

Abstract
Members from Colletotrichum genus adopt a diverse range of lifestyles during infection of plants and represent a group of agriculturally devastating pathogens. In this study, we present the draft genome of Colletotrichum incanum from the spaethianum clade of Colletotrichum and the comparative analyses with five other Colletotrichum species from distinct lineages. We show that the C. incanum strain, originally isolated from Japanese daikon radish, is able to infect both eudicot plants, such as certain ecotypes of the eudicot Arabidopsis, and monocot plants, such as lily. Being closely related to Colletotrichum species both in the graminicola clade, whose members are restricted strictly to monocot hosts, and to the destructivum clade, whose members are mostly associated with dicot infections, C. incanum provides an interesting model system for comparative genomics to study how fungal pathogens adapt to monocot and dicot hosts. Genus-wide comparative genome analyses reveal that Colletotrichum species have tailored profiles of their carbohydrate-degrading enzymes according to their infection lifestyles. In addition, we show evidence that positive selection acting on secreted and nuclear localized proteins that are highly conserved may be important in adaptation to specific hosts or ecological niches.
Keywords: comparative genomics, evolutionary biology, genome assembly, Colletotrichum, hemibiotrophic fungi, plant pathogen
Introduction
The genus Colletotrichum is of considerable interest in studies of plant–pathogen interactions due to its diversity as well as the commercial impact of its various members (Crouch et al. 2014). This has led to it being named one of the top ten most important fungi in a recent survey of plant pathologists in terms of scientific importance (Dean et al. 2012). Within the genus, considerable variation exists, with many known hosts, including important crop species, such as maize, which is infected by Colletotrichum graminicola (Crouch and Beirn 2009), fruits like strawberries, citrus fruits, and bananas (Cannon et al. 2012), as well as model plants, such as Arabidopsis, which is a host of Colletotrichum higginsianum (Narusaka et al. 2004; O’Connell et al. 2004).
In addition, different members adopt a variety of infection lifestyles, even though most members are identified as hemibiotrophic plant pathogens, some have been categorized as endophytes (Gangadevi and Muthumary 2008; Sharma et al. 2011; Mejía et al. 2014). Fungi within this genus classified as hemibiotrophs undergo different phases of infection which have previously been characterized by the expression of distinct classes of genes at different stages (Kleemann et al. 2012; O’Connell et al. 2012; Gan et al. 2013). This lifestyle includes a biotrophic phase of infection in living plant cells, followed by a necrotrophic phase, in which there is massive cell death of host cells, similar to other commercially important pathogens, such as Magnaporthe oryzae.
In the past few years, resources to study the molecular mechanisms underlying the diversity of different lifestyles adopted by different members of this genus have taken off with the genome sequences of different strains being sequenced representing distinct lineages within the genus (O’Connell et al. 2012; Alkan et al. 2013; Gan et al. 2013; Baroncelli, Sanz-Martín, et al. 2014; Baroncelli, Sreenivasaprasad, et al. 2014). Phylogenetic studies have shown that the genus can be divided into distinct lineages (fig. 1; Cannon et al. 2012), with specific characteristics. Among the different clades, members of the graminicola lineage, including C. graminicola, stand out as being exclusively associated with graminaceous monocots, whereas members of the other sequenced species are associated with infection of dicotyledonous plants (Crouch et al. 2014). The mechanisms underlying the adaptation of the graminicola lineage as a monocot-specific pathogen is still largely unknown.
Fig. 1.—
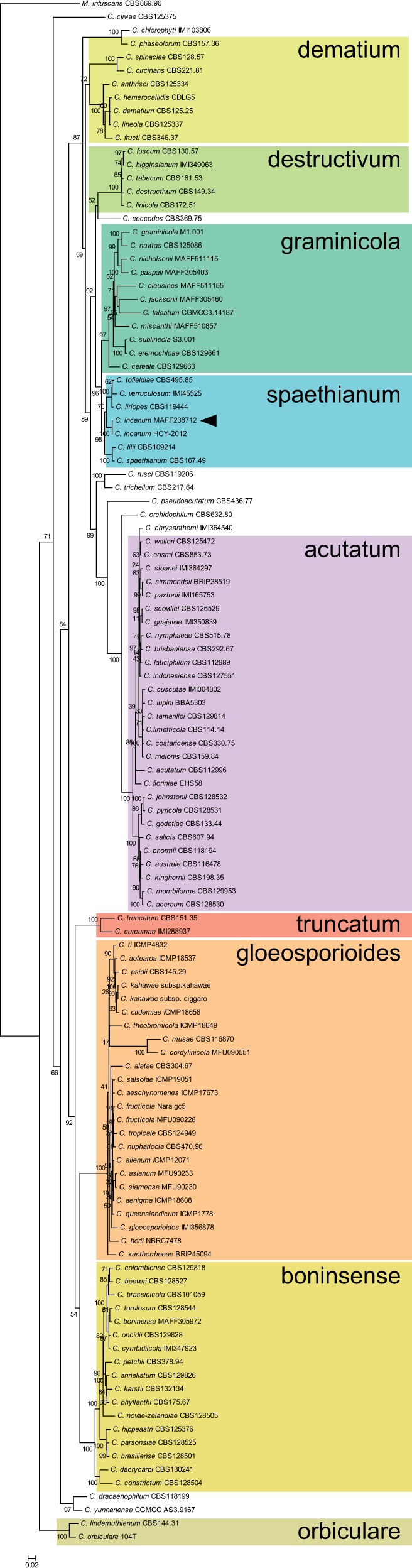
Phylogenetic tree showing relationship between Colletotrichum incanum and other known Colletotrichum species based on the combined alignment of chitin synthase, actin, internal transcribed spacer (ITS), and tubulin sequences. Arrowhead indicates sequenced C. incanum strain. Values at the branch points represent bootstrap support values out of 1,000 replicates.
Here we present the draft genome of Colletotrichum incanum, a member of the spaethianum clade, a group with no previously sequenced member within the Colletotrichum genus. Colletotrichum incanum belongs to a distinct group that is closely related to the graminicola and destructivum clades. While graminicola clade members are graminicolous, destructivum clade members are mostly associated with eudicots (Crouch et al. 2014), although at least one recent study has reported the isolation of Colletotrichum destructivum as an asymptomatic endophyte on orchid (Tao et al. 2013). In contrast, several members of the spaethianum clade have been reported to infect both dicot and nongraminaceous monocot plants (Crouch et al. 2014). In this study, we sequenced a strain of C. incanum and show that it is able to infect both monocot and dicot plants, providing a new and unique model for studying host specificities in plant–fungal interactions. We performed genus-wide analyses and found potential pathogenic lifestyle-specific expansions and contractions of gene families, particularly in carbohydrate-degrading enzymes. Interestingly, secreted proteins of members from the gloeosporioides and acutatum clades, important postharvest pathogens with many phenotypic similarities, were found to be more conserved to one another despite their phylogenetic separation. Furthermore, analysis of positively selected sequences conserved throughout the genus indicated that genes encoding proteins which are targeted for secretion or to the nucleus may undergo higher levels of diversifying selection in a lineage-specific manner compared with those that are targeted to other localizations.
Materials and Methods
Fungal Culture and Infection Conditions
All fungal cultures were maintained on potato dextrose agar (Becton, Dickinson and Company, Franklin Lakes, NJ) at 24 °C under 12 h black light fluorescent bulb (BLB) light/12 h dark conditions. Conidia were harvested after 6 days and sprayed onto plants at a concentration of 1 × 106 conidia/ml. Arabidopsis plants were grown on mixed Supermix A (Sakata Seed Corp., Yokohama, Japan) and vermiculite soil at 21 °C under short day conditions (8 h light/16 h dark) in 70% relative humidity and transplanted 8 days-post-germination (DPG). Col-0 plants with the eds1-2 null mutation, Col-0 eds1-2 (Falk et al. 1999; Bartsch et al. 2006) were also grown as described. Plants at 30–35 DPG were sprayed with Colletotrichum conidia and incubated in 100% relative humidity. Hyphae were observed by a confocal microscope TCP SP5 (Leica microsystems, Wetzlar, Germany). For infections on maize and lily, leaves were inoculated with 5 µl droplets of conidia at a concentration of 5 × 105 conidia/ml. Maize was transplanted to vermiculite soil at 3 DPG and inoculated at 10 DPG and grown at 24 °C under 12 h light/12 h dark. After incubation for 3 days at 4 °C in the dark, Brachypodium distachyon Bd3-1 seeds were transferred to pots of a 1:1 mix of perlite:vermiculite and maintained at 22 °C under long day conditions (16 h light/8 h dark), and leaves were inoculated at 4 weeks after germination. Lily plants were grown at 25 °C under long day conditions. Mature leaves were detached from “casa blanca” lily plants after budding and maintained under 100% humidity at 25 °C under long day conditions during infection. Transformation of C. incanum for expression of green fluorescent protein (GFP) under control of the translation elongation factor (TEF) promoter and scd1 terminator was carried out on protoplasts using the polyethylene glycol transformation protocol as described below (Kubo et al. 1991). For rice infections, detached leaves from 5-week-old plants grown in a rhizotron as described (Mutuku et al. 2015). Rice leaves were inoculated with 5 µl droplets of conidia at a concentration of 1 × 106 conidia/ml and then maintained in 16 h light (28 °C)/8 h dark (23 °C).
Genome Sequencing and Assembly
All cultures were maintained on potato dextrose agar plates or broth at 24 °C. Genomic DNA was extracted using CTAB and QIAgen genomic tips as described for the 1000 fungal genomes project. Paired-end 100 bp sequencing was performed on 150 and 500 bp insert libraries prepared using the Illumina TruSeq PCR-free DNA sample prep kit (Illumina) with an Illumina HiSeq2000 (RIKEN Omics Science Center, Yokohama, Japan). Jellyfish was utilized to calculate k-mer multiplicity for genome size estimation. Reads were trimmed using trimmomatic with the options “LEADING:15 TRAILING:15 MINLEN:36” (Bolger et al. 2014) and assembled using the 127mer version of SOAPdenovo (version 1.05) (Luo et al. 2012) with map_len = 32 and k-mer values of 69. The sequences were deposited at DDBJ/EMBL/GenBank under the accession JTLR00000000. The version described in this article is version JTLR01000000. Files containing all predicted proteins, transcripts, and annotations are available for download at https://sites.google.com/site/colletotrichumgenome/ (last accessed May 2, 2016). The Core Eukaryotic Genes Mapping Approach (CEGMA) pipeline to identify a conserved set of eukaryotic genes was utilized to assess coverage of gene coding regions in the assembly using default settings (Parra et al. 2007).
Gene Prediction and Annotations
The MAKER pipeline (Cantarel et al. 2008) was utilized for gene prediction to combine annotations from the ab initio gene predictors Augustus (Stanke et al. 2006), GeneMark-ES (Ter-Hovhannisyan et al. 2008), and SNAP (available from http://korflab.ucdavis.edu/software.html, last accessed May 2, 2016) using additional evidence from proteins from C. higginsianum. GeneMark-ES was automatically trained on the C. incanum 9503 genome, while Augustus was trained on Scipio (Keller et al. 2008) gene structures derived from the CEGMA gene set from C. incanum using the optimize_augustus.pl script included with Augustus. In addition, SNAP was trained using MAKER combined gene evidence derived using SNAP and alignments from C. higginsianum proteins. Proteins from C. graminicola were also mapped to the genome using exonerate within the MAKER pipeline and used to help improve annotations when manually assessing the gene models. Gene ontology (GO) terms were assigned to predicted proteins by InterProScan5 (Jones et al. 2014), which matches sequences to InterPro protein signatures, and enrichment of GO terms associated with specific gene sets was assessed using hypergeometric tests with the GOStats (Falcon and Gentleman 2007) package in R. For analyses of GO terms associated with orthoMCL clusters, GO terms were assigned to specific clusters only when present in at last half of the Colletotrichum sequences within each cluster. Carbohydrate active enzymes (CAzymes) were classified using dbCAN release 3 (Yin et al. 2012). In order to identify lineage-specific expansions, sequences that were identified as GH43 enzymes were aligned in CLCGenomicsWorkbench8 (CLC Bio) and alignments were used to draw a phylogenetic tree using FastTree (Price et al. 2010) using the JTT + CAT model. The output tree was displayed using the iTol web-based tool (Letunic and Bork 2011). Transporters were classified by performing BLASTP with an E-value cutoff of 1 × 10−5 using all Colletotrichum sequences with at least one predicted transmembrane domain as predicted by TMHMM (Krogh et al. 2001) against all Transporter Classification Database (TCDB) sequences (Saier et al. 2014). Proteases were classified at the MEROPS database (Rawlings et al. 2012) using the batch BLAST search service available on the web. Only complete hits with all active sites conserved that were predicted to be secreted were included in the counts. Secondary metabolite clusters were classified using version 3 of the antismash program setting a threshold of a minimum of five genes to form a cluster (Blin et al. 2013). The localizations of secreted proteins were predicted using SignalP 4.1 (Petersen et al. 2011) to predict proteins with signal peptides, which target proteins to the secretory pathway, removing proteins that were predicted to have transmembrane domains according to TMHMM (Krogh et al. 2001) or glycosylphosphatidylinositol (GPI) anchors according to fungal BigPI (Eisenhaber et al. 2004). Proteins were classified as being targeted to membranes if they were predicted to have signal peptides and transmembrane domains according to SignalP (Petersen et al. 2011) and TMHMM (Krogh et al. 2001) analysis. In addition, NLStradamus (Ba et al. 2009) was utilized to predict proteins with nuclear localization signals. In each case, the default setting of each program was utilized. The cysteine contents of predicted protein sequences was assessed using the pepstats package from the EMBOSS 6.4.0.0 suite (Rice et al. 2000). Repeats were identified using Repeatmasker (Smit et al. 1996) using a custom repeat library generated by Repeatscout (Price et al. 2005) after filtering out identified genomic repeats of less than 50 bp and occurring less than ten times in the C. incanum genome assembly. Hierarchical clustering of species according to their secreted protein profiles in supplementary figure S7, Supplementary Material online, was performed and visualized using the pheatmap package (Kolde 2015) within R (R Core Team 2012).
Comparative Genomics
From the BROAD Institute, Neurospora crassa or74a version 12, M. oryzae 70-15 version 6, Fusarium oxysporum f. sp. Lycopersici 4287 version 2, Fusarium graminearum PH-1 version 3, Aspergillus nidulans fgsg a4 1, C. graminicola, C. higginsianum, Sclerotinia sclerotiorum version 2, Botrytis cinerea B04.10 from the JGI Institute, protein sequences from Nectria haematococca version 2 (Coleman et al. 2009), Trichoderma virens Gv29-8 version 2 (Kubicek et al. 2011), Metarhizium robertsii ARSEF 23 (Gao et al. 2011), Verticillium dahliae version 1 (Klosterman et al. 2011), Chaetomium globosum version 1 (Berka et al. 2011), Podospora anserina S mat+ (EspagneĪ et al. 2008), Eutypa lata UCREL1 (Blanco-Ulate et al. 2013), Taphrina deformans (Cissé et al. 2013), Saccharomyces cerevisiae M3707 (Brown et al. 2013), Leptosphaeria maculans version 1 (Rouxel et al. 2011), and sequences from Colletotrichum fructicola Nara gc5 (ANPB00000000.1), Colletotrichum orbiculare 104-T (AMCV00000000.1), Colletotrichum gloeosporioides Cg-14 (Alkan et al. 2013; AMYD01000001.1), Colletotrichum sublineola (JMSE00000000.1), and Colletotrichum fioriniae (JARH00000000.1) were utilized for various analyses. OrthoMCL with an inflation value of 1.5 was performed to identify orthogroups between 22 different fungi (Li et al. 2003) with a blastp E-value cutoff of 1 × 10−5. For analysis of secreted protein families, Colletotrichum proteins grouped by orthoMCL were analyzed using numbers for secreted Colletotrichum proteins only. DAGchainer was utilized to identify syntenic regions with minimum chain length equal to five colinear genes that were a maximum of ten genes apart using BLASTP results as an input to identify matching protein-encoding genes (Haas et al. 2004).
Similarly, sequences from 874 single gene orthogroups identified by orthoMCL analysis of the 22 fungi were aligned using MAFFT and trimmed using trimAl (Katoh et al. 2002; Capella-Gutiérrez et al. 2009). Trimmed alignments concatenated in the commercially available program CLCGenomicsWorkbench8 (CLC bio) resulted in a data set of 14909 positions that was partitioned according to gene and was then utilized to draw a maximum-likelihood species phylogeny with RAxML using WAG as a model for substitution and the autoMRE setting to determine the appropriate number of bootstrap samples and specifying the basal ascomycete species Ta. deformans as the outgroup. The best tree (supplementary fig. S8, Supplementary Material online) was then converted into an ultrametric chronogram using the r8s (Sanderson 2003) program applying the nonparametric rate smoothing approach. Divergence times were then estimated using previously derived estimates from Beimforde et al. (2014) of 443–695 Myr for the divergence between Pezizomycotina–Saccharomycotina, 400–583 Myr for the Pezizomycotina crown group, 267–430 Myr for Leotiomycetes–Sordariomycetes, 207–339 Myr for divergence of Sordariomycetes, 487–773 Myr for Ascomycete crown (Beimforde et al. 2014), and on the previously estimated divergence time of 47 Myr between C. graminicola and C. higginsianum (O’Connell et al. 2012). The branch including members from Colletotrichum and V. dahliae was used as the input for the CAFE program version 3 (De Bie et al. 2006) to identify OrthoMCL-defined gene families experiencing gain/loss with P ≤ 0.01 using the filter function to exclude families that are inferred to have no genes at the root of the tree. For this analysis, only families with at least one member present in the analyzed taxa were used. The phylogenetic tree to classify C. incanum (fig. 1) was drawn using previously identified sequences from other Colletotrichum species (Cannon et al. 2012), C. incanum (Yang et al. 2014), and Monilochaetes infuscans as an outgroup (O’Connell et al. 2012). In brief, sequences from actin (ACT), tubulin-2 (TUB2), internal transcribed spacer (ITS), and chitin synthase I (CHS-1) were aligned using MAFFT and trimmed with trimAL with the automated1 settings (Katoh et al. 2002; Capella-Gutiérrez et al. 2009). The concatenated trimmed alignments were then utilized to estimate the maximum-likelihood species phylogeny with RAxML version 8.2.4 (Stamatakis 2014) using the GTRGAMMA model with 1,000 bootstrap replicates.
Positive Selection
To test for positive selection, Colletotrichum sequences conserved in C. incanum, C. graminicola, C. higginsianum, C. fructicola, C. fioriniae, and C. orbiculare were aligned using PRANK (Löytynoja and Goldman 2005). Protein alignments were trimmed using trimAl (Capella-Gutiérrez et al. 2009) to remove regions where more than 70% of the sequences were gapped. Trees were generated based on the trimmed alignments using the PhyML program (Guindon et al. 2010). The ETE toolkit (Huerta-Cepas et al. 2010) was utilized to automatically label branches being tested under the branch-site model of CodeML (Yang 2007). Likelihood-ratio tests were carried out on PRANK (http://wasabiapp.org/software/prank/, last accessed May 2, 2016) nucleotide alignments using the branch-site model of CodeML using the “cleandata” option to remove any sites with gaps in at least one sequence (Yang 2007). To test for branch-site diversifying selection, likelihood-ratio tests comparing the null hypothesis, where dN/dS was fixed at 1 across all branches and sites (model A1), and the alternative hypothesis, where dN/dS was allowed to vary across the branches and sites (model A), were carried out. P-values corresponding to the chi-square values were obtained and False Discovery Rate (FDR) estimates were computed using the Benjamini–Hochberg procedure. Positive selection was considered significant when FDR ≤ 0.05. One-sided Fisher’s exact tests were carried out in R (R Core Team 2012) to test for enrichment of positively selected genes according to predicted localizations.
Results
Colletotrichum incanum Is Able to Infect Arabidopsis but Not Maize
Colletotrichum incanum (MAFF238712, strain 9503) was originally isolated from Japanese daikon radish and was previously identified as a strain of Colletotrichum dematium from the dematium clade (Sato et al. 2005). However, molecular phylogenetic analysis with sequences from CHS, ACT, ITS, and TUB genes indicated that it is a member of the spaethianum clade (fig. 1) and that it is in fact a strain of the C. incanum species, a recently described species that has been shown to infect soybean (Yang et al. 2014).
Given that the strain was isolated from Japanese daikon radish, a member of the Brassicaceae family, we thought that C. incanum may also be able to infect the model plant, Arabidopsis. To this end, 30 accessions of Arabidopsis were tested as potential hosts for infection, with results showing distinct virulence phenotypes in different ecotype accessions (fig. 2 and supplementary table S1, Supplementary Material online). Host susceptibility profiles differed from that of the known Colletotrichum pathogen of Arabidopsis, C. higginsianum (Narusaka et al. 2004, 2009) which belongs to the destructivum clade, indicating that there are fungal strain-specific factors that determine host compatibility between the different species rather than a general resistance against fungal infection. Intriguingly, nonsusceptible Arabidopsis plants lacking a functional copy of the eds1 gene (Bartsch et al. 2006), which is required for the function of many plant resistance proteins, still did not show any increase in susceptibility to C. incanum infection (supplementary fig. S1, Supplementary Material online).
Fig. 2.—

Infection phenotypes of Colletotrichum incanum on various host plants. The ability of C. incanum to infect (A, B) Arabidopsis, (C) lily, and (D) maize. (A) Trypan blue staining of Arabidopsis leaves infected by C. incanum at 6 days post infection (dpi). The growth of intracellular hyphae during infection of C. incanum transformed with GFP during leaf infection of (B) the susceptible Arabidopsis accession Bay-0 at 2 dpi and (C) lily at 5 dpi. (D) Infection of maize leaves by (I) C. incanum and (II) Colletotrichum graminicola at 8 dpi. Necrotic lesions were observed on drop-inoculated C. graminicola-infected but not C. incanum-infected leaves. White arrowheads: conidia showing site of initial infection; unfilled arrowheads: host cell wall.
In addition, because members of the spaethianum clade are known to associate with both dicot and monocot hosts, specifically lilies, we tested if C. incanum could also infect lily plants. Lily leaves were shown to be compatible to infection allowing for growth of intracellular hyphae and subsequent invasion of neighboring cells from the primary site of infection (fig. 2C). In comparison, at the same time point, C. higginsianum showed the formation of appressoria but no further hyphal growth. It was also tested if C. incanum could infect maize, because it is relatively closely related to C. graminicola. However, C. incanum 9503 was not found to be able to infect maize, Brachypodium, or rice under the conditions tested (fig. 2D and supplementary fig. S2, Supplementary Material online).
Assembly of the Colletotrichum incanum Genome
The genome of C. incanum strain 9503 was sequenced using Illumina HiSeq paired-end reads. After filtering of low quality reads, reads were assembled into 1,036 scaffolds to a final assembly of 53.25 Mb size with an estimated 153× coverage and a scaffold N50 of 292 kb (table 1). The size of the genome was estimated to be 58.92 Mb according to k-mer analysis, indicating that most of the genome is included in the assembly. Furthermore, according to CEGMA analysis, where the presence of a set of conserved eukaryotic genes is assessed, 97.98% full/99.19% partial copies of these highly conserved genes were present in the assembly, indicating that gene-encoding regions are well represented. This level of gene coverage was comparable with previously sequenced Colletotrichum genomes. For example, CEGMA assessed coverage was 91.9% for C. higginsianum, 98.8% for C. graminicola, 97.98% for C. orbiculare, and 96.37% complete in the case of C. fructicola (previously reported as C. gloeosporioides) assemblies, respectively (O’Connell et al. 2012; Gan et al. 2013). A total of 11,852 protein-coding genes were predicted in the C. incanum 9503 genome, which is lower compared with the Arabidopsis-infecting Colletotrichum species, C. higginsianum, which possesses 16,172 protein-coding genes. However, the number of predicted genes in C. incanum is comparable with that of the C. graminicola and C. sublineola genomes (O’Connell et al. 2012; Baroncelli, Sanz-Martín, et al. 2014), which have 12,006 and 12,699 protein-coding genes, respectively. GO terms could be assigned to 6967, representing 58.8% of the predicted coding sequences using Interproscan5. In addition, 8878 (74.9%) genes could be assigned a PFAM domain. Approximately 5.86% of the assembly was indicated to be repeat sequences.
Table 1.
Genome Assembly Statistics of Colletotrichumincanum Strain 9503
| Assembly size | 53,254,579 bp |
| Genome estimated size | 58.92 Mb |
| Estimated coverage | 153× |
| G + C% | 52.15 |
| Scaffold N50 | 292,512 bp |
| Contig N50 | 139,052 bp |
| Number of sequencesa | 1,036 |
| Max scaffold size | 1,056,626 bp |
| Max contig size | 790,451 bp |
| Number of genes | 11,852 |
| Number of secreted proteins | 1,002 |
| CEGMA coverage | 97.98% (complete)/99.19% (partial) |
Number of sequences greater than 200 bp.
Gene Family Expansion/Contraction Relative to Other Colletotrichum Genomes
OrthoMCL was used to group proteins from the C. incanum strain with that of proteins from other Colletotrichum species and nonplant pathogenic fungi, S. cerevisiae, N. crassa, P. anserina, and A. nidulans, as well as the more distantly related plant-interacting fungi, M. oryzae, F. graminearum, B. cinerea, E. lata, Ta. deformans, T. virens, V. dahliae, Ch. globosum, Nec. haematococca, and the insect-pathogenic fungi Metarhizium anisopliae. In total, 19,281 orthogroups were identified between the different species. A total of 7,306 of these were found to be conserved in all Colletotrichum species, representing core Colletotrichum genes, with 491 of these including at least one C. incanum predicted secreted protein. According to GO analysis using all C. incanum genes as a reference, noncore genes in C. incanum were enriched in oxidoreductases. In addition, 2,421 gene families were identified, which were specific to members of the Colletotrichum genus. However, only 234 of these Colletotrichum-specific orthogroups were conserved in all members of Colletotrichum. Significantly over-represented GO terms associated with C. incanum genes within these conserved Colletotrichum-specific orthogroups included ion binding, ribonuclease activity, oxidoreductase activity, peptidase and carbohydrate binding, and transport, indicating the function of groups that may have been expanded specifically in the Colletotrichum lineage. The majority of Colletotrichum genes were found to be single-copy genes within individual genomes (supplementary fig. S3, Supplementary Material online).
Out of 11,263 C. incanum proteins to be included in the various orthogroups, only 60 proteins, belonging to 20 groups, were predicted to be coded by genes specific to C. incanum, indicating that the majority of genes predicted are conserved in other members within the genus. More genes were found to be shared with C. higginsianum than any other species tested as may be expected from the close evolutionary distance between the two fungi and overlapping host range. Among the C. incanum-specific orthogroups (supplementary table S2, Supplementary Material online) were 5 with homology to transposable elements, 1 with homology to kinase-like proteins, 1 with homology to alcohol dehydrogenase-like domain-containing proteins, 1 with homology to superoxide dismutase, and 11 groups encoding hypothetical proteins.
Analysis using the program CAFE (De Bie et al. 2006) indicated that the number of gene families experiencing gene loss is greater in Colletotrichum members than the corresponding numbers of families undergoing expansions at each speciation event with the exception of C. higginsianum where gene family expansion appeared to be more dominant and C. graminicola where there were equal numbers of gene families experiencing gain and loss (fig. 3). A total of 20 gene families were expanded and 316 contracted in the graminicola-clade strains C. sublineola, which infects sorghum, and C. graminicola, which infects maize, relative to their most recent common ancestor (MRCA) with C. incanum (fig. 3). Out of the 336 orthogroups undergoing changes in copy number at P < 0.01, 82 gene families experiencing rapid evolution in the graminicola clade were identified using the Viterbi algorithm in the CAFE program (Han et al. 2013) to assign P-values to the expansions/contractions experienced at each branch and using a cutoff of P < 0.05. GO terms over-represented among proteins in the orthogroups reduced in C. graminicola but present in C. incanum include ATPase activity, hydrolase activity, transporter activity, and chitinase activity (supplementary table S3, Supplementary Material online).
Fig. 3.—

Comparison of gene family sizes in Colletotrichum incanum relative to related fungi. Maximum-likelihood tree constructed from 1,697 single-copy gene families. Divergence dates were estimated using the r8s program. +: numbers of gene families estimated to have experienced expansions, -: numbers of gene families estimated to have experienced contractions at each node.
In order to gain insights about the mechanism underlying the gene losses experienced by the graminicola lineage, the genomic context of genes from gene families experiencing significant gene loss or gain in the graminicola lineage was assessed within the C. incanum genome. Thus, the number of genes in C. incanum in regions with synteny to other Colletotrichum genomes was assessed (supplementary table S4, Supplementary Material online). A total of 8,990 (75.9%) and 7,851 (66.2%) genes out of the 11,852 predicted genes in C. incanum were found to be in 297 and 683 regions of conserved gene order of at least 5 genes or more in comparison with C. graminicola and C. sublineola, respectively. Relative to C. graminicola, the largest region with synteny included 151 genes. In contrast, only 1,428 (12.0%) genes were found to be syntenic to genes in the C. higginsianum genome assembly. However, 7,838 (66.1%), 7,096 (59.9%), and 6,514 (55.0%) genes were found in regions with synteny to the more distantly related species, C. fioriniae from the acutatum clade (Baroncelli, Sreenivasaprasad, et al. 2014), C. orbiculare from the orbiculare clade, and C. fructicola from the gloeosporioides clade, indicating that the lack of synteny with C. higginsianum genome is unusual, and that in general the majority of Colletotrichum genes among different members of the genus are in conserved order. This analysis also indicated that C. incanum is likely to have a genome organization that is similar to C. graminicola despite the difference in host range.
In order to further characterize the genomic regions associated with genes from gene families that were significantly changing in terms of copy number, it was investigated if these genes were being lost or inserted within regions with synteny to C. graminicola or from nonsyntenic regions, which may represent regions that may be experiencing genomic rearrangements or high rates of mutation. Out of the 214 genes in C. incanum associated with gene families that were found to be rapidly evolving in the graminicola lineage in terms of changes to gene copy number, a total of 20 (9.3%) of the genes were identified at the borders of the regions with synteny to C. graminicola making up 2.6% of the genes that fell into this category. A total of 50 of these genes were found within the regions with synteny to C. graminicola making up a small proportion (0.6%) of genes within these syntenic regions. This meant that the majority (69.6%) of genes associated with rapidly evolving gene families were in the regions that were outside of these syntenic regions, despite the fact that genes outside of these regions represent only 16.8% of predicted protein sequences.
In contrast to these rapidly changing gene families, genes encoding secondary metabolite synthesis proteins that are normally found in clusters of conserved gene order (Keller et al. 2005) were analyzed. Analysis of the presence of secondary metabolite backbone synthesis genes in C. incanum indicated the presence of 63 potential clusters, making them similar in number compared with previously sequenced members of Colletotrichum (supplementary table S5, Supplementary Material online). In this case, 41% of genes were found to be conserved in a syntenic region in C. graminicola. Similarly, 37.9% of genes were found to be in some region of synteny with the more distantly related C. orbiculare genome.
Carbohydrate Active Enzymes
CAzymes are among the gene families which are known to be important for virulence in plant pathogens. Graminaceous monocots have lower pectin contents in their primary cell walls compared with dicots and gymnosperms (McNeil et al. 1984). As C. incanum is able to infect monocot (Lilium sp.) plants as well as dicots, the CAzyme complement of C. incanum was investigated. The number of potential plant cell wall hemicellulose and pectin-degrading enzymes in the C. incanum genome was most similar to that of C. higginsianum despite the lower number of genes in the C. incanum genome. In C. incanum, expansions were noted in GH10 (hemicellulose) and PL1, PL3, PL4, PL9, and GH28 pectin-degrading enzyme-encoding genes relative to C. graminicola and C. sublineola (fig. 4). The graminaceous monocot-specific Colletotrichum members showed CAzyme profiles that were distinctly different from all the other Colletotrichum species analyzed. This indicates that gene loss from members of the graminicola clade may have been a consequence of their monocot-specific lifestyle. Interestingly, C. fioriniae, C. gloeosporioides, and C. fructicola were also noted to cluster together with expansions in GH43-encoding genes despite belonging to phylogenetically separate branches of the Colletotrichum lineage with C. fructicola and C. gloeosporioides belonging to the gloeosporioides clade, and C. fioriniae belonging to the acutatum clade (figs. 1 and 3). Importantly, all three species are important postharvest pathogens that exhibit phenotypic similarities, implying the relation of GH43-encoding gene expansions to their postharvest infection strategy. Phylogenetic analysis of all identified Colletotrichum GH43 proteins show lineage-specific expansions of GH43 members within the acutatum and gloeosporioides genomes, with duplications of specific genes within the respective lineages (supplementary fig. S4, Supplementary Material online).
Fig. 4.—

Comparative analysis of selected families of Colletotrichum incanum carbohydrate-active enzymes which may be involved during plant infection listed according to common substrates (Zhao et al. 2013).
Transporters
Given that GO terms associated with transporters were also enriched among gene families experiencing significant changes in copy number in the graminicola clade relative to other Colletotrichum genomes, gene families encoding transporters identified as experiencing significant gene gain/loss compared with other Colletotrichum species were assessed. In most cases, these gene families were associated with major facilitator superfamily (MFS) type transporters, including those encoding myo-inositol transporters (2.A.1.1.8), general glucose transporters (2.A.1.1.11), and several family members from the anion:cation symporter (ACS) family (2.A.1.14.17, 2.A.1.14.11, and 2.A.1.14.3). Global analysis of all MFS transporters encoded in the genomes showed that indeed the graminicola clade members clustered separately from that of the other Colletotrichum genome with the exception of C. incanum. As mentioned, C. incanum can infect monocot plants as well as dicot plants; thus, the result might suggest a possible link between MFS transporter specification with monocot plant infection. However, even compared with C. incanum, the graminicola clade members showed reduced numbers of myo-inositol transporters (2.A.1.1.8), glucose transporters (2.A.1.1.11 and 2.A.1.1.68), and selected ACS family transporters (2.A.1.14.11 and 2.A.1.14.17) (supplementary fig. S5, Supplementary Material online). Potentially, these could be dispensable for a graminicolous infection lifestyle.
Proteases
The protease profiles of various members of Colletotrichum were analyzed. In contrast to the CAzyme genes, the protease-encoding gene profile of C. incanum showed more similarities to that of graminicola clade members, C. sublineola, and C. graminicola despite the difference in host range between C. incanum and the graminicola clade members. However, as observed for CAzymes, C. fioriniae and C. fructicola, which belong to the acutatum and gloeosporioides clades respectively, clustered together based on their secreted protease profiles rather than together with Colletotrichum species to which they are more closely related (supplementary fig. S6, Supplementary Material online). In both these fungi, expansions were noted among the S10 serine carboxypeptidases, which have broad substrate specificities and are active in acidic environments, in contrast to the other serine proteases that are normally active in neutral/alkaline environments (Laskar et al. 2012), indicating that these enzymes may be important for their common infection lifestyle. In addition, a search for homologs of subtilisins (S08A proteases), which were previously shown to be more closely related to plant than fungal proteins (Armijos Jaramillo et al. 2013; Gan et al. 2013), revealed the presence of two related sequences in the genome, indicating that they are also conserved in C. incanum.
Secreted Proteins
A total of 1,002 genes (8.2% of predicted protein-encoding genes) were predicted to encode secreted proteins in C. incanum. Out of these, 972 were assigned to 840 orthogroups, with only 32 predicted secreted proteins that were C. incanum-specific. GO terms over-represented among these secreted proteins included those associated with hydrolase activity including peptidase and carbohydrate-degrading activity. In addition, a number of homologs to known effectors from other plant pathogenic fungi were identified including AvrPi54, MC69 from M. oryzae, and secreted in xylem 6 from Fusarium spp. (supplementary table S6, Supplementary Material online). Homologs of AvrPi54 are also conserved in other Colletotrichum fungi although its expression has not been detected. Notably, no homolog of DN3, which was found to be essential for suppression of the conserved effector NIS1 (Yoshino et al. 2012), was identified in C. incanum, as well as in the C. sublineola and C. graminicola assemblies, despite conservation of the NIS1 gene.
Gene families identified by orthoMCL that were predicted to include secreted proteins were analyzed based on their conservation in members from representatives of the six major sequenced groups within the Colletotrichum genus (fig. 5). The majority of secreted proteins which were present in orthogroups consisting of two or more secreted proteins are not lineage-specific. The gene families associated with higher copy numbers were found to be widely conserved and the majority of these were found to consist of proteins that could be associated with known PFAM domains. It was also noted that proteins that were less widely conserved (in four species or less) were enriched in cysteine residues relative to more widely conserved proteins (fig. 5). Further, among these, fewer proteins could be assigned PFAM domains, indicating an enrichment of genes with putatively unknown function among the less conserved proteins (fig. 5). The C. fructicola secreted protein profile showed more similarity to C. fioriniae than to the more closely related C. orbiculare in agreement with the findings on CAzymes and proteases described above (supplementary fig. S7, Supplementary Material online). Significantly, this similarity was not noted when clustering all proteases identified within the genomes rather than only those predicted to be secreted (supplementary fig. S6, Supplementary Material online).
Fig. 5.—

Conservation of secreted protein clusters in Colletotrichum across six major clades of the Colletotrichum genus. Ticks indicate number of clusters containing two or more genes in each category. Single-copy genes were not included in this diagram. Tracks represent heatmaps indicating the numbers of genes from each cluster present in the genomes of the following: Ci: Colletotrichum incanum, Ch: Colletotrichum higginsianum, Cg: Colletotrichum graminicola, Cfi: Colletotrichum fioriniae, Cfr: Colletotrichum fructicola, Co: Colletotrichum orbiculare. Len: Scatter plot indicating average length of proteins in each gene family where each line represents 200 amino acids (aa) and green: 0–240 aa, orange: <480 aa, purple: <720 aa, red: <960 aa, light green: <1200 aa, yellow: ≥1440 aa; Ann: Dark blue marks denote gene families where at least half the members can be annotated with a known PFAM domain. Cys %: Average cysteine content of predicted proteins in orthogroup where the line represents 10% average cysteine content and where green: 0–1.6 Cys%, purple: <3.1%, orange: <4.7%, yellow: <6.3%, blue: <7.8%, red: <9.4%, brown: <10.9%, gray: ≤12.5%.
Comparisons between C. incanum and the C. graminicola genome indicated that 58 orthogroups containing secreted proteins were absent in C. graminicola and C. sublineola but present in all other Colletotrichum species analyzed, and may represent genes that are dispensable for infection of graminaceous monocots (supplementary table S2, Supplementary Material online). These orthogroups included PL1, PL3, and GH28 pectin-degrading enzymes, glucooligosaccharide oxidases discussed above, as well as families of three potential effectors EC16, EC20, and EC34, which were previously identified by Kleeman et al. (2012) in C. higginsianum.
Positive Selection
Apart from changes in gene family copy number, mutations in coding sequences are also important for adaptation to specific hosts. Signatures of positive selection with higher levels of nonsynonymous to synonymous mutations indicate genes that are under diversifying selection. Secreted protein-encoding gene sequences were assessed using PAML for positive selection. Because analysis using less than six sequences that are closely related reduces the accuracy of predicting positively selected sites (Anisimova et al. 2002), only gene families with a 1:1 orthology conserved in six sequenced genomes, representing each of the major clades sequenced, were assessed allowing for more reliable assessment of selection. These proteins were then divided based on the predicted localizations of C. incanum homologs. In addition, rather than analyzing dN/dS ratios over whole protein sequences, branch-site models were used, allowing for the detection of positive selection that acts on only certain sites of a full protein and in specified lineages, to identify sequences important for lineage-specific differences. In these tests, only conserved regions were tested for positive selection.
Out of the 5,940 single-gene orthogroups that were tested, 310 were predicted to include a secreted protein from the C. incanum genome, 1,010 were predicted membrane proteins, and 1,367 were predicted to have a nuclear localization signal. Out of the secreted protein orthogroups, only ten sequences were predicted to have experienced positive selection specifically in the C. incanum lineage (FDR ≤ 0.05). Furthermore, it was estimated that in C. graminicola, seven sequences (supplementary table S7, Supplementary Material online) were under some significant levels of positive selection. Notably, GLRG_09110 is a homolog of the cas1 appressorium-specific protein and GLRG_05601 and GLRG_04689 are hypothetical proteins.
Although signatures of positive selection could be detected in proteins localized to all compartments predicted even among these highly conserved single-copy genes, higher proportions of genes encoding secreted proteins were predicted to be under positive selection relative to those that targeted to other localizations in C. graminicola, C. higginsianum, C. fioriniae, and C. fructicola (P < 0.05; fig. 6). In addition, in C. fructicola, C. fioriniae, and C. orbiculare, genes predicted to encode proteins with nuclear localization signals were also enriched with positively selected sequences relative to those targeted to other localizations (P < 0.05). In contrast, no such enrichment was observed for membrane localized proteins. This indicates that the diversification of both secreted and nuclear localized proteins could play an important role in adaption to lineage-specific infection lifestyles.
Fig. 6.—
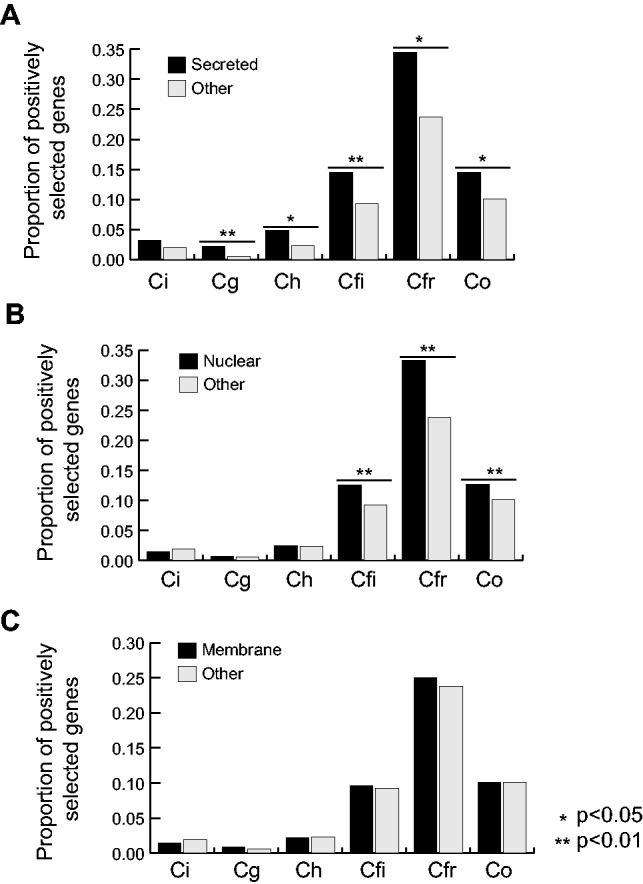
Analysis of positive selection in all single-copy genes identified in Colletotrichum according to predicted localizations. Proportions of lineage-specific positively selected genes among (A) secreted, (B) nuclear, or (C) membrane-localized genes relative to all other proteins. Fisher’s exact test was used to test for significant differences among the proportions within each species. Ci: Colletotrichum incanum, Ch: Colletotrichum higginsianum, Cg: Colletotrichum graminicola, Cfi: Colletotrichum fioriniae, Cfr: Colletotrichum fructicola, Co: Colletotrichum orbiculare.
Discussion
With the inclusion of the C. incanum genome presented in this study, the genomes of six major clades within the genus are now available (O’Connell et al. 2012; Gan et al. 2013; Baroncelli, Sreenivasaprasad, et al. 2014), enabling analysis of evolution between different members of Colletotrichum at a genus-wide level. Members of the spaethianum clade including C. incanum are interesting in that they are able to associate with a range of hosts, from dicots such as Arabidopsis, to the monocot lily, as shown in this study.
Interestingly, C. incanum shows a slightly different host range on different Arabidopsis accessions compared with that of the closely related species C. higginsianum, which has been widely utilized as a model for hemibiotrophic fungal infection in the model plant Arabidopsis (Narusaka et al. 2004; O’Connell et al. 2004). In many plant–pathogen interactions, it is thought that pathogen host range may be determined by the presence or absence of specific pathogen effector proteins that promote virulence on specific hosts. In some cases, effectors may be recognized by cognate host plant resistance proteins, leading to pathogen death. In the case of the Col-0 Arabidopsis accession, that is susceptible to C. higginsianum but not C. incanum, we show that plants that were lacking the eds1 gene, a key signaling component for a large number of plant resistance proteins, were not compromised in resistance to C. incanum. Based on this result, it is not clear that resistance of Arabidopsis to C. incanum is mediated by host resistance proteins. Future analyses of crosses between susceptible and resistant Arabidopsis lines may provide further information regarding the molecular mechanism of C. incanum resistance in Arabidopsis.
Gene family expansions and contractions have been shown to be important for evolution as an adaption to new ecological niches (Lespinet et al. 2002). Indeed, gene losses were associated with the change in host range of the smut fungus Melanopsichium pennyslvanicum, a dicot-infecting fungus that evolved from an ancestral monocot-infecting fungus (Sharma et al. 2014). This study shows that gene family losses are more common than gene family expansions during the evolution of Colletotrichum species. One possible explanation of this is that genes required for infection of different host plants possibly existed in ancestral Colletotrichum species and that losses from existing families occurred after host specialization. However, it is noted that the CAFE analysis used in this case is limited to the birth–death model of gene family evolution which may result in an overestimation of families with members present in the MRCA of the analyzed taxa (De Bie et al. 2006) and thus an increase in the number of families estimated to have experienced losses. Also, such a model is limited because it simplifies the nature of gene gain and loss that occurs in nature. For example, it does not take into account more complex gene gain or loss mechanisms such as horizontal gene transfer between two phylogenetically distinct taxa (Ames et al. 2012; Librado et al. 2012).
Of the Colletotrichum species studied, only C. higginsianum showed a greater number of gene families experiencing expansions compared with contractions. The C. higginsianum genome assembly is relatively fragmented relative to that of the other analyzed Colletotrichum species, consisting of more than 10,000 scaffolds of greater than 1 kb in length. Thus, it is possible that some gene families may be artificially inflated in number if two or more partial gene models corresponding to the same transcript are split onto different scaffolds. It was due to this as well that synteny and conservation of potential secondary metabolite biosynthesis clusters in C. higginsianum relative to that of other Colletotrichum species could not be accurately assessed.
An important feature that has emerged from the comparative analyses has been that the CAzyme complement of closely related fungi within the genus was shown to differ based on pathogen lifestyle rather than according to their phylogenetic relationship. It has previously been shown that the dicot-infecting C. higginsianum encodes more pectin-degrading enzymes compared with the monocot-adapted pathogen C. graminicola (O’Connell et al. 2012). In keeping with the hypothesis that the types and numbers of fungal CAzyme proteins are influenced by their host ranges, C. incanum, which can infect both Arabidopsis and the monocot lily, and C. higginsianum, which infects Arabidopsis, show CAzyme profiles that are more similar to that of other dicot-infecting Colletotrichum species, despite being more closely related to the monocot-specific graminicola clade members. Interestingly, it was also observed that members of the gloeosporioides clade showed more similarities in terms of their plant cell wall-degrading CAzymes to that of C. fioriniae, rather than to more closely related species such as C. orbiculare. Lineage-specific expansions especially in GH43 were noted in both C. fioriniae and C. fructicola indicating that expansions are likely to have occurred after the two lineages diverged and may have occurred independently within the genus. Together, this genus-wide analysis indicates that gene loss and gene gain are important mechanisms for Colletotrichum to tailor their genes according to their specific pathogenic lifestyles.
Intriguingly, both C. fructicola and C. fioriniae also showed a similar clustering when grouped according to the number and types of predicted secreted protease-encoding genes, especially with expansions in S10 proteases. Members from both the gloeosporioides and the acutatum clades are noted for broad host ranges and their ability to infect fruit, causing fruit rot on a variety of plants. Indeed, until molecular methods were developed, they were difficult to distinguish by traditional taxonomical methods and strains from one group were often confused for the other (Wharton and Diéguez-Uribeondo 2004). The genetic similarities observed indicate that similar molecular mechanisms may underlie the phenotypic similarities observed between the two clades of fungi despite their phylogenetic separation.
In addition, in our analysis of secreted proteins, it was observed that groups of less conserved protein-encoding genes were associated with higher average cysteine contents and lack of PFAM domain assignments (fig. 5). This is interesting given that hallmarks of effector proteins that are known to contribute to infection in other plant pathogenic fungi include lack of known protein domains and higher cysteine contents, which may be important for structural stabilization of these proteins (Stergiopoulos and de Wit 2009; Sperschneider et al. 2015). Because some effectors are also recognized by host immune components that differ from host to host, the reduced conservation of these proteins among different members of Colletotrichum may be due to effector diversification or loss to avoid recognition by different hosts.
In addition, specific gene families associated with transmembrane transport were also found to be reduced among graminicola clade members relative to that of other Colletotrichum species. Among transporter families with reduced numbers were those encoding myo-inositol transporters. Interestingly, there is evidence that the presence of exogenous myo-inositol can differentially affect monocot plants, such as perennial ryegrass, and the dicot plant Arabidopsis (Zhang et al. 2013). Differences include lignin and starch accumulation in the presence of exogenous myo-inositol and the reduction of defense responses in its absence in monocots, which were not detected in Arabidopsis (Zhang et al. 2013). It is possible that the reduced number of myo-inositol transporters in monocot-specific Colletotrichum may have occurred during host specialization although this has yet to be explored.
Another important mechanism of adaptation to new ecological niches and lifestyles has been functional mutation of genes. A recent study analyzing genome sequence evolution among eight different C. graminicola strains indicated that noncoding and coding sequences associated with effectors as well as genes upregulated during infection were shown to have higher levels of polymorphisms (Rech et al. 2014). In this study, we searched for signatures of positive selection in specific lineages with the hypothesis that genes under positive selection would be rapidly evolving in response to different ecological niches. Interestingly, even among highly conserved proteins, it was noted that genes predicted to encode secreted proteins were enriched for lineage-specific positively selected genes. In addition, it was found that in C. orbiculare, C. fioriniae, and C. fructicola, genes encoding proteins with potential nuclear localization sequences were also enriched for positively selected proteins to a similar degree as the secreted proteins. Conceivably, the diversification of transcription regulators may also be important in adaptation to different lifestyles.
Supplementary Material
Supplementary tables S1–S7 and figures S1–S8 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work was supported in part by the Council for Science, Technology and Innovation, Cross-ministerial Strategic Innovation Promotion Program, “Technologies for creating next-generation agriculture, forestry and fisheries” (Funding agency: Bio-oriented Technology Research Advancement Institution, NARO), the Science and Technology Research Promotion Program for the Agriculture, Forestry, Fisheries, and Food industry to Y.N., Y.T., and K.S., and grant-in-aid for Scientific Research (KAKENHI) (24228008 to K.S, 21380031 to Y.K.). Computations were partially performed on the NIG Supercomputer at ROIS National Institute of Genetics.
Literature Cited
- Alkan N, et al. 2013. Global aspects of pacC regulation of pathogenicity genes in Colletotrichum gloeosporioides as revealed by transcriptome analysis. Mol Plant Microbe Interact. 26:1345–1358. [DOI] [PubMed] [Google Scholar]
- Ames RM, Money D, Ghatge VP, Whelan S, Lovell SC. 2012. Determining the evolutionary history of gene families. Bioinformatics 28:48–55. [DOI] [PubMed] [Google Scholar]
- Anisimova M, Bielawski JP, Yang Z. 2002. Accuracy and power of Bayes prediction of amino acid sites under positive selection. Mol Biol Evol. 19:950–958. [DOI] [PubMed] [Google Scholar]
- Armijos Jaramillo VD, Vargas WA, Sukno SA, Thon MR. 2013. Horizontal transfer of a subtilisin gene from plants into an ancestor of the plant pathogenic fungal genus Colletotrichum. PLoS One 8:e59078.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ba ANN, Pogoutse A, Provart N, Moses AM. 2009. NLStradamus: a simple Hidden Markov Model for nuclear localization signal prediction. BMC Bioinformatics 10:202.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroncelli R, Sanz-Martín JM, Rech GE, Sukno SA, Thon MR. 2014. Draft genome sequence of Colletotrichum sublineola, a destructive pathogen of cultivated sorghum. Genome Announc. 2:e00540–e00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroncelli R, Sreenivasaprasad S, Sukno SA, Thon MR, Holub E. 2014. Draft genome sequence of Colletotrichum acutatum sensu lato (Colletotrichum fioriniae). Genome Announc 2:e00112–e00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch M, et al. 2006. Salicylic acid-independent ENHANCED DISEASE SUSCEPTIBILITY1 signaling in Arabidopsis immunity and cell death is regulated by the monooxygenase FMO1 and the nudix hydrolase NUDT7. Plant Cell 18:1038–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beimforde C, et al. 2014. Estimating the Phanerozoic history of the Ascomycota lineages: combining fossil and molecular data. Mol Phylogenet Evol. 78:386–398. [DOI] [PubMed] [Google Scholar]
- Berka RM, et al. 2011. Comparative genomic analysis of the thermophilic biomass-degrading fungi Myceliophthora thermophila and Thielavia terrestris. Nat Biotechnol. 29:922–927. [DOI] [PubMed] [Google Scholar]
- Blanco-Ulate B, Rolshausen PE, Cantu D. 2013. Draft genome sequence of the grapevine dieback fungus Eutypa lata UCR-EL1. Genome Announc 1:e00228–e00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin K, et al. 2013. antiSMASH 2.0—a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 41:W204–W212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SD, et al. 2013. Genome sequences of industrially relevant Saccharomyces cerevisiae strain M3707, isolated from a sample of distillers yeast and four haploid derivatives. Genome Announc. 1:e00323–e00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon PF, Damm U, Johnston PR, Weir BS. 2012. Colletotrichum—current status and future directions. Stud Mycol. 73:181–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel BL, et al. 2008. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18:188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cissé OH, et al. 2013. Genome sequencing of the plant pathogen Taphrina deformans, the causal agent of peach leaf curl. MBio 4:e00055-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman JJ, et al. 2009. The genome of Nectria haematococca: contribution of supernumerary chromosomes to gene expansion. PLoS Genet. 5:e1000618.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch JA, Beirn LA. 2009. Anthracnose of cereals and grasses. Fungal Divers. 39:19–44. [Google Scholar]
- Crouch J, et al. 2014. The genomics of Colletotrichum In: Dean RA, Lichens-Park A, Kole C, editors. Genomics of plant-associated fungi: monocot pathogens. Berlin (Germany: ): Springer; p. 69–102. [Google Scholar]
- De Bie T, Cristianini N, Demuth JP, Hahn MW. 2006. CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22:1269–1271. [DOI] [PubMed] [Google Scholar]
- Dean R, et al. 2012. The top 10 fungal pathogens in molecular plant pathology. Mol Plant Pathol. 13:414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhaber B, Schneider G, Wildpaner M, Eisenhaber F. 2004. A sensitive predictor for potential GPI lipid modification sites in fungal protein sequences and its application to genome-wide studies for Aspergillus nidulans, Candida albicans, Neurospora crassa, Saccharomyces cerevisiae and Schizosaccharomyces pombe. J Mol Biol. 337:243–253. [DOI] [PubMed] [Google Scholar]
- EspagneĪ E, et al. 2008. The genome sequence of the model ascomycete fungus Podospora anserina. Genome Biol. 9:R77.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon S, Gentleman R. 2007. Using GOstats to test gene lists for GO term association. Bioinformatics 23:257–258. [DOI] [PubMed] [Google Scholar]
- Falk A, et al. 1999. EDS1, an essential component of R gene-mediated disease resistance in Arabidopsis has homology to eukaryotic lipases. Proc Natl Acad Sci U S A. 96:3292–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan P, et al. 2013. Comparative genomic and transcriptomic analyses reveal the hemibiotrophic stage shift of Colletotrichum fungi. New Phytol. 197:1236–1249. [DOI] [PubMed] [Google Scholar]
- Gangadevi V, Muthumary J. 2008. Isolation of Colletotrichum gloeosporioides, a novel endophytic taxol-producing fungus from the leaves of a medicinal plant, Justicia gendarussa. Mycol Balcanica. 5:1–4. [Google Scholar]
- Gao Q, et al. 2011. Genome sequencing and comparative transcriptomics of the model entomopathogenic fungi Metarhizium anisopliae and M. acridum. PLoS Genet. 7:e1001264.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, et al. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 59:307–321. [DOI] [PubMed] [Google Scholar]
- Haas BJ, Delcher AL, Wortman JR, Salzberg SL. 2004. DAGchainer: a tool for mining segmental genome duplications and synteny. Bioinformatics 20:3643–3646. [DOI] [PubMed] [Google Scholar]
- Han MV, Thomas GW, Lugo-Martinez J, Hahn MW. 2013. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol Biol Evol. 30:1987–1997. [DOI] [PubMed] [Google Scholar]
- Huerta-Cepas J, Dopazo J, Gabaldón T. 2010. ETE: a python environment for tree exploration. BMC Bioinformatics 11:24.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, et al. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 30:1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment on fast Fourier transform. Nucleic Acids Res. 30:3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller NP, Turner G, Bennett JW. 2005. Fungal secondary metabolism—from biochemistry to genomics. Nat Rev Microbiol. 3:937–947. [DOI] [PubMed] [Google Scholar]
- Keller O, Odronitz F, Stanke M, Kollmar M, Waack S. 2008. Scipio: using protein sequences to determine the precise exon/intron structures of genes and their orthologs in closely related species. BMC Bioinformatics 9:278.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleemann J, et al. 2012. Sequential delivery of host-induced virulence effectors by appressoria and intracellular hyphae of the phytopathogen Colletotrichum higginsianum. PLoS Pathog. 8:e1002643.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klosterman SJ, et al. 2011. Comparative genomics yields insights into niche adaptation of plant vascular wilt pathogens. PLoS Pathog. 7:e1002137.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolde R. 2015. pheatmap: Pretty Heatmaps. R package version 1.0. 2. [cited 2016 Jan 19]. https://CRAN.R-project.org/package=pheatmap.
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 305:567–580. [DOI] [PubMed] [Google Scholar]
- Kubicek CP, et al. 2011. Comparative genome sequence analysis underscores mycoparasitism as the ancestral life style of Trichoderma. Genome Biol. 12:R40.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo Y, Nakamura H, Kobayashi K, Okuno T, Furusawa I. 1991. Cloning of a melanin biosynthetic gene essential for appressorial penetration of Colletotrichum lagenarium. Mol Plant Microbe Interact. 4:440–445. [DOI] [PubMed] [Google Scholar]
- Laskar A, Chatterjee A, Chatterjee S, Rodger EJ. 2012. Three-dimensional molecular modeling of a diverse range of SC clan serine proteases. Mol Biol Int. 2012:e580965.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lespinet O, Wolf YI, Koonin EV, Aravind L. 2002. The role of lineage-specific gene family expansion in the evolution of eukaryotes. Genome Res. 12:1048–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Bork P. 2011. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 39:W475–W478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ, Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13:2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P, Vieira FG, Rozas J. 2012. BadiRate: estimating family turnover rates by likelihood-based methods. Bioinformatics 28:279–281. [DOI] [PubMed] [Google Scholar]
- Löytynoja A, Goldman N. 2005. An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci U S A. 102:10557–10562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R, et al. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1:18.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil M, Darvill AG, Fry SC, Albersheim P. 1984. Structure and function of the primary cell walls of plants. Annu Rev Biochem. 53:625–663. [DOI] [PubMed] [Google Scholar]
- Mejía LC, et al. 2014. Pervasive effects of a dominant foliar endophytic fungus on host genetic and phenotypic expression in a tropical tree. Front Microbiol. 5:479.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutuku JM, et al. 2015. The WRKY45-dependent signaling pathway is required for resistance against Striga hermonthica parasitism. Plant Physiol. 168:1152–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narusaka M, Kubo Y, Shiraishi T, Iwabuchi M, Narusaka Y. 2009. A dual resistance gene system prevents infection by three distinct pathogens. Plant Signal Behav. 4:954–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narusaka Y, et al. 2004. RCH1, a locus in Arabidopsis that confers resistance to the hemibiotrophic fungal pathogen Colletotrichum higginsianum. Mol Plant Microbe Interact. 17:749–762. [DOI] [PubMed] [Google Scholar]
- O’Connell R, et al. 2004. A novel Arabidopsis-Colletotrichum pathosystem for the molecular dissection of plant-fungal interactions. Mol Plant Microbe Interact. 17:272–282. [DOI] [PubMed] [Google Scholar]
- O’Connell RJ, et al. 2012. Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat Genet. 44:1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra G, Bradnam K, Korf I. 2007. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23:1061–1067. [DOI] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 8:785–786. [DOI] [PubMed] [Google Scholar]
- Price AL, Jones NC, Pevzner PA. 2005. De novo identification of repeat families in large genomes. Bioinformatics 21:i351–i358. [DOI] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP. 2010. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2012. R: a language and environment for statistical computing. Vienna (Austria: ): R Foundation for Statistical Computing [Google Scholar]
- Rawlings ND, Barrett AJ, Bateman A. 2012. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 40:D343–D350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rech GE, Sanz-Martín JM, Anisimova M, Sukno SA, Thon MR. 2014. Natural selection on coding and noncoding DNA sequences is associated with virulence genes in a plant pathogenic fungus. Genome Biol Evol. 6:2368–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. 2000. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 16:276–277. [DOI] [PubMed] [Google Scholar]
- Rouxel T, et al. 2011. Effector diversification within compartments of the Leptosphaeria maculans genome affected by Repeat-Induced Point mutations. Nat Commun. 2:202.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saier MH, Reddy VS, Tamang DG, Vastermark A. 2014. The transporter classification database. Nucleic Acids Res. 42:D251–D258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson MJ. 2003. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19:301–302. [DOI] [PubMed] [Google Scholar]
- Sharma KR, Bhagya N, Sheik S, Samhitha M. 2011. Isolation of endophytic Colletotrichum gloeosporioides Penz. from Salacia chinensis and its antifungal sensitivity. J Phytol. 3:20–22. [Google Scholar]
- Sharma R, Mishra B, Runge F, Thines M. 2014. Gene loss rather than gene gain is associated with a host jump from monocots to dicots in the smut fungus Melanopsichium pennsylvanicum. Genome Biol Evol. 6:2034–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AF, Hubley R, Green P. 1996–2010. RepeatMasker Open-3.0. http://www.repeatmasker.org.
- Sperschneider J, et al. 2015. Advances and challenges in computational prediction of effectors from plant pathogenic fungi. PLoS Pathog. 11:e1004806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Schöffmann O, Morgenstern B, Waack S. 2006. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stergiopoulos I, de Wit PJGM. 2009. Fungal effector proteins. Annu Rev Phytopathol. 47:233–263. [DOI] [PubMed] [Google Scholar]
- Tao G, Liu ZY, Liu F, Gao YH, Cai L. 2013. Endophytic Colletotrichum species from Bletilla ochracea (Orchidaceae), with descriptions of seven new speices. Fungal Divers. 61:139–164. [Google Scholar]
- Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. 2008. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 18:1979–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wharton PS, Diéguez-Uribeondo J. 2004. The biology of Colletotrichum acutatum. Ann Jardín Bot Madrid. 61:3–22. [Google Scholar]
- Yang HC, Haudenshield JS, Hartman GL. 2014. Colletotrichum incanum sp. nov., a curved-conidial species causing soybean anthracnose in USA. Mycologia 106:32–42. [DOI] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24:1586–1591. [DOI] [PubMed] [Google Scholar]
- Yin Y, et al. 2012. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 40:W445–W451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino K, et al. 2012. Cell death of Nicotiana benthamiana is induced by secreted protein NIS1 of Colletotrichum orbiculare and is suppressed by a homologue of CgDN3. Mol Plant Microbe Interact. 25:625–636. [DOI] [PubMed] [Google Scholar]
- Zhang WJ, Dewey RE, Boss W, Phillippy BQ, Qu R. 2013. Enhanced Agrobacterium-mediated transformation efficiencies in monocot cells is associated with attenuated defense responses. Plant Mol Biol. 81:273–286. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Liu H, Wang C, Xu JR. 2013. Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genomics 14:274.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.