

Abstract
Many proteins depend on an interaction with molecular chaperones in order to fold into a functional tertiary structure. Previous studies showed that protein interaction with the GroEL/GroES chaperonine and Hsp90 chaperone can buffer the impact of slightly deleterious mutations in the protein sequence. This capacity of GroEL/GroES to prevent protein misfolding has been shown to accelerate the evolution of its client proteins. Whether other bacterial chaperones have a similar effect on their client proteins is currently unknown. Here, we study the impact of DnaK (Hsp70) chaperone on the evolution of its client proteins. Evolutionary parameters were derived from comparison of the Escherichia coli proteome to 1,808,565 orthologous proteins in 1,149 proteobacterial genomes. Our analysis reveals a significant positive correlation between protein binding frequency with DnaK and evolutionary rate. Proteins with high binding affinity to DnaK evolve on average 4.3-fold faster than proteins in the lowest binding affinity class at the genus resolution. Differences in evolutionary rates of DnaK interactor classes are still significant after adjusting for possible effects caused by protein expression level. Furthermore, we observe an additive effect of DnaK and GroEL chaperones on the evolutionary rates of their common interactors. Finally, we found pronounced similarities in the physicochemical profiles that characterize proteins belonging to DnaK and GroEL interactomes. Our results thus implicate DnaK-mediated folding as a major component in shaping protein evolutionary dynamics in bacteria and supply further evidence for the long-term manifestation of chaperone-mediated folding on genome evolution.
Keywords: genome evolution, GroEL, microbial evolution, chaperones
Introduction
Molecular chaperones (Ellis 1987), also called heat-shock proteins (HSPs), are essential in all living cells as they assist protein folding, prevent protein misfolding and aggregation, and play a crucial role in survival under stress conditions (Hartl 2011). Chaperones typically operate by covering the hydrophobic regions in non-native polypeptides to avoid aggregation (Hartl and Hayer-Hartl 2002) and to stabilize them energetically (Netzer and Hartl 1997). Certain types of chaperones assist the folding of their client proteins as early as these interactors are emerging from the ribosome cavity (e.g., DnaK) while others operate down the line of the protein folding pathway (e.g., GroEL/GroES) (Hartl and Hayer-Hartl 2002). Studies of chaperone function at the organismal level revealed that their key role in canalizing the genetic information encoded in genes into functional proteins is of a significant evolutionary importance. The long-term effects of chaperones were studied mainly in the GroEL/GroES chaperonine system. The GroEL forms a barrel shaped complex with the co-chaperone GroES providing the lid (Xu et al. 1997) that assists protein folding by isolating the substrate polypeptide from the cytoplasmic environment in a cage-like compartment (Hartl 2011). GroEL constitutes a major hub in the Escherichia coli protein–protein interaction network (Arifuzzaman et al. 2006).
The evolutionary importance of GroEL/GroES was highlighted in experimental evolution studies where it was shown to compensate for fitness reduction following high mutational loads in E. coli (Fares et al. 2002; Sabater-Muñoz et al. 2015). Interestingly, Salmonella typhimurium lines that evolve under high mutational loads adapt an increased expression level of the GroEL/GroES and DnaK chaperones, which was suggested to contribute to antagonistic epistasis (Maisnier-Patin et al. 2005). In eukaryotes, Hsp90 has been shown to contribute to phenotypic stability in Drosophila melanogaster (Rutherford and Lindquist 1998) and Arabidopsis thaliana (Queitsch et al. 2002). This led to the suggestion that chaperones are capacitors of phenotypic variation that serve as fitness modulators in a changing environment by allowing for a wide spectrum of genetic variants to be maintained within the population (Queitsch et al. 2002; Rutherford 2003).
The function of chaperones in relaxing the intensity of selection against slightly deleterious mutations has been observed also at the molecular level. Directed enzyme evolution in vitro revealed that proteins interacting with the GroEL/GroES chaperonine are less prone to the effects of destabilizing mutations (Tokuriki and Tawfik 2009). Interaction with the chaperonine leads to a doubled accumulation of mutations and can increase the rate of new function acquisition (Tokuriki and Tawfik 2009) Phylogenetic studies showed that the capability of GroEL/GroES to increase the evolutionary rate of their client proteins has consequences for long-term protein evolution and is imprinted in genomes. A comparative analysis of bacterial genomes revealed a significant correlation between protein dependency upon GroEL/GroES for folding and substrate evolutionary rate. Thus, obligatory interaction with GroEL/GroES accelerates the evolution of its client proteins (Bogumil and Dagan 2010; Williams and Fares 2010). Similarly, a phylogenetic study of kinase evolution in mammals showed a significant positive correlation between the protein evolutionary rate and binding affinity to Hsp90 (Lachowiec et al. 2015). This indicates that interaction with the Hsp90 chaperone can lead to accelerated evolutionary rate as well. Furthermore, an analysis of the substrate–chaperone interaction network in Saccharomyces cerevisiae revealed significant differences in evolutionary rates between distinct groups of proteins interacting with different chaperone combinations (Bogumil et al. 2012). Thus, proteins that interact with similar chaperones share the functional constraints that are inherent to chaperone-mediated folding as well as similar relaxation of selection intensity against the accumulation of slightly deleterious mutation during evolution.
A recent survey of proteins that interact with the DnaK chaperone in E. coli (Calloni et al. 2012) enables to investigate whether interaction with that chaperone entails a relaxation of selection on the primary structure of its client proteins. The E. coli DnaK is the most studied Hsp70 chaperone and is a major hub in the E. coli chaperone network (Arifuzzaman et al. 2006; Calloni et al. 2012). DnaK functions with the assistance of two co-chaperones: DnaJ that determines the DnaK substrate specificity and GrpE that catalyzes ATP re-binding and releases the interacting protein (Hartl and Hayer-Hartl 2009). DnaK substrate specificity is determined by a hydrophobic core of four to five residues enriched with Leucine and flanking regions enriched with basic residues (Rüdiger et al. 1997). Furthermore, DnaK clients are characterized by slow folding dynamics (Sekhar et al. 2012). The DnaK interactome in E. coli was estimated by applying a pull-down assay followed by liquid chromatography mass spectrometry (LC-MS) (Calloni et al. 2012), and identified 674 DnaK interactors. The level of protein dependency upon DnaK for folding was estimated by the frequency in which the interaction between each protein and DnaK was observed, relative to the protein abundance in the cytosol. Based on this relative binding frequency, the DnaK interactome was divided into three dependency classes comprising proteins whose interaction with DnaK was either rarely, occasionally, or frequently observed. The three classes of protein dependency on DnaK for folding were verified by the propensity of their member proteins to form aggregates in a ΔdnaK E. coli strain (Calloni et al. 2012). Here, we test for DnaK-mediated accelerated protein evolution by comparing the evolutionary rates of proteins in the three DnaK-dependency classes. In addition, we test for an additive effect of DnaK and GroEL/GroES on the evolutionary rate of common substrates.
Methods
Data
Here, we used the E. coli K12 MG1655 strain (NC_000931) as a reference genome. Data of protein relative binding frequency with DnaK were obtained from Calloni et al. (2012). Data of GroEL dependency classes were obtained from Kerner et al. (2005). Protein accession numbers in both datasets were matched with the E. coli K12 MG1655 accessions according to gene annotations using Uniprot (Bairoch et al. 2005). Genes in the E. coli K12 MG1655 genome were classified into DnaK-dependency classes using the thresholds of relative DnaK-binding frequency as previously described (Calloni et al. 2012). Class IDnaK includes 136 substrates showing the lowest DnaK-dependency. Class IIDnaK includes 329 substrates of medium DnaK-dependency and Class IIIDnaK includes 199 substrates showing the highest dependency upon the DnaK for folding. A total of 3,476 E. coli genes remain unclassified with regards to their interaction with DnaK. Genes that were found to interact with GroEL were divided into three GroEL-dependency classes as previously described (Kerner et al. 2005) and matched to the reference genome. Class IGroEL includes 37 casual interactors whose folding in vitro can be independent of GroEL. Class IIGroEL comprises 123 proteins that depend on GroEL-mediated folding in a temperature-dependent manner and Class IIIGroEL includes 83 obligatory GroEL clients. The remaining 3,897 E. coli genes are unclassified with regard to their interaction with GroEL.
A total of 1,150 complete proteobacterial genomes were downloaded from the NCBI RefSeq database (Tatusova et al. 2014) (version of August 2014). Protein expression levels for E. coli K12 MG1566 proteome were obtained from Lu et al. (2007). Protein expression data are available only for a subset of chaperone interactors and is listed here according to class: IDnaK:32, IIDnaK:114, IIIDnaK:102, IGroEL:28, IIGroEL:31, IIIGroEL:18.
Comparative Evolutionary Analysis
Orthologs to the primary E. coli proteome were identified using a reciprocal best BLAST hit procedure (BLAST version 2.2.29+) (Tatusov et al. 1997; Wolf and Koonin 2012) using an e-value threshold of 1 × 10−10. This yielded 1,809,891 orthologous pairs, including 458,645 orthologs to E. coli DnaK interactors and 168,586 orthologs to E. coli GroEL interactors. The comparative analyses we perform are “star” analyses—one species (E. coli) against all other proteobacterial species. This minimal formulation circumvents the biases that are frequently encountered in the alignment and phylogenetic reconstruction of protein families including hundreds of sequences. Pairwise alignments of E. coli proteins with their orthologs were generated using MAFFT (version 7) (Katoh and Standley 2013). Protein alignments were reverse-translated into nucleotide alignments using PAL2NAL (Suyama et al. 2006). Rates of nonsynonymous nucleotide substitutions were calculated using CODEML (Yang 2007). Amino acid replacement rates were calculated using PROTDIST from the PHYLIP package (version 3.695) (Felsenstein 2005), with the JTT substitution matrix (Jones et al. 1992). Codon adaptation index (CAI) (Sharp and Li 1987) for each gene was calculated using the EMBOSS package (Rice et al. 2000), the codon usage table was constructed using 27 highly expressed housekeeping genes (Sharp and Li 1987) from E. coli K12 MG1566.
Protein Properties
The following protein properties were calculated using in-house PERL scripts: GC content, amino acid usage, molecular weight, protein length, the grand average of hydropathy score (Kyte and Doolittle 1982), and the proportions of: hydrophobic amino acids, positively charged amino acids, negatively charged amino acids, polar amino acids, large amino acids, and rare amino acids. The hypothetical isoelectric point (PI) was inferred using the ExPASy server (Gasteiger et al. 2003). The composition of protein secondary structure (coiled coils, alpha helices, and beta sheets) was determined using PSIPRED software (McGuffin et al. 2000). All statistical tests were performed using MatLab statistical toolbox.
Results
DnaK-Dependent Evolutionary Rates
Here, we test for the footprints of protein interaction with DnaK across 1,149 completely sequenced genomes of Proteobacteria, considered in four taxonomic distances from E. coli: genus, order, class, and phylum. Comparing the evolutionary rates of proteins in the three DnaK-dependency classes revealed significant differences in their number of nonsynonymous substitutions per site as well as the number of amino acid replacements per site in all four taxonomic depths (table 1). For a given genome comparison, the three class-specific mean rates were plotted against the mean rate of all comparisons (i.e., genes) between the pair of genomes; this compensates for genome- and lineage-specific differences in substitution rate and nucleotide bias (fig. 1). Further post hoc comparisons of the mean evolutionary rate among the three classes revealed that in most comparisons Class IIIDnaK shows the highest evolutionary rate, followed by Class IIDnaK and Class IDnaK proteins for which the slowest rates were observed (i.e., Class IIIDnaK > Class IIDnaK > Class IDnaK). The comparison at the genus level is an exception as the evolutionary rate of substrates in classes IDnaK and IIDnaK is not significantly different (α = 0.05, using Tukey’s post hoc test), probably due to the low sequence divergence among Escherichia strains. These results uncover a clear and significant correlation between protein dependency upon DnaK for folding and the protein’s evolutionary rate.
Table 1.
Comparison of Evolutionary Rates Among DnaK-Dependency Classes
| Taxonomic depth | Homogeneity of medians (P value)a | Class orderb | |
|---|---|---|---|
| dN | Genus: Escherichia | <2.2 × 10−16 | III > II = I |
| Order: Enterobacteriales | <2.2 × 10−16 | III > II > I | |
| Class: Gammaproteobacteria | <2.2 × 10−16 | III > II > I | |
| Phylum: Proteobacteria | <2.2 × 10−16 | III > II > I | |
| Protein distance | Genus: Escherichia | <2.2 × 10−16 | III > II = I |
| Order: Enterobacteriales | <2.2 × 10−16 | III > II > I | |
| Class: Gammaproteobacteria | <2.2 × 10−16 | III > II > I | |
| Phylum: Proteobacteria | <2.2 × 10−16 | III > II > I |
aUsing Friedman Mack–Skilling test (Mack and Skillings 1980).
bOrder of the DnaK-dependency classes (IDnaK, IIDnaK, IIIDnaK) sorted by the mean of the relevant measure (α = 0.05, using Tukey’s post hoc test).
Fig. 1.—

Nonsynonymous substitution rates and amino acid replacement rates of DnaK-dependency classes in proteobacteria. Each data point represents the mean of distances of all class members in one genome from their orthologs in E. coli K12. Comparisons are shown in four taxonomic depths: genus (Escherichia), order (Enterobacteriales), class (Gammaproteobacteria), and phylum (Proteobacteria). The taxonomic depth samples are mutually exclusive.
To quantify the effect of DnaK-mediated folding on the evolution of its client proteins we examined the relative evolutionary rates of interactors in the three classes. The ratio of Class IIIDnaK and Class IDnaK calculated from the dN and protein distance at the genus depth reveals that proteins in the highest dependency class evolve, on average, 4-fold faster than proteins in the lowest dependency class (table 2). The ratio of Class IIDnaK and Class IDnaK rates shows that Class IIDnaK proteins evolve, on average, 2-fold faster than Class IDnaK proteins. The comparison of evolutionary rates at the order depth shows that an average of 2-fold rate increase in Class IIIDnaK proteins in comparison to Class IDnaK proteins is still observed. Further comparisons of protein evolutionary rates among the classes at increasing taxonomic depth reveal smaller ratios than the ones observed at the genus and order levels (table 2). These results are explained by the increasing distance between the compared genomes in the class and phylum depths and the reference genome of E. coli as well as possible differences in the DnaK interactome among proteobacteria.
Table 2.
Mean Ratio of Class Rates for DnaK (left) and GroEL (right) Dependency Classes
| Genus | Order | Class | Phylum | |
|---|---|---|---|---|
| dN | ||||
| Class III/I | 4.3; 2.8 | 2.4; 1.5 | 1.4; 1.3 | 1.2; 1.2 |
| Class II/I | 2.4; 1.4 | 1.6; 1.3 | 1.3; 1.1 | 1.1; 1.1 |
| Protein distance | ||||
| Class III/I | 3.9; 2.6 | 2.4; 1.5 | 1.5; 1.2 | 1.2; 1.1 |
| Class II/I | 2.2; 1.2 | 1.6; 1.3 | 1.3; 1.1 | 1.1; 1 |
Note.—The taxonomic depth samples are mutually exclusive.
DnaK-Dependency Classes Correlate with Protein Expression Level
Protein evolutionary rate and expression level are known to be negatively correlated (Zhang and Yang 2015). Indeed, the absolute protein expression levels measured in E. coli K12 MG1655 (Lu et al. 2007) are significantly different among the three DnaK-dependency classes (P = 3.77 × 10−06, using Kruskal–Wallis test). Furthermore, post hoc comparisons show that the mean expression level is negatively correlated with the DnaK-dependency so that proteins in Class IIIDnaK have significantly lower levels of protein expression than Class IIDnaK members and those are significantly less expressed in comparison to Class IDnaK proteins (i.e., Class IDnaK > Class IIDnaK > Class IIIDnaK, α = 0.05, using Tukey’s post hoc test; fig. 2A). We further tested for a correlation between DnaK-dependency and CAI that is known to be positively correlated with protein expression (Sharp and Li 1987; Drummond and Wilke 2008). Our analysis reveals that the CAI is significantly different among the three DnaK-dependency classes, where the following order is observed: Class IDnaK > Class IIDnaK > Class IIIDnaK (α = 0.05, using Tukey’s post hoc test; fig. 2B). Thus, CAI is significantly negatively correlated with the dependency on DnaK for folding.
Fig. 2.—
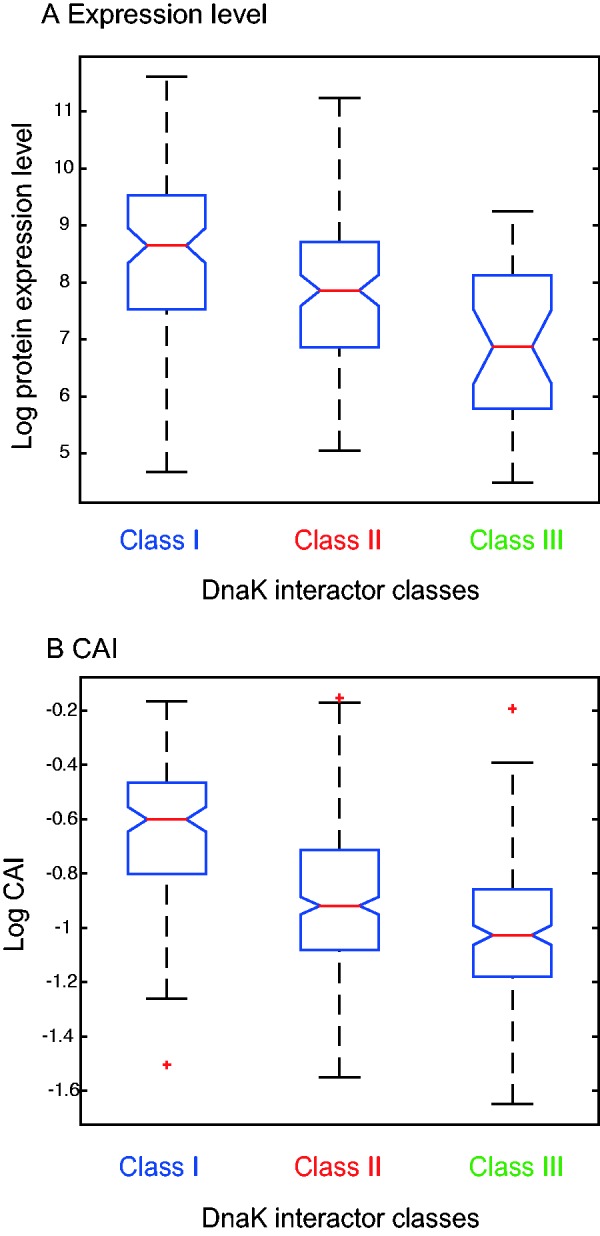
Expression level and CAI of the DnaK-dependency classes in E. coli. Expression data are available for only 248 of the substrate proteins, necessitating the use of CAI as a proxy for expression. All classes are significantly different from each other for both expression level and CAI (α = 0.05, using Kruskal–Wallis and Tukey’s post hoc tests).
Our analysis so far shows that differences in evolutionary rates, expression level, and CAI among the DnaK-dependency classes correspond the long-known correlations among these three measures. This raises the possibility that the observed positive correlation between DnaK-dependency and evolutionary rate stems from differing expression levels of the class members. To exclude that possibility, we tested whether the observed differences in evolutionary rates among the DnaK-dependency classes are still significant when adjusting for protein expression level or CAI. For the application of analysis of covariance (ANCOVA) we first tested the first assumption of that test, namely, that the response variable and covariate are linearly correlated (Zar 1999). All ANCOVA combinations, except those where the linearity assumption could not be validated, resulted in a rejection of the null hypothesis (table 3). This means that after adjusting for differences in the response variable (dN or protein distance) caused by the covariate (protein expression level or CAI), there are still significant differences in the evolutionary rates among the three DnaK-dependency classes.
Table 3.
Analysis of Covariance
| Response variable | Covariate | Taxonomic depth | Pooled regressiond | Homogeneity of slopes among classese | Homogeneity of intercepts among classesf |
|---|---|---|---|---|---|
| dNa | Protein expression level | Genus | 0.349 | n.a. | n.a. |
| Order | 0.001** | 0.15 | 3.5 × 10−6 ** | ||
| Class | 0.009** | 0.6 | 1.01 × 10−7 ** | ||
| Phylum | 0.23 | n.a. | n.a. | ||
| Protein distanceb | Protein expression level | Genus | 0.23 | n.a. | n.a. |
| Order | 0.009* | 0.602 | 1.01 × 10−7** | ||
| Class | 0.049* | 0.54 | 3.22 × 10−6** | ||
| Phylum | 1.29 × 10−4** | 0.08 | 3.63 × 10−7** | ||
| dNa | CAI | Genus | 0.76 | n.a. | n.a. |
| Order | 9.73 × 10−11** | 0.62 | 3.1 × 10−30 ** | ||
| Class | 1.14 × 10−27** | 0.25 | 4.37 × 10−16 ** | ||
| Phylum | 3.26 × 10−13 ** | 0.56 | 1.42 × 10−15 ** | ||
| Protein distancec | CAI | Genus | 0.37 | n.a. | n.a. |
| Order | 9.73 × 10−11 ** | 0.63 | 3.1 × 10−30 ** | ||
| Class | 1.14 × 10−27 ** | 0.26 | 4.37 × 10−16 ** | ||
| Phylum | 3.26 × 10−19 ** | 0.56 | 1.42 × 10−15 ** |
Note.—The ANCOVA test and its underlying assumptions (Zar 1999). The analysis in each taxonomic depth was performed using a single representative genome while maximizing the sample size for the test. The best linear models were obtained when both responses and covariates were transformed logarithmically. Note that the pooled regression hypothesis was rejected in all combinations at the genus depth. This may be due to the small range of rates measured within the Escherichia genomes.
aRepresentative genomes: Genus: E. coli BW2952; Order: Klebsiella oxytoca E718; Class: Aeromonas hydrophila ATCC 7966; Phylum: Burkholderia pseudomallei K96243.
bRepresentative genomes: Genus: E. coli SE15; Order: Klebsiella pneumoniae NTUH_K2044; Class: A. hyrophila ATCC_7966; Phylum: B. pseudomallei K96243.
cRepresentative genomes: Genus: E. coli K12_W3110; Order: Shigella nonnei 53G; Class: A. hyrophila ML09_119; Phylum: Burkholderia sp. 383.
dP value of an F-test with the null hypothesis H0: the response and covariate variables are linearly correlated.
eP value of an F-test for equality of slopes among the classes.
fP value of an F-test for equality of intercepts among the classes.
*P value < 0.05.
**P value <0.01.
Additive Effect of DnaK and GroEL
The observed correlations between DnaK-dependency and protein evolutionary rates correspond to earlier reports of GroEL-dependent accelerated evolutionary rates in prokaryotes (Bogumil and Dagan 2010). Because DnaK and GroEL have a different mode of action, folding assistance by these two chaperones might be compensating for different sets of slightly deleterious mutations in the protein sequence. We therefore probed the possibility of an additive effect of both DnaK and GroEL chaperones on the evolutionary rate of their common substrates. The GroEL interactome has been documented in E. coli by Kerner et al. (2005) to comprise 243 interactors. Those were classified into three classes of dependency including casual (Class IGroEL), temperature dependent (Class IIGroEL), and obligatory (Class IIIGroEL) interactors. An intersection of the DnaK interactome in our dataset with the GroEL interactome yielded 116 proteins that interact with both chaperones.
To test for additive effect of both chaperones we define three combined dependency classes. Proteins that rarely bind with DnaK (Class IDnaK) and are obligatory for GroEL (Classes IIGroEL and IIIGroEL) are classified into Class Gd. This class accounts for 17 proteins that are assumed to depend on the GroEL folding assistance more than that of DnaK. A total of 15 proteins were found to casually bind to GroEL (Class IGroEL) and often bind to DnaK (Classes IIDnaK and IIIDnaK). Those are assumed to depend on the folding assistance of DnaK more than that of GroEL, termed Class Dg. Proteins that were found to interact frequently with DnaK and have an obligatory requirement for the interaction with GroEL are assumed to depend on both chaperones in order to gain a functional conformation. Those were classified into Class DG that includes 14 members. A comparison of the three combined dependency classes reveals that their member proteins are significantly different in the rate of nonsynonymous substitutions and amino acid replacements at all taxonomic depths (fig. 3; table 4). Higher evolutionary rates are observed for proteins in class DG in comparison to the other two dependency classes. Furthermore, most comparisons reveal elevated evolutionary rates of Dg proteins in comparison to members of the Gd class. This indicates that proteins having high dependency upon both chaperones evolve significantly faster than proteins that depend only on DnaK or GroEL. Hence, the folding assistance supplied by DnaK and GroEL chaperones confers an additive effect on the long-term evolutionary rate of their common interactors. Furthermore, our results indicate that DnaK-mediated folding has a stronger impact on protein evolutionary rate in comparison to GroEL-mediated folding.
Fig. 3.—

Evolutionary rates of combined DnaK and GroEL dependency classes. Each data point represents the mean of distances of all class members in one genome from their orthologs in E. coli K12. Comparisons are shown in four taxonomic depths: genus (Escherichia), order (Enterobacteriales), class (Gammaproteobacteria), and phylum (Proteobacteria). The taxonomic depth samples are mutually exclusive.
Table 4.
Comparison of Evolutionary Rates Among the Three Combined Classes
| Taxonomic depth | Equality of medians (P value)a | Class orderb | |
|---|---|---|---|
| dN | Genus | 4.9 × 10−8 | Dg > Gd |
| Order | <2.2 × 10−16 | DG > dG > Dg | |
| Class | <2.2 × 10−16 | DG > Dg > dG | |
| Phylum | <2.2 × 10−16 | DG > Dg > dG | |
| Protein distance | Genus | 1.2 × 10−5 | DG > dG > Dg |
| Order | <2.2 × 10−16 | DG > dG | |
| Class | <2.2 × 10−16 | DG > Dg > dG | |
| Phylum | <2.2 × 10−16 | DG > Dg > dG |
aUsing Friedman Mack–Skilling test (Mack and Skillings 1980).
bα = 0.05, using Tukey’s post hoc test of mean evolutionary rates of the class within each species.
Physiochemical Properties of DnaK and GroEL Interactors
Proteins classified into the DnaK-dependency classes are significantly different in numerous physiochemical properties including solubility, molecular weight, average hydropathy, and aggregation propensity (Calloni et al. 2012). Similar properties were found to be significantly different among proteins in the GroEL-dependency classes (Bogumil and Dagan 2010; Fujiwara et al. 2010). We examined the commonalities and differences between the DnaK and GroEL interactomes by means of the physicochemical, structural, and sequence properties that are characteristic of the dependency classes. A total of 39 properties were compared between the DnaK and GroEL dependency classes. To identify protein properties that are related to an increased dependency upon the chaperones for folding, we compared the property enrichment in class III versus class I for all properties in both chaperones. After correcting for multiple comparisons we find that similar properties are enriched in the highest or lowest dependency classes of both chaperones (fig. 4; supplementary table S1, Supplementary Material online). For example, the usage of large amino acids is enriched in the highest dependency classes of both chaperones. Considering specific amino acid composition of the proteins, we find that Leucine, Cysteine, Methionine, Serine, Tryptophan, Proline, Histidine, Glutamine, and Arginine are enriched in dependency class III of both DnaK and GroEL. In contrast, the usage of Valine, Alanine, Glutamate, Glycine, and Lysine is enriched in dependency class I of both chaperones. Hence, the increased dependency upon the chaperone for folding is characterized by similar trends of amino acid usage in interactors of both chaperones.
Fig. 4.—

Enrichment of physiochemical properties in chaperone dependency classes. Enrichment ratios are calculated as the mean value of Class III substrates divided by the mean value of Class I substrates. Ratios for DnaK are in red and for GroEL in blue. Kruskal–Wallis test significant P values are marked with *P < 0.05 and **P < 0.01.
Discussion
Accumulating evidence suggests that molecular chaperones have a cumulative impact on the evolutionary rate of their client proteins (Bogumil and Dagan 2012). Here, we show that the dependency on DnaK for folding is positively correlated with protein evolutionary rate in multiple taxonomic depths. Notably, the evolutionary rate of proteins that were not found by Calloni et al. (2012) to bind with DnaK is significantly higher than those observed for the three dependency classes (P < 2.2 × 10−16, using Friedman Mack–Skilling test) (Mack and Skillings 1980), (α = 0.05, using Tukey’s post hoc test). A similar observation has been made for proteins that were not found by Kerner et al. (2005) to bind with GroEL (Williams and Fares 2010). These findings can be explained by differences in the expression level of classified versus unclassified chaperone clients. A comparison of the two groups reveals that unclassified proteins have a significantly lower expression level in comparison to the classified proteins (P = 1.66 × 10−10, using Kruskal–Wallis test). This finding is not surprising, considering the known negative correlation between evolutionary rate and expression level. The enrichment of highly expressed proteins in the chaperone dependency classes is no doubt a result of the experimental approach that is used to survey chaperone interactomes. Pull-down experiments using a chaperone bait are highly dependent on the interactor abundance in the cytosol and are therefore biased toward highly expressed interactors.
Our analysis demonstrates that protein expression level is not the source of these correlations. The difference in evolutionary rate observed at the highest DnaK-dependency class reaches a maximum of 4-fold increase in comparison to the lowest DnaK-dependency class. This raises the possibility that DnaK overexpression can be used to facilitate heterologous protein expression and to promote directed enzyme evolution, in a manner similar to GroEL overexpression (Tokuriki and Tawfik 2009). Our findings should be contrasted against the impact of GroEL on the evolutionary rate of its client proteins. Previous reports showed that GroEL obligatory clients (Class IIIGroEL) evolve faster than casual GroEL clients (Class IGroEL) (Bogumil and Dagan 2010) (Table 2). Comparing to the correlations presented here, we may conclude that the impact of DnaK-mediated folding on protein evolution exceeds that of GroEL. This observation is supported by our analysis of the combined dependency classes, where a greater dependency on DnaK entails higher evolutionary rates.
Our analysis thus indicates that DnaK-mediated folding represents a significant mechanism of buffering for slightly deleterious mutations. Proteins that interact frequently with DnaK have fewer constraints on their sequence evolution and consequently a larger freedom to probe the sequence space during evolution. Consequently, protein interaction with DnaK has long-term consequences for genome evolution.
Supplementary Material
Supplementary tables S1 and S2 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors acknowledge support from the European Research Council (Grant No. 281357). A.S.K. is a member of the International Max-Planck Research School (IMPRS) for Evolutionary Biology at the Christian-Albrechts University of Kiel.
Literature Cited
- Arifuzzaman M, et al. 2006. Large-scale identification of protein-protein interaction of Escherichia coli K-12. Genome Res. 16:686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A, et al. 2005. The universal protein resource (UniProt). Nucleic Acids Res. 33:D154–D159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogumil D, Dagan T. 2010. Chaperonin-dependent accelerated substitution rates in prokaryotes. Genome Biol Evol. 2:602–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogumil D, Dagan T. 2012. Cumulative impact of chaperone-mediated folding on genome evolution. Biochemistry 51:9941–9953. [DOI] [PubMed] [Google Scholar]
- Bogumil D, Landan G, Ilhan J, Dagan T. 2012. Chaperones divide yeast proteins into classes of expression level and evolutionary rate. Genome Biol Evol. 4:618–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calloni G, et al. 2012. DnaK functions as a central hub in the E. coli chaperone network. Cell Rep. 1:251–264. [DOI] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO. 2008. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134:341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis J. 1987. Proteins as molecular chaperones. Nature 328:378–379. [DOI] [PubMed] [Google Scholar]
- Fares MA, Ruiz-González MX, Moya A, Elena SF, Barrio E. 2002. Endosymbiotic bacteria: groEL buffers against deleterious mutations. Nature 417:398–398.. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. 2005. PHYLIP (Phylogeny Inference Package) version 3.6. Distributed by the author. Department of Genome Sciences, University of Washington, Seattle.
- Fujiwara K, Ishihama Y, Nakahigashi K, Soga T, Taguchi H. 2010. A systematic survey of in vivo obligate chaperonin-dependent substrates. EMBO J. 29:1552–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E, et al. 2003. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31:3784–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. 2002. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295:1852–1858. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. 2009. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 16:574–581. [DOI] [PubMed] [Google Scholar]
- Hartl FU. 2011. Chaperone-assisted protein folding: the path to discovery from a personal perspective. Nat Med. 17:1206–1210. [DOI] [PubMed] [Google Scholar]
- Jones DT, Taylor WR, Thornton JM. 1992. The rapid generation of mutation data matrices from protein sequences. Bioinformatics 8:275–282. [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerner MJ, et al. 2005. Proteome-wide analysis of chaperonin-dependent protein folding in Escherichia coli. Cell 122:209–220. [DOI] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. 1982. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 157:105–132. [DOI] [PubMed] [Google Scholar]
- Lachowiec J, Lemus T, Borenstein E, Queitsch C. 2015. Hsp90 promotes kinase evolution. Mol Biol Evol. 32:91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Vogel C, Wang R, Yao X, Marcotte EM. 2007. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat Biotechnol. 25:117–124. [DOI] [PubMed] [Google Scholar]
- Mack GA, Skillings JH. 1980. A Friedman-type rank test for main effects in a two-factor ANOVA. J Am Stat Assoc. 75:947–951. [Google Scholar]
- Maisnier-Patin S, et al. 2005. Genomic buffering mitigates the effects of deleterious mutations in bacteria. Nat Genet. 37:1376–1379. [DOI] [PubMed] [Google Scholar]
- McGuffin LJ, Bryson K, Jones DT. 2000. The PSIPRED protein structure prediction server. Bioinformatics 16:404–405. [DOI] [PubMed] [Google Scholar]
- Queitsch C, Sangster TA, Lindquist S. 2002. Hsp90 as a capacitor of phenotypic variation. Nature 417:618–624. [DOI] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European molecular biology open software suite. Trends Genet. 16:276–277. [DOI] [PubMed] [Google Scholar]
- Rüdiger S, Germeroth L, Schneider-Mergener J, Bukau B. 1997. Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. Embo J. 16:1501–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford SL, Lindquist S. 1998. Hsp90 as a capacitor for morphological evolution. Nature 396:336–342. [DOI] [PubMed] [Google Scholar]
- Rutherford SL. 2003. Between genotype and phenotype: protein chaperones and evolvability. Nat Rev Genet. 4:263–274. [DOI] [PubMed] [Google Scholar]
- Sabater-Muñoz B, et al. 2015. Fitness trade-offs determine the role of the molecular chaperonin GroEL in buffering mutations. Mol Biol Evol. 32:2681–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekhar A, Lam HN, Cavagnero S. 2012. Protein folding rates and thermodynamic stability are key determinants for interaction with the Hsp70 chaperone system. Protein Sci. 21:1489–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Li W-H. 1987. The codon adaptation index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15:1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama M, Torrents D, Bork P. 2006. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34:W609–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. 1997. A genomic perspective on protein families. Science 278:631–637. [DOI] [PubMed] [Google Scholar]
- Tatusova T, Ciufo S, Fedorov B, O'Neill K, Tolstoy I. 2014. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res. 42:D553–D559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuriki N, Tawfik DS. 2009. Protein dynamism and evolvability. Science 324:203–207. [DOI] [PubMed] [Google Scholar]
- Williams TA, Fares MA. 2010. The effect of chaperonin buffering on protein evolution. Genome Biol Evol. 2:609–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Koonin EV. 2012. A tight link between orthologs and bidirectional best hits in bacterial and archaeal genomes. Genome Biol Evol. 4:1286–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Horwich AL, Sigler PB. 1997. The crystal structure of the asymmetric GroEL-GroES-(ADP)7 chaperonin complex. Nature 388:741–750. [DOI] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24:1586–1591. [DOI] [PubMed] [Google Scholar]
- Zar JH. 1999. Biostatistical analysis 4th ed. Upper Saddle River: Prentice-Hall. [Google Scholar]
- Zhang J, Yang J-R. 2015. Determinants of the rate of protein sequence evolution. Nat Rev Genet. 16:409–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.