

Autosomal recessively inherited mutations in WNT1 were recently identified as a cause of severe osteogenesis imperfecta (OI).1–5 This finding does not address the critical role for Wnt1 in mid-hindbrain development that is well described in model organisms.7,8 Severe intellectual and motor deficits were noted in 4 of 16 families in the initial reports, but few details were provided. We reviewed developmental outcomes and brain-imaging studies for one new and five previously reported individuals with WNT1-associated OI. All six have brain malformations, with prominent brainstem and cerebellar hypoplasia in five of these six individuals.
Homozygous or compound heterozygous mutations in WNT1 were recently described as a novel cause for severe autosomal recessive OI in 25 individuals from 16 families in a series of six papers.1–6 Brain-imaging studies in two individuals were reported to show unilateral cerebellar hypoplasia,2,4 and another was reported to have Chiari malformation type 1.5 However, only limited data were presented regarding the brain and neurological phenotypes, including only a single MRI image. This is an important issue to address, as the WNT family of secreted signaling proteins play key roles in many developmental and homeostatic processes.9 Indeed, prominent defects in early brain development were described in two mouse lines with Wnt1 mutations long before WNT1 mutations were identified as a cause of bone fragility in humans.7,8
To examine the human brain phenotype associated with mutations in WNT1, we reviewed all available brain-imaging studies from one new and 5 previously reported individuals including one sibling pair,1–5 which consisted of 5 brain MRI (figure 1) and one cranial CT scan (see online supplementary figure S1). We found significant malformations in all 6 individuals (table 1). Hippocampal malformations were found in three affected individuals for whom coronal MRI sequences were available (figure 1D,H,L). The midbrain especially the tectum was small in 5 of 6 individuals, which we rated as moderate in two (figure 1E and online supplementary figure S1) and severe in three (figure 1A,I,M) of five individuals. Notably, few human disorders have been reported with specific hypoplasia of the midbrain. Cerebellar vermis hypoplasia, always most severe in the uvula and pyramis lobules, was rated as mild in one (figure 1E) and severe in three (figure 1A,I,M) of five individuals.
Figure 1. Brain MRI with WNT1 mutations.
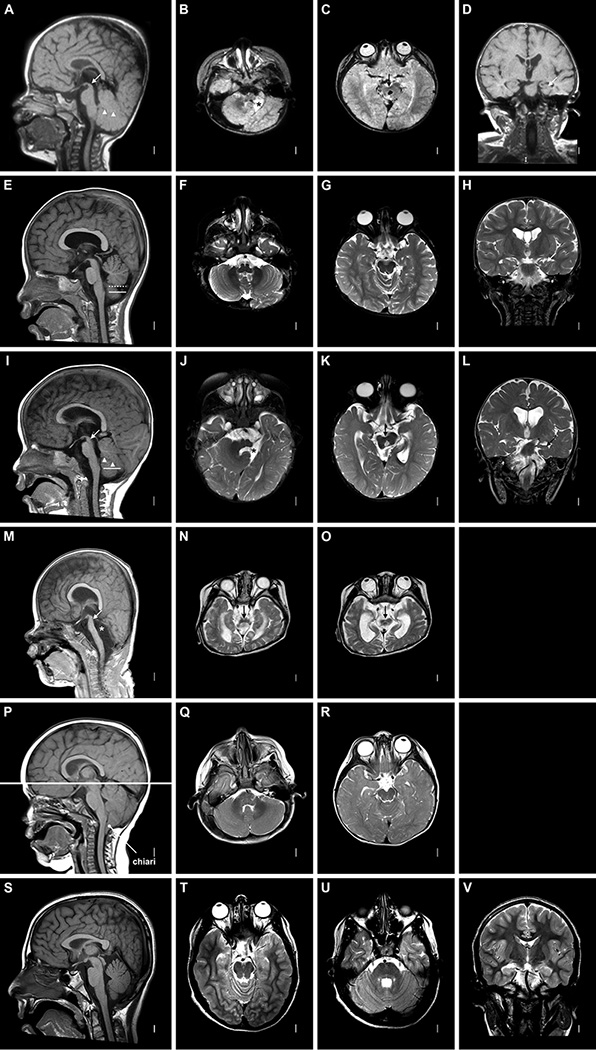
The images shown are T1-weighted mid-sagittal, T2-weighted axial images through the low and high brainstem, and T2-weighted coronal images through the hippocampus. These images show variable brainstem hypoplasia that is typically most severe in the midbrain especially the tectum. Imaging patterns include a normal brainstem (P–R), normal (A, E) or mildly small (I) pons and medulla with very small midbrain (A–C, I–K with arrows in A and I), and diffuse severe hypoplasia of the entire brainstem (M–O with arrow in M). The cerebellar vermis varies from normal (P) to small (mild in E, moderate in A and I) to completely absent (asterisk in M). In E, the dashed line marks the observed lower limit of the vermis and the solid line the expected lower limit of the vermis. The cerebellar hemispheres may appear normal (F, Q) or small (right hemispheres in B, J). Here the left cerebellar hemispheres are absent in two patients (asterisks in B, J), and the entire cerebellum is missing in one (M–O). The hippocampi are small and malformed (arrows in D, H, L). The gyral pattern and cortex appear normal. Images of a typically developing individual are shown for reference (S–V).
Table 1.
Brain and developmental characteristics in affected individuals with WNT1 mutations
| Individual | LR12-457a2 | LR13-235a1 | LR13-235a2 | LR13-243 | LR13-327 | LR14-042 |
| Reference | Laine 2013 Family 2 |
Pyott 2013 Family 2 |
Pyott 2013 Family 2 |
Faqeih 2013 | This report | Pyott 2013 Family 4 |
| Ethnicity | Hmong | Hmong | Hmong | Saudi | Chinese | Caucasian |
| Sex | F | M | M | M | M | F |
| WNT1 cDNA | c.884C>A | c.884C>A | c.884C>A | c.990C>A | c.184C>T c.677C>T |
c.287_300del14 |
| WNT1 protein | p.Ser295* | p.Ser295* | p.Ser295* | p.Cys220* | p.Gln62* p.Ser226Leu |
p.Gln96Profs*54 |
| Inheritance | Homozygous | Homozygous | Homozygous | Homozygous | Compound Heterozygous |
Homozygous |
| MRI/CT* | A-D | E-H | I-L | S1 | M-O | P-R |
| brachyceph | moderate | moderate | normal | na | moderate | normal |
| HIPD | moderate | moderate | moderate | na | na | na |
| BSH-MIDB | severe | moderate | severe | normal | severe | normal |
| BSH-POMED | normal | normal | mild | normal | severe | normal |
| R CBLH | normal | normal | normal | moderate | severe | normal |
| CBVH | severe | normal | severe | severe | severe | normal |
| L CBLH | severe | normal | severe | normal | severe | normal |
| CBTE chiari | absent | absent | absent | absent | absent | moderate |
| Development | ||||||
| Age last seen | 23y | 18m | 10y | 3y | 7y | 7y |
| OFC | 51.5 (−2.5 SD) |
47.5 (−1.5 SD) |
50.5 (−1 SD) | NA | 47 (−3 SD) | 48.5 (−1.5 SD) |
| ID | profound | severe | severe | severe | severe | Normal IQ 109 |
| Walk | no | no | no | no | no | yes |
| Talk | no | no | no | no | no | yes |
| Feed | no | no | no | no | no | yes |
| Seizures | no | na | no | yes | yes | no |
| Autism | no | yes | yes | no | no | yes |
| Other | Unil Ptosis | Optic chiasm hypoplasia |
Profound ID following hyperthermia and asphixia |
Unil Ptosis | Unil Ptosis | |
| Age at death | – | – | – | 3.5y | 7y | – |
The panel in figure 1 showing the individual’s MRI or online supplementary figure S1 showing CT.
Abbreviations: M, male; F, female; brachceph, brachychephaly; HIPD, hippocampal dysplasia; BSH-MIDB, brainstem midbrain hypoplasia; BSH-POMED, brainstem pons medulla hypoplasia; R CBLH, right cerebellar hemisphere hypoplasia; CBVH, cerebellar vermis hypoplasia; L CBLH, left cerebellar hemisphere hypoplasia; CBTE chiari, cerebellar tonsilar ectopia; NA, imaging not available to make an assessment; ID, intellectual disability; Unil, unilateral; OFC, occipitofrontal circumfrence
The cerebellar hemispheres were also small in the four individuals with vermis hypoplasia. Unexpectedly, this was unilateral in three of four subjects with cerebellar hypoplasia, involving the right hemisphere in 1 (see online supplementary figure S1) and the left hemisphere in 2 (figure 1B,J) individuals. The last patient had complete (bilateral) cerebellar agenesis (figure 1N,O). The only individual with normal size of the brainstem and cerebellum had severe Chiari malformation type 1 (figure 1P–R). Thus, the brainstem and cerebellar hypoplasia varied from normal to unilateral hypoplasia to complete absence.
For one severely involved patient, LR13-235a2 previously Family 2, proband II-6,5 our interpretation differed from the published report. We found unilateral cerebellar hypoplasia on the left (not right) and did not find schizencephaly. We agree with other changes reported, finding hypoplasia of the anterior commissure, optic chiasm, hypothalamus, tectum, pons and right cerebellar hemisphere, and absent vermis.
We also reviewed the developmental features for these six affected individuals (table 1). Severe to profound intellectual disability (ID) was noted in five out of six of these individuals. The sixth patient (with Chiari malformation) was diagnosed with mild autism at 3 years, but by 7 years her Full Scale IQ was 109. The head circumference was below the mean (−1 to −3 SDs) in 5 of 5 individuals with data available; the smallest head size was found in the severely affected individual with total cerebellar agenesis. Five individuals had ocular problems including four with unilateral ptosis and another with an unspecified eye movement disorder. Care for these patients was challenging due to their profound disabilities and multiple fractures. Two patients died at 3.5 and 7 years due to a chest infectin and sepsis followed by respiratory failure, respectively. Another patient has no useful neurological function after sustaining a profound brain injury at 28 months following an episode of severe hyperthermia (T 42.8 °C), respiratory failure, shock and multiorgan failure.
In summary, we found cerebellar hypoplasia in five of six individuals that varied from mild hypoplasia to complete agenesis of the cerebellum, with frequent asymmetry. The brainstem and cerebellar hypoplasia fit well with the brain phenotypes reported for two mouse lines with Wnt1 mutations.7,8 Both have severe developmental defects of the midbrain, pons and cerebellum that vary from severe midbrain and pontine hypoplasia with complete cerebellar agenesis to anterior hypoplasia of the same structures. The knockout mutants typically die at birth, while the hypomorphic Wnt1sw/sw mice have ataxia, but often live to adulthood. Wnt1 is expressed in a rostral-caudal gradient beginning in the developing midbrain and spreading to the cerebellum and pons. In the cerebellum, Wnt1 is primarily expressed in progenitor cells in the upper rhombic lip that contribute to glutamatergic neurons.10 The skeletal phenotype was not examined in the Wnt1 mutants in the original reports, but spontaneous fractures and severe osteopenia were recently reported in Wnt1sw/sw mice.11
The most unexpected feature is the asymmetry seen in several patients. Asymmetric cerebellar hypoplasia with cerebellar clefts has been reported as an isolated anomaly presumed to be caused by prenatal posterior fossa or cerebellar haemorrhage,12,13 and asymmetric hemispheric hypoplasia is sometimes seen with Dandy-Walker malformation.14 Interestingly, the complete cerebellar agenesis observed in one patient with mutations in WNT1 resembles the brain phenotype seen in individuals with homozygous PTF1A mutations.15,16 However, such striking asymmetry is rare among known genetic types of cerebellar hypoplasia.
The previously reported WNT1 mutations include truncation and missense mutations, but the patients in our cohort include five homozygous truncations (usually in the last exon) and one compound heterozygote with a truncation and a missense mutation (table 1). Interestingly, affected individuals in two unrelated Hmong families were homozygous for the same truncation, suggesting a possible founder mutation. In this small series, we observed no clear genotype-phenotype correlation. In one family (LR12-457) with two affected sisters, the younger sister had severe ID and profound disabilities, while the older sister, who declined brain imaging, was intellectually normal. These six patients may not be representative of all individuals with WNT1-associated OI, as all had clinical brain-imaging studies performed to evaluate neurologic abnormalities. Thus there may be a bias of ascertainment for more severely affected individuals since typically developing individuals would not usually undergo brain imaging. While 6 of 25 patients in six papers focusing on the OI phenotype had reportedly normal development, we found severe ID in five of six individuals in whom brain imaging was performed, and mild autism in the sixth. We suspect a relationship between severity of the ID and brainstem-cerebellar hypoplasia, but cannot evaluate this statistically given the lack of brain scans in other reported patients. We also could not correlate the severity of the OI phenotype with the neurodevelopmental deficits or cerebellar malformations, but suggest that this analysis take place as additional patients are examined. From this limited dataset, we recommend that brain imaging be performed in any individual with WNT1-associated OI who also has developmental delay or any neurological deficits. However, our data is not sufficient to support brain imaging in affected individuals with normal neurologic status.
Supplementary Material
{kind=link}
Acknowledgments
Funding Research reported in this publication was supported by the National Institute of Neurological Disorders and Stroke (NINDS), the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), the National Institute of Dental and Craniofacial Research (NIDCR) and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) of the National Institutes of Health (NIH) under award numbers 1R01NS092772 to WBD, P01HD070394 to DHC, R01DE019567 to DHC and R01AR062651 to DHC, and by the SK Yee Medical Foundation and Orthopaedic Hospital Research Center at UCLA.
Footnotes
Contributors KAA, WBD and CJC participated in the design of the study. NJM, BHYC, WZ, DHC, BF, FSA and CJC collected and/or generated data. KAA, WBD and CJC analysed and interpreted the data. KAA, WBD, DHC and CJC drafted the manuscript. All coauthors read and approved the final manuscript.
Competing interests None declared.
Ethics approval The Institutional Review Board at Seattle Children’s Hospital, Seattle, Washington, USA.
Provenance and peer review Not commissioned; externally peer reviewed.
REFERENCES
- 1.Fahiminiya S, Majewski J, Mort J, et al. Mutations in WNT1 are a cause of osteogenesis imperfecta. J Med Genet. 2013;50(5):345–348. doi: 10.1136/jmedgenet-2013-101567. [DOI] [PubMed] [Google Scholar]
- 2.Faqeih E, Shaheen R, Alkuraya FS. WNT1 mutation with recessive osteogenesis imperfecta and profound neurological phenotype. J Med Genet. 2013;50(7):491–492. doi: 10.1136/jmedgenet-2013-101750. [DOI] [PubMed] [Google Scholar]
- 3.Keupp K, Beleggia F, Kayserili H, et al. Mutations in WNT1 cause different forms of bone fragility. Am J Hum Genet. 2013;92(4):565–574. doi: 10.1016/j.ajhg.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laine CM, Joeng KS, Campeau PM, et al. WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N Engl J Med. 2013;368(19):1809–1816. doi: 10.1056/NEJMoa1215458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pyott SM, Tran TT, Leistritz DF, et al. WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. Am J Hum Genet. 2013;92(4):590–597. doi: 10.1016/j.ajhg.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stephen J, Girisha KM, Dalal A, Shukla A, Shah H, Srivastava P, Kornak U, Phadke SR. Mutations in patients with osteogenesis imperfecta from consanguineous Indian families. Eur J Med Genet. 2015;58:21–27. doi: 10.1016/j.ejmg.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 7.McMahon AP, Bradley A. The Wnt-1 (int-1) proto-oncogene is required for development of a large region of the mouse brain. Cell. 1990;62(6):1073–1085. doi: 10.1016/0092-8674(90)90385-r. [DOI] [PubMed] [Google Scholar]
- 8.Thomas KR, Musci TS, Neumann PE, et al. Swaying is a mutant allele of the proto-oncogene Wnt-1. Cell. 1991;67(5):969–976. doi: 10.1016/0092-8674(91)90369-a. [DOI] [PubMed] [Google Scholar]
- 9.Coombs GS, Covey TM, Virshup DM. Wnt signaling in development, disease and translational medicine. Curr Drug Targets. 2008;9(7):513–531. doi: 10.2174/138945008784911796. [DOI] [PubMed] [Google Scholar]
- 10.Hagan N, Zervas M. Wnt1 expression temporally allocates upper rhombic lip progenitors and defines their terminal cell fate in the cerebellum. Mol Cell Neurosci. 2012;49(2):217–229. doi: 10.1016/j.mcn.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joeng KS, Lee YC, Jiang MM, et al. The swaying mouse as a model of osteogenesis imperfecta caused by WNT1 mutations. Hum Mol Genet. 2014;23(15):4035–4042. doi: 10.1093/hmg/ddu117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boltshauser E, Steinlin M, Martin E, Deonna T. Unilateral cerebellar aplasia. Neuropediatrics. 1996;27:50–53. doi: 10.1055/s-2007-973748. [DOI] [PubMed] [Google Scholar]
- 13.Poretti A, Leventer RJ, Cowan FM, Rutherford MA, Steinlin M, Klein A, Scheer I, Huisman TA, Boltshauser E. Cerebellar cleft: a form of prenatal cerebellar disruption. Neuropediatrics. 2008;39:106–112. doi: 10.1055/s-2008-1081460. [DOI] [PubMed] [Google Scholar]
- 14.Patel S, Barkovich AJ. Analysis and classification of cerebellar malformations. AJNR Am J Neuroradiol. 2002;23:1074–1087. [PMC free article] [PubMed] [Google Scholar]
- 15.Al-Shammari M, Al-Husain M, Al-Kharfy T, Alkuraya FS. A novel PTF1A mutation in a patient with severe pancreatic and cerebellar involvement. Clin Genet. 2011;80:196–198. doi: 10.1111/j.1399-0004.2010.01613.x. [DOI] [PubMed] [Google Scholar]
- 16.Sellick GS, Barker KT, Stolte-Dijkstra I, Fleischmann C, Coleman RJ, Garrett C, Gloyn AL, Edghill EL, Hattersley AT, Wellauer PK, Goodwin G, Houlston RS. Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat Genet. 2004;36:1301–1305. doi: 10.1038/ng1475. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.