redesign a dipeptide-based labeling strategy that exploited the reduced removal at the fourth position on the stem peptide but improved incorporation efficiency.
The lack of haptens displaying dipeptide incorporation prompted us to develop a two-step labeling method. In this strategy, a small chemically reactive side chain can be integrated into the dipeptide to reduce steric hindrance. Upon epitope display on the bacterial cell surface, cells are treated with a complementary reaction handle modified with the DNP hapten (Figure 3A). The choice of DNP was based on its small size and an endogenous reservoir of anti-DNP IgG antibodies in human serum within the general population.39−44 For a model reaction scheme, we used sulfhydryl and maleimide handles as complementary reactive partners. Thiol–maleimide chemistry provided a moderately selective and facile method to monitor modification of bacterial cell surfaces. We envisioned two distinct benefits of decoupling hapten conjugation from MurF-mediated metabolic labeling. First, incorporation of the dipeptide is expected to increase due to better mimicking of the endogenous d-Ala-d-Ala substrate. Second, in a two-step strategy, it is possible to install linkers of lengths that may not be enzymatically tolerated but would provide improved cell surface hapten protrusion. Haptens imbedded within the peptidoglycan become unavailable for interaction and binding with antibodies. By utilizing tethers modified with hapten, it becomes possible to rescue epitopes that may be sterically occluded within the mesh network of the peptidoglycan (Figure 3B).
Figure 3.

Antibody recruitment to bacterial surfaces. (A) Utilizing a dipeptide that contains a reactive chemical handle, bacterial peptidoglycan surfaces can be remodeled. Bacterial cells incubated in the presence of a hapten-containing linker can then react with the chemical handle to yield a hapten-modified bacterial cell surface that can recruit antibodies to the cell surface. (B) A “one-step” labeling strategy is limited by protrusion of haptens from the peptidoglycan and therefore generates lower levels of antibody opsonization. Utilizing a “two-step” dipeptide strategy in which the hapten is conjugated to a linker molecule allows for greater protrusion of the antibody recruitment molecule from the peptidoglycan, thus resulting in greater bacterial cell opsonization.
To test whether this two-step dipeptide strategy would provide greater cell surface modification, B. subtilis wild type cells were incubated in the presence of thiol -containing dipeptides and subsequently reacted with a maleimide-modified NBD fluorophore to quantify labeling levels (Figure 4A). We synthesized two cysteine-containing dipeptides, d-Cys-d-Ala (4) and d-Ala-d-Cys (5), to provide a thiol-reactive handle at the fourth or fifth position of the stem peptide, respectively. As we had predicted, cells treated with dipeptide 4 displayed higher levels of fluorescence relative to the dipeptide 5 and single amino acid d-Cys (6). These results illustrate that a properly designed dipeptide can lead to higher levels of surface remodeling compared to the single amino acid strategy (Figure 4B and Figure S3). Control dipeptides (Figure 4C) demonstrated stereochemistry to be critical in surface remodeling as treatment of cells with neither the diastereomer (7) nor the enantiomer (8) of dipeptide 4 leads to any significant increase in fluorescence compared to untreated cells, thus lending support to the MurF-based metabolic pathway.
Figure 4.

(A) Installation of thiol-containing amino acids followed by reaction with maleimide fluorophores endows bacterial cell surface labeling through a two-step process. (B) Flow cytometry analysis of B. subtilis cells incubated overnight with 1 mM 4, 5, 6, 7, or 8 was followed by incubation with 50 μM FL0. Data are represented as the mean ± SD (n = 3). (C) Chemical structures sulfhydryl-containing dipeptides used for two-step labeling of bacterial surfaces are shown.
Having established that dipeptide 4 provided superior cell surface labeling relative to single d-amino acids, we sought to optimize the tether length connecting the stem peptide modification to the displayed epitopes. Cross-linking within the peptidoglycan layer creates pores that range from 5 to 25 nm in diameter, potentially reducing the recruitment of antibodies beyond surface-exposed haptens due to impeded permeation.45 We hypothesized that elongated tethers may facilitate antibody recruitment from stem peptides beyond the immediate surface. We constructed a panel of fluorophore–linker (FL) conjugates containing both a maleimide handle and an NBD handle with the goal of determining permeability of tethers within the peptidoglycan (Figure 5A). These molecular tethers were built with a central polar/flexible linker composed of various units of (Gly-Gly-Ser) (GGS linker), where n = 0–4 (Figure 5B). To evaluate the effect of tether length, B. subtilis cells were incubated overnight in the presence of dipeptide 4, subsequently labeled with each variant (FL0–FL4) and analyzed for cellular fluorescence via flow cytometry. It is evident that FL0 and FL1 labeled bacterial cell surfaces to similar levels, an indication that smaller tethers can readily penetrate the peptidoglycan pores. Elongation of the tethers revealed diminished labeling efficiency in a length-dependent manner, with the longest tether FL4 resulting in approximately half of the fluorescence levels of FL0 (Figure 5C). Although these results serve as an integral part for the design of hapten molecular tethers, we also realize that they can potentially be a facile assay for peptidoglycan permeability. We anticipate that based on the mechanism of incorporation, d-Cys epitopes are evenly dispersed throughout the entire peptidoglycan. Therefore, labeling levels can be interpreted as a readout on the permeability of biomolecules from the extracellular space into the peptidoglycan network.
Figure 5.

(A) Chemical structure of fluorescent-linkers; (B) predicted length of fluorescent linkers; (C) flow cytometry analysis of B. subtilis cells incubated overnight with 1 mM dipeptide 4, followed by incubation with 50 μM FL0-4. Data are represented as the mean ± SD (n = 3).
Finally, we set out to demonstrate this strategy could be used to induce a recruitment of endogenous anti-DNP antibodies to the bacterial cell surface. The series FL0–FL4 served to inform us about biomolecule permeation through the cell surface. However, labeling levels were measured on the basis of total cellular fluorescence and may not necessarily reflect epitope availability for antibody binding. To optimize labeling levels concomitantly with availability to antibody binding, we built a second series of GGS-based tethered molecules. For this DNP-linker (DL) series, a DNP antigen was installed on the N-terminus in place of NBD (Figure 6A). B. subtilis cells incubated with dipeptide 4 were subsequently treated with the various DNP linker conjugates and incubated in the presence of FITC-labeled anti-DNP IgG antibodies. Interestingly, flow cytometry analysis showed a sharp contrast in opsonization levels compared with the FL series (Figure 6B). Cells treated with DL1 led to the highest levels of anti-DNP antibody recruitment. On the basis of the extensive structural similarities between the FL and DL series, it is reasonable to expect that bacterial cell surfaces were remodeled with DL0 to a similar level as DL1. Yet, DL0-treated cells led to >2-fold lower recruitment levels than DL1. Likewise, the much longer DL5 led to similar antibody recruitment levels as the shorter DL0. These results illustrate that to optimize bacterial cell opsonization using our strategy, it is necessary to balance tether permeation with hapten binding on the cell surface. Most importantly, we showed that by decoupling the MurF-mediated metabolic labeling from hapten conjugation it may be possible to increase the availability of haptens on the cell surface for antibody binding by adjusting the tether length.
Figure 6.
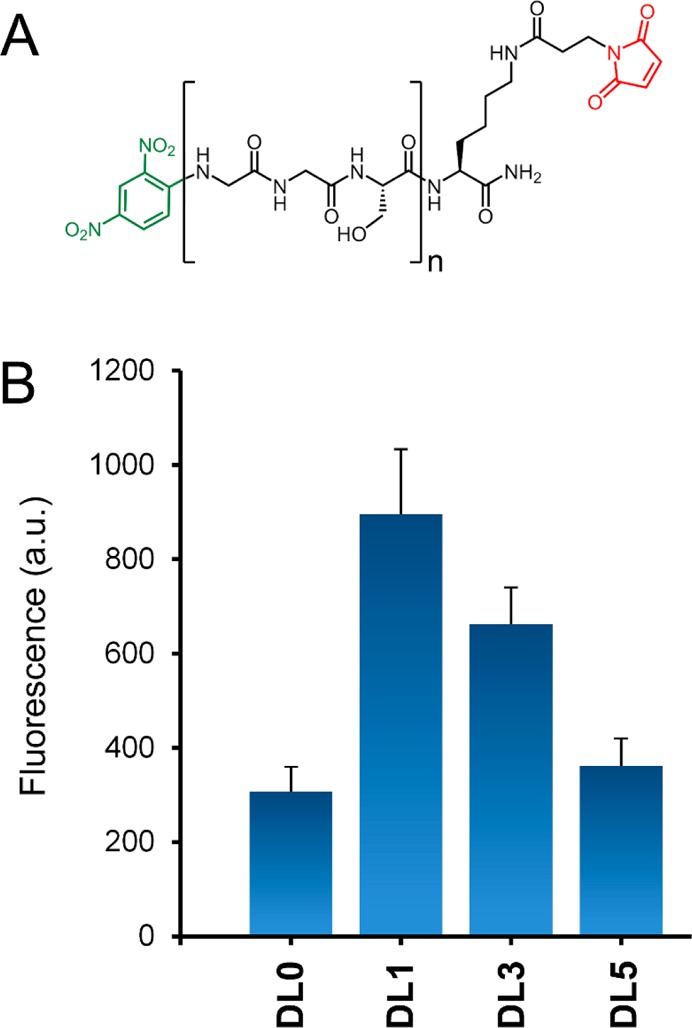
(A) Chemical structure of DNP-linker (DL) series; (B) flow cytometry analysis of B. subtilis cells incubated overnight with 1 mM dipeptide 4, 50 μM DNP-linker, and Alexa Fluor 488 anti-DNP IgG antibody displaying an increase in opsonization. Data are represented as the mean ± SD (n = 3).
In conclusion, we have demonstrated that a dipeptide-based metabolic labeling strategy led to higher levels of remodeling compared to single d-amino acids. MurF promiscuity was exploited to enable intracellular hijacking of the peptidoglycan biosynthetic pathway. By designing and synthesizing a dipeptide to install modifications onto the fourth position of the stem peptide, it became possible to improve the retention of unnatural surface-bound epitopes. Subsequent modification with heterobifunctional tethers of various lengths results in improved display of epitopes for extracellular interactions. Through the evaluation of various fluorescently tagged tethers, pore size constraints for remodeling bacterial peptidoglycan were established. On the basis of tether length restrictions, we were able to successfully install haptens onto bacterial cell surfaces and induce high levels of bacterial cell opsonization. We anticipate that based on the strategy disclosed here, it should be possible to use unnatural dipeptides displaying bio-orthogonal handles to label bacterial cell surfaces for imaging and therapeutic applications. More importantly, we propose that the combination of elevated metabolic labeling levels and the use of tethers to endow bacterial cells with readily accessible epitopes could provide the basis for improved immunomodulation strategies to combat bacterial infections.
Methods
Materials
Amino acids were purchased from Chem-Impex. Antibody reagents were purchased from Life-Technologies. All other organic reagents were purchased from Fisher Scientific and used without further purification. B. subtilis ΔdacA and B. subtilis NCIB3610 were the strains of bacteria used for experiments.
Cell Surface Modification
B. subtilis NCIB 3610 and B. subtilis ΔdacA were grown in LB medium at 37 °C with shaking. Cells were incubated overnight at 37 °C with shaking in LB medium that was supplemented with 1 mM compound 1, 2, or 3 and protected from light. The following morning, the cells were washed with phosphate-buffered saline (PBS) three times. A 4% formaldehyde fixation solution was prepared in PBS and used to fixate the cells prior to performing flow cytometry analysis and fluorescence microscopy. Flow cytometry analysis was performed with a BD Facs Canto II (BD Biosciences, San Jose, CA, USA) equipped with a 488 nm argon laser and a 530/30 band-pass filter (FL1). A minimum of 10,000 events were collected for each data set. The data were analyzed using FACSDiva version 6.1.1 software. The fluorescence data are expressed as mean arbitrary fluorescence units and were gated to include all healthy bacteria. Fluorescence microscopy was conducted using a Nikon Eclipse TE-2000-U microscope with a 480/30 excitation band-pass filter and a 530/30 emission band-pass filter at 100× magnification.
Two-Step Dipeptide Cell Surface Modification
B. subtilis NCIB 3610 and B. subtilis ΔdacA were grown in LB medium at 37 °C with shaking. These cells were then incubated overnight at 37 °C with shaking in LB medium that was supplemented with 1 mM compounds 4–8. The following morning the cells were washed three times with 5 mM dithiothreitol (DTT) to reverse any thiol oxidation that may have occurred. The cells were then subsequently washed five times with PBS to remove any residual DTT. Following PBS washing, the cells were then incubated with 50 μM FL0 for 30 min at 37 °C and protected from light. The cells were then fixated in a 4% formaldehyde solution prior to performing flow cytometry analysis and fluorescence microscopy as previously stated.
Peptidoglycan Pore Size Exclusion Determination
B. subtilis NCIB 3610 was grown in LB medium at 37 °C with shaking. These cells were then incubated overnight at 37 °C with shaking in LB medium that was supplemented with 1 mM d-Cys-d-Ala. The following morning the cells were washed three times with 5 mM DTT to reverse any thiol oxidation that may have occurred. The cells were then subsequently washed five times with PBS to remove any residual DTT. Following washing, the cells were then incubated with 50 μM FL0–FL4 for 30 min at 37 °C and protected from light. The cells were washed three more times with PBS and were then fixated in a 4% formaldehyde solution prior to performing flow cytometry analysis as previously stated. Linker lengths were calculated using PyMol.
Anti-DNP IgG Antibody Bacterial Cell Opsonization
B. subtilis ΔdacA was grown in LB medium at 37 °C with shaking. These cells were then incubated overnight at 37 °C with shaking in LB medium that was supplemented with 1 mM d-Cys-d-Ala. The following morning the cells were washed three times with 5 mM DTT to reverse any thiol oxidation that may have occurred. The cells were then subsequently washed five times with PBS to remove any residual DTT. Following washing, the cells were then incubated with 50 μM DL0–DL5 for 30 min at 37 °C and protected from light. The cells were then incubated for 1 h at 37 °C in 100 μL of a PBS solution containing 10% fetal bovine serum (FBS) and 0.02 mg/mL Alexa Fluor 488 conjugated rabbit anti-DNP IgG fraction KLH (Life-Technologies, catalog no. A11097). The cells were analyzed for bacterial cell opsonization using flow cytometry as previously stated.
Acknowledgments
This study was supported by Lehigh University (M.P.). We thank D. Kearns (Indiana Univesity) for the kind gifts of B. subtilis ΔdacA and B. subtilis NCIB 3610.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.6b00007.
Additional figures and experimental details (synthesis of small molecules) (PDF)
Author Contributions
∥ J.M.F. and S.E.P. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Davies J.; Davies D. (2010) Origins and Evolution of Antibiotic Resistance. Microbiol. Mol. Biol. Rev. 74, 417–433. 10.1128/MMBR.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham E. P.; Chain E. (1940) An Enzyme from Bacteria Able to Destroy Penicillin. Nature 146, 837. 10.1038/146837a0. [DOI] [PubMed] [Google Scholar]
- Payne D. J.; Gwynn M. N.; Holmes D. J.; Pompliano D. L. (2007) Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discovery 6, 29–40. 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- Levy S. B.; Marshall B. (2004) Antibacterial resistance worldwide: causes, challenges and responses. Nat. Med. 10, S122–S129. 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- Spellberg B.; Guidos R.; Gilbert D.; Bradley J.; Boucher H. W.; Scheld W. M.; Bartlett J. G.; Edwards J.; Amer I. D. S. (2008) The epidemic of antibiotic-resistant infections: a call to action for the medical community from the Infectious Diseases Society of America. Clin. Infect. Dis. 46, 155–164. 10.1086/524891. [DOI] [PubMed] [Google Scholar]
- Alanis A. J. (2005) Resistance to antibiotics: are we in the post-antibiotic era?. Arch. Med. Res. 36, 697–705. 10.1016/j.arcmed.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Carlet J.; Collignon P.; Goldmann D.; Goossens H.; Gyssens I. C.; Harbarth S.; Jarlier V.; Levy S. B.; N’Doye B.; Pittet D.; Richtmann R.; Seto W. H.; van der Meer J. W. M.; Voss A. (2011) Society’s failure to protect a precious resource: antibiotics. Lancet 378, 369–371. 10.1016/S0140-6736(11)60401-7. [DOI] [PubMed] [Google Scholar]
- Walsh C. (2000) Molecular mechanisms that confer antibacterial drug resistance. Nature 406, 775–781. 10.1038/35021219. [DOI] [PubMed] [Google Scholar]
- Vollmer W.; Blanot D.; de Pedro M. A. (2008) Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 32, 149–167. 10.1111/j.1574-6976.2007.00094.x. [DOI] [PubMed] [Google Scholar]
- Lovering A. L.; Safadi S. S.; Strynadka N. C. J. (2012) Structural Perspective of Peptidoglycan Biosynthesis and Assembly. Annu. Rev. Biochem. 81, 451–478. 10.1146/annurev-biochem-061809-112742. [DOI] [PubMed] [Google Scholar]
- Hayhurst E. J.; Kailas L.; Hobbs J. K.; Foster S. J. (2008) Cell wall peptidoglycan architecture in Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 105, 14603–14608. 10.1073/pnas.0804138105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvage E.; Kerff F.; Terrak M.; Ayala J. A.; Charlier P. (2008) The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 32, 556–556. 10.1111/j.1574-6976.2008.00115.x. [DOI] [PubMed] [Google Scholar]
- Lock R. L.; Harry E. J. (2008) Cell-division inhibitors: new insights for future antibiotics. Nat. Rev. Drug Discovery 7, 324–338. 10.1038/nrd2510. [DOI] [PubMed] [Google Scholar]
- Cava F.; de Pedro M. A.; Lam H.; Davis B. M.; Waldor M. K. (2011) Distinct pathways for modification of the bacterial cell wall by non-canonical d-amino acids. EMBO J. 30, 3442–3453. 10.1038/emboj.2011.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds P. E.; Courvalin P. (2005) Vancomycin resistance in enterococci due to synthesis of precursors terminating in d-alanyl-d-serine. Antimicrob. Agents Chemother. 49, 21–25. 10.1128/AAC.49.1.21-25.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez L.; Espaillat A.; Hermoso J. A.; de Pedro M. A.; Cava F. (2014) Peptidoglycan remodeling by the coordinated action of multispecific enzymes. Microb. Drug Resist. 20, 190–198. 10.1089/mdr.2014.0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegrist M. S.; Whiteside S.; Jewett J. C.; Aditham A.; Cava F.; Bertozzi C. R. (2013) d-Amino Acid Chemical Reporters Reveal Peptidoglycan Dynamics of an Intracellular Pathogen. ACS Chem. Biol. 8, 500–505. 10.1021/cb3004995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuru E.; Hughes H. V.; Brown P. J.; Hall E.; Tekkam S.; Cava F.; de Pedro M. A.; Brun Y. V.; VanNieuwenhze M. S. (2012) In Situ Probing of Newly Synthesized Peptidoglycan in Live Bacteria with Fluorescent d-Amino Acids. Angew. Chem., Int. Ed. 51, 12519–12523. 10.1002/anie.201206749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebar M. D.; May J. M.; Meeske A. J.; Leiman S. A.; Lupoli T. J.; Tsukamoto H.; Losick R.; Rudner D. Z.; Walker S.; Kahne D. (2014) Reconstitution of Peptidoglycan Cross-Linking Leads to Improved Fluorescent Probes of Cell Wall Synthesis. J. Am. Chem. Soc. 136, 10874–10877. 10.1021/ja505668f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuru E.; Tekkam S.; Hall E.; Brun Y. V.; Van Nieuwenhze M. S. (2015) Synthesis of fluorescent d-amino acids and their use for probing peptidoglycan synthesis and bacterial growth in situ. Nat. Protoc. 10, 33–52. 10.1038/nprot.2014.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh P.; Siegrist M. S.; Cullen A. J.; Bertozzi C. R. (2014) Imaging bacterial peptidoglycan with near-infrared fluorogenic azide probes. Proc. Natl. Acad. Sci. U. S. A. 111, 5456–5461. 10.1073/pnas.1322727111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidgeon S. E.; Fura J. M.; Leon W.; Birabaharan M.; Vezenov D.; Pires M. M. (2015) Metabolic Profiling of Bacteria by Unnatural C-terminated d-Amino Acids. Angew. Chem., Int. Ed. 54, 6158–6162. 10.1002/anie.201409927. [DOI] [PubMed] [Google Scholar]
- Pidgeon S. E.; Pires M. M. (2015) Metabolic remodeling of bacterial surfaces via tetrazine ligations. Chem. Commun. 51, 10330–10333. 10.1039/C5CC01693B. [DOI] [PubMed] [Google Scholar]
- Lupoli T. J.; Tsukamoto H.; Doud E. H.; Wang T. S. A.; Walker S.; Kahne D. (2011) Transpeptidase-Mediated Incorporation of d-Amino Acids into Bacterial Peptidoglycan. J. Am. Chem. Soc. 133, 10748–10751. 10.1021/ja2040656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweppe D. K.; Harding C.; Chavez J. D.; Wu X.; Ramage E.; Singh P. K.; Manoil C.; Bruce J. E. (2015) Host-Microbe Protein Interactions during Bacterial Infection. Chem. Biol. 22, 1521–1530. 10.1016/j.chembiol.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kershaw M. H.; Westwood J. A.; Darcy P. K. (2013) Gene-engineered T cells for cancer therapy. Nat. Rev. Cancer 13, 525–541. 10.1038/nrc3565. [DOI] [PubMed] [Google Scholar]
- Pardoll D. M. (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264. 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubrovska A.; Kim C.; Elliott J.; Shen W. J.; Kuo T. H.; Koo D. I.; Li C.; Tuntland T.; Chang J.; Groessl T.; Wu X.; Gorney V.; Ramirez-Montagut T.; Spiegel D. A.; Cho C. Y.; Schultz P. G. (2011) A Chemically Induced Vaccine Strategy for Prostate Cancer. ACS Chem. Biol. 6, 1223–1231. 10.1021/cb200222s. [DOI] [PubMed] [Google Scholar]
- Jakobsche C. E.; Parker C. G.; Tao R. N.; Kolesnikova M. D.; Douglass E. F.; Spiegel D. A. (2013) Exploring Binding and Effector Functions of Natural Human Antibodies Using Synthetic Immunomodulators. ACS Chem. Biol. 8, 2404–2411. 10.1021/cb4004942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; You F.; Vlahov I.; Westrick E.; Fan M.; Low P. S.; Leamon C. P. (2007) Folate-targeted dinitrophenyl hapten immunotherapy: effect of linker chemistry on antitumor activity and allergic potential. Mol. Pharmaceutics 4, 695–706. 10.1021/mp070050b. [DOI] [PubMed] [Google Scholar]
- Carlson C. B.; Mowery P.; Owen R. M.; Dykhuizen E. C.; Kiessling L. L. (2007) Selective tumor cell targeting using low-affinity, multivalent interactions. ACS Chem. Biol. 2, 119–127. 10.1021/cb6003788. [DOI] [PubMed] [Google Scholar]
- Fura J. M.; Sabulski M. J.; Pires M. M. (2014) d-Amino Acid Mediated Recruitment of Endogenous Antibodies to Bacterial Surfaces. ACS Chem. Biol. 9, 1480–1489. 10.1021/cb5002685. [DOI] [PubMed] [Google Scholar]
- Fura J. M.; Pires M. M. (2015) d-Amino Carboxamide-Based Recruitment of Dinitrophenol Antibodies to Bacterial Surfaces via Peptidoglycan Remodeling. Biopolymers 104, 351–359. 10.1002/bip.22618. [DOI] [PubMed] [Google Scholar]
- Typas A.; Banzhaf M.; Gross C. A.; Vollmer W. (2012) From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat. Rev. Microbiol. 10, 123–136. 10.1038/nrmicro2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popham D. L.; Gilmore M. E.; Setlow P. (1999) Roles of low-molecular-weight penicillin-binding proteins in Bacillus subtilis spore peptidoglycan synthesis and spore properties. J. Bacteriol. 181, 126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liechti G. W.; Kuru E.; Hall E.; Kalinda A.; Brun Y. V.; VanNieuwenhze M.; Maurelli A. T. (2014) A new metabolic cell-wall labelling method reveals peptidoglycan in Chlamydia trachomatis. Nature 506, 507–510. 10.1038/nature12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenshein A. L., Hoch J. A., and Losick R. (1993) Bacillus subtilis and Other Gram-Positive Bacteria: Biochemistry, Physiology, And Molecular Genetics, American Society for Microbiology, Washington, DC, USA. [Google Scholar]
- Fura J. M., Kearns D., Pires M. M.. d-Amino Acid Probes for Penicillin Binding Protein-Based Bacterial Surface Labeling. J. Biol. Chem. 2015, DOI: 10.1074/jbc.M115.683342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farah F. S. (1973) Natural antibodies specific to the 2,4-dinitrophenyl group. Immunology 25, 217–226. [PMC free article] [PubMed] [Google Scholar]
- Murelli R. P.; Zhang A. X.; Michel J.; Jorgensen W. L.; Spiegel D. A. (2009) Chemical control over immune recognition: a class of antibody-recruiting small molecules that target prostate cancer. J. Am. Chem. Soc. 131, 17090–17092. 10.1021/ja906844e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker C. G.; Domaoal R. A.; Anderson K. S.; Spiegel D. A. (2009) An antibody-recruiting small molecule that targets HIV gp120. J. Am. Chem. Soc. 131, 16392–16394. 10.1021/ja9057647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karjalainen K.; Makela O. (1976) Concentrations of three hapten-binding immunoglobulins in pooled normal human serum. Eur. J. Immunol. 6, 88–93. 10.1002/eji.1830060204. [DOI] [PubMed] [Google Scholar]
- Jakobsche C. E.; Parker C. G.; Tao R. N.; Kolesnikova M. D.; Douglass E. F. Jr.; Spiegel D. A. (2013) Exploring binding and effector functions of natural human antibodies using synthetic immunomodulators. ACS Chem. Biol. 8, 2404–2411. 10.1021/cb4004942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan R. T.; Hudon J.; Hank J. A.; Sondel P. M.; Kiessling L. L. (2014) Rhamnose glycoconjugates for the recruitment of endogenous anti-carbohydrate antibodies to tumor cells. ChemBioChem 15, 1393–1398. 10.1002/cbic.201402019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meroueh S. O.; Bencze K. Z.; Hesek D.; Lee M.; Fisher J. F.; Stemmler T. L.; Mobashery S. (2006) Three-dimensional structure of the bacterial cell wall peptidoglycan. Proc. Natl. Acad. Sci. U. S. A. 103, 4404–4409. 10.1073/pnas.0510182103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.