

Abstract
Yessotoxin and adriatoxin are members of the polycyclic ether family of marine natural products. Outlined in this article is our synthetic approach to two subunits of these targets. Central to our strategy is a coupling sequence that employs an olefinic-ester cyclization reaction. As outlined, this sequence was used in two coupling sequences. First it was used to merge the A,B- and E,F-bicyclic precursors and in the process generate the C, D-rings. Second it was used to couple the F- and I-rings while building the eight-membered G-ring and subsequently the H-ring pyran.
Keywords: ciguatera, polycyclic ether, yessotoxin, adriatoxin, ring-closing metathesis, glycal
Introduction
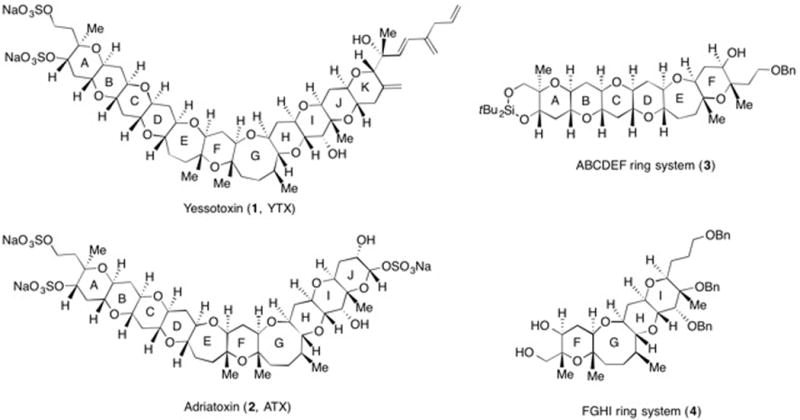
Yessotoxin (1, YTX), a disulfated polycyclic ether natural product was first reported in 1987 by Murate, Yasumoto, and co-workers from the digestive gland of the scallop Patinopecten yessoensis (Figure 1).1 It was subsequently discovered that YTX is produced by the dinoflagellates Protoceratium reticulatum, Lingulodinium polyedrum, and Gonyaulax spinifera.2–4 YTX is acutely toxic to mice when administered intraperitoneally (LD50 = 286 μg/Kg) but not orally (up to 54 mg/Kg).5–9 The bioactivity of YTX ranges from modulating cytosolic calcium homeostasis,10 disrupting the degradation pathway of E-cadherin,11,12 inducing apoptotic cell death through the activation of caspases,13 to inducing lysosomal damage.14 Adriatoxin (2, ATX), a trisulfated analogue of YTX, was first isolated in 1998 from the digestive gland of the mussel Mytilus galloprovincialis collected in the Adriatic Sea.15 Structurally, YTX and ATX share the same A-J ring system, with YTX bearing an additional pyran (K-ring in YTX) and a hydrophobic side chain. Both compounds have attracted considerable interest from the organic synthesis community because of their challenging structures and intriguing properties.16–27 We have previously reported the syntheses of the AB, EF, and IJ ring systems of ATX.28 Our approach to the total synthesis of marine polycyclic ether natural products has largely focused on a convergent strategy that is centered around an olefinic-ester cyclization reaction that pairs cyclic ether subunits and leads to the generation of two additional rings.29–31 Herein, we describe our convergent synthesis of the ABCDEF and FGHI ring systems of YTX and ATX (3 and 4).
Figure 1.
Results and Discussion
The synthesis of the AB bicyclic coupling precursor is depicted in Scheme 1. From known tricyclic substrate 5,32 TBS ether formation and ozonolysis gave 7 after conversion of the aldehyde into the corresponding carboxylic acid.33
Scheme 1.
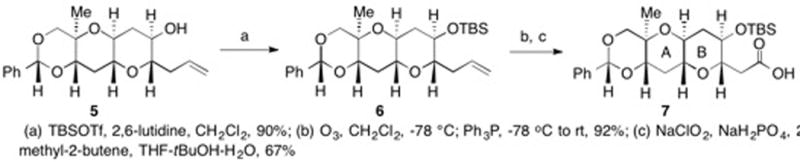
Outlined in Scheme 2 is our synthesis of the EF bicyclic coupling precursor. From tricycle 8,32 PMB ether formation, ozonolysis and reduction of the resulting aldehyde gave primary alcohol 9. Benzyl ether generation and acetal hydrolysis gave diol 10. A single flask conversion of the 1° alcohol in 10 into the corresponding triflate and the 2° alcohol into the corresponding TBS ether gave 11. Displacement of the triflate in 11 by lithium trimethylsilylacetylide and removal of the TBS group afforded 12.34 Partial hydrogenation provided EF coupling precursor 13.35
Scheme 2.
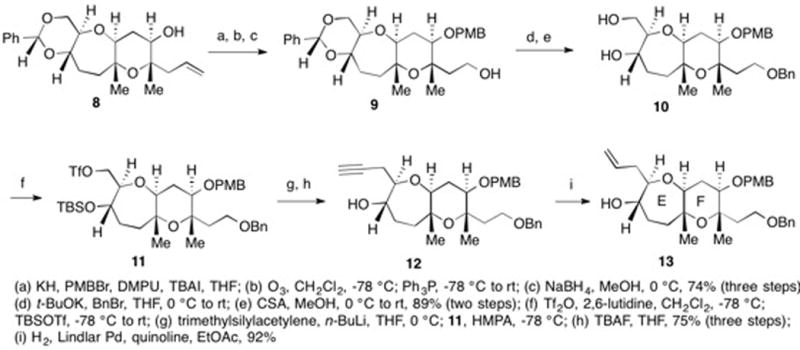
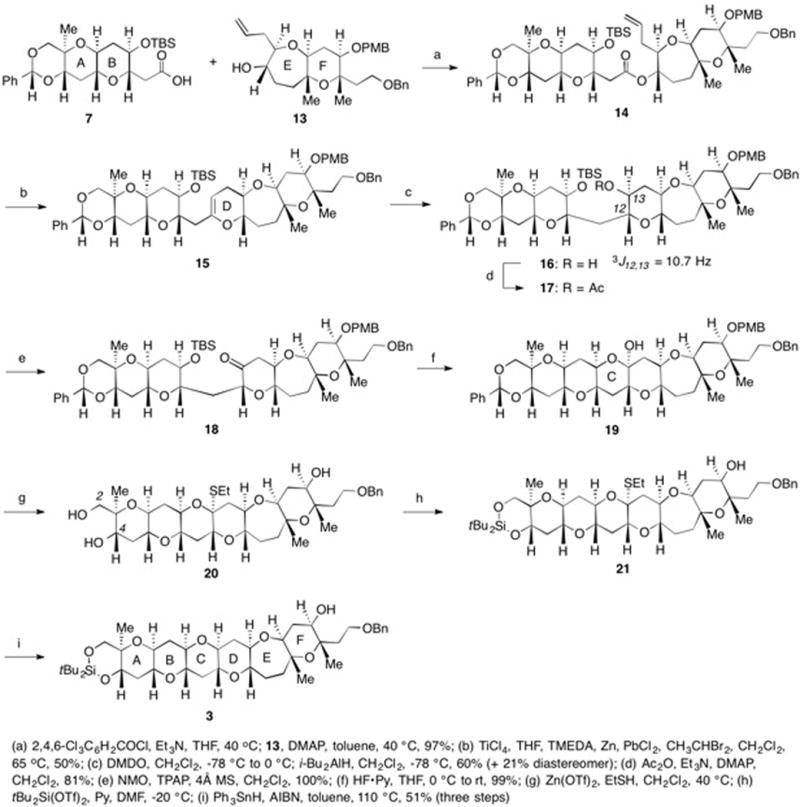
With 7 and 13 in hand, esterification using Yamaguchi’s conditions gave 14 (Scheme 3).36 When subjected to our modified Takai-Utimoto reaction conditions, 14 underwent a smooth olefinic ester cyclization to afford D ring cyclic enol ether 15 as the only product in 50% yield.37 Based on our inability to isolate by-products, we believe that the modest yield was due to the instability of 15 and not the inefficiency of the reaction. Treatment of 15 with DMDO followed by reduction of the resulting epoxide with iBu2AlH generated secondary alcohol 16 in 60% yield as a 3:1 mixture of diastereomers.38 The stereochemistry at C12 and C13 in 16 was established using 1H NMR and the 3J value between C12 and C13 following the conversion of 16 into the corresponding acetate 17 (TYX and ATX numbering). Oxidation of the mixture of diastereomers to ketone 18 followed by removal of the TBS group afforded hemiketal 19. Treatment of 19 with Zn(OTf)2 and EtSH gave the corresponding O,S-mixed ketal as a single diastereomer with concomitant removal of the benzylidene and the PMB groups. The C2 and C4 hydroxyl groups of triol 20 were selectively converted into cyclic silylene 21. When 21 was subjected to Ph3SnH and AIBN in refluxing toluene, the O,S-mixed ketal was reduced to give 3 containing the A-F ring system of both YTX and ATX.39
Scheme 3.
Having completed the synthesis of the A-F ring system, we turned our attention to the F-I subunit. Depicted in Scheme 4 is the synthesis of the F-ring coupling precursor 30. From known alcohol 22,40 acetylation followed by olefinic ester cyclization gave cyclic enol ether 24. The C19 angular methyl group helped to direct a stereoselective epoxidation of 24 with mCPBA. In situ epoxide opening with MeOH and allylation of the resulting alcohol generated 25. The treatment of 25 with PPTS, pyridine, and heat initiated a Claisen rearrangement giving ketone 26 as a single diastereomer.28,41 The stereochemistry of the newly installed angular methyl group was confirmed through the indicated nOe correlations. Reduction of the ketone and PMB ether formation afforded 27. While the extension of the propenyl side chain in 27 into the pentenyl side chain present in 29 was relatively inefficient, it could be carried out on a reasonable scale. Removal of the benzylidene and PMB groups followed by selective C-18, C-20 PMB acetal formation provide the F-ring coupling precursor 30.
Scheme 4.
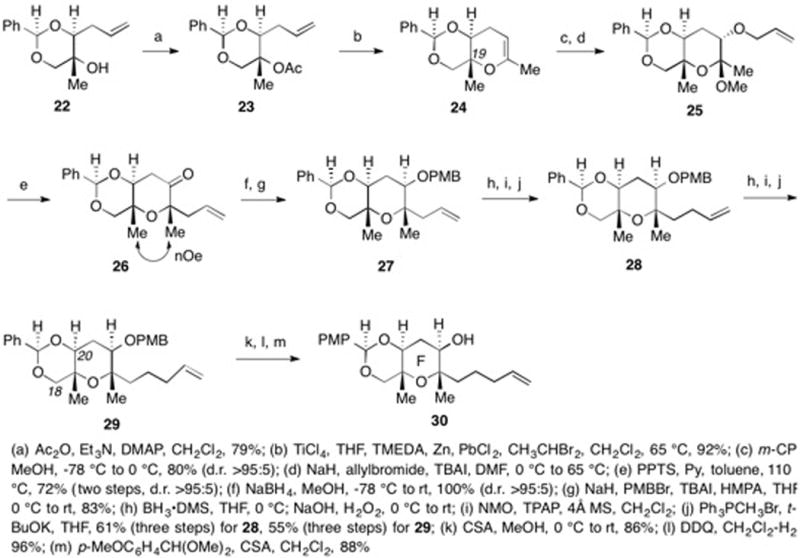
The I-ring subunit 39 was synthesized according to the route outlined in Scheme 5. From the previously reported alcohol 31,42 tertiary alcohol formation, removal of PMB group and oxidation gave 32. Formation of the TMS ether and reduction gave alcohol 33. Hydroboration and oxidative work-up provided the 1° alcohol while simultaneously removing the TMS group to give 34. Tris-benzyl ether formation and removal of the silylene group generated diol 35. Primary triflate formation and secondary TES ether formation gave 36. The coupling of 36 with lithium acetylide gave 37 after removal of the silyl protecting groups. TES ether formation and partial hydrogenation of the alkyne followed by ozonolysis and oxidation afforded the I-ring coupling precursor 39.
Scheme 5.
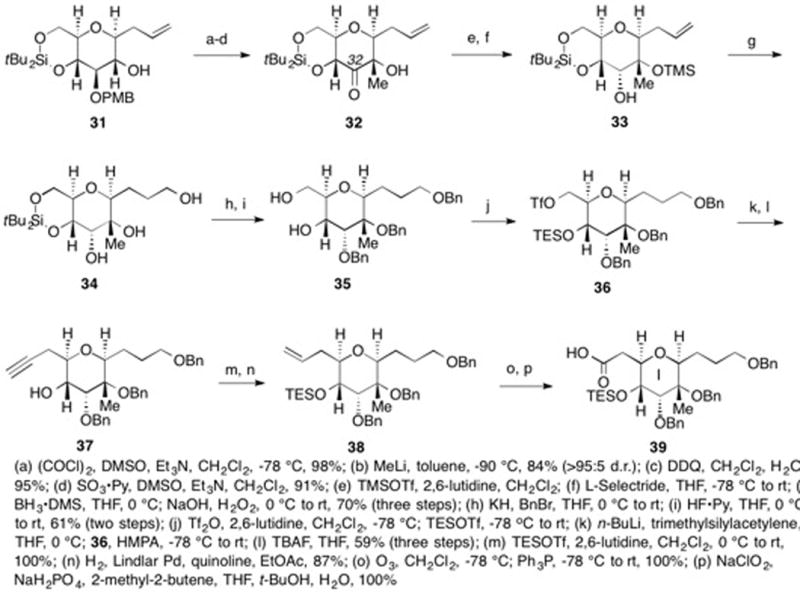
Yamaguchi esterification using alcohol 30 and acid 39 gave ester 40 (Scheme 6).36 Olefinic-ester cyclization gave eight-membered G-ring cyclic enol ether 41 in 40% yield. We are unaware of any other reports of eight-membered cyclic enol ether formation using either olefinic-ester cyclization or olefinic enol ether cyclization conditions.
Scheme 6.
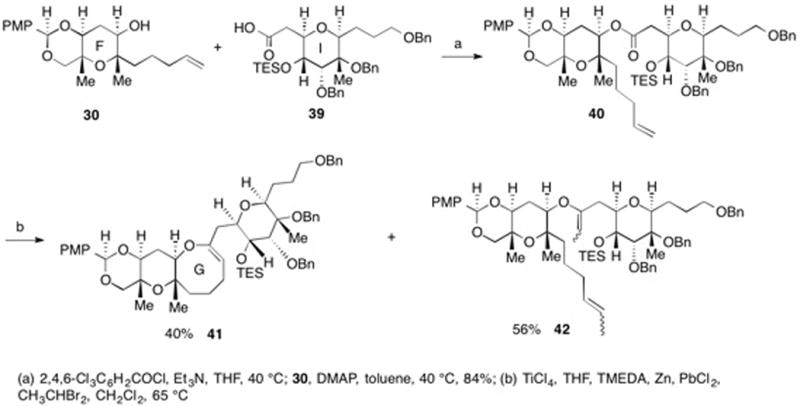
Having access to 41, we next examined the incorporation of the H-ring and the C-26 methyl group (Scheme 7). The oxidation of 41 with DMDO was followed by the in situ reduction of the resulting epoxide with iBu2AlH giving secondary alcohol 43 as a single diastereomer. Oxidation provided ketone 44 whose stereochemistry was determined from the indicated nOe enhancements. The incorporation of the C-26 methyl group was accomplished through alkylation of the enolate from 44 to give 45. Reductive cyclization of the TES-protected hydroxy ketone 45 using TMSOTf and Et3SiH resulted in the generation of the G-ring. This transformation also resulted in the removal of the PMB acetal. Interestingly, when a benzylidene acetal rather than a p-methoxy benzylidene actal was used as the protecting group on the F-ring, it could not be cleanly removed under the reductive cyclization conditions, giving instead a mixture of benzyl protected alcohols. We were able to ascertain the relative stereochemistry of 4 using the indicated nOe enhancements.
Scheme 7.
In summary, we have achieved the convergent syntheses of both the ABCDEF and FGHI ring system of YTX and ATX using our olefinic ester cyclization strategy. We have also demonstrated that eight-membered cyclic enol ethers can be generated using an olefinic ester cyclization reaction. Further studies towards the total syntheses of YTX and ATX are currently in progress in our laboratory.
Experimental Details
NMR spectra were recorded on Varian Inova-400 MHz, Varian Inova-500 MHz or Varian VXR-500 MHz spectrometers. Chemical shifts were reported in δ, parts per million (ppm), relative to benzene (7.16), chloroform (7.27), or dichloromethane (5.32) as internal standards. Coupling constants, J, were reported in Hertz (Hz) and refer to apparent peak multiplicities and not true coupling constants. Mass spectra were recorded at the Mass Spectrometry Facility at the Department of Chemistry of the University of Utah at Salt Lake City on a Finnigan MAT 95 mass spectrometer. IR spectra were recorded on a Bruker Tensor 27 FT-IR spectrometer. Optical rotations were obtained on a Perkin-Elmer Model 343 polarimerter (Na D line) using a microcell with 1 dm path length. Solvents were purified according to the guidelines in Purification of Common Laboratory Chemicals (Perrin, Armarego, and Perrin: Oxford, 1966). Dichloromethane, 2,6-lutidine, triethylamine, TMEDA, chlorobenzene and pyridine were distilled from CaH2. Tetrahydrofuran and diethyl ether were dried from the sodium ketyl of benzophenone and distilled before use. Zinc dust (<10 μm, Aldrich) was activated by washing with 5% hydrochloric acid, H2O, methanol, and ether and dried in vacuo overnight. All other reagents were used without further purification. Unless otherwise noted, all reactions were performed under a nitrogen atmosphere in flame-dried glassware using standard syringe, cannula, and septa apparatus. Concentration refers to removal of solvent under reduced pressure (house vacuum at ca. 20mm Hg). Analytical thin–layer chromatography (TLC) was performed on silica gel plates (0.25 mm) precoated with a fluorescent indicator. Flash chromatography was performed using 40–63 μm silica gel (200 × 400 mesh).
(((2R,4aR,5aS,7R,8S,9aR,10aS)-8-allyl-4a-methyl-2-phenyloctahydro-4H-pyrano[2′,3′:5,6]pyrano[3,2-d][1,3]dioxin-7-yl)oxy)(tert-butyl)dimethylsilane (6). To a solution of 5 (25 mg, 0.072 mmol) and 2,6-lutidine (25 μL, 0.22 mmol) in CH2Cl2 (10 mL) was added TBSOTf (25 μL, 0.11 mmol). The reaction mixture was stirred for 2 h after which the reaction was quenched with sat. NaHCO3 (5 mL). The aqueous phase was extracted with CH2Cl2 (3 × 5 mL) and the extracts were combined, dried (Na2SO4), and concentrated. Flash chromatography (8:1 hexanes: ethyl acetate) provided 30 mg of 6 (90%) as a colorless oil. Rf 0.60 (5:1 hexanes:ethyl acetate); [α]20D = −48.9° (c = 0.47, THF); 1H NMR (500 MHz, C6D6) δ 7.60 (d, J = 7.3 Hz, 2H), 7.19 (t, J = 7.3 Hz, 2H), 7.13 (t, J = 7.3 Hz, 1H), 6.14 (dddd, J = 17.1, 10.2, 6.8, 6.8 Hz, 1H), 5.40 (s, 1H), 5.22 (bd, J = 17.1 Hz, 1H), 5.13 (bd, J = 10.2 Hz, 1H), 3.92 (d, J = 10.0 Hz, 1H), 3.53 (d, J = 10.0 Hz, 1H), 3.47 (ddd, J = 10.7, 7.3, 4.9 Hz, 1H), 3.38 (ddd, J = 10.2, 9.3, 3.4 Hz, 1H), 3.35 (dd, J = 12.5, 3.5 Hz, 1H), 3.26 (ddd, J = 8.8, 8.8, 2.9 Hz, 1H), 2.94 (ddd, J = 10.2, 9.8, 4.9 Hz, 1H), 2.75−2.69 (m, 1H), 2.35 (ddd, J = 7.3, 7.3, 7.3 Hz, 1H), 2.31 (ddd, J = 11.7, 4.4, 4.4 Hz, 1H), 2.19 (ddd, J = 11.2, 4.4, 4.4 Hz, 1H), 1.88 (ddd, J = 11.2, 11.2, 11.2 Hz, 1H), 1.43 (s, 3H), 0.93 (s, 9H), 0.01 (s, 3H), −0.01 (s, 3H); 13C NMR (125 MHz, C6D6) δ 138.5, 135.3, 128.8, 128.2, 126.7, 116.8, 102.8, 82.6, 79.9, 78.2, 76.4, 70.3, 69.9, 69.6, 40.1, 36.4, 30.9, 25.8, 18.0, 15.2, −4.0, −4.8; IR (neat) 2955, 2933, 2859, 1463, 1380, 1331, 1254, 1092 cm−1; ESI/MS (m/z) calcd for C26H41O5Si 461.3 (M+H+), found 461.3.
2-((2R,4aR,5aS,7R,8S,9aR,10aS)-7-((tert-butyldimethylsilyl)oxy)-4a-methyl-2-phenyloctahydro-4H-pyrano[2′,3′:5,6]pyrano[3,2-d][1,3]dioxin-8-yl)acetic acid (7). O3 was bubbled through a solution of 6 (30 mg, 0.065 mmol) in CH2Cl2 (20 mL) at −78 °C until the reaction was a light blue color. Excess O3 was purged from the reaction mixture by bubbling N2 through it until the light blue color completely faded away. Triphenylphosphine (51 mg, 0.19 mmol) was then added and the reaction mixture was allowed to slowly warm to rt. After 12 h, the solution was concentrated under reduced pressure. Flash chromatography (50:1 to 5:1 hexanes:ethyl acetate) provided the corresponding aldehyde (28 mg, 92%) as a colorless oil.
To a solution of aldehyde obtained above (28 mg, 0.061 mmol) in THF (3 mL) was successively added tBuOH (3 mL), H2O (3 mL), 2-Me-2-butene (0.6 mL), NaH2PO4 (36 mg, 0.30 mmol) and NaClO2 (27 mg, 0.30 mmol). The resulting mixture was stirred at rt for 2 h after which the reaction was quenched with H2O (3 mL). The reaction mixture was extracted with ethyl acetate (3 × 5 mL) and the organic extracts were combined, dried (Na2SO4) and concentrated. Flash chromatography (gradient of 1:1 hexanes:ethyl acetate to ethyl acetate) provided acid 7 (19 mg, 67%) as a colorless oil. Rf 0.30 (2:1 hexanes:ethyl acetate); [α]20D = −52.2° (c = 0.39, THF); 1H NMR (500 MHz, CD2Cl2) δ 7.51−7.47 (m, 2H), 7.41−7.37 (m, 3H), 5.58 (s, 1H), 3.92 (d, J = 10.2 Hz, 1H), 3.69−3.60 (m, 3H), 3.55 (ddd, J = 10.7, 9.3, 4.9 Hz, 1H), 3.48 (ddd, J = 11.2, 9.3, 3.9 Hz, 1H), 3.22 (ddd, J = 11.2, 9.8, 4.4 Hz, 1H), 2.89 (dd, J = 15.6, 2.9 Hz, 1H), 2.42 (dd, J = 15.6, 9.3 Hz, 1H), 2.27 (ddd, J = 11.7, 4.4, 4.4 Hz, 1H), 2.20 (ddd, J = 11.2, 4.4, 4.4 Hz, 1H), 1.78 (ddd, J = 11.7, 11.7, 11.7 Hz, 1H), 1.53 (ddd, J = 11.7, 11.7, 11.7 Hz, 1H), 1.52 (s, 3H), 0.92 (s, 9H), 0.14 (s, 3H), 0.13 (s, 3H); 13C NMR (125 MHz, CD2Cl2) δ 177.2, 138.1, 129.2, 128.4, 126.5, 103.1, 79.8, 79.6, 78.2, 76.4, 70.3, 70.0, 69.3, 39.5, 37.7, 30.6, 25.7, 17.9, 15.1, −4.1, −4.9; IR (neat) 3259, 2954, 2861, 1714, 1463, 1377, 1256, 1093 cm−1; ESI/MS (m/z) calcd for C25H37O7Si 477.3 (M−H+), found 477.4.
2-((2R,4aR,5aS,7R,8S,9aR,11aS)-7-((4-methoxybenzyl)oxy)-8,9a-dimethyl-2-phenyldecahydro-[1,3]dioxino[5,4-b]pyrano[2,3-f]oxepin-8-yl)ethan-1-ol (9). To a solution of 8 (28 mg, 0.075 mmol) in THF (5 mL) at rt was added KH (30 mg 30% dispersion in mineral oil, 0.23 mmol). After being allowed to stir for 10 min., DMPU (4.5 μL, 0.038 mmol), PMBBr (54 μL, 0.38 mmol) and a catalytic amount of TBAI were added to the reaction mixture. After stirring overnight, the reaction mixture was quenched with sat. NH4Cl (aq., 5 mL). The aqueous phase was extracted with CH2Cl2 (3 × 5 mL) and the combined organic extracts were dried (Na2SO4), and concentrated. Flash chromatography (10:1 hexanes:ethyl acetate) gave a colorless oil which was used in the next step without further purification.
O3 was bubbled through a solution of the PMB ether from the procedure outlined above in CH2Cl2 (20 mL) at −78 °C until the reaction mixture was a light blue color. Excess O3 was purged from the reaction mixture by bubbling N2 through it until the light blue color completely faded (ca. 10 min). Triphenylphosphine (59 mg, 0.23 mmol) was then added and the reaction mixture was allowed to slowly warm to rt. After stirring for 12 h, the resulting solution was concentrated under reduced pressure. Flash chromatography (50:1 to 5:1 hexanes:ethyl acetate) provided the corresponding aldehyde as a colorless oil. The aldehyde was used in the subsequent transformation without additional purification.
To a solution of the aldehyde obtained above in MeOH (5 mL) at 0 °C was added NaBH4 (8.5 mg, 0.23 mmol). The reaction mixture was stirred for 2 h after which the reaction was quenched with acetone (3 mL). The mixture was concentrated and the residue was purified using flash chromatography (4:1 hexanes:ethyl acetate) to give 9 as a colorless oil (28 mg, 74% over 3 steps). Rf 0.50 (2:1 hexanes:ethyl acetate); [α]20D = −15.5° (c = 0.45, THF); 1H NMR (500 MHz, CD2Cl2) δ 7.54−7.51 (m, 2H), 7.44−7.38 (m, 3H), 7.29 (d, J = 8.8 Hz, 2H), 6.94 (d, J = 8.3 Hz, 2H), 5.45 (s, 1H), 4.64 (d, J = 11.2 Hz, 1H), 4.41 (d, J = 11.2 Hz, 1H), 4.33 (dd, J = 10.2, 4.9 Hz, 1H), 3.83 (s, 3H), 3.84−3.78 (partially obscured m, 1H), 3.61 (dd, J = 10.3, 10.3 Hz, 1H), 3.54 (ddd, J = 9.8, 9.8, 4.9 Hz, 1H), 3.50 (dd, J = 11.7, 4.4 Hz, 1H), 3.37 (dd, J = 12.2, 3.4 Hz, 1H), 3.15 (br s, 1H), 2.30−2.17 (m, 2H), 1.96−1.85 (m, 4H), 1.79 (ddd, J = 12.2, 12.2, 12.2 Hz, 1H), 1.74 (ddd, J = 14.7, 7.8, 3.9 Hz, 1H), 1.34 (s, 3H), 1.33 (s, 3H); 13C NMR (125 MHz, CD2Cl2) δ 159.6, 138.3, 130.7, 129.5, 129.1, 128.4, 126.4, 113.9, 100.9, 83.2, 80.4, 79.2, 78.9, 78.1, 74.8, 70.8, 69.7, 59.3, 55.5, 43.5, 39.5, 28.7, 28.5, 21.9, 21.0; IR (neat) 3448, 2946, 2870, 1613, 1513, 1458, 1377, 1248, 1096 cm−1; ESI/MS (m/z) calcd for C29H38O7Na 521.2 (M+Na+), found 521.3.
(2S,3R,4aS,6R,7S,9aR)-2-(2-(benzyloxy)ethyl)-6-(hydroxymethyl)-3-((4-methoxybenzyl)oxy)-2,9a-dimethyloctahydro-2H-pyrano[3,2-b]oxepin-7-ol (10). To a solution of 9 (0.126 g, 0.253 mmol) in THF (5 mL) at rt was added tBuOK (0.50 mL of 1.0 M solution in THF, 0.50 mmol). The mixture was stirred for 30 min and then cooled to 0 °C. BnBr (0.12 mL, 1.01 mmol) was added slowly and the reaction mixture was stirred for 3 h before the reaction was quenched with sat. NH4Cl (aq., 5 mL). The aqueous phase was extracted with CH2Cl2 (3 × 5 mL) and the combined organic extracts were dried (Na2SO4), and concentrated.
The residue from above was dissolved in MeOH (15 mL) and the mixture was cooled to 0 °C. CSA (15 mg, 0.065 mmol) was added and the reaction mixture was allowed to slowly warm to rt. The reaction was quenched with sat. NaHCO3 (aq., 10 mL) after 5h. The aqueous phase was extracted with ethyl acetate (3 × 10 mL) and the combined organic extracts were dried (Na2SO4), and concentrated. Flash chromatography (1:1 hexanes:ethyl acetate) gave 10 as a colorless oil (0.112 g, 89% over 2 steps). Rf 0.60 (ethyl acetate); [α]20D = −19.0° (c = 0.51, THF); 1H NMR (500 MHz, CD2Cl2) δ 7.40−7.30 (m, 5H), 7.28 (d, J = 8.3 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H), 4.59 (d, J = 11.2 Hz, 1H), 4.48 (s, 2H), 4.38 (d, J =10.7 Hz, 1H), 3.87 (br d, J = 5.5 Hz, 1H), 3.82 (s, 3H), 3.71 (br s, 1H), 3.64−3.60 (m, 2H), 3.55−3.48 (m, 3H), 3.44 (dd, J = 11.2, 4.2 Hz, 1H), 2.70 (br s, 2H), 2.17 (ddd, J = 11.7, 3.9, 3.9 Hz, 1H), 1.95−1.76 (m, 4H), 1.76−1.65 (m, 2H), 1.54 (ddd, J = 13.7, 6.3, 2.2 Hz, 1H), 1.27 (s, 6H); 13C NMR (125 MHz, CD2Cl2) δ 159.4, 139.2, 131.1, 129.4, 128.5, 127.9, 127.6, 113.9, 85.7, 79.6, 79.3, 76.5, 75.5, 73.0, 71.7, 70.8, 66.7, 64.5, 55.4, 41.7, 36.2, 28.8, 27.9, 22.3, 20.2; IR (neat) 3383, 2929, 2870, 1613, 1455, 1375, 1248, 1090 cm−1; ESI/MS (m/z) calcd for C29H40O7Na 523.3 (M+Na+), found 523.3.
(2S,3R,4aS,6R,7S,9aR)-2-(2-(benzyloxy)ethyl)-3-((4-methoxybenzyl)oxy)-2,9a-dimethyl-6-(prop-2-yn-1-yl)octahydro-2H-pyrano[3,2-b]oxepin-7-ol (12). To a solution of 10 (0.112 g, 0.224 mmol) and 2,6-lutidine (0.104 mL, 0.893 mmol) in CH2Cl2 (20 mL) at −78 °C was added trifluoromethanesulfonic anhydride (40.0 μL, 0.234 mmol). After 30 min TBSOTf (77.0 μL, 0.335 mmol) was added to the reaction mixture. The reaction mixture was allowed to warm slowly to rt before it was quenched with sat. NaHCO3 (aq., 10 mL). The aqueous phase was extracted with CH2Cl2 (3 × 10 mL). The combined organic phase was dried (Na2SO4) and concentrated to give a yellow oil. The oil was dissolved in a 10:1 mixture of hexanes:ethyl acetate, filtered through a plug of silica gel (10:1), and concentrated. The resulting colorless oil was used in the next transformation without additional purification.
To a solution of trimethylsilylacetylene (0.168 mL, 1.19 mmol) in THF (10 mL) at 0 °C was added nBuLi (0.474 mL of 2.50 M solution in hexanes, 1.19 mmol). The reaction mixture was stirred at 0 °C for 30 min and then cooled to −78 °C. A solution of 11 obtained from above and HMPA (0.207 mL, 1.19 mmol) in THF (5 mL) was transferred via cannula into the acetylide containing reaction mixture. The reaction mixture was stirred for 2 h after which the reaction was quenched with sat. NH4Cl (aq., 10 mL). The aqueous phase was extracted with CH2Cl2 (3 × 10 mL). The combined organic phase was dried (Na2SO4) and concentrated to give a dark brown oil which was taken up in THF (10 mL). To the resulting solution was added TBAF (1.12 mL of 1.0 M solution in THF, 1.12 mmol) and the mixture was stirred at rt overnight before the reaction was quenched with sat. NH4Cl (aq., 10 mL). The aqueous phase was extracted with CH2Cl2 (3 × 10 mL), the organic phases were combined, dried (Na2SO4), and concentrated. Flash chromatography (4:1 hexanes:ethyl acetate) gave 12 as a colorless oil (85.7 mg, 75% 3 steps). Rf 0.50 (2:1 hexanes:ethyl acetate); [α]20D = −21.6° (c = 0.42, THF); 1H NMR (500 MHz, CD2Cl2) δ 7.40−7.28 (m, 5H), 7.27 (d, J = 8.8 Hz, 2H), 6.90 (d, J = 8.3 Hz, 2H), 4.58 (d, J = 10.8 Hz, 1H), 4.48 (s, 2H), 4.38 (d, J =11.2 Hz, 1H), 4.08 (br s, 1H), 3.81 (s, 3H), 3.79 (partially obscured ddd, J = 7.3, 5.4, 1.5 Hz, 1H), 3.63−3.59 (m, 2H), 3.52 (dd, J = 12.2, 4.2 Hz, 1H), 3.42 (dd, J = 11.7, 3.9 Hz, 1H), 3.41 (ddd, J = 16.6, 5.4, 2.4 Hz, 1H), 2.33 (ddd, J = 17.1, 7.8, 2.7 Hz, 1H), 2.18−2.10 (partially obscured m, 1H), 2.13 (ddd, J = 12.2, 3.9, 3.9 Hz, 1H), 2.10 (t, J = 2.4 Hz, 1H), 1.94−1.75 (m, 5H), 1.68 (ddd, J = 12.2, 12.2, 12.2 Hz, 1H), 1.51 (ddd, J = 13.2, 5.9, 2.9 Hz, 1H), 1.25 (s, 6H); 13C NMR (125 MHz, CD2Cl2) δ 159.4, 139.3, 131.2, 129.4, 128.5, 127.8, 127.6, 113.8, 83.3, 81.2, 79.5, 78.5, 76.5, 75.4, 73.3, 73.0, 70.8, 70.1, 66.7, 55.4, 41.7, 35.9, 28.7, 26.7, 24.6, 22.2, 20.1; IR (neat) 3419, 2929, 2869, 2339, 1613, 1454, 1374, 1248, 1089 cm−1; ESI/MS (m/z) calcd for C31H40O6Na 531.3 (M+Na+), found 531.3.
(2S,3R,4aS,6R,7S,9aR)-6-allyl-2-(2-(benzyloxy)ethyl)-3-((4-methoxybenzyl)oxy)-2,9a-dimethyloctahydro-2H-pyrano[3,2-b]oxepin-7-ol (13) To a solution of 12 (85.7 mg, 0.169 mmol) in ethyl acetate (10 mL) was added quinoline (10 μL, 0.08 mmol) and Lindlar’s Pd catalyst (10 mg). The mixture was stirred under 1 H2 (1 atm) for 2 h before it was passed through a Celite plug with ethyl acetate. The filtrate was concentrated and flash chromatography (4:1 hexanes:ethyl acetate) gave 13 as a colorless oil (78.8 mg, 92%). Rf 0.55 (2:1 hexanes:ethyl acetate); [α]20D = −14.9° (c = 0.43, THF); 1H NMR (500 MHz, CD2Cl2) δ 7.38−7.28 (m, 5H), 7.26 (d, J = 8.8 Hz, 2H), 6.90 (d, J = 8.3 Hz, 2H), 5.89 (dddd, J = 17.1, 10.3, 6.8, 6.8 Hz, 1H), 5.14−5.07 (m, 2H), 4.58 (d, J = 11.2 Hz, 1H), 4.47 (s, 2H), 4.37 (d, J = 11.2 Hz, 1H), 3.89 (br d, J = 5.9 Hz, 1H), 3.80 (s, 3H), 3.68 (ddd, J = 6.4, 6.4, 1.4 Hz, 1H), 3.62−3.58 (m, 2H), 3.47 (dd, J = 12.2, 4.2 Hz, 1H), 3.41 (dd, J = 11.7, 3.9 Hz, 1H), 2.27−2.15 (m, 2H), 2.10 (ddd, J = 12.2, 4.4, 3.9 Hz, 1H), 1.93−1.80 (m, 4H), 1.78−1.69 (m, 2H), 1.65 (ddd, J = 12.2, 12.2, 12.2 Hz, 1H), 1.49 (ddd, J = 13.2, 5.3, 2.7 Hz, 1H), 1.24 (s, 6H); 13C NMR (125 MHz, CD2Cl2) δ 159.4, 139.3, 135.4, 131.2, 129.4, 128.5, 127.8, 127.5, 116.9, 113.8, 84.7, 79.6, 78.5, 76.5, 75.4, 73.7, 72.9, 70.7, 66.7, 55.4, 41.7, 39.4, 35.9, 28.8, 26.7, 22.3, 20.1; IR (neat) 3423, 2929, 1613, 1513, 1454, 1374, 1248, 1087 cm−1; ESI/MS (m/z) calcd for C31H42O6Na 533.3 (M+Na+), found 533.3.
(2S,3R,4aS,6R,7S,9aR)-6-allyl-2-(2-(benzyloxy)ethyl)-3-((4-methoxybenzyl)oxy)-2,9a-dimethyloctahydro-2H-pyrano[3,2-b]oxepin-7-yl 2-((2R,4aR,5aS,7R,8S,9aR,10aS)-7-hydroxy-4a-methyl-2-phenyloctahydro-4H-pyrano[2′,3′:5,6]pyrano[3,2-d][1,3]dioxin-8-yl)acetate (14). To a solution of acid 7 (36.0 mg, 0.0753 mmol) in THF (8 mL) was added triethylamine (45.9 μL, 0.330 mmol) and 2,4,6-trichlorobenzoyl chloride (35.3 μL, 0.223 mmol). The reaction mixture was heated to 40 °C and stirred for 2 h before being concentrated. A solution of alcohol 13 (36.8 mg, 0.0722 mmol) in toluene (10 mL) was transferred via cannula to the resulting residue. DMAP (42.3 mg, 0.347 mmol) was then added and the reaction mixture was heated at 40 °C for 2 h after which the reaction was quenched with sat. NaHCO3 (aq., 10 mL). The aqueous phase was extracted with CH2Cl2 (3 × 10 mL) and the organic extracts were combined, dried (Na2SO4) and concentrated. Flash chromatography (10:1 hexanes:ethyl acetate) provided ester 14 (67.8 mg, 97%) as a colorless oil. Rf 0.25 (5:1 hexanes:ethyl acetate); [α]20D = −14.8° (c = 0.30, THF); 1H NMR (500 MHz, CD2Cl2) δ 7.46−7.43 (m, 2H), 7.38−7.34 (m, 7H), 7.32−7.27 (m, 1H), 7.22 (d, J = 8.3 Hz, 2H), 6.86 (d, J = 8.8 Hz, 2H), 5.88 (dddd, J = 17.1, 10.3, 6.8, 6.8 Hz, 1H), 5.52 (s, 1H), 5.13 (dd, J = 18.1, 1.0 Hz, 1H), 5.09 (d, J = 10.7 Hz, 1H), 4.99 (d, J = 6.3 Hz, 1H), 4.57 (d, J = 10.7 Hz, 1H), 4.49 (d, J = 11.7 Hz, 1H), 4.45 (d, J = 11.7 Hz, 1H), 4.36 (d, J = 10.7 Hz, 1H), 3.89 (d, J = 10.3 Hz, 1H), 3.79 (s, 3H), 3.75 (dd, J = 6.2, 6.2 Hz, 1H), 3.72−3.58 (m, 5H), 3.55 (ddd, J = 9.8, 9.8, 4.4 Hz, 1H), 3.48 (partially obscured ddd, J =10.8, 9.3, 3.9 Hz, 1H), 3.44 (partially obscured dd, J =11.7, 4.4 Hz, 1H), 3.36 (dd, J = 12.2, 3.9 Hz, 1H), 3.19 (ddd, J = 10.7, 9.3, 4.4 Hz, 1H), 2.87 (dd, J = 14.9, 2.7 Hz, 1H), 2.39 (dd, J = 14.9, 9.5 Hz, 1H), 2.31−2.23 (m, 3H), 2.16 (ddd, J = 11.2, 3.9, 3.9 Hz, 1H), 2.12 (ddd, J = 12.2, 3.9, 3.9 Hz, 1H), 1.97−1.64 (m, 8H), 1.59−1.48 (m, 1H), 1.50 (s, 3H), 1.25 (s, 3H), 1.24 (s, 3H), 0.92 (s, 9H), 0.13 (s, 3H), 0.11 (s, 3H); 13C NMR (125 MHz, CD2Cl2) δ 170.8, 159.4, 139.3, 138.0, 134.8, 131.1, 129.3, 129.2, 128.5, 128.4, 127.8, 127.6, 126.6, 117.2, 113.8, 103.1, 82.6, 80.5, 80.1, 79.9, 78.9, 78.2, 77.1, 76.3, 76.2, 75.4, 73.0, 71.0, 70.4, 70.0, 69.4, 66.8, 55.4, 42.0, 39.6, 39.2, 38.3, 36.6, 30.7, 28.9, 25.8, 23.6, 22.0, 19.8, 18.0, 15.1, −4.1, −4.8; IR (neat) 2932, 2859, 1735, 1613, 1514, 1456, 1378, 1250, 1092 cm−1; ESI/MS (m/z) calcd for C56H78O12SiNa 993.5 (M+Na+), found 993.5.
(2R,4aR,5aS,7R,8S,9aR,10aS)-8-(((4aR,5aS,7R,8S,9aR,11aS)-8-(2-(benzyloxy)ethyl)-7-((4-methoxybenzyl)oxy)-8,9a-dimethyl-4,4a,5a,6,7,8,9a,10,11,11a-decahydrodipyrano[3,2-b:2′,3′-f]oxepin-2-yl)methyl)-4a-methyl-2-phenyloctahydro-4H-pyrano[2′,3′:5,6]pyrano[3,2-d][1,3]dioxin-7-ol (15). To a solution of TiCl4 (0.245 mL, 2.23 mmol) in CH2Cl2 (15 mL) at 0 °C was added THF (1.18 mL, 13.4 mmol) dropwise. To the resulting yellow solution was added TMEDA (2.03 mL, 13.4 mmol) dropwise; the solution turned to a red-brown color. The ice bath was then removed and the mixture was allowed to warm for 15 min. Activated Zn dust (330 mg, 5.08 mmol) and PbCl2 (74.0 mg, 0.266 mmol) were then added. The resulting mixture went through a series of color changes from brown to green to purple and finally to blue-green over the course of 3–5 min. To the slurry was transferred a solution of ester 14 (67.8 mg, 0.0699 mmol) and CH3CHBr2 (0.200 mL, 2.23 mmol) in CH2Cl2 (5 mL) via cannula. The reaction mixture was then heated to reflux for 30 min before it was cooled to 0 °C. The reaction was quenched by adding sat. K2CO3 (aq., 1.0 mL) for 30 min and filtering the mixture. Concentration and flash chromatography (10:1 hexanes:ethyl acetate) gave 15 as a colorless oil (32.9 mg, 50%). Rf 0.20 (5:1 hexanes:ethyl acetate); [α]20D = −40.3° (c = 0.29, THF); 1H NMR (500 MHz, CD2Cl2) δ 7.48−7.45 (m, 2H), 7.40−7.32 (m, 7H), 7.30−7.26 (m, 1H), 7.24 (d, J = 8.3 Hz, 2H), 6.86 (d, J = 8.8 Hz, 2H), 5.56 (s, 1H), 4.56 (d, J = 11.2 Hz, 1H), 4.52 (br d, J = 3.4 Hz, 1H), 4.47 (d, J = 11.7 Hz, 1H), 4.42 (d, J = 11.7 Hz, 1H), 4.37 (d, J = 11.2 Hz, 1H), 3.90 (d, J = 10.2 Hz, 1H), 3.78 (s, 3H), 3.67−3.60 (m, 2H), 3.60−3.52 (m, 2H), 3.50−3.40 (m, 5H), 3.37 (dd, J = 12.2, 3.9 Hz, 1H), 3.31 (ddd, J = 9.3, 9.3, 2.0 Hz, 1H), 3.12 (ddd, J =11.2, 9.3, 4.4 Hz, 1H), 2.54 (d, J = 15.1 Hz, 1H), 2.28 (br d, J = 16.6 Hz, 1H), 2.21 (ddd, J = 11.2, 4.4, 4.4 Hz, 1H), 2.16 (ddd, J = 11.7, 3.9, 3.9 Hz, 1H), 2.13 (ddd, J = 12.2, 3.9, 3.9 Hz, 1H), 2.05−1.66 (m, 10H), 1.50 (s, 3H), 1.47 (ddd, J = 10.7, 10.7, 10.7 Hz, 1H), 1.24 (s, 3H), 1.19 (s, 3H), 0.90 (s, 9H), 0.10 (s, 3H), 0.09 (s, 3H); 13C NMR (125 MHz, CD2Cl2) δ 159.4, 151.6, 139.3, 138.1, 131.1, 129.4, 129.2, 128.5, 128.4, 1279, 127.6, 126.5, 113.8, 103.0, 94.8, 80.6, 80.0, 79.9, 79.7, 79.1, 78.8, 78.0, 77.0, 76.4, 75.6, 73.0, 70.8, 70.7, 69.9, 69.5, 66.5, 55.4, 41.9, 40.0, 39.9, 35.9, 30.7, 29.7, 29.4, 28.8, 25.8, 23.5, 22.5, 18.0, 15.1, −4.1, −4.8; IR (neat) 2933, 2859, 1651, 1513, 1458, 1377, 1250, 1093 cm−1; ESI/MS (m/z) calcd for C55H76O11SiNa 963.5 (M+Na+), found 963.6.
(2R,4aR,5aS,7R,8S,9aR,11aS)-8-(2-(benzyloxy)ethyl)-2-(((2R,4aR,5aS,7R,8S,9aR,10aS)-7-hydroxy-4a-methyl-2-phenyloctahydro-4H-pyrano[2′,3′:5,6]pyrano[3,2-d][1,3]dioxin-8-yl)methyl)-7-((4-methoxybenzyl)oxy)-8,9a-dimethyldecahydrodipyrano[3,2-b:2′,3′-f]oxepin-3(2H)-one (18). To a solution of 15 (28.6 mg, 0.0304 mmol) in CH2Cl2 (5 mL) at −78 °C was added a solution of dimethyl dioxirane (0.61 mL of 0.10 M solution in CH2Cl2, 0.061 mmol) dropwise. The reaction mixture was warmed to 0 °C and then concentrated. The resulting residue was dissolved in CH2Cl2 (5 mL) and cooled to −78 °C. To this was added a solution of DIBALH (0.304 mL of 1.0 M solution in THF, 0.30 mmol). After stirring for 2 h, the reaction was quenched with sat. NH4Cl (aq., 3 mL) and allowed to warm to rt. A solution of sat. potassium sodium tartrate solution (aq, 10 mL) was added and the resulting mixture was stirred for 30 min. The aqueous phase was extracted with CH2Cl2 (3 × 10 mL) and the combined organic phase was dried (Na2SO4) and concentrated. Flash chromatography (5:1 hexanes:ethyl acetate) provided major alcohol 16 (17.5 mg, 60%) and minor alochol (6.1 mg, 21%) both as colorless oil.
To a solution of 16 (17.5 mg, 0.0183 mmol) in CH2Cl2 (5 mL) was added activated 4Å MS (20 mg), NMO (21.4 mg, 0.183 mmol) and TPAP (1 mg, 0.003 mmol). The reaction mixture was stirred at rt for 2 h before being concentrated. Flash chromatography (5:1 hexanes:ethyl acetate) gave ketone 18 as a colorless oil (17.4 mg, 100%). Rf 0.55 (3:1 hexanes:ethyl acetate); [α]20D = −42.0° (c = 0.10, THF); 1H NMR (500 MHz, C6D6) δ 7.60 (d, J = 7.8 Hz, 2H), 7.33 (d, J = 7.8 Hz, 2H), 7.26−7.06 (m, 9H), 6.79 (d, J = 7.8 Hz, 2H), 5.39 (s, 1H), 4.44 (d, J = 11.2 Hz, 1H), 4.41 (d, J = 11.2 Hz, 1H), 4.36 (d, J = 11.7 Hz, 1H), 4.25 (d, J = 11.2 Hz, 1H), 4.08 (dd, J = 7.3, 3.4 Hz, 1H), 3.92 (d, J = 10.2 Hz, 1H), 3.78−3.70 (m, 2H), 3.66−3.60 (m, 1H), 3.56−3.50 (partially obscured m, 1H), 3.53 (partially obscured d, J = 10.2 Hz, 1H), 3.45−3.25 (m, 4H), 3.28 (s, 3H), 3.16 (ddd, J = 8.3, 8.3, 4.4 Hz, 1H), 3.08−3.00 (m, 2H), 2.97 (dd, J = 15.6, 6.4 Hz, 1H), 2.76 (ddd, J =13.2, 7.8, 2.4 Hz, 1H), 2.36−2.22 (m, 3H), 2.16−2.00 (m, 4H), 2.00−1.88 (m, 2H), 1.86−1.62 (m, 6H), 1.43 (s, 3H), 1.33 (s, 3H), 1.18 (s, 3H), 0.98 (s, 9H), 0.06 (s, 3H), 0.02 (s, 3H); 13C NMR (125 MHz, C6D6) δ 205.1, 159.6, 139.4, 138.4, 131.0, 129.2, 128.9, 128.4, 128.2, 127.4, 126.7, 114.0, 102.8, 81.0, 80.2, 80.1, 79.8, 79.5, 79.4, 78.9, 78.3, 76.9, 76.4, 75.7, 73.0, 71.7, 70.6, 69.9, 69.5, 66.5, 54.6, 46.1, 42.2, 40.2, 39.6, 36.7, 33.3, 31.0, 30.1, 29.2, 28.8, 25.8, 24.8, 22.8, 22.6, 18.0, 15.1, −4.1, −4.8; IR (neat) 2931, 2858, 1727, 1513, 1460, 1377, 1250, 1092 cm−1; ESI/MS (m/z) calcd for C55H78O12SiK 995.5 (M+K+), found 995.5.
(2R,3S,4aR,5aS,7R,8S,9aR,11aS)-8-(2-(benzyloxy)ethyl)-2-(((2R,4aR,5aS,7R,8S,9aR,10aS)-7-hydroxy-4a-methyl-2-phenyloctahydro-4H-pyrano[2′,3′:5,6]pyrano[3,2-d][1,3]dioxin-8-yl)methyl)-7-((4-methoxybenzyl)oxy)-8,9a-dimethyldodecahydrodipyrano[3,2-b:2′,3′-f]oxepin-3-yl acetate (17). To a solution of 16 (25.6 mg, 0.0267 mmol) in CH2Cl2 (8 mL) was added Et3N (37.2 μL, 0.267 mmol), Ac2O (12.6 μL, 0.133 mmol), and DMAP (3.3 mg, 0.027 mmol). The reaction mixture was stirred for 2 h before the reaction was quenched with sat. NaHCO3 (aq., 5 mL). After separation the aqueous phase was extracted with CH2Cl2 (3 × 5 mL) and the combined organic phase was dried (Na2SO4) and concentrated. Flash chromatography (5:1 hexanes:ethyl acetate) provided 17 as a colorless oil (21.7 mg, 81%). Rf 0.60 (3:1 hexanes:ethyl acetate); [α]20D = −24.9° (c = 0.24, THF); 1H NMR (500 MHz, C6D6) δ 7.60 (d, J = 7.3 Hz, 2H), 7.33 (d, J = 7.3 Hz, 2H), 7.24−7.08 (m, 8H), 6.79 (d, J = 7.8 Hz, 2H), 5.39 (s, 1H), 4.99 (ddd, J = 10.7, 10.7, 4.4 Hz, 1H), 4.52 (d, J = 11.2 Hz, 1H), 4.42 (d, J = 11.2 Hz, 1H), 4.37 (d, J = 13.2 Hz, 1H), 4.34 (d, J = 12.2 Hz, 1H), 3.92 (d, J = 9.8 Hz, 1H), 3.76 (dd, J = 15.6, 7.3 Hz, 1H), 3.68−3.50 (m, 6H), 3.50−3.37 (m, 2H), 3.30 (s, 3H), 3.20−3.01 (m, 4H), 2.77 (ddd, J = 11.7, 4.4, 4.4 Hz, 1H), 2.44 (ddd, J =12.2, 3.9, 3.9 Hz, 1H), 2.38−2.30 (m, 2H), 2.16−2.02 (m, 3H), 2.02−1.94 (m, 2H), 1.90 (ddd, J = 11.7, 11.7, 11.7 Hz, 1H), 1.85−1.76 (m, 3H), 1.75 (s, 3H), 1.72−1.53 (m, 3H), 1.45 (s, 3H), 1.32 (s, 3H), 1.20 (s, 3H), 0.98 (s, 9H), 0.09 (s, 3H), 0.03 (s, 3H); 13C NMR (125 MHz, C6D6) δ 169.2, 159.6, 139.4, 138.5, 131.2, 129.3, 128.8, 128.4, 128.2, 127.4, 126.7, 114.0, 102.8, 81.5, 81.1, 79.9, 79.7, 79.3, 79.1, 78.3, 77.0, 76.4, 75.7, 73.0, 71.1, 70.6, 69.9, 69.6, 66.6, 54.7, 42.2, 40.4, 39.8, 37.7, 34.5, 30.9, 30.1, 29.5, 29.0, 25.9, 22.7, 22.6, 20.7, 18.0, 15.2, −3.9, −4.6; IR (neat) 2931, 2858, 1740, 1614, 1513, 1458, 1375, 1245, 1090 cm−1; ESI/MS (m/z) calcd for C57H80O13SiNa 1023.5 (M+Na+), found 1023.5.
(2R,4aR,5aS,6aR,7aS,8aR,9aS,11R,12S,13aR,15aS,16aR,17aS,18aR,19aS)-12-(2-(benzyloxy)ethyl)-11-((4-methoxybenzyl)oxy)-4a,12,13a-trimethyl-2-phenylicosahydro-[1,3]dioxino[4″″,5″″:5‴,6‴]pyrano[2‴,3‴:5″″,6″″]pyrano[2″″,3″″:5′,6′]pyrano[2′,3′:5,6]pyrano[3,2-b]pyrano[2,3-f]oxepin-7a(4H)-ol (19). To a solution of 18 (17.7 mg, 0.0185 mmol) in THF (10 mL) at 0 °C was slowly added HF•Py (0.60 mL, 33 mmol). The reaction mixture was allowed to warm to rt and stirred for 2 d before it was quenched with sat. NaHCO3 (aq., 50 mL). The aqueous phase was extracted with ethyl acetate (3 × 10 mL) and the combined organic phase was dried (Na2SO4) and concentrated. Flash chromatography (2:1 hexanes:ethyl acetate) provided 19 as a colorless oil (15.5 mg, 99%). Rf 0.35 (2:1 hexanes:ethyl acetate); [α]20D = −15.8° (c = 0.22, THF); 1H NMR (500 MHz, CD2Cl2) δ 7.48−7.45 (m, 2H), 7.39−7.24 (m, 8H), 7.23 (d, J = 8.3 Hz, 2H), 6.86 (d, J = 8.3 Hz, 2H), 5.56 (s, 1H), 4.55 (d, J = 11.2 Hz, 1H), 4.45 (d, J = 11.7 Hz, 1H), 4.41 (d, J = 12.2 Hz, 1H), 4.36 (d, J = 10.7 Hz, 1H), 4.08 (br d, J = 6.8 Hz, 1H), 3.92 (d, J = 9.8 Hz, 1H), 3.80−3.74 (m, 1H), 3.78 (s, 3H), 3.68−3.62 (m, 2H), 3.59−3.44 (m, 4H), 3.42 (partially obscured dd, J = 11.7, 4.4 Hz, 1H), 3.33−3.27 (m, 1H), 3.22 (ddd, J = 11.7, 2.4, 1.5 Hz, 1H), 3.17 (ddd, J = 12.7, 9.3, 3.4 Hz, 1H), 3.13 (ddd, J = 11.2, 9.3, 3.9 Hz, 1H), 2.23−2.00 (m, 4H), 1.93−1.88 (m, 2H), 1.86−1.73 (m, 3H), 1.73−1.65 (m, 2H), 1.63−1.52 (m, 2H), 1.50 (s, 3H), 1.45 (ddd, J = 11.7, 11.7, 11.7 Hz, 1H), 1.41−1.25 (m, 2H), 1.22 (s, 3H), 1.18 (s, 3H), 1.16−1.95 (m, 1H); 13C NMR (125 MHz, CD2Cl2) δ 138.0, 131.0, 129.3, 129.2, 128.4, 127.8, 127.5, 126.5, 113.8, 103.0, 93.9, 82.2, 81.9, 79.8, 79.6, 79.3, 78.6, 77.6, 77.0, 76.3, 75.6, 72.9, 70.8, 70.2, 68.9, 66.5, 55.4, 49.2, 43.2, 41.8, 39.7, 35.3, 34.1, 30.6, 30.2, 29.2, 28.8, 25.8, 25.2, 22.4, 22.2, 15.0; IR (neat) 3323, 3055, 2927, 1624, 1433, 1265, 1113 cm−1; ESI/MS (m/z) calcd for C49H62O12Na 865.4 (M+Na+), found 865.4.
(4aR,5aS,6aR,7aS,8aR,9aS,11R,12S,13aR,15aS,16aR,17aS,18aR,19aS)-12-(2-(benzyloxy)ethyl)-4a,12,13a-trimethyldocosahydro-[1,3,2]dioxasilino[4″″,5″″:5‴,6‴]pyrano[2‴,3‴:5″,6″]pyrano[2″,3″:5′,6′]pyrano[2′,3′:5,6]pyrano[3,2-b]pyrano[2,3-f]oxepin-11-ol (3). To a solution of 19 (15.9 mg, 0.0189 mmol) in CH2Cl2 (6 mL) and EtSH (2 mL) was added Zn(OTf)2 (140 mg, 0.385 mmol). The reaction mixture was heated to reflux and stirred for 12 h before it was cooled to rt and the reaction quenched with sat. NaHCO3 (aq., 5 mL). The aqueous phase was extracted with CH2Cl2 (3 × 5 mL) and the combined organic phase was dried (Na2SO4) and concentrated. Flash chromatography (20:1 dichloromethane:methanol) provided triol 20 as a colorless oil.
To the solution of the triol obtained above in DMF (10 mL) at −20 °C was added tBu2Si(OTf)2 (14.0 μL, 0.0384 mmol). The reaction mixture was stirred at −20 °C for 1 h before pyridine (6.2 mL, 0.077 mmol) was added. The mixture was stirred for another 5 min and the reaction was quenched with sat. NaHCO3 (aq., 10 mL). The aqueous phase was extracted with Et2O (3 × 10 mL) and the combined organic phase was dried (Na2SO4) and concentrated. Flash chromatography (3:1 hexanes:ethyl acetate) provided a colorless oil which was taken on to the next transformation without additional purification.
To a solution of the colorless oil obtained above in toluene (10 mL) was added Ph3SnH (200 mg, 0.570 mmol). The reaction mixture was heated to reflux and AIBN (0.0046 M solution in toluene, 2.0 mL, 9.2 μmol) was added via syringe pump over 2 h. The mixture was then cooled to rt and solvent was removed under reduced pressure. Flash chromatography (2:1 hexanes:ethyl acetate) provided 3 as a colorless oil (7.3 mg, 51% 3 steps). Rf 0.20 (2:1 hexanes:ethyl acetate); [α]20D = −7.6° (c = 0.10, THF); 1H NMR (500 MHz, CD2Cl2) δ 7.39−7.27 (m, 5H), 4.50 (s, 2H), 4.07 (dd, J = 12.2, 4.4 Hz, 1H), 3.88 (d, J = 10.2 Hz, 1H), 3.75 (d, J = 10.2 Hz, 1H), 3.59 (dd, J = 6.4, 4.4 Hz, 1H), 3.54 (dd, J = 12.2, 3.9 Hz, 1H), 3.40−3.35 (m, 1H), 3.33 (dd, J = 11.7, 3.9 Hz, 1H), 3.26 (ddd, J = 10.8, 9.3, 4.4 Hz, 1H), 3.19 (ddd, J = 12.7, 8.8, 4.4 Hz, 1H), 3.13−2.95 (m, 6H), 2.32−2.24 (m, 2H), 2.18−2.10 (m, 2H), 2.00−1.68 (m, 10H), 1.65 (ddd, J = 11.7, 11.7, 11.7 Hz, 1H), 1.42 (s, 3H), 1.41−1.34 (m, 1H), 1.23 (s, 3H), 1.16 (s, 3H), 1.10 (s, 9H), 1.03 (s, 9H); 13C NMR (125 MHz, CD2Cl2) δ 138.4, 129.0, 128.4, 128.4, 81.9, 81.4, 80.4, 78.6, 77.8, 77.7, 77.6, 77.4, 76.8, 75.0, 74.9, 74.3, 73.7, 73.2, 69.8, 67.1, 43.8, 40.5, 37.4, 35.9, 35.7, 34.0, 32.3, 29.9, 29.3, 28.0, 23.9, 20.4, 20.1, 15.6; IR (neat) 3417, 2925, 2855, 1652, 1457, 1375, 1080 cm−1; FAB/MS (m/z) calcd for C42H66O10SiAg 865.3476 (M+Ag+), found 865.3475.
(2R,4S,5R)-4-allyl-5-methyl-2-phenyl-1,3-dioxan-5-yl acetate (23). To a solution of 22 (1.24 g, 5.30 mmol) in CH2Cl2 (25 mL) was added Et3N (3.70 mL, 26.6 mmol), Ac2O (1.50 mL, 15.6 mmol), and DMAP (0.646 g, 5.30 mmol). The resulting mixture was stirred for 2 h before the reaction was quenched with sat. NaHCO3 (aq., 20 mL). The phases were separated and the aqueous phase was extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were dried (Na2SO4) and concentrated. Flash chromatography (5:1 hexanes:ethyl acetate) provided 23 as a colorless oil (1.15 g, 79%). Rf 0.60 (4:1 hexanes:ethyl acetate); [α]20D = −71.2° (c = 0.32, THF); 1H NMR (500 MHz, C6D6) δ 7.59 (d, J = 7.3 Hz, 2H), 7.18 (t, J = 7.3 Hz, 2H), 7.12 (t, J = 7.3 Hz, 1H), 5.96 (dddd, J = 17.6, 10.3, 7.3, 6.3 Hz, 1H), 5.34 (s, 1H), 5.14 (d, J = 17.1 Hz, 1H), 5.08 (d, J = 10.3 Hz, 1H), 4.99 (d, J = 10.7 Hz, 1H), 3.73 (dd, J = 10.1, 2.7 Hz, 1H), 3.64 (d, J = 10.7 Hz, 1H), 2.40−2.33 (m, 1H), 2.29−2.21 (m, 1H), 1.69 (s, 3H), 1.56 (s, 3H); 13C NMR (125 MHz, C6D6) δ 168.8, 138.5, 135.2, 128.8, 128.2, 126.5, 116.8, 101.7, 81.6, 75.7, 74.2, 33.5, 21.1, 16.1; IR (neat) 2985, 2859, 1737, 1643, 1452, 1367, 1237, 1099 cm−1; ESI/MS (m/z) calcd for C16H20O4Na 299.1 (M+Na+), found 299.1.
(2R,4aR,8aS)-4a,6-dimethyl-2-phenyl-4,4a,8,8a-tetrahydropyrano[3,2-d][1,3]dioxine (24). To a solution of TiCl4 (7.37 mL, 67.2 mmol) in CH2Cl2 (440 mL) at 0 °C was added THF (35.5 mL, 0.404 mol) dropwise. To the resulting yellow solution was added TMEDA (60.9 mL, 0.404 mol) dropwise. The solution turned a red-brown color. The ice bath was removed and the mixture was allowed to stir for 15 min after which activated Zn dust (9.83 g, 0.151 mol) and PbCl2 (2.22 g, 7.98 mmol) were added. The resulting mixture went through a series of color changes from brown to green to purple and finally to blue-green over the course of 10 min. To the resulting slurry was transferred a solution of ester 23 (1.16 g, 4.20 mmol) and CH3CHBr2 (6.02 mL, 67.2 mmol) in CH2Cl2 (20 mL) via cannula. The reaction mixture was then heated to reflux for 2 h before it was cooled to 0 °C. The reaction was quenched by adding sat. K2CO3 (aq., 50 mL). The resulting mixture was stirred for 30 min, filtered and the filtrate was concentrated. Flash chromatography (15:1 hexanes:ethyl acetate) gave 24 as a colorless oil (0.949 g, 92%). Rf 0.70 (5:1 hexanes:ethyl acetate); [α]20D = +23.0° (c = 0.43, THF); 1H NMR (500 MHz, C6D6) δ 7.52 (d, J = 7.3 Hz, 2H), 7.17 (t, J = 7.3 Hz, 2H), 7.12 (t, J = 7.3 Hz, 1H), 5.37 (s, 1H), 4.22 (d, J = 4.4 Hz, 1H), 3.94 (d, J = 9.8 Hz, 1H), 3.65 (dd, J = 10.7, 6.4 Hz, 1H), 3.60 (d, J = 10.3 Hz, 1H), 2.12−1.98 (m, 2H), 1.61 (s, 3H), 1.34 (s, 3H); 13C NMR (125 MHz, C6D6) δ 148.5, 138.5, 128.8, 128.1, 126.6, 102.3, 92.9, 77.1, 75.7, 69.6, 24.0, 19.9, 15.3; IR (neat) 2921, 2859, 1673, 1453, 1378, 1312, 1150, 1093 cm−1; ESI/MS (m/z) calcd for C15H18O3Na 269.1 (M+Na+), found 269.1.
(2R,4aR,6S,8aS)-6-allyl-4a,6-dimethyl-2-phenyltetrahydropyrano[3,2-d][1,3]dioxin-7(6H)-one (26). To a solution of 24 (0.576 g, 2.34 mmol) in MeOH (30 mL) at −78 °C was added mCPBA (77%, 1.57 g, 7.00 mmol). The reaction mixture was slowly warmed up to 0 °C, at which point sat. NaHCO3 (aq., 30 mL) was added. The mixure was extracted with CH2Cl2 (3 × 15 mL) and the combined organic extracts were dried (Na2SO4) and concentrated. Flash chromatography (3:1 hexanes:ethyl acetate) provided a white solid (0.550 g, 80%) which was used in the next transformaiton without additional purification.
To a solution of the material obtained above in THF (30 mL) was added NaH (88.0 mg, 3.67 mmol), allyl bromide (0.800 mL, 9.19 mmol), and TBAI (50.0 mg, 0.135 mmol). The resulting reaction mixture was heated to reflux overnight. After cooling to rt, the reaction was quenched with sat. NH4Cl (aq., 20 mL). The aqueous phase was extracted with CH2Cl2 (3 × 15 mL) and the organic extracts were combined, dried (Na2SO4) and concentrated. The resulting residue was passed through a short silica gel plug (5:1 hexanes:ethyl acetate) and concentrated to give a colorless oil 25 which was taken into the subsequent transformation without additional purification.
To the colorless oil obtained above in toluene (50 mL) was added pyridine (7.40 mL, 91.6 mmol) and PPTS (2.76 g, 11.0 mmol). The resulting reaction mixture was heated to reflux for 16 h. The mixture was cooled to rt and the reaction was quenched with sat. NaHCO3 (aq., 30 mL). The aqueous phase was extracted with CH2Cl2 (3 × 20 mL) and the combined organic extracts were dried (Na2SO4) and concentrated. Flash chromatography (5:1 hexanes:ethyl acetate) provided 26 (0.409 g, 72% 2 steps). Rf 0.70 (3:1 hexanes:ethyl acetate); [α]20D = +11.2° (c = 0.43, THF); 1H NMR (500 MHz, C6D6) δ 7.50 (d, J = 7.3 Hz, 2H), 7.20 (t, J = 7.3 Hz, 2H), 7.14 (t, J = 7.3 Hz, 1H), 5.77 (dddd, J = 17.6, 10.3, 7.3, 7.3 Hz, 1H), 5.31 (s, 1H), 5.03 (d, J = 10.3 Hz, 1H), 4.99 (d, J = 17.6 Hz, 1H), 3.85 (d, J = 10.3 Hz, 1H), 3.81 (dd, J = 12.2, 7.0 Hz, 1H), 3.47 (d, J = 10.3 Hz, 1H), 2.61 (dd, J = 18.6, 6.8 Hz, 1H), 2.41 (dd, J = 18.6, 11.7 Hz, 1H), 2.30 (dd, J = 13.2, 7.5 Hz, 1H), 2.14 (dd, J = 13.7, 7.5 Hz, 1H), 1.25 (s, 3H), 1.19 (s, 3H); 13C NMR (100 MHz, C6D6) δ 208.8, 138.1, 133.0, 128.9, 128.2, 126.5, 119.1, 101.8, 83.8, 76.3, 68.3, 46.4, 39.4, 26.5, 17.1; IR (neat) 2982, 2865, 1716, 1521, 1456, 1372, 1143, 1114 cm−1; ESI/MS (m/z) calcd for C18H22O4Na 325.2 (M+Na+), found 325.1.
Summary of COSY spectrum for 26
Proton at 5.77 ppm (C-25) shows cross peaks with protons at 5.03 ppm (C-26), 4.99 ppm (C-26′), 2.30 ppm (C-24) and 2.14 (C-24′).
Proton at 3.85 ppm (C-18) shows cross peaks with proton at 3.47 ppm (C-18′).
Proton at 3.81 ppm (C-20) shows cross peaks with protons at 2.61 ppm (C-21) and 2.41 ppm (C-21′).
Proton at 2.61 ppm (C-21) shows cross peaks with proton at 2.41 ppm (C-21′).
Proton at 2.30 ppm (C-24) shows cross peaks with proton at 2.14 ppm (C-24′).
Summary of 1D nOe spectrum for 26
Irradiation at 1.25 ppm (C-23 methyl) resulted in enhancement at 1.19 ppm (C-19 methyl) and 2.30 ppm (C-24) and 2.14 ppm (C-24′).
(2R,4aR,6S,7R,8aS)-6-allyl-7-((4-methoxybenzyl)oxy)-4a,6-dimethyl-2-phenylhexahydropyrano[3,2-d][1,3]dioxine (27). To a solution of 26 (378 mg, 1.25 mmol) in MeOH (25 mL) at 0 °C was added NaBH4 (190 mg, 5.0 mmol). The reaction was quenched with acetone (10 mL) after the reaction mixture had stirred for 2. The solvent was then removed under reduced pressure and the residue was purified using flash chromatography (3:1 hexanes:ethyl acetate) to give the corresponding alcohol as a colorless oil (382 mg, 100%).
To a solution of the alcohol obtained above (382 mg, 1.25 mmol) in THF (20 mL) at 0 °C was added NaH (82.4 mg, 3.43 mmol), PMBBr (0.82 mL, 5.69 mmol), HMPA (0.220 mL, 1.15 mmol), and TBAI (50 mg, 0.135 mmol). The reaction mixture was stirred at rt for 12 h before the reaction was quenched with sat. NH4Cl (aq., 20 mL). The mixure was extracted with CH2Cl2 (3 × 15 mL) and the combined organic extracts were dried (Na2SO4) and concentrated. Flash chromatography (10:1 hexanes:ethyl acetate) gave 27 as a colorless oil (442 mg, 83%). Rf 0.55 (5:1 hexanes:ethyl acetate); [α]20D = −32.8° (c = 0.30, THF); 1H NMR (500 MHz, C6D6) δ 7.64 (d, J = 7.8 Hz, 2H), 7.27−7.12 (m, 5H), 6.82 (d, J = 7.8 Hz, 2H), 6.07 (dddd, J = 17.1, 9.8, 7.3, 7.3 Hz, 1H), 5.39 (s, 1H), 5.14 (dd, J = 9.8, 1.3 Hz, 1H), 5.04 (dd, J = 17.6, 1.0 Hz, 1H), 4.44 (d, J = 11.2 Hz, 1H), 4.24 (d, J = 11.7 Hz, 1H), 3.85 (d, J = 9.7 Hz, 1H), 3.44 (dd, J = 11.2, 4.4 Hz, 1H), 3.40 (d, J = 9.7 Hz, 1H), 3.32 (s, 3H), 3.23 (dd, J = 12.7, 3.4 Hz, 1H), 2.46 (dd, J = 13.7, 6.4 Hz, 1H), 2.33 (dd, J = 14.2, 7.8 Hz, 1H), 2.18 (ddd, J = 11.7, 3.9, 3.9 Hz, 1H), 1.85 (ddd, J = 11.7, 11.7, 11.7 Hz, 1H), 1.53 (s, 3H), 1.34 (s, 3H); 13C NMR (125 MHz, C6D6) δ 159.7, 138.7, 135.0, 130.8, 129.3, 129.3, 128.8, 128.2, 126.7, 117.4, 114.0, 102.7, 80.6, 78.6, 77.7, 76.9, 71.5, 70.3, 68.9, 54.7, 46.6, 26.8, 23.3, 19.1; IR (neat) 2947, 2864, 1611, 1512, 1461, 1376, 1247, 1144, 1086 cm−1; ESI/MS (m/z) calcd for C26H32O5Na 447.2 (M+Na+), found 447.2.
(2R,4aR,6S,7R,8aS)-6-(but-3-en-1-yl)-7-((4-methoxybenzyl)oxy)-4a,6-dimethyl-2-phenylhexahydropyrano[3,2-d][1,3]dioxine (28). To a solution of 27 (0.245 g, 0.578 mmol) in THF (15 mL) at 0 °C was added BH3•DMS (0.87 mL of 2.0 M solution in THF, 1.7 mmol). After stirring for 2 h H2O (1.0 mL), NaOH (2.0 mL of 3.0 M aq. solution), and H2O2 (5.0 mL of 30% aq. solution) were added to the reaction mixture. The mixture was warmed to rt and stirred overnight. The aqueous phase was extracted with ethyl acetate (3 × 10 mL). The combined organic extracts were dried (Na2SO4) and concentrated.
To the product obtained above in CH2Cl2 (15 mL) was added 4Å MS (0.60 g), NMO (0.680 g, 5.78 mmol), and TPAP (5 mg, 14.2 μmol). The resulting mixture was stirred at rt for 2 h and then concentrated. The residue was filtered through a short sílica gel plug (5:1 hexanes:ethyl acetate) to give the crude aldehyde.
To a slurry of methyltriphenylphosphonium bromide (1.03 g, 2.88 mmol) in THF (10 mL) was added tBuOK (2.9 mL of 1.0 M solution in THF, 2.9 mmol). After stirring at rt for 30 min, the resulting solution was transferred to a solution of the crude aldehyde from above in THF (10 mL). After 2 h the reaction was quenched with NH4Cl (aq., 10 mL). The aqueous phase was extracted with CH2Cl2 (3 × 10 mL). The organic extracts were combined, dried (Na2SO4) and concentrated. Flash chromatography (10:1 hexanes:ethyl acetate) gave 28 as a colorless oil (0.154 g, 61% for 3 steps). Rf 0.60 (5:1 hexanes:ethyl acetate); [α]20D = −55.4° (c = 0.37, THF); 1H NMR (500 MHz, C6D6) δ 7.64 (d, J = 7.3 Hz, 2H), 7.22 (t, J = 7.3 Hz, 2H), 7.18−7.12 (m, 3H), 6.81 (d, J = 8.3 Hz, 2H), 5.85 (dddd, J = 17.1, 10.2, 6.8, 6.3 Hz, 1H), 5.42 (s, 1H), 5.09 (dd, J = 17.1, 1.5 Hz, 1H), 5.00 (dd, J = 10.2, 1.5 Hz, 1H), 4.44 (d, J = 11.2 Hz, 1H), 4.20 (d, J = 11.7 Hz, 1H), 3.84 (d, J = 9.8 Hz, 1H), 3.39 (d, J = 10.8 Hz, 1H), 3.36 (dd, J = 11.2, 4.9 Hz, 1H), 3.31 (s, 3H), 3.24 (dd, J = 12.2, 3.4 Hz, 1H), 2.24−2.12 (m, 3H), 1.86 (ddd, J = 11.7, 11.7, 11.7 Hz, 1H), 1.81 (ddd, J = 13.2, 11.2, 5.4 Hz, 1H), 1.64 (ddd, J = 13.7, 11.2, 5.4 Hz, 1H), 1.53 (s, 3H), 1.33 (s, 3H); 13C NMR (125 MHz, C6D6) δ 160.2, 140.0, 139.2, 131.1, 129.9, 129.3, 128.7, 127.2, 114.6, 114.5, 103.2, 81.3, 79.5, 77.9, 77.5, 70.7, 69.3, 55.2, 42.1, 28.3, 27.1, 23.5, 19.6; IR (neat) 2943, 2863, 1639, 1513, 1460, 1375, 1248, 1086 cm−1; ESI/MS (m/z) calcd for C27H34O5K 477.2 (M+K+), found 477.2.
(2R,4aR,6S,7R,8aS)-7-((4-methoxybenzyl)oxy)-4a,6-dimethyl-6-(pent-4-en-1-yl)-2-phenylhexahydropyrano[3,2-d][1,3]dioxine (29). To a solution of 28 (74.6 mg, 0.170 mmol) in THF (5 mL) at 0 °C was added BH3•DMS (0.340 mL of 2.0 M solution in THF, 0.680 mmol). The reaction mixture was stirred for 2 before it was quenched with H2O (1.0 mL), NaOH (1.0 mL of 3.0 M aq. solution), and H2O2 (3.0 mL of 30% aq. solution). The reaction mixture was then warmed to rt and stirred overnight. The aqueous phase was extracted with ethyl acetate (3 × 10 mL). The combined organic phase was dried (Na2SO4) and concentrated.
To the crude product obtained from above in CH2Cl2 (10 mL) was added 4Å MS (0.20 g), NMO (0.200 g, 1.71 mmol), and TPAP (2.0 mg, 5.7 μmol). After stirring for 2 h the reaction mixture was concentrated. The resulting residue was filtered through a short silica gel plug (5:1 hexanes:ethyl acetate) to give the crude aldehyde.
To a slurry of methyltriphenylphosphonium bromide (300. mg, 0.840 mmol) in THF (5 mL) was added tBuOK (0.84 mL of 1.0 M solution in THF, 0.84 mmol). After stirring at rt for 30 min, the solution was transferred to a solution of the crude aldehyde from above in THF (5 mL). The reaction mixture was stirred for an additional 2 h before the reaction was quenched with NH4Cl (aq., 10 mL). The aqueous phase was extracted with CH2Cl2 (3 × 10 mL). The organic extracts were combined, dried (Na2SO4) and concentrated. Flash chromatography (15:1 hexanes:ethyl acetate) gave 29 as a colorless oil (42.1 mg, 55% for 3 steps). Rf 0.60 (5:1 hexanes:ethyl acetate); [α]20D = −24.2° (c = 0.24, THF); 1H NMR (500 MHz, C6D6) δ 7.66 (d, J = 7.3 Hz, 2H), 7.22 (t, J = 7.3 Hz, 2H), 7.20−7.12 (m, 3H), 6.82 (d, J = 8.8 Hz, 2H), 5.83 (dddd, J = 16.6, 9.8, 6.3, 6.3 Hz, 1H), 5.41 (s, 1H), 5.08 (dd, J = 17.1, 1.5 Hz, 1H), 5.02 (d, J = 10.2 Hz, 1H), 4.46 (d, J = 11.7 Hz, 1H), 4.20 (d, J = 11.7 Hz, 1H), 3.85 (d, J = 9.8 Hz, 1H), 3.40 (d, J = 10.3 Hz, 1H), 3.37 (dd, J = 11.2, 4.9 Hz, 1H), 3.31 (s, 3H), 3.25 (dd, J = 12.2, 3.4 Hz, 1H), 2.20 (ddd, J = 9.7, 3.9, 3.9 Hz, 1H), 2.02−1.96 (m, 3H), 1.88 (ddd, J = 11.7, 11.7, 11.7 Hz, 1H), 1.73−1.66 (m, 1H), 1.55 (s, 3H), 1.55−1.45 (m, 3H), 1.35 (s, 3H); 13C NMR (125 MHz, C6D6) δ 159.7, 139.1, 138.7, 130.7, 129.5, 128.9, 128.2, 126.7, 114.5, 114.0, 102.7, 80.8, 78.9, 77.7, 77.1, 70.2, 68.8, 54.7, 42.0, 34.6, 26.6, 23.1, 22.6, 19.2; IR (neat) 2943, 2864, 1639, 1513, 1461, 1375, 1248, 1086 cm−1; ESI/MS (m/z) calcd for C28H36O5Na 475.3 (M+Na+), found 475.2.
(2R,4aR,6S,7R,8aS)-2-(4-methoxyphenyl)-4a,6-dimethyl-6-(pent-4-en-1-yl)hexahydropyrano[3,2-d][1,3]dioxin-7-ol (30). To a solution of 29 (42.1 mg, 93.1 μmol) in MeOH (5 mL) at 0 °C was added CSA (6.5 mg, 28 μmol). The reaction mixture was warmed to rt and stirred for 5 h before the reaction was quenched with sat. NaHCO3 (aq., 5 mL). The aqueous phase was extracted with ethyl acetate (3 × 5 mL). The combined organic extracts were dried (Na2SO4), and concentrated. Flash chromatography (3:1 hexanes:ethyl acetate) gave the corresponding diol as a colorless oil (29.3 mg, 86%).
To a solution of the diol obtained above in CH2Cl2 (4.5 mL) was added pH 7 buffer (0.5 mL) and DDQ (36.5 mg, 0.161 mmol). The reaction mixture was stirred at rt for 2 h before H2O (5 mL) was added. The aqueous phase was extracted with ethyl acetate (3 × 5 mL) and the combined organic extracts were dried (Na2SO4), and concentrated. Flash chromatography (2:1 hexanes:ethyl acetate) gave the corresponding triol as a colorless oil (18.9 mg, 96%).
To a solution of the triol obtained from the procedure outlined in the previous paragraph in CH2Cl2 (5 mL) was added anisaldehyde dimethylacetal (19.8 μL, 0.116 mmol) and CSA (9.0 mg, 39 μmol). The reaction mixture was stirred at rt overnight before the reaction was quenched with NaHCO3 (aq., 5 mL). The aqueous phase was extracted with ethyl acetate (3 × 5 mL) and the combined organic extracts were dried (Na2SO4), and concentrated. Flash chromatography (3:1 hexanes:ethyl acetate) gave 30 as a colorless oil (24.8 mg, 88%). Rf 0.25 (3:1 hexanes:ethyl acetate); [α]20D = −9.3° (c = 0.15, THF); 1H NMR (500 MHz, C6D6) δ 7.57 (d, J = 8.3 Hz, 2H), 6.83 (d, J = 8.3 Hz, 2H), 5.84 (dddd, J = 17.1, 10.3, 6.8, 6.8 Hz, 1H), 5.42 (s, 1H), 5.08 (d, J = 17.6 Hz, 1H), 5.02 (d, J = 10.3 Hz, 1H), 3.86 (d, J = 9.8 Hz, 1H), 3.42 (d, J = 9.8 Hz, 1H), 3.38 (dd, J = 12.2, 4.9 Hz, 1H), 3.30−3.26 (partially obscured m, 1H), 3.26 (s, 3H), 2.05−2.00 (m, 2H), 1.87 (ddd, J = 11.2, 4.4, 4.4 Hz, 1H), 1.80 (ddd, J = 11.7, 11.7, 11.7 Hz, 1H), 1.64−1.58 (m, 2H), 1.51 (s, 3H), 1.53−1.41 (m, 2H), 1.15 (s, 3H); 13C NMR (125 MHz, C6D6) δ 160.9, 139.6, 131.6, 128.5, 115.1, 114.2, 103.3, 81.3, 78.6, 77.5, 73.7, 69.4, 55.1, 42.6, 35.1, 31.4, 23.1, 22.2, 19.6; IR (neat) 3483, 2943, 2867, 1615, 1517, 1465, 1377, 1250, 1069 cm−1; ESI/MS (m/z) calcd for C21H30O5Na 385.2 (M+Na+), found 385.2.
(4aR,6S,7S,8aR)-6-allyl-7-hydroxy-7-methyltetrahydropyrano[3,2-d][1,3,2]dioxasilin-8(4H)-one (32). To a solution of (COCl)2 (0.53 mL, 6.2 mmol) in CH2Cl2 (40 mL) at −78 °C was added DMSO (0.58 mL, 8.2 mmol) dropwise. To this was added a solution of 31 (1.90 g, 4.08 mmol) in CH2Cl2 (10 mL) via cannula. After stirring at −78 °C for 45 min, Et3N (2.28 mL, 16.4 mmol) was added and the reaction mixture was stirred for another 5 min. The reaction was quenched with sat. NH4Cl (aq., 50 mL) and allowed to warm to rt. The aqueous phase was extracted with CH2Cl2 (3 × 50 mL) and the combined organic extracts were dried (Na2SO4) and concentrated. Flash chromatography (20:1 hexanes:ethyl acetate) provided the corresponding ketone (1.85 g, 98%) as a colorless oil.
To a solution of the ketone obtained from the protocol outlined in the previous paragraph (1.80 g, 4.17 mmol) in 80 mL toluene at −90 °C was added MeLi (18 mL of 1.6 M in diethyl ether, 29 mmol) dropwise. After stirring the reaction mixture for 1 h at −90 °C, the reaction was quenched with sat. NH4Cl (aq., 50 mL) and allowed to warm to rt. The aqueous phase was extracted with CH2Cl2 (3 × 20 mL) and the combined organic extracts were dried (Na2SO4) and concentrated. Flash chromatography (20:1 hexanes:ethyl acetate) provided the corresponding tertiary alcohol (1.56 g, 84%) as a colorless oil.
To a solution of the tertiary alcohol from above (0.80 g, 1.67 mmol) in CH2Cl2 (27 mL) was added pH 7 buffer (3 mL) and DDQ (0.76 g, 3.4 mmol). The reaction mixture was stirred at rt for 2 h before H2O (20 mL) was added. The aqueous phase was extracted with ethyl acetate (3 × 15 mL) and the combined organic extracts were dried (Na2SO4), and concentrated. Flash chromatography (2:1 hexanes:ethyl acetate) gave the corresponding diol as a colorless oil (0.57 g, 95%).
To a solution of the diol from above (0.11 g, 0.31 mmol) in a mixture of CH2Cl2 (6 mL) and DMSO (3 mL) were added Et3N (0.21 mL, 1.65 mmol) and SO3•Py (0.24 g, 1.65 mmol). The reaction mixture was stirred for 3 h at rt before the reaction was quenched with H2O (10 mL). The aqueous phase was extracted with CH2Cl2 (3 × 5 mL) and the combined organic extracts were dried (Na2SO4), and concentrated. Flash chromatography (10:1 hexanes:ethyl acetate) gave 32 as a colorless oil (0.10 g, 91%). Rf 0.60 (5:1 hexanes:ethyl acetate); [α]20D = −54.2° (c = 0.57, THF); 1H NMR (500 MHz, CDCl3) δ 5.72 (dddd, J = 17.0, 10.0, 6.6, 6.6 Hz, 1H), 4.98 (dd, J = 17.0, 1.3 Hz, 1H), 4.93 (d, J = 10.0 Hz, 1H), 4.56 (d, J = 9.7 Hz, 1H), 4.14 (dd, J = 10.2, 4.6 Hz, 1H), 3.98 (s, 1H), 3.89 (dd, J = 10.1, 10.1 Hz, 1H), 3.34 (ddd, J = 9.8, 9.8, 4.6 Hz, 1H), 3.18 (dd, J = 9.9, 2.1 Hz, 1H), 2.32 (dd, J = 14.4, 7.1 Hz, 1H), 2.13 (ddd, J = 16.1, 9.8, 6.4 Hz, 1H), 1.22 (s, 3H), 0.93 (s, 9H), 0.87 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 206.4, 134.8, 117.2, 85.9, 78.4, 77.9, 77.7, 66.9, 32.9, 27.4, 27.1, 22.8, 20.2, 19.9; IR (neat) 3482, 2937, 2863, 1736, 1473, 1390, 1162, 1112 cm−1; ESI/MS (m/z) calcd for C18H32O5SiNa 379.2 (M+Na+), found 379.2.
(4aR,6S,7R,8R,8aS)-6-(3-hydroxypropyl)-7-methylhexahydropyrano[3,2-d][1,3,2]dioxasiline-7,8-diol (34). To a solution of 32 (1.56 g, 4.38 mmol) in CH2Cl2 (40 mL) at 0 °C was added 2,6-lutidine (3.10 mL, 26.6 mmol) and TMSOTf (2.38 mL, 13.3 mmol). The reaction mixture was allowed to slowly warm up to rt over 2 h before it was quenched with H2O (20 mL). The aqueous phase was extracted with CH2Cl2 (3 × 10 mL). The combined organic phase was dried (Na2SO4) and concentrated. Flash chromatography (20:1 hexanes:ethyl acetate)gave the TMS ether as a colorless oil (1.90 g, 100%).
To a solution of TMS ether (1.90 g, 4.44 mmol) in THF (40 mL) at −78 °C was added L-Selectride (6.7 mL of 1.0 M solution in THF, 6.7 mmol). The reaction mixture was allowed to slowly warm to rt over 2 h before the reaction was quenched with sat. NH4Cl (aq., 20 mL). The aqueous phase was extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts wer dried (Na2SO4) and concentrated. The resulting residue was passed through a plug of silica gel (10:1 hexanes:ethyl acetate) and concentrated giving a colorless oil that was used in the subsequente transformation without additional purification.
To a solution of the colorless oil obtained from the above procedure in THF (15 mL) at 0 °C was added BH3•DMS (2.0 M solution in THF, 11 mL, 22 mmol). H2O (1.0 mL), NaOH (25 mL of 3.0 M aq. solution), and H2O2 (30 mL of 30% aq. solution) were added after 2 h. The reaction mixture was warmed to rt and stirred overnight. The aqueous phase was extracted with ethyl acetate (3 × 20 mL). The combined organic exgtracts were dried (Na2SO4) and concentrated. Flash chromatography gave 34 as a colorless oil (1.17 g, 70% over 2 steps). Rf 0.35 (1:1 hexanes:ethyl acetate); [α]20D = −39.3° (c = 0.29, THF); 1H NMR (500 MHz, CDCl3) δ 4.10 (dd, J = 9.5, 4.6 Hz, 1H), 3.78 (dd, J = 9.3, 2.7 Hz, 1H), 3.73 (dd, J = 10.2, 10.2 Hz, 1H), 3.71−3.63 (m, 2H), 3.58−3.53 (m, 2H), 3.43 (d, J = 10.2 Hz, 1H), 3.40 (br s, 1H), 3.16 (br s, 1H), 3.07 (br s, 1H), 1.76 (ddd, J = 14.2, 7.1, 7.1 Hz, 1H), 1.71−1.62 (m, 1H), 1.60−1.50 (m, 1H), 1.29−1.21 (m, 1H), 1.08 (s, 3H), 0.98 (s, 9H), 0.95 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 78.9, 75.8, 74.3, 71.8, 70.9, 67.0, 62.5, 30.1, 27.6, 27.4, 24.6, 22.9, 20.4, 18.8; IR (neat) 3414, 2936, 2893, 2862, 1472, 1393, 1157, 1102 cm−1; ESI/MS (m/z) calcd for C18H36O6SiNa 399.2 (M+Na+), found 399.3.
(2R,3R,4R,5S,6S)-4,5-bis(benzyloxy)-6-(3-(benzyloxy)propyl)-2-(hydroxymethyl)-5-methyltetrahydro-2H-pyran-3-ol (35). To a solution of 34 (1.17 g, 3.11 mmol) in THF (40 mL) was added KH (3.0 g of 30% dispersion in mineral oil, 22 mmol). The mixture was stirred at rt for 30 min and then cooled to 0 °C. To the resulting mixture was added BnBr (4.22 mL, 35.5 mmol) dropwise. The reaction mixture was warmed to rt and stirred overnight before the reaction was quenched with sat. NH4Cl (aq., 20 mL). The aqueous phase was extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were dried (Na2SO4) and concentrated to give a yellow oil which was taken to the next step without additional purification.
To the residue from above in THF (20 mL) at 0 °C was added HF•Py (1.0 mL, 55 mmol). The resulting reaction mixture was allowed to warm up to rt overnight. The reaction was quenched with sat. NaHCO3 (aq., 20 mL) and the aqueous phase was extracted with ethyl acetate (3 × 20 mL). The organic extracts were combined, dried (Na2SO4) and concentrated. Flash chromatography (1:1 hexanes:ethyl acetate) provided diol 35 (0.98 g, 61% over 2 steps) as a colorless oil. Rf 0.40 (1:1 hexanes:ethyl acetate); [α]20D = −31.4° (c = 0.18, THF); 1H NMR (500 MHz, CDCl3) δ 7.50−7.35 (m, 15H), 5.02 (d, J = 11.2 Hz, 1H), 4.80 (d, J = 11.2 Hz, 1H), 4.72 (d, J = 11.5 Hz, 1H), 4.66 (d, J = 11.5 Hz, 1H), 4.62 (s, 2H), 4.08 (d, J = 3.9 Hz, 1H), 3.98 (d, J = 10.2 Hz, 1H), 3.95 (partially obscured br d, J = 14.6 Hz, 1H), 3.88−3.76 (m, 2H), 3.70 (ddd, J = 10.0, 3.4, 3.4 Hz, 1H), 3.66−3.62 (m, 2H), 3.14−3.04 (m, 2H), 2.10−1.98 (m, 2H), 1.90−1.78 (m, 1H), 1.60−1.48 (m, 1H), 1.38 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 139.2, 139.1, 139.0, 128.8, 128.7, 128.0, 127.9, 127.8, 127.7, 127.3, 81.2, 78.1, 76.9, 76.8, 75.4, 73.1, 70.8, 68.0, 63.9, 63.2, 27.3, 25.3, 16.6; IR (neat) 3421, 2930, 2874, 1496, 1454, 1380, 1210, 1106 cm−1; ESI/MS (m/z) calcd for C31H38O6Na 529.3 (M+Na+), found 529.3.
(2R,3R,4R,5S,6S)-4,5-bis(benzyloxy)-6-(3-(benzyloxy)propyl)-5-methyl-2-(prop-2-yn-1-yl)tetrahydro-2H-pyran-3-ol (37). To a solution of 35 (0.57 g, 1.13 mmol) and 2,6-lutidine (0.495 mL, 4.25 mmol) in CH2Cl2 (20 mL) at −78 °C was added trifluoromethanesulfonic anhydride (0.192 mL, 0.114 mmol). After stirring for 30 min TESOTf (0.288 mL, 0.127 mmol) was added to the reaction mixture. After warming to rt the reaction was quenched with sat. NaHCO3 (aq., 10 mL). The aqueous phase was extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were dried (Na2SO4) and concentrated to give a yellow oil. The oil was passed through a short plug of silica gel (10:1 hexanes:ethyl acetate) and concentrated to give triflate 36.
To a solution of trimethylsilylacetylene (0.690 mL, 4.88 mmol) in THF (20 mL) at 0 °C was added nBuLi (1.94 mL of 2.5 M solution in hexanes, 4.85 mmol). The reaction mixture was stirred for 30 min and then cooled to −78 °C. To the resulting solution of acetylide was added a solution of 36 and HMPA (0.900 mL, 5.17 mmol) in THF (10 mL) via cannula. The reaction mixture was warmed to rt before the reaction was quenched with sat. NH4Cl (aq., 10 mL). The aqueous phase was extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were dried (Na2SO4) and concentrated to give a dark brown oil which was taken up in THF (15 mL). To the solution was added TBAF (4.90 mL of 1.0 M solution in THF, 4.90 mmol) and the mixture was stirred at rt overnight before it was quenched with sat. NH4Cl (aq., 10 mL). The aqueous phase was extracted with CH2Cl2 (3 × 10 mL). The combined organic phase was dried (Na2SO4) and concentrated. Flash chromatography (4:1 hexanes:ethyl acetate) gave 37 as a colorless oil (0.34 g, 59% over 3 steps). Rf 0.20 (4:1 hexanes:ethyl acetate); [α]20D = −21.6° (c = 0.45, THF); 1H NMR (500 MHz, CDCl3) δ 7.42−7.27 (m, 15H), 5.01 (d, J = 11.0 Hz, 1H), 4.72 (d, J = 11.5 Hz, 1H), 4.65 (d, J = 11.5 Hz, 1H), 4.56 (s, 2H), 4.54 (d, J = 11.5 Hz, 1H), 4.02 (d, J = 1.7 Hz, 1H), 3.85 (d, J = 9.8 Hz, 1H), 3.64−3.58 (m, 4H), 2.67 (ddd, J = 17.1, 2.4, 2.4 Hz, 1H), 2.47 (ddd, J = 17.1, 5.9, 2.7 Hz, 1H), 2.30−2.23 (m, 1H), 2.01 (t, J = 2.7 Hz, 1H), 2.00−1.90 (m, 2H), 1.81−1.70 (m, 1H), 1.50−1.40 (m, 1H), 1.34 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 139.1, 138.9, 138.7, 128.8, 128.6, 128.5, 128.2, 128.0, 127.8, 127.6, 127.5, 127.1, 81.3, 81.2, 78.3, 77.0, 75.5, 75.2, 73.0, 70.8, 69.9, 64.0, 27.1, 25.1, 22.4, 16.2; IR (neat) 3420, 2928, 2873, 2119, 1606, 1496, 1454, 1378, 1209, 1104 cm−1; ESI/MS (m/z) calcd for C33H38O5K 553.3 (M+K+), found 553.2.
(((2R,3R,4R,5S,6S)-2-allyl-4,5-bis(benzyloxy)-6-(3-(benzyloxy)propyl)-5-methyltetrahydro-2H-pyran-3-yl)oxy)triethylsilane (38). To a solution of 37 (600 mg, 1.17 mmol) in CH2Cl2 (20 mL) at 0 °C was added 2,6-lutidine (0.272 mL, 2.34 mmol) and TESOTf (0.343 mL, 1.52 mmol). The reaction mixture was allowed to slowly warm to rt over 3 h before the reaction was quenched with sat. NaHCO3 (aq., 10 mL). The aqueous phase was extracted with CH2Cl2 (3 × 10 mL) and the combined organic extracts were dried (Na2SO4) and concentrated. Flash chromatography (5:1 hexanes:ethyl acetate) provided the corresponding TES ether (732 mg, 100%) as a colorless oil.
To a solution of the TES ether obtained from above in ethyl acetate (30 mL) was added quinoline (70 μL, 0.59 mmol) and Lindlar’s Pd catalyst (50 mg). The reaction mixture was stirred H2 (1 atm) for 2 h before being passed through a Celite plug using ethyl acetate. The filtrate was concentrated and flash chromatography (10:1 hexanes:ethyl acetate) gave 38 as a colorless oil (638 mg, 87%). Rf 0.50 (15:2 hexanes:ethyl acetate); [α]20D = +1.2° (c = 0.45, THF); 1H NMR (500 MHz, C6D6) δ 7.44 (d, J = 7.3 Hz, 2H), 7.33 (d, J = 7.3 Hz, 2H), 7.28 (d, J = 7.3 Hz, 2H), 7.20−7.04 (m, 9H), 6.08 (dddd, J = 17.1, 11.2, 6.8, 6.8 Hz, 1H), 5.19 (dd, J = 17.1, 1.9 Hz, 1H), 5.11 (d, J = 6.8 Hz, 1H), 5.08 (dd, J = 8.8, 1.9 Hz, 1H), 4.68 (d, J = 11.2 Hz, 1H), 4.42 (d, J = 10.7 Hz, 1H), 4.38 (d, J = 11.3 Hz, 1H), 4.29 (s, 2H), 4.05−4.00 (m, 2H), 3.82 (d, J = 2.4 Hz, 1H), 3.61 (dd, J = 9.3, 2.0 Hz, 1H), 3.39−3.31 (m, 2H), 2.68 (dd, J = 13.2, 6.8 Hz, 1H), 2.24 (ddd, J = 15.6, 7.8, 7.8 Hz, 1H), 2.06−1.95 (m, 2H), 1.70−1.60 (m, 1H), 1.56−1.48 (m, 1H), 1.19 (s, 3H), 1.00 (t, J = 7.8 Hz, 9H), 0.64 (q, J = 7.8 Hz, 6H), 0.30 (s, 3H); 13C NMR (125 MHz, C6D6) δ 140.3, 140.1, 139.9, 136.2, 128.9, 128.8, 128.8, 128.0, 127.9, 127.8, 127.8, 127.7, 117.0, 83.0, 78.0, 77.7, 76.4, 76.0, 74.7, 73.1, 71.1, 63.9, 37.3, 27.7, 26.0, 17.7, 7.7, 5.9; IR (neat) 2955, 2876, 1454, 1379, 1239, 1107, 1027 cm−1; ESI/MS (m/z) calcd for C39H54O5SiNa 653.4 (M+Na+), found 653.4.
2-((2R,3R,4R,5S,6S)-4,5-bis(benzyloxy)-6-(3-(benzyloxy)propyl)-5-methyl-3-((triethylsilyl)oxy)tetrahydro-2H-pyran-2-yl)acetic acid (38). O3 was bubbled through a solution of 38 (638 mg, 1.01 mmol) in CH2Cl2 (60 mL) at −78 °C until the reaction mixture remained light blue in color. The excess O3 was purged from the reaction mixture by bubbling N2 through it until the light blue color completely dissipated (ca. 10 min). Triphenylphosphine (1.33 g, 5.08 mmol) was then added and the mixture was allowed to slowly warm to rt. After 12 h, the solution was concentrated under reduced pressure. Flash chromatography (50:1 to 5:1 hexanes:ethyl acetate) provided the aldehyde (600. mg, 94%) as a colorless oil.
To a solution of the aldehyde from the ozonolysis (600. mg, 0.949 mmol) in THF (10 mL) at 0 °C was added tBuOH (10 mL), H2O (10 mL), 2-Me-2-butene (2.0 mL), NaH2PO4 (570 mg, 0.475 mmol) and NaClO2 (430 mg, 0.48 mmol). The reaction was quenched after 30 min by adding H2O (30 mL). The resulting mixture was extracted with ethyl acetate (3 × 15 mL) and the organic extracts were combined, dried (Na2SO4) and concentrated. Flash chromatography (1:1 hexanes:ethyl acetate to ethyl acetate) provided acid 39 (615 mg, 100%) as a colorless oil. Rf 0.50 (2:1 hexanes:ethyl acetate); [α]20D = −9.4° (c = 0.27, THF); 1H NMR (500 MHz, C6D6) δ 11.7 (br s, 1H), 7.44 (d, J = 7.8 Hz, 2H), 7.32 (d, J = 7.8 Hz, 2H), 7.29 (d, J = 7.4 Hz, 2H), 7.19−7.03 (m, 9H), 5.09 (d, J = 11.2 Hz, 1H), 4.65 (d, J = 11.2 Hz, 1H), 4.52 (ddd, J = 8.8, 8.8, 3.4 Hz, 1H), 4.41 (d, J = 11.2 Hz, 1H), 4.37 (d, J = 11.2 Hz, 1H), 4.31 (s, 2H), 4.11 (d, J = 10.7 Hz, 1H), 3.82 (d, J = 2.0 Hz, 1H), 3.73 (dd, J = 9.2, 2.0 Hz, 1H), 3.40−3.32 (m, 2H), 2.86 (dd, J = 15.4, 3.2 Hz, 1H), 2.54 (dd, J = 15.4, 9.0 Hz, 1H), 2.02−1.92 (m, 2H), 1.72−1.62 (m, 1H), 1.50 (ddd, J = 9.3, 9.3, 9.3 Hz, 1H), 1.16 (s, 3H), 0.96 (t, J = 7.8 Hz, 9H), 0.58 (q, J = 7.8 Hz, 6H); 13C NMR (125 MHz, C6D6) δ 178.3, 140.1, 140.0, 139.9, 128.9, 128.8, 128.8, 128.1, 127.9, 127.8, 127.8, 127.7, 82.8, 78.0, 77.9, 76.0, 73.8, 73.8, 73.0, 70.8, 63.9, 38.2, 27.4, 25.7, 17.6, 7.6, 5.7; IR (neat) 3330, 2956, 2877, 1713, 1454, 1241, 1111 cm−1; ESI/MS (m/z) calcd for C38H52O7SiNa 671.4 (M+Na+), found 671.3.
(2R,4aR,6S,7R,8aS)-2-(4-methoxyphenyl)-4a,6-dimethyl-6-(pent-4-en-1-yl)hexahydropyrano[3,2-d][1,3]dioxin-7-yl 2-((2R,3R,4R,5S,6S)-4,5-bis(benzyloxy)-6-(3-(benzyloxy)propyl)-5-methyl-3-((triethylsilyl)oxy)tetrahydro-2H-pyran-2-yl)acetate (40). To a solution of acid 39 (53.4 mg, 82.2 μmol) in THF (8 mL) was added triethylamine (0.100 mL, 0.719 mmol) and 2,4,6-trichlorobenzoyl chloride (77.0 μL, 0.493 mmol). The reaction mixture was heated at 40 °C for 2 h before the solvent was removed in vacuo. To the resulting residue was transfered a solution of alcohol 30 (24.8 mg, 68.5 μmol) in toluene (8 mL) followed by DMAP (0.100 g, 0.820 mmol). The resulting reaction mixture was heated at 40 °C for 2 h and then the reaction was quenched with sat. NaHCO3 (aq., 10 mL). The aqueous phase was extracted with CH2Cl2 (3 × 10 mL), the organic extracts were combined, dried (Na2SO4) and concentrated. Flash chromatography (10:1 hexanes:ethyl acetate) provided ester 40 (57.4 mg, 84%) as a colorless oil. Rf 0.70 (4:1 hexanes:ethyl acetate); [α]20D = −26.2° (c = 0.29, THF); 1H NMR (500 MHz, C6D6) δ 7.55 (d, J = 8.8 Hz, 2H), 7.44 (d, J = 7.3 Hz, 2H), 7.35 (d, J = 7.3 Hz, 2H), 7.29 (d, J = 7.8 Hz, 2H), 7.20−7.05 (m, 9H), 6.82 (d, J = 8.3 Hz, 2H), 5.84 (dddd, J = 17.1, 10.3, 6.8, 6.8 Hz, 1H), 5.32 (s, 1H), 5.22 (dd, J = 11.2, 4.9 Hz, 1H), 5.11 (d, J = 11.2 Hz, 1H), 5.08 (dd, J = 15.6, 1.5 Hz, 1H), 5.02 (dd, J = 10.3, 1.0 Hz, 1H), 4.68 (d, J = 11.2 Hz, 1H), 4.49 (ddd, J = 10.3, 7.3, 3.4 Hz, 1H), 4.43 (d, J = 11.2 Hz, 1H), 4.38 (d, J = 11.2 Hz, 1H), 4.33 (s, 2H), 4.13 (d, J = 9.8 Hz, 1H), 3.99 (dd, J = 9.8, 2.0 Hz, 1H), 3.88 (d, J = 2.0 Hz, 1H), 3.80 (d, J = 9.8 Hz, 1H), 3.42−3.34 (m, 3H), 3.27 (s, 3H), 3.28−3.24 (partially obscured m, 1H), 2.86 (dd, J = 14.7, 3.4 Hz, 1H), 2.62 (dd, J = 14.7, 7.3 Hz, 1H), 2.29 (ddd, J = 11.2, 4.4, 3.9 Hz, 1H), 2.08−1.96 (m, 5H), 1.80−1.55 (m, 6H), 1.49 (s, 3H), 1.30 (s, 3H), 1.22 (s, 3H), 1.00 (t, J = 8.3 Hz, 9H), 0.67 (q, J = 7.8 Hz, 3H), 0.66 (q, J = 8.3 Hz, 3H); 13C NMR (125 MHz, C6D6) δ 170.7, 160.8, 140.2, 140.0, 139.8, 139.5, 131.6, 128.9, 128.8, 128.8, 128.5, 128.0, 127.9, 127.9, 127.9, 127.8, 127.7, 115.1, 114.1, 103.1, 82.6, 80.5, 78.1, 78.0, 77.3, 77.1, 76.0, 74.3, 74.0, 73.2, 73.0, 71.0, 69.5, 63.9, 55.1, 42.4, 37.8, 34.9, 30.5, 28.2, 27.8, 26.1, 23.6, 23.0, 19.5, 17.6, 7.6, 5.8; IR (neat) 2954, 2876, 1741, 1496, 1379, 1249, 1106 cm−1; ESI/MS (m/z) calcd for C59H80O11SiNa 1015.6 (M+Na+), found 1015.5.
(2R,3R,4R,5S,6S)-4,5-bis(benzyloxy)-6-(3-(benzyloxy)propyl)-2-(((2R,4aR,5aS,11aR,12aS,Z)-2-(4-methoxyphenyl)-4a,5a-dimethyl-4a,5a,6,7,8,11a,12,12a-octahydro-4H-[1,3]dioxino[4′,5′:5,6]pyrano[3,2-b]oxocin-10-yl)methyl)-5-methyltetrahydro-2H-pyran-3-ol (41). To a solution of TiCl4 (0.230 mL, 2.09 mmol) in CH2Cl2 (13 mL) at 0 °C was added THF (1.10 mL, 12.5 mmol) dropwise. TMEDA (1.90 mL, 12.5 mmol) was added dropwise to the resulting yellow solution. The solution turned red-brown and the ice bath was removed. Activated Zn dust (304 mg, 4.68 mmol) and PbCl2 (69.0 mg, 0.248 mmol) were added after the red-brown reaction mixture had stirred for 15 min. The resulting slurry went through a series of color changes from brown to green to purple and finally to blue-green over the course of 3–5 min. A solution of ester 40 (32.3 mg, 32.5 μmol) and CH3CHBr2 (0.190 mL, 2.12 mmol) in CH2Cl2 (3 mL) was transferred to the reaction mixture via cannula. The reaction mixture was then heated to reflux for 2 h before it was cooled to 0 °C and the reaction was quenched with sat. K2CO3 (aq., 1.0 mL). After stirring for 30 min at 0 °C, the resulting mixture was filtered. The filtrate was concentrated and flash chromatography (10:1 hexanes:ethyl acetate) gave 41 as a colorless oil (12.5 mg, 40%). Rf 0.55 (5:1 hexanes:ethyl acetate); [α]20D = −14.1° (c = 0.28, THF); 1H NMR (500 MHz, C6D6) δ 7.61 (d, J = 8.8 Hz, 2H), 7.50 (d, J = 7.3 Hz, 2H), 7.38 (d, J = 7.3 Hz, 2H), 7.30 (d, J = 7.3 Hz, 2H), 7.21−7.06 (m, 9H), 6.83 (d, J = 8.3 Hz, 2H), 5.48 (s, 1H), 5.16 (d, J = 11.2 Hz, 1H), 4.90 (dd, J = 8.3, 2.9 Hz, 1H), 4.71 (d, J = 11.2 Hz, 1H), 4.47 (d, J = 11.2 Hz, 1H), 4.41 (d, J = 11.2 Hz, 1H), 4.31 (s, 2H), 4.14 (d, J = 9.8 Hz, 1H), 3.98 (dd, J = 9.8, 4.9 Hz, 1H), 3.89 (d, J = 6.8 Hz, 1H), 3.88 (s, 1H), 3.69 (dd, J = 9.8, 2.2 Hz, 1H), 3.48 (d, J = 9.8 Hz, 1H), 3.44−3.38 (m, 2H), 3.27 (s, 3H), 3.28−3.22 (partially obscured m, 1H), 2.91 (d, J = 14.6 Hz, 1H), 2.30−1.97 (m, 8H), 1.84−1.70 (m, 2H), 1.69−1.56 (m, 3H), 1.55 (s, 3H), 1.36 (s, 3H), 1.24 (s, 3H), 1.01 (t, J = 7.8 Hz, 9H), 0.64 (q, J = 7.8 Hz, 6H); 13C NMR (100 MHz, C6D6) δ 160.8, 155.7, 140.3, 140.1, 140.0, 131.8, 128.9, 128.8, 128.5, 128.0, 127.9, 127.9, 127.8, 127.7, 114.1, 108.1, 103.2, 84.0, 83.1, 81.3, 78.5, 78.0, 77.9, 77.8, 76.1, 74.9, 74.5, 73.1, 71.1, 69.4, 63.9, 55.1, 46.3, 40.2, 33.6, 31.0, 30.5, 28.5, 27.8, 25.9, 28.5, 27.8, 25.9, 22.4, 22.1, 19.8, 17.8, 7.6, 5.8; IR (neat) 2928, 2876, 1615, 1496, 1378, 1250, 1108 cm−1; ESI/MS (m/z) calcd for C58H78O10SiK 1001.6 (M+K+), found 1001.5.
(2R,4aR,5aS,10S,11aR,12aS)-10-(((2R,3R,4R,5S,6S)-4,5-bis(benzyloxy)-6-(3-(benzyloxy)propyl)-3-hydroxy-5-methyltetrahydro-2H-pyran-2-yl)methyl)-2-(4-methoxyphenyl)-4a,5a-dimethyloctahydro-4H-[1,3]dioxino[4′,5′:5,6]pyrano[3,2-b]oxocin-9(10H)-one (44). To a solution of 41 (19.7 mg, 20.4 μmol) in CH2Cl2 (5 mL) at −78 °C was added a solution of dimethyl dioxirane (0.42 mL of 0.10 M solution in CH2Cl2, 0.042 mmol) dropwise. The reaction mixture was warmed to 0 °C and the solvent was removed in vacuo. The residue was dissolved in CH2Cl2 (5 mL) and the reaction mixture was cooled to −78 °C and a solution of i-Bu2AlH (0.21 mL of 1.0 M solution in THF, 0.21 mmol) was added. After stirring for 2 h at −78 °C, the reaction was quenched with sat. NH4Cl (aq., 3 mL) and allowed to warm to rt. A solution of Saturated potassium sodium tartrate (10 mL) was added and the mixture was stirred violently for 30 min. The aqueous phase was extracted with CH2Cl2 (3 × 10 mL) and the combined organic extracts were dried (Na2SO4) and concentrated to give alcohol 43.
To a solution of the alcohol obtained from above in CH2Cl2 (5 mL) was added activated 4Å MS (20 mg), NMO (25.0 mg, 0.213 mmol) and TPAP (1 mg, 0.003 mmol). The reaction mixture was stirred at rt for 2 h before being concentrated. Flash chromatography (5:1 hexanes:ethyl acetate) gave ketone 44 as a colorless oil (12.2 mg, 61% 2 steps). Rf 0.55 (3:1 hexanes:ethyl acetate); [α]20D = −20.2° (c = 0.18, THF); 1H NMR (500 MHz, C6D6) δ 7.58 (d, J = 8.8 Hz, 2H), 7.46 (d, J = 7.3 Hz, 2H), 7.36 (d, J = 7.8 Hz, 2H), 7.30 (d, J = 7.8 Hz, 2H), 7.21−7.06 (m, 9H), 6.83 (d, J = 8.3 Hz, 2H), 5.36 (s, 1H), 5.08 (d, J = 11.2 Hz, 1H), 4.70 (d, J = 11.2 Hz, 1H), 4.50 (dd, J = 9.3, 6.3 Hz, 1H), 4.44 (d, J = 11.2 Hz, 1H), 4.40 (d, J = 11.2 Hz, 1H), 4.32 (s, 2H), 4.12 (ddd, J = 9.8, 6.8, 2.9 Hz, 1H), 4.09 (dd, J = 10.3, 1.5 Hz, 1H), 3.85 (d, J = 2.0 Hz, 1H), 3.81 (d, J = 9.8 Hz, 1H), 3.79 (dd, J = 9.8, 2.4 Hz, 1H), 3.44−3.33 (m, 4H), 3.27 (s, 3H), 3.14 (dd, J = 12.2, 3.0 Hz, 1H), 2.87 (ddd, J = 14.2, 7.8, 4.9 Hz, 1H), 2.31−2.06 (m, 5H), 2.04−1.94 (m, 3H), 1.84−1.70 (m, 2H), 1.64−1.54 (m, 3H), 1.52 (s, 3H), 1.34 (s, 3H), 1.24 (s, 3H), 1.03 (t, J = 7.8 Hz, 9H), 0.69 (q, J = 7.8 Hz, 6H); 13C NMR (100 MHz, C6D6) δ 213.9, 160.9, 140.2, 140.1, 139.9, 131.6, 128.9, 128.8, 128.5, 128.0, 127.9, 127.9, 127.8, 127.7, 114.1, 103.1, 85.4, 83.3, 82.9, 81.4, 78.0, 77.9, 77.4, 76.0, 73.9, 73.2, 73.2, 71.1, 69.7, 63.9, 55.1, 44.0, 40.2, 35.4, 28.9, 27.8, 26.0, 21.5, 21.1, 20.2, 17.6, 7.6, 5.9; IR (neat) 2934, 2876, 1704, 1616, 1496, 1378, 1249, 1105 cm−1; ESI/MS (m/z) calcd for C58H78O11SiK 1017.6 (M+K+), found 1017.5.
Summary of COSY spectrum for 44
Proton at 4.50 ppm (C-28) shows cross peaks with proton at 2.28 ppm (C-29).
Proton at 4.12 ppm (C-30) shows cross peaks with proton at 3.79 ppm (C-31).
Proton at 4.09 ppm (C-34) shows cross peaks with protons at 1.62 ppm (C-35).
Proton at 3.81 ppm (C-18) shows cross peaks with proton at 3.40 ppm (C-18′).
Proton at 2.06 ppm (C-21) shows cross peaks with proton at 3.36 ppm (C-22) and 3.14 (C-20).
Summary of 1D nOe spectrum for 44
Irradiation at 4.50 ppm (C-28) resulted in enhancement at 3.36 ppm (C-22).
Irradiation at 3.14 ppm (C-20) resulted in enhancement at 5.36 ppm (C-a), 3.40 ppm (C-18′), and 3.36 ppm (C-22).
(2R,4aR,5aS,8S,10S,11aR,12aS)-10-(((2R,3R,4R,5S,6S)-4,5-bis(benzyloxy)-6-(3-(benzyloxy)propyl)-3-hydroxy-5-methyltetrahydro-2H-pyran-2-yl)methyl)-2-(4-methoxyphenyl)-4a,5a,8-trimethyloctahydro-4H-[1,3]dioxino[4′,5′:5,6]pyrano[3,2-b]oxocin-9(10H)-one (45). To a solution of 44 (5.0 mg, 5.1 μmol) in THF (4 mL) at −78 °C was added LiHMDS (0.102 mL of 1.0 M solution in THF, 0.10 mmol). After 10 min, MeI (16 μL, 0.254 mmol) was added and the reaction mixture was allowed to slowly warm up to −22 °C before the reaction was quenched with NH4Cl (aq., 5 mL). The aqueous phase was extracted with CH2Cl2 (3 × 5 mL) and the combined organic extracts were dried (Na2SO4) and concentrated. Flash chromatography (5:1 hexanes:ethyl acetate) provided 45 (4.7 mg, 93%) as a colorless oil. Rf 0.55 (3:1 hexanes:ethyl acetate). [a]20D = −22.3° (c = 0.14, THF); 1H NMR (400 MHz, CD2Cl2) δ 7.36 (d, J = 8.8 Hz, 2H), 7.32−7.19 (m, 15H), 6.87 (d, J = 8.3 Hz, 2H), 5.44 (s, 1H), 4.96 (d, J = 10.7 Hz, 1H), 4.58 (d, J = 11.2 Hz, 1H), 4.54 (d, J = 11.2 Hz, 1H), 4.48 (d, J = 11.2 Hz, 1H), 4.46 (s, 2H), 4.32 (dd, J = 9.8, 5.9 Hz, 1H), 3.85 (d, J = 1.5 Hz, 1H), 3.79 (s, 3H), 3.74 (d, J = 9.8 Hz, 1H), 3.68−3.52 (m, 3H), 3.52−3.40 (m, 5H), 2.99 (ddd, J = 7.8, 7.8, 7.8 Hz, 1H), 2.60−2.49 (m, 1H), 2.35−2.19 (m, 2H), 2.13 (ddd, J = 11.7, 3.9, 3.9 Hz, 1H), 1.95−1.50 (m, 8H), 1.50 (s, 3H), 1.36 (s, 3H), 1.28 (s, 3H), 1.20 (d, J = 7.3 Hz, 3H), 1.03 (t, J = 7.8 Hz, 9H), 0.71 (q, J = 7.8 Hz, 6H); 13C NMR (100 MHz, C6D6) δ 214.2, 160.4, 139.6, 139.5, 131.1, 128.4, 128.3, 128.0, 127.5, 127.4, 127.3, 127.2, 113.6, 102.6, 85.4, 83.5, 82.4, 80.5, 77.4, 77.4, 77.0, 76.9, 75.5, 74.8, 73.1, 72.6, 70.5, 69.2, 63.4, 57.6, 54.6, 49.9, 44.7, 35.8, 30.0, 29.2, 28.4, 27.3, 25.5, 21.6, 21.3, 19.4, 17.2, 7.1, 5.3; IR (neat) 2956, 2876, 1718, 1616, 1496, 1377, 1249, 1104 cm−1; ESI/MS (m/z) calcd for C59H80O11SiNa 1015.6 (M+Na+), found 1015.5.
(2S,3R,4aS,7S,7aR,8aR,9R,10S,11S,12aR,13aS,14aR)-9,10-bis(benzyloxy)-11-(3-(benzyloxy)propyl)-3-(hydroxymethyl)-3,4a,7,10-tetramethylhexadecahydropyrano[3,2-b]pyrano[2′,3′:5,6]pyrano[2,3-g]oxocin-2-ol (4). To a solution of 45 (4.7 mg, 4.7 μmol) in CH2Cl2 (3 mL) at −78 °C were added Et3SiH (76 μL, 0.47 mmol) and TMSOTf (8.6 μL, 47.0 μmol). After stirring for 1 h, the reaction mixture was slowly warmed to −50 °C, at which temperature the reaction was quenched with NaHCO3 (aq., 5 mL). The aqueous phase was extracted with CH2Cl2 (3 × 5 mL) and the combined organic extracts were dried (Na2SO4) and concentrated. Flash chromatography (2:1 hexanes:ethyl acetate) provided 4 (2.8 mg, 80%) as a colorless oil. Rf 0.45 (1:1 hexanes:ethyl acetate); [α]20D = −1.1° (c = 0.05, THF); 1H NMR (500 MHz, CD2Cl2) δ 7.40−7.20 (m, 15H), 4.96 (d, J = 11.2 Hz, 1H), 4.56 (d, J = 11.2 Hz, 1H), 4.45 (d, J = 11.2 Hz, 1H), 4.43 (s, 2H), 4.40 (d, J = 11.2 Hz, 1H), 4.04 (d, J = 2.0 Hz, 1H), 3.82 (br d, J = 11.7 Hz, 1H), 3.71−3.64 (m, 2H), 3.49−3.46 (m, 2H), 3.41−3.35 (m, 2H), 3.29 (d, J = 10.7 Hz, 1H), 3.22 (ddd, J = 11.7, 8.8, 4.4 Hz, 1H), 3.12 (dd, J = 9.8, 2.0 Hz, 1H), 2.76 (dd, J = 9.3, 9.3 Hz, 1H), 2.30 (ddd, J = 11.2, 4.4, 4.4 Hz, 1H), 2.06−2.00 (m, 2H), 1.96 (ddd, J = 10.7, 4.4, 4.4 Hz, 1H), 1.95−1.35 (m, 11H), 1.28 (s, 3H), 1.22 (s, 3H), 1.14 (s, 3H), 1.08 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, C6D6) δ 140.1, 128.9, 128.8, 128.0, 127.9, 127.8, 127.7, 88.1, 86.2, 83.8, 80.8, 78.8, 78.7, 77.7, 77.2, 76.6, 75.0, 73.2, 71.1, 71.0, 69.1, 68.7, 63.8, 46.3, 40.7, 40.0, 33.9, 32.1, 30.5, 27.9, 26.0, 22.7, 20.3, 18.8, 17.9; IR (neat) 3500, 3028, 2925, 1599, 1494, 1451, 1264, 1077 cm−1; ESI/MS (m/z) calcd for C45H60O9K 783.4 (M+K+), found 783.3.
Summary of COSY spectrum for 4
Proton at 4.04 ppm (C-32) shows cross peaks with proton at 3.12 ppm (C-31).
Proton at 3.12 ppm (C-31) shows cross peaks with proton at 3.66 ppm (C-30).
Proton at 1.08 ppm (C-26 methyl) shows cross peaks with protons at 1.70 ppm (C-26).
Proton at 1.70 ppm (C-26) shows cross peaks with proton at 2.76 ppm (C-27).
Proton at 2.76 ppm (C-27) shows cross peaks with proton at 3.22 ppm (C-28).
Proton at 3.22 ppm (C-28) shows cross peaks with protons at 2.30 ppm (C-29) and 1.50 ppm (C-29′).
protons at 2.30 ppm (C-29) and 1.50 ppm (C-29′) both show cross peaks with proton at 3.66 ppm (C-30).
Summary of 1D nOe spectrum for 4
Irradiation at 3.22 ppm (C-28) resulted in enhancement at 3.66 ppm (C-30), 3.40 ppm (C-22), 2.30 ppm (C-29), and 1.70 ppm (C-26).
Irradiation at 2.76 ppm (C-27) resulted in enhancement at 3.12 ppm (C-31), 1.50 ppm (C-29′), and 1.08 ppm (C-26 methyl).
Supplementary Material
Acknowledgments
The authors would like to thank the National Institutes of Health, General Medical Sciences (GM56677) for their support of this work. We are grateful to the staff at the University of Utah, especially Dr. Peter Flynn and Dr. Dennis Edwards (NMR) and Dr. Jim Muller (mass spectrometry).
Footnotes
In honor of Professor Amos B. Smith, III and his 50 years of dedication to the Organic Chemistry community
References
- 1.Murata M, Kumagai M, Masanori K, Lee JS, Yasumoto T. Isolation and structure of yessotoxin, a novel polyether compound implicated in diarrhetic shellfish poisoning. Tetrahedron Lett. 1987;28:5869–5872. [Google Scholar]
- 2.Satake M, MacKenzie L, Yasumoto T. Identification of Protoceratium reticulatum as the biogenetic origin of yessotoxin. Nat Toxins. 1997;5:164–167. doi: 10.1002/nt.7. [DOI] [PubMed] [Google Scholar]
- 3.Paz B, Riobó P, Fernández LM, Fraga S, Franco JM. Production and release of yessotoxins by the dinoflagellates Protoceratium reticulatum and Lingulodinium polyedrum in culture. Toxicon. 2004;44:251–258. doi: 10.1016/j.toxicon.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 4.Rhodes L, McNabb P, de Salas M, Briggs L, Beuzenberg V, Gladstone M. Yessotoxin production by Gonyaulax spinifera. Harmful Algae. 2006;5:148–155. [Google Scholar]
- 5.Tubaro A, Dell’Ovo V, Sosa S, Florio C. Yessotoxins: A toxicological overview. Toxicon. 2010;56:163–172. doi: 10.1016/j.toxicon.2009.07.038. [DOI] [PubMed] [Google Scholar]
- 6.Terao K, Ito E, Oarada M, Murata M, Yasumoto T. Histopathological studies on experimental marine toxin poisoning-5. The effects in mice of yessotoxin isolated from Patinopecten yessoensis and of a desulfated derivative. Toxicon. 1990;28:1095–1104. doi: 10.1016/0041-0101(90)90148-z. [DOI] [PubMed] [Google Scholar]
- 7.Tubaro A, Bandi E, Sosa S, Soranzo MR, De Ninis V, Yasumoto T, Lorenzon P. Effects of yessotoxin (YTX) on the skeletal muscle: an update. Food Addit Contam. 2008;25:1095–1100. doi: 10.1080/02652030802130017. [DOI] [PubMed] [Google Scholar]
- 8.Tubaro A, Sosa S, Altinier G, Soranzo MR, Satake M, Yasumoto T. Short-term oral toxicity of homoyessotoxins, yessotoxin and okadaic acid in mice. Toxicon. 2004;43:439–445. doi: 10.1016/j.toxicon.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 9.Tubaro A, Giangaspero A, Ardizzone M, Soranzo MR, Vita F, Yasumoto T, Maucher JM, Ramsdell JS, Sosa S. Ultrastructural damage to heart tissue from repeated oral exposure to yessotoxin resolves in three months. Toxicon. 2008;51:1225–1235. doi: 10.1016/j.toxicon.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 10.De la Rosa LA, Alfonso A, Vilariño N, Vieytes MR, Botana LM. Modulation of cytosolic calcium levels of human lymphocytes by yessotoxin, a novel marine phycotoxin. Biochem Pharmacol. 2001;61:827–833. doi: 10.1016/s0006-2952(01)00549-4. [DOI] [PubMed] [Google Scholar]
- 11.Ronzitti G, Callegari F, Malaguti C, Rossini GP. Selective disruption of the E-cadherin-catenin system by an algal toxin. Br J Cancer. 2004;90:1100–1107. doi: 10.1038/sj.bjc.6601640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ronzitti G, Rossini GP. Yessotoxin induces the accumulation of altered E-cadherin dimers that are not part of adhesive structures in intact cells. Toxicology. 2008;244:145–156. doi: 10.1016/j.tox.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 13.Korsnes MS, Espenes A. Yessotoxin as an apoptotic inducer. Toxicon. 2011;57:947–958. doi: 10.1016/j.toxicon.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 14.Malagoli D, Marchesini E, Ottaciani E. Lysosomes as the target of yessotoxin in invertebrate and vertebrate cell lines. Toxicol Lett. 2006;167:75–83. doi: 10.1016/j.toxlet.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 15.Ciminiello P, Fattorusso E, Forino M, Magno S, Poletti R, Viviani R. Isolation of adriatoxin, a new analog of yessotoxin from mussels of the Adriatic Sea. Tetrahedron Lett. 1998;39:8897–8900. [Google Scholar]
- 16.Suzuki K, Nakata T. Convergent Synthesis of the ABCDEF-Ring System of Yessotoxin and Adriatoxin. Org Lett. 2002;4:3943–3946. doi: 10.1021/ol026804w. [DOI] [PubMed] [Google Scholar]
- 17.Mori Y, Nogami K, Hayashi H, Noyori R. Sulfonyl-Stabilized Oxiranylithium-Based Approach to Polycyclic Ethers. Convergent Synthesis of the ABCDEF-Ring System of Yessotoxin and Adriatoxin. J Org Chem. 2003;68:9050–9060. doi: 10.1021/jo035145d. [DOI] [PubMed] [Google Scholar]
- 18.Hiramatsu N, Mori Y. Synthesis of the 37-epi-HIJ ring system of adriatoxin. Heterocycles. 2006;69:437–446. [Google Scholar]
- 19.Kadota I, Ueno H, Yamamoto Y. Convergent synthesis of the A-F ring segment of yessotoxin and adriatoxin. Tetrahedron Lett. 2003;44:8935–8938. [Google Scholar]
- 20.Kadota I, Ueno H, Sato Y, Yamamoto Y. Convergent synthesis of the FGHI ring segment of yessotoxin. Tetrahedron Lett. 2006;47:89–92. [Google Scholar]
- 21.Kadota I, Abe T, Sato Y, Kabuto C, Yamamoto Y. Stereocontrolled synthesis of the IJK ring segment of yessotoxin. Tetrahedron Lett. 2006;47:6545–6548. [Google Scholar]
- 22.Watanabe K, Suzuki M, Murata M, Oishi T. Convergent synthesis of the FGHI ring system of yessotoxin: stereoselective construction of the G ring. Tetrahedron Lett. 2005;46:3991–3995. [Google Scholar]
- 23.Oishi T, Suzuki M, Watanabe K, Murata M. Convergent synthesis of the CDEF ring fragment of yessotoxin via α-cyano ethers. Heterocycles. 2006;69:91–98. [Google Scholar]
- 24.Oishi T, Suzuki M, Watanabe K, Murata M. Synthesis of the ABC and IJ ring fragments of yessotoxin. Tetrahedron Lett. 2006;47:3975–3978. [Google Scholar]
- 25.Watanabe K, Minato H, Murata M, Oishi T. Synthesis of the JK ring fragments of yessotoxin and 42,43,44,45,46,47,55-heptanor-41-oxoyessotoxin. Heterocycles. 2007;72:207–212. [Google Scholar]
- 26.Torikai K, Watanabe K, Minato H, Imaizumi T, Murata M, Oishi T. Convergent synthesis of the A-J ring system of yessotoxin. Synlett. 2008:2368–2372. [Google Scholar]
- 27.Oishi T, Imaizumi T, Murata M. Reductive etherification under microfluidic conditions: application to practical synthesis of the FGHIJ-ring system of yessotoxin. Chem Lett. 2010;39:108–109. [Google Scholar]
- 28.Akoto CO, Rainier JD. Harnessing glycal-epoxide rearrangements: the generation of the AB, EF, and IJ rings of adriatoxin. Angew Chem, Int Ed. 2008;47:8055–8058. doi: 10.1002/anie.200803791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson HWB, Majumder U, Rainier JD. The Total Synthesis of Gambierol. J Am Chem Soc. 2005;127:848–849. doi: 10.1021/ja043396d. [DOI] [PubMed] [Google Scholar]
- 30.Johnson HWB, Majumder U, Rainier JD. Total synthesis of gambierol: subunit coupling and completion. Chem Eur J. 2006;12:1747–1753. doi: 10.1002/chem.200500994. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y, Rainier JD. Two-Directional Olefinic-Ester Ring-Closing Metathesis using Reduced Ti Alkylidenes. A Rapid Entry into Polycyclic Ether Skeletons. Org Lett. 2009;11:237–239. doi: 10.1021/ol8025439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Rohanna J, Zhou J, Iyer K, Rainier JD. Total Synthesis of Brevenal. J Am Chem Soc. 2011;133:3208–3216. doi: 10.1021/ja200089f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bal BS, Childers WE, Pinnick HW. Oxidation of α,β-unsaturated aldehydes. Tetrahedron. 1981;37:2091–2096. [Google Scholar]
- 34.Clark JS, Kimber MC, Robertson J, McErlean CSP, Wilson C. Rapid two-directional synthesis of the F-J fragment of the gambieric acids by iterative double ring-closing metathesis. Angew Chem, Int Ed. 2005;44:6157–6162. doi: 10.1002/anie.200501925. [DOI] [PubMed] [Google Scholar]
- 35.Lindlar H. A new catalyst for selective hydrogenations. Helv Chim Acta. 1952;35:446–448. [Google Scholar]
- 36.Inanaga J, Hirata K, Saeki T, Katusuki T, Yamaguchi M. A rapid esterification by mixed anhydride and its application to large-ring lactonization. Bull Chem Soc Jpn. 1979;52:1989–1993. [Google Scholar]
- 37.Iyer K, Rainier JD. Olefinic Ester and Diene Ring-Closing Metathesis Using a Reduced Titanium Alkylidene. J Am Chem Soc. 2007;129:12604–12605. doi: 10.1021/ja073880r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roberts SW, Rainier JD. Synthesis of an A-E Gambieric Acid Subunit with Use of a C-Glycoside Centered Strategy. Org Lett. 2007;9:2227–2230. doi: 10.1021/ol0707970. [DOI] [PubMed] [Google Scholar]
- 39.Cox JM, Rainier JD. C-Glycosides to Fused Polycyclic Ethers. An Efficient Entry into the A-D Ring System of Gambierol. Org Lett. 2001;3:2919–2922. [PubMed] [Google Scholar]
- 40.Nicolaou KC, Prasad CVC, Hwang CK, Duggan ME, Veale CA. Cyclizations of hydroxyl dithioketals. New synthetic technology for the construction of oxocenes and related medium0ring systems. J Am Chem Soc. 1989;111:5321–5330. [Google Scholar]
- 41.Rainier JD, Allwein SP, Cox JM. C-glycosides to Fused Polycyclic Ethers. A Formal Synthesis of (±)-Hemibrevetoxin B. J Org Chem. 2001;66:1380–1386. doi: 10.1021/jo001514j. [DOI] [PubMed] [Google Scholar]
- 42.Clark JS, Hamelin O. Synthesis of polycyclic ethers by two-directional ring-closing metathesis. Angew Chem Int Ed. 2000;39:372–374. doi: 10.1002/(sici)1521-3773(20000117)39:2<372::aid-anie372>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.