

Abstract
Introduction
In bone and joint infections (BJIs), bacterial toxins are major virulence factors: Panton—Valentine leukocidin (PVL) expression leads to severe local damage, including bone distortion and abscesses, while α-hemolysin (Hla) production is associated with severe sepsis-related mortality. Recently, other toxins, namely phenol-soluble modulins (PSMs) expressed by community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA) strain USA300 (LAC WT) were shown to have ex vivo intracellular cytotoxic activity after S. aureus invasion of osteoblasts, but their in vivo contribution in a relatively PVL-sensitive osteomyelitis model remains poorly elucidated.
Materials and Methods
We compared the outcomes of experimental rabbit osteomyelitises induced with pvl+hla+psms+ LAC WT and its isogenic Δpsm derivatives (LAC Δpsmα and LAC Δpsmαβhld) using an inoculum of 3 × 108 CFUs. Mortality, hematogenous spread (blood culture, spleen and kidney), lung and bone involvements were assessed in two groups (non-survivors of severe sepsis and survivors sacrificed on day (D) 14).
Results
Severe sepsis-related mortality tended to be lower for Δpsm derivatives (Kaplan—Meier curves, P = .06). Non-survivors’ bone LAC-Δpsmα (6.9 log10 CFUs/g of bone, P = .04) or -Δpsmαβhld (6.86 log10 CFUs/g of bone, P = .014) densities were significantly higher than LAC WT (6.43 log10 CFUs/g of bone). Conversely, lung Δpsmαβhld CFUs were significantly lower than LAC WT (P = .04). LAC Δpsmα, Δpsmαβhld and WT induced similar bone damage in D14 survivors, with comparable bacterial densities (respectively: 5.89, 5.91, and 6.15 log10 CFUs/g of bone). Meanwhile, pulmonary histological scores of inflammation were significantly higher for LAC Δpsmα- and Δpsmαβhld-infected rabbits compared to LAC WT (P = .04 and .01, respectively) but with comparable lung bacterial densities.
Conclusion
Our experimental results showed that deactivating PSM peptides significantly limited bacterial dissemination from bone during the early phase of infection, but did not affect local severity of USA300 rabbit osteomyelitis.
Introduction
Since 1990, extensive spread in the United States of the community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA) USA300 clone has been responsible for severe infections, including bone and joint infections (BJIs), especially in children [1]. BJIs represent up to 38% of pediatric CA-MRSA infections in the United States [2]. The global severity of CA-MRSA BJIs has been linked to local musculoskeletal involvement, including extraosseous abscesses, and the frequent need for surgical management [3,4], and systemic complications including severe sepsis and dissemination to the lungs [5,6].
Numerous CA-MRSA virulence factors have been identified [7], some of which appeared to play a specific role in the course of osteomyelitis. Panton—Valentine leukocidin (PVL), a phage-borne pore-forming toxin highly prevalent in CA-MRSA, was able to induce local complications, e.g. bone deformation and muscular abscesses in a rabbit osteomyelitis model [8]. PVL involvement in this context was further supported by the epidemiological association of PVL expression and clinical methicillin-susceptible S. aureus BJI severity [1]. CA-MRSA overexpression of another pore-forming toxin, α-hemolysin (Hla), was also shown to contribute to BJI pathogenesis [9]. We previously showed that Hla was associated with systemic complications, like severe sepsis-related mortality in CA-MRSA rabbit osteomyelitis [10].
In addition to PVL and Hla, secreted peptides called phenol-soluble modulins (PSMs) have been identified as key virulence factors that are also strongly expressed in staphylococci and CA-MRSA [11]. PSM-encoding genes appear in three distinct loci on the S. aureus chromosome. PSMs encoded by the first two loci have been designated PSMα and β. The third PSM locus encodes the δ-toxin; its open-reading frame is part of the RNAIII effector of the staphylococcal accessory-gene regulator (agr), a major two-component system coupled to a density-sensing cassette controlling the expression of most S. aureus virulence factors. All PSM-encoding genes are under agr control, either through the AgrA-mediated regulation pathway for psmα and β, or as a consequence of the co-transcription with RNAIII, as for hld [12]. Thus, PSM expression is tightly coupled with staphylococcal quorum sensing through agr.
Several PSM biological functions impact pathogenesis and, possibly, the course of S. aureus osteomyelitis. PSMs are small peptides with amphipathic properties, allowing them to destabilize lipid bilayers at high concentrations. This activity has been linked to receptor-independent cytotoxicity to host cells, including neutrophils and osteoblasts, the bone-forming cells [13,14]. Moreover, receptor-dependent proinflammatory activation of neutrophils by PSMs was found, resulting from PSM detection by the neutrophil formyl-peptide receptor 2 (FRP2) [15]. Finally, PSMs can assemble into amyloid-like fibrillae, which contribute to stabilizing staphylococcal biofilms [16,17] and induce a tolerogenic phenotype in dendritic cells, contributing to bacterial interference [12] and cell-cycle disruption [18]. These cytotoxic, proinflammatory and biofilm-enhancing properties could suggest PSM involvement in the in vivo outcome of osteomyelitis.
In previous in vivo studies on PSMs using a model of skin-and-soft-tissue infection, the LAC psmα-deleted strain, but not the other psm-deleted strains, was significantly less able to cause skin lesions in mice [11]. Also, Kobayashi et al demonstrated that PSMα and Hla contributed to the pathogenesis of USA300 skin infections in rabbits [19], whereas PSMα peptides had no impact in a PVL-negative ST72 CA-MRSA strain in a mouse model of skin infection [20]. Moreover, PSMs have been shown to facilitate dissemination from an infected catheter in a mouse model of biofilm-associated infection [21]. Furthermore, in a rabbit model of experimental endocarditis, Spaulding et al showed that deactivating PSMs delayed lethal sepsis but did not prevent mortality or valve lesions [22] and, thus, that they do not play a major role in infective endocarditis.
Concerning BJIs, PSMα expression was associated with extensive bone damage inducing the death of infected human osteoblasts in an ex vivo model of intracellular infection [14], whereas in a murine osteomyelitis model, PSMs enhanced cortical bone destruction [23]. Addressing in vivo PSM BJIs requires using a model sufficiently close to the human situation in terms of PVL susceptibility. In particular, murine models used in previous studies of PSM involvement in CA-MRSA osteomyelitis could not account for the PVL effect, because murine immune cells respond poorly to PVL [24], unlike those of rabbits that are highly PVL-sensitive [25]. Hence, disease outcomes as a function of PVL may differ between these species.
In this context, we sought to determine the contribution of PSMs to local and systemic osteomyelitis severity in a PVL-sensitive rabbit model of acute BJI, using the highly virulent USA300 CA-MRSA strain LAC, which expresses PSMs, Hla and PVL, and its isogenic derivatives lacking PSM expression.
Materials and Methods
We used the clinical S. aureus Los Angeles County wild-type strain (LAC-WT), and its isogenic Δpsm derivatives (LAC Δpsmα and LAC Δpsmαβhld, respectively), all kindly provided by Frank Deleo, as for our previous studies. The strains were created and originated in Dr. Michael Otto’s laboratory and were previously described [11].
The Δpsm derivatives were constructed via allelic replacement with a spectinomycin-resistance cassette of the psmα and psmβ operons, as previously described [26], and by disrupting codon usage within the hld gene inside RNAIII to disable δ-toxin. Technical difficulties and cost prevented us from verifying modifications/confirming plasmid transfer with a gene-recomplementation assay.
Microorganisms were stored at –80°C until use. Prior to the experiments, bacteria were cultured in casein hydrolysate and yeast extract medium (CCY) at 37°C with shaking for 18 h. After centrifugation, the supernatants were passed through 0.22-μm filters, and the pellets were washed and resuspended in phosphate-buffered saline (PBS). All inocula were quantified by optical density (OD), then serial dilutions were plated on tryptic soy agar (bioMérieux, Paris, France).
Norden’s method [27] was used to induce osteomyelitis in female New Zealand white rabbits, weighing between 2 and 3 kg, housed in individual cages with ad libitum access to food and water, in compliance with French legislation on animal experimentation and with the approval of the Animal Use Committee of Maisons-Alfort Veterinary School. Rabbits were anesthetized by intramuscular injection of 25 mg/kg each of ketamine (Virbac, Carros, France) and 2% Xylazine (Bayer Santé, Division Santé Animal, Loos, France). An 18-gauge needle was inserted percutaneously through the right tibial metaphysis into the medullary cavity to aspirate 0.4 mL of bone marrow. Infection was induced by direct injection of 0.1 mL of a sclerosing agent (3% sodium-tetradecyl sulfate), followed by 0.2 mL of inoculum (3 × 108 colony-forming units (CFUs)) and 0.1 mL of saline to rinse the syringe. Fentanyl-patch analgesia was given for 7 days following surgery.
Animals were assigned to receive either LAC Δpsmα (n = 24) or LAC Δpsmαβhld (n = 24) to evaluate the in vivo impact of PSMs. Nineteen LAC-WT—infected rabbits served as controls.
Macroscopic Appearance and Bacterial Densities in Bone and Lungs
Animals were monitored daily for general and local signs of osteomyelitis (mobility, leg appearance) and were weighed on inoculum and sacrifice days. Moribund animals (immobile, unable to be aroused from a recumbent position and unable to access food or water) were euthanized by rapid intravenous injection of pentobarbital. Otherwise, rabbits were observed until day (D)14 post-infection to assess PSM impact on the osteomyelitis time course.
Before death, venous blood was drawn for culture and serum samples were stored at –20°C for later antibody-profile determination.
On the day of death, lungs, right leg, spleen and kidneys were removed. Both lungs were weighed and visually examined. Left lungs were stored at –80°C until determination of bacterial densities and right lungs were embedded in paraffin for histological examination (scored 0–4: none, minimal, mild, moderate, severe, respectively) for diffusion, edema, congestion, hemorrhages, thrombi, inflammation, megakaryocytes, infarcts, abscesses, pleural involvement and bacterial density. Spleen and kidneys were crushed and cultured on blood agar to determine their infection status. Macroscopic appearance of the right leg was noted and photographed, and the upper third of the tibia was frozen for subsequent quantitative culture, as previously described [10].
Serum-Antibody Assay
Anti-PVL and -Hla antibodies were detected with specific enzyme-linked immunosorbent assays (ELISAs). Antibody levels are expressed as arbitrary units per mL (AU/mL), as previously described [10]. Anti-PSM antibodies could not be quantified.
Statistical Analyses
Percentages of hematogenous spread (positive blood, kidney and/or spleen cultures), bone deformations and abscesses were compared using Student’s t-test. Non-parametric Mann—Whitney U-tests were used to compare tibial bacterial counts and 2-way ANOVA with Tukey’s multiple comparisons tests for histological scores.
The Kaplan—Meier method was used to estimate survival mortality, with inoculation as time 0 and censoring at sacrifice on D14. Percentages of survivors were compared with the log-rank (Mantel—Cox) test (Fig 1).
Fig 1. Kaplan—Meier survival curves compared with the log-rank (Mantel—Cox) test.
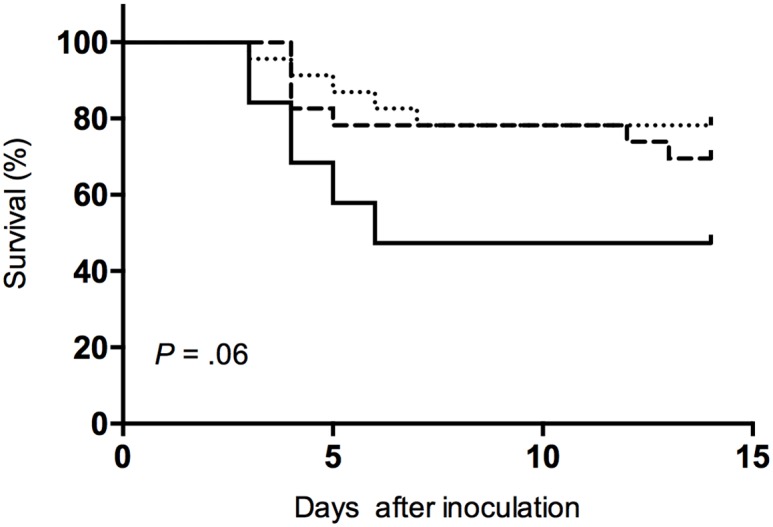
Severe sepsis-related mortality tended to be lower for Δpsmα (··) and Δpsmαβhld (- -) (P = .06) vs. LAC WT (–).
Survivors’ antibody-titer changes between D0 and D14 were analyzed with paired Welch’s t-test after log10 transformation.
Results
We inoculated 48 rabbits with 3 × 108 Δpsmα (n = 24) or Δpsmαβhld (n = 24) CFUs; the two that died immediately post-anesthesia (1 each in Δpsmα or Δpsmαβhld group) were excluded. Nineteen LAC-WT—inoculated rabbits served as controls.
D1 blood cultures were positive for 50% of the LAC-WT—infected rabbits tested vs. 65% of Δpsmα-infected (P = .47) and 61% of Δpsmαβhld-infected animals (P = .72).
Ten (52%) LAC-WT—infected rabbits died of severe sepsis with disseminated infection between D0 and D7 (median: 4 days) compared to five (22%) of the Δpsmα and seven (30%) of Δpsmαβhld groups (P = .03 and .11, respectively), with respective median survival of 5 and 4 days (P = .53, non-significant (NS)). The lack of significantly different mortality rates between Δpsmαβhld and WT groups, as opposed to the significantly different Δpsmα group, was likely attributable to random fluctuation and small sample size rather than to a real difference between Δpsmα and Δpsmαβhld groups. Indeed, Kaplan—Meier survival curves (Fig 1) showed a trend towards lower mortality of Δpsmα- and Δpsmαβhld-infected rabbits (P = .06).
Most non-survivors had positive spleen (80% of Δpsmα vs. 100% of Δpsmαβhld and LAC WT, NS) and kidney (100% of all groups) cultures. Macroscopic examination of all non-survivors’ lungs found red and congestive lesions, as in the LAC-WT group. Their lung histological scores confirmed the comparability of Δpsmα and Δpsmαβhld vs. LAC-WT infections. However, LAC-WT bacterial lung densities were higher (8.19 (interquartile range (IQR), 7.55–8.23) log10 CFUs/g than Δpsmαβhld (6.39 (IQR, 6.27–6.48) log10 CFUs/g P = .04), but comparable to Δpsmα (6.79 (IQR 5.62–7.10) log10 CFUs/g, P = .25).
All non-survivors had infected bones, with median bacterial densities of 6.9 (IQR 6.85–6.96) log10 Δpsmα CFUs/g and 6.86 (IQR, 6.81–7.38) Δpsmαβhld log10 CFUs/g vs. 6.43 (IQR, 6.29–6.58) log10 LAC WT CFUs/g of bone (P = .04 and .014, respectively) (Fig 2A). As for LAC-WT—infected rabbits, rare microabscesses were found at the inoculation site (0% Δpsmα, 20% Δpsmαβhld vs. 20% LAC WT, NS) but no cortical deformation (NS).
Fig 2. Comparisons of osteomyelitis parameters observed in Δpsmα (●○)-, Δpsmαβhld ▲△)- and with LAC-WT (◆◇)–infected non-survivors (black) and D14 survivors (white).
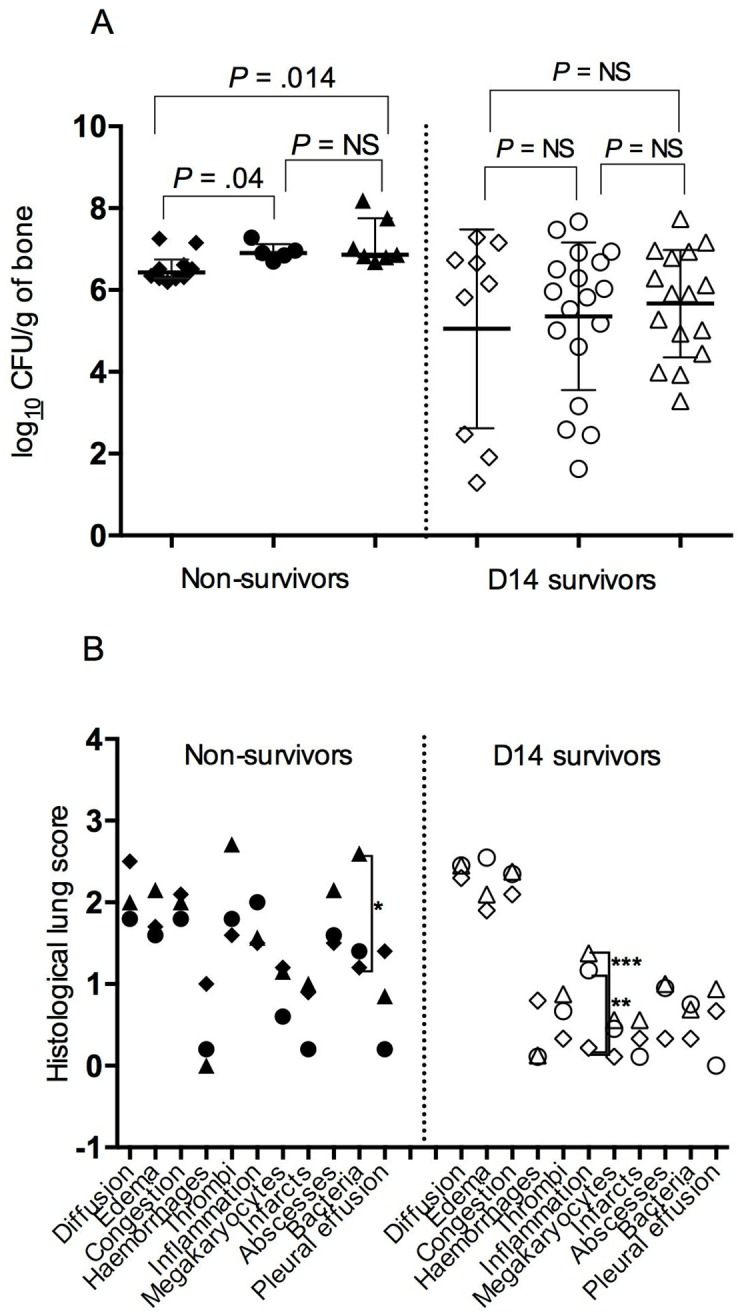
(A) Median bacterial bone densities, expressed in log10 CFUs/g of bone. The Δpsmα- or Δpsmαβhld-infected non-survivors differed significantly from those infected with LAC WT. (B) Mean histological scores (0–4: none, minimal, mild, moderate, severe) for lung involvement. The bacterial densities in Δpsmαβhld-infected non-survivors differed significantly from those infected with LAC WT (*P = .017), whereas inflammation was more severe in Δpsmαβhld- and Δpsmα-infected D14 survivors than those infected with LAC WT (**P = .04 and ***.01, respectively).
Survivors were sacrificed on D14 to evaluate bones and organs (Fig 3).
Fig 3. Macroscopic findings after challenge with a high inoculum of CA-MRSA USA300.

(A) Pulmonary hemorrhages demarcated by hyperemic regions in both lungs. (B) Abscesses in the left lung indicating disseminated infection. (C) White circled muscle abscess in the right leg. (D) Bone marrow filled with pus indicating osteomyelitis. (E) Splenomegaly with necrosis observed after disseminated sepsis.
D14 Δpsmα- and Δpsmαβhld-infected survivors did not differ from LAC-WT—inoculated rabbits for hematogenous dissemination: 17%, 19% vs. 11% spleen-positive cultures (NS) and 34%, 44% vs. 11% positive kidney cultures (NS). Histological scores were comparable except for higher lung inflammation in Δpsmα (P = .04) and Δpsmαβhld (P = .01) than LAC-WT infections (Fig 2B), while mean (±SD) lung bacterial densities were comparable for the three groups (2.31 ± 1.04, 2.21 ± 0.39 vs. 2.03 ± 1.18 log10 CFUs/g, respectively; NS).
Unexpectedly, Δpsmα and Δpsmαβhld vs. LAC-WT strains, respectively, induced similar bone deformities (39% and 43% vs. 67%; P = .73 and .41), muscle abscesses (83% and 75% vs. 89%; NS), and bone bacterial density (5.89 (IQR, 4.71–6.63) log10 CFUs/g and 5.91 (IQR, 4.82–6.83) log10 CFUs/g) vs. (6.15 (IQR, 2.47–6.73) log10 CFUs/g) (P = .95 and .82) (Fig 2A).
D14 survivor’s titers were significantly higher than on D0 for anti-PVL (6.4-fold increase, 95% CI 2.9, 14.1; P = 3.5 × 10−5, paired t-test of log10 values) and anti-Hla antibodies (41.3-fold increase, 95% CI 18.1, 96.8; P = 2.9 × 10−10), thereby confirming a significant immune response towards the two toxins. Survivors’ and non-survivors’ D0 anti-PVL and anti-Hla antibody titers were similar (Fig 4).
Fig 4. Serum anti-PVL—and -Hla—antibody titers in a rabbit model of S. aureus LAC experimental osteomyelitis.

Survivors’ and non-survivors’ Initial anti-PVL (A) and -Hla titers (C) did not differ significantly. Survivors had significantly higher anti-PVL (B) and -Hla (D) antibody levels between D0 (inoculation) and D14 (sacrifice) (P < .001). Welch’s t-test analyses of paired (B, D) or unpaired (A, C) log10 values.
Discussion
Our experimental model closely reproduces features of severe acute osteomyelitis seen in children with about half of the LAC-WT—infected rabbits dying within 7 days of severe sepsis, bacteremia, and high bone and lung bacterial densities associated with histological lung lesions, while D14 survivors developed severe bone infections with deformation and abscesses.
Our main results demonstrated that, during the early first phase of acute disseminated severe sepsis, deactivating PSMs tended to limit sepsis-attributable mortality, albeit not significantly. Δpsmαβhld-infected non-survivors had more bone CFUs than LAC-WT—infected non-survivors. In contrast, the former non-survivors had lower lung CFUs. This result can appear to be inconsistent with the bacterial lung histological score. However, bacterial enumeration by CFU counting is more reliable because (i) based on a larger sample size (tissue homogenate of a large piece of tissue) and (ii) assessing living but not dead cells.
These findings agree with the previously described role of PSMs in facilitating bacterial dissemination from an infected catheter in a mouse model [21, 28], a biofilm-associated infection like BJI. Conversely, during the second phase of subacute osteomyelitis (D14), deactivating PSMs did not modify bone CFUs, abscesses or deformities, in contrast to what could have been anticipated based on previous in vitro and ex vivo observations of PSMs’ osteoblast cytotoxicities [14].
The impact of PSMs (mainly PSMα) during the early phase of severe sepsis could be due to elevated production of agr-dependent toxins, including Hla and PSMs [29], especially by the LAC-WT strain. Although PSM deletion resulted in lower mortality, the effect was not significant, in contrast to previous observations with Hla [10].
Alternatively, the absence of PSMs’ effect on bone damage in D14 survivors could be explained by two hypotheses. First, agr might be less expressed during this late phase of osteomyelitis, considered a localized biofilm infection [29]. That possibility underlines the need to study the impact of virulence factors in a subacute osteomyelitis model (D14) with long-term animal survival [14]. Second, a potential PSM effect could have been masked by the impact of PVL, which plays a major role by enhancing rapid local spread of rabbit osteomyelitis with extraosseous infection extension, especially muscle abscesses [8], or by other toxins, e.g. Hla, which has been associated with severe sepsis-related mortality [10]. PSMs are by far the most abundant protein secreted by S. aureus (70–80%), much more than Hla or PVL [30]. Our results do not exclude a possible PSM effect on osteomyelitis caused by PVL-negative S. aureus isolates. A double-mutant strain Δpvl–Δpsmαβhld would have helped test this hypothesis, but we were unsuccessful in constructing one.
Our results do not corroborate those of Cassat et al, who showed that PSMα significantly limited bone remodeling in mice after creation of a cortical defect in the femur and local inoculation of 1 × 106 LAC-WT or Δpsmα1–4 CFUs [23]. However, it should be stressed that their model of post-traumatic localized osteomyelitis is far from what was encountered in children with primary CA-MRSA osteomyelitis [1,2,4,5,31].
Finally, because PSMs were shown to play a proinflammatory role by inducing neutrophil activation and cytokine release through the through human formyl-peptide receptor-2 (FPR2) pathway, the higher pulmonary inflammation scores for Δpsmα and Δpsmαβhld groups were also unexpected [15]. Our results cannot be explained by the comparable lung bacterial densities of Δpsmαβhld and LAC-WT groups. FPR2 is known to be expressed by several epithelial tissues including human, mouse and rat lungs. However, FPR expressed by rabbits is only 68% homologous to FPR2. Also, FPR2 can be triggered by different ligands and induce pro- or anti-inflammatory responses [32]. Therefore, we think the pulmonary inflammation seen in experimental rabbit osteomyelitis might involve other pathways and cytokines, which would support our results.
In conclusion, our results showed that deactivating PSMs prevented bacterial dissemination from bone during the early stage of the infection, but did not impact the local severity of rabbit USA300 osteomyelitis during the later stage. In agreement with our earlier work on PVL and Hla involvements in CA-MRSA osteomyelitis [8,10], these new findings confirmed that PVL expression in a susceptible host, e.g. rabbit, is the major cause of abscesses and bone damage, potentially masking PSM effects. Nevertheless, PSMs and their modulation by the staphylococcal PSM-degrading protease aureolysin might remain important bone-damaging factors in PVL-insensitive hosts [23] and perhaps in PVL-negative S. aureus. This strong dependence of staphylococcal osteomyelitis pathophysiology on host susceptibility factors and the infecting strain’s toxin-gene content are exemplary of the difficulties that must be overcome to improve our understanding of this disease. Beyond the pathogenic role of toxins, other bacterial factors related to quorum sensing and hypoxic response in poorly oxygenated bone tissue are likely equally important to establish bone infection, as recently demonstrated by comparisons of S. aureus genes essential to infection in abscess and osteomyelitis models [33,34]. Among the intricate array of staphylococcal virulence factors known to contribute to osteomyelitis so far, Hla is the only one that: measurably influences local and systemic outcomes, even in the presence of PVL [10]; is common to virtually all S. aureus lineages [35]; and is already targeted by a monoclonal antibody being tested in a clinical trial (study identifier NCT02296320) to treat S. aureus infection [36]. In this context, it seems reasonable to designate Hla as a more promising target than PSMs to treat severe CA-MRSA osteomyelitis, in agreement with recent conclusions drawn from a mice model of skin and soft tissue infection [20].
Acknowledgments
The authors thank the Raymond Poincaré University Hospital and Infectious Diseases Colleagues: Aurore Lagrange and Aurélien Dinh for their support and Janet Jacobson for editorial assistance.
Data Availability
All relevant data are within the paper.
Funding Statement
The authors have no support or funding to report.
References
- 1.1. Dohin B, Gillet Y, Kohler R, Lina G, Vandenesch F, Vanhems P, et al. Pediatric bone and joint infections caused by Panton-Valentine leukocidin-positive Staphylococcus aureus. Pediatr Infect Dis J. November 2007;26(11):1042–8. [DOI] [PubMed] [Google Scholar]
- 2.Iwamoto M, Mu Y, Lynfield R, Bulens SN, Nadle J, Aragon D, et al. Trends in invasive methicillin-resistant Staphylococcus aureus infections. Pediatrics. October 2013;132(4):e817–824. 10.1542/peds.2013-1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martínez-Aguilar G, Avalos-Mishaan A, Hulten K, Hammerman W, Mason EO, Kaplan SL. Community-acquired, methicillin-resistant and methicillin-susceptible Staphylococcus aureus musculoskeletal infections in children. Pediatr Infect Dis J. août 2004;23(8):701‑6. [DOI] [PubMed] [Google Scholar]
- 4.Arnold SR, Elias D, Buckingham SC, Thomas ED, Novais E, Arkader A, et al. Changing patterns of acute hematogenous osteomyelitis and septic arthritis: emergence of community-associated methicillin-resistant Staphylococcus aureus. J Pediatr Orthop. Déc 2006;26(6):703‑8. [DOI] [PubMed] [Google Scholar]
- 5.Gonzalez BE, Martinez-Aguilar G, Hulten KG, Hammerman WA, Coss-Bu J, Avalos-Mishaan A, et al. Severe Staphylococcal sepsis in adolescents in the era of community-acquired methicillin-resistant Staphylococcus aureus. Pediatrics. mars 2005;115(3):642‑8. [DOI] [PubMed] [Google Scholar]
- 6.Gonzalez BE, Hulten KG, Dishop MK, Lamberth LB, Hammerman WA, Mason EO, et al. Pulmonary manifestations in children with invasive community-acquired Staphylococcus aureus infection. Clin Infect Dis Off Publ Infect Dis Soc Am. 1 September 2005;41(5):583‑90. [DOI] [PubMed] [Google Scholar]
- 7.Otto M. Basis of virulence in community-associated methicillin-resistant Staphylococcus aureus. Annu Rev Microbiol. 2010;64:143‑62. 10.1146/annurev.micro.112408.134309 [DOI] [PubMed] [Google Scholar]
- 8.Crémieux A-C, Dumitrescu O, Lina G, Vallee C, Côté J-F, Muffat-Joly M, et al. Panton-valentine leukocidin enhances the severity of community-associated methicillin-resistant Staphylococcus aureus rabbit osteomyelitis. PloS One. 2009;4(9):e7204 10.1371/journal.pone.0007204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bubeck Wardenburg J, Bae T, Otto M, Deleo FR, Schneewind O. Poring over pores: alpha-hemolysin and Panton-Valentine leukocidin in Staphylococcus aureus pneumonia. Nat Med. Déc 2007;13(12):1405‑6. [DOI] [PubMed] [Google Scholar]
- 10.Crémieux A-C, Saleh-Mghir A, Danel C, Couzon F, Dumitrescu O, Lilin T, et al. α-Hemolysin, not Panton-Valentine leukocidin, impacts rabbit mortality from severe sepsis with methicillin-resistant Staphylococcus aureus osteomyelitis. J Infect Dis. 1 juin 2014;209(11):1773‑80. 10.1093/infdis/jit840 [DOI] [PubMed] [Google Scholar]
- 11.Wang R, Braughton KR, Kretschmer D, Bach T-HL, Queck SY, Li M, et al. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med. Déc 2007;13(12):1510‑4. [DOI] [PubMed] [Google Scholar]
- 12.Peschel A, Otto M. Phenol-soluble modulins and staphylococcal infection. Nat Rev Microbiol. October 2013;11(10):667‑73. 10.1038/nrmicro3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Surewaard BGJ, de Haas CJC, Vervoort F, Rigby KM, DeLeo FR, Otto M, et al. Staphylococcal alpha-phenol soluble modulins contribute to neutrophil lysis after phagocytosis. Cell Microbiol. août 2013;15(8):1427‑37. 10.1111/cmi.12130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rasigade J-P, Trouillet-Assant S, Ferry T, Diep BA, Sapin A, Lhoste Y, et al. PSMs of hypervirulent Staphylococcus aureus act as intracellular toxins that kill infected osteoblasts. PloS One. 2013;8(5):e63176 10.1371/journal.pone.0063176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kretschmer D, Gleske A-K, Rautenberg M, Wang R, Köberle M, Bohn E, et al. Human formyl peptide receptor 2 senses highly pathogenic Staphylococcus aureus. Cell Host Microbe. 25 juin 2010;7(6):463‑73. 10.1016/j.chom.2010.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwartz K, Syed AK, Stephenson RE, Rickard AH, Boles BR. Functional amyloids composed of phenol soluble modulins stabilize Staphylococcus aureus biofilms. PLoS Pathog. 2012;8(6):e1002744 10.1371/journal.ppat.1002744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Periasamy S, Chatterjee SS, Cheung GYC, Otto M. Phenol-soluble modulins in staphylococci: What are they originally for? Commun Integr Biol. 1 mai 2012;5(3):275‑7. 10.4161/cib.19420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deplanche M, Filho RAE-A, Alekseeva L, Ladier E, Jardin J, Henry G, et al. Phenol-soluble modulin α induces G2/M phase transition delay in eukaryotic HeLa cells. FASEB J Off Publ Fed Am Soc Exp Biol. mai 2015;29(5):1950‑9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi SD, Malachowa N, Whitney AR, Braughton KR, Gardner DJ, Long D, et al. Comparative analysis of USA300 virulence determinants in a rabbit model of skin and soft tissue infection. J Infect Dis. 15 September 2011;204(6):937‑41. 10.1093/infdis/jir441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Y, Yeh AJ, Cheung GYC, Villaruz AE, Tan VY, Joo H-S, et al. Basis of virulence in a Panton-Valentine leukocidin-negative community-associated methicillin-resistant Staphylococcus aureus strain. J Infect Dis. 1 févr 2015;211(3):472‑80. 10.1093/infdis/jiu462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Periasamy S, Joo H-S, Duong AC, Bach T-HL, Tan VY, Chatterjee SS, et al. How Staphylococcus aureus biofilms develop their characteristic structure. Proc Natl Acad Sci U S A. 24 janv 2012;109(4):1281‑6. 10.1073/pnas.1115006109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spaulding AR, Satterwhite EA, Lin Y-C, Chuang-Smith ON, Frank KL, Merriman JA, et al. Comparison of Staphylococcus aureus strains for ability to cause infective endocarditis and lethal sepsis in rabbits. Front Cell Infect Microbiol. 2012;2:18 10.3389/fcimb.2012.00018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cassat JE, Hammer ND, Campbell JP, Benson MA, Perrien DS, Mrak LN, et al. A secreted bacterial protease tailors the Staphylococcus aureus virulence repertoire to modulate bone remodeling during osteomyelitis. Cell Host Microbe. 12 juin 2013;13(6):759‑72. 10.1016/j.chom.2013.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spaan AN, Henry T, van Rooijen WJM, Perret M, Badiou C, Aerts PC, et al. The staphylococcal toxin Panton-Valentine Leukocidin targets human C5a receptors. Cell Host Microbe. 15 mai 2013;13(5):584‑94. 10.1016/j.chom.2013.04.006 [DOI] [PubMed] [Google Scholar]
- 25.Li M, Cheung GYC, Hu J, Wang D, Joo H-S, DeLeo FR, et al. Comparative Analysis of Virulence and Toxin Expression of Global Community-Associated Methicillin-Resistant Staphylococcus aureus Strains. J Infect Dis. 15 Déc 2010;202(12):1866‑76. 10.1086/657419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vuong C, Götz F, Otto M. Construction and Characterization of an agr Deletion Mutant of Staphylococcus epidermidis. Infect Immun. mars 2000;68(3):1048‑53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Norden CW. Experimental osteomyelitis. I. A description of the model. J Infect Dis. November 1970;122(5):410‑8. [DOI] [PubMed] [Google Scholar]
- 28.Otto M. Staphylococcal infections: mechanisms of biofilm maturation and detachment as critical determinants of pathogenicity. Annu Rev Med. 2013;64:175‑88. 10.1146/annurev-med-042711-140023 [DOI] [PubMed] [Google Scholar]
- 29.Kong K-F, Vuong C, Otto M. Staphylococcus quorum sensing in biofilm formation and infection. Int J Med Microbiol IJMM. avr 2006;296(2‑3):133‑9. [DOI] [PubMed] [Google Scholar]
- 30.Periasamy S, Chatterjee SS, Cheung GYC, Otto M. Phenol-soluble modulins in staphylococci. Commun Integr Biol. 1 mai 2012;5(3):275‑7. 10.4161/cib.19420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vardakas KZ, Kontopidis I, Gkegkes ID, Rafailidis PI, Falagas ME. Incidence, characteristics, and outcomes of patients with bone and joint infections due to community-associated methicillin-resistant Staphylococcus aureus: a systematic review. Eur J Clin Microbiol Infect Dis Off Publ Eur Soc Clin Microbiol. juin 2013;32(6):711‑21. [DOI] [PubMed] [Google Scholar]
- 32.Horewicz VV, Crestani S, de Sordi R, Rezende E, Assreuy J. FPR2/ALX activation reverses LPS-induced vascular hyporeactivity in aorta and increases survival in a pneumosepsis model. Eur J Pharmacol. 5 janv 2015;746:267‑73. 10.1016/j.ejphar.2014.11.026 [DOI] [PubMed] [Google Scholar]
- 33.Valentino MD, Foulston L, Sadaka A, Kos VN, Villet RA, Santa Maria J, et al. Genes contributing to Staphylococcus aureus fitness in abscess- and infection-related ecologies. mBio. 2014;5(5):e01729–1714. 10.1128/mBio.01729-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilde AD, Snyder DJ, Putnam NE, Valentino MD, Hammer ND, Lonergan ZR, et al. Bacterial Hypoxic Responses Revealed as Critical Determinants of the Host-Pathogen Outcome by TnSeq Analysis of Staphylococcus aureus Invasive Infection. PLoS Pathog. Déc 2015;11(12):e1005341 10.1371/journal.ppat.1005341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vandenesch F, Lina G, Henry T. Staphylococcus aureus hemolysins, bi-component leukocidins, and cytolytic peptides: a redundant arsenal of membrane-damaging virulence factors? Front Cell Infect Microbiol. 2012;2:12 10.3389/fcimb.2012.00012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hua L, Cohen TS, Shi Y, Datta V, Hilliard JJ, Tkaczyk C, et al. MEDI4893* Promotes Survival and Extends the Antibiotic Treatment Window in a Staphylococcus aureus Immunocompromised Pneumonia Model. Antimicrob Agents Chemother. 2015. August; 59(8):4526‑32. 10.1128/AAC.00510-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.