

Abstract
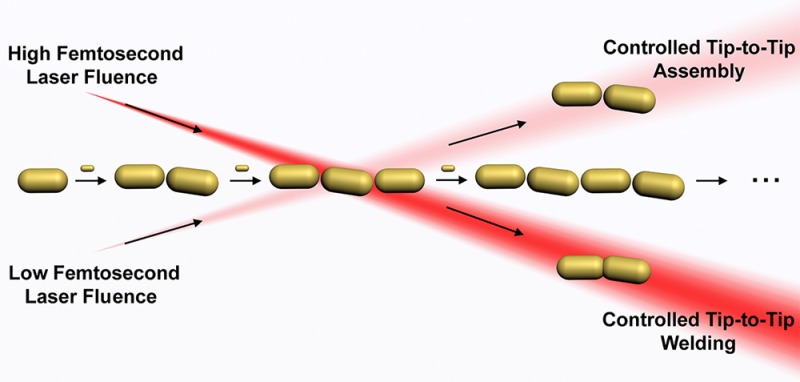
Directed assembly of gold nanorods through the use of dithiolated molecular linkers is one of the most efficient methodologies for the morphologically controlled tip-to-tip assembly of this type of anisotropic nanocrystals. However, in a direct analogy to molecular polymerization synthesis, this process is characterized by difficulties in chain-growth control over nanoparticle oligomers. In particular, it is nearly impossible to favor the formation of one type of oligomer, making the methodology hard to use for actual applications in nanoplasmonics. We propose here a light-controlled synthetic procedure that allows obtaining selected plasmonic oligomers in high yield and with reaction times in the scale of minutes by irradiation with low fluence near-infrared (NIR) femtosecond laser pulses. Selective inhibition of the formation of gold nanorod n-mers (trimers) with a longitudinal localized surface plasmon in resonance with a 800 nm Ti:sapphire laser, allowed efficient trapping of the (n – 1)-mers (dimers) by hot spot mediated photothermal decomposition of the interparticle molecular linkers. Laser irradiation at higher energies produced near-field enhancement at the interparticle gaps, which is large enough to melt gold nanorod tips, offering a new pathway toward tip-to-tip welding of gold nanorod oligomers with a plasmonic response at the NIR. Thorough optical and electron microscopy characterization indicates that plasmonic oligomers can be selectively trapped and welded, which has been analyzed in terms of a model that predicts with reasonable accuracy the relative concentrations of the main plasmonic species.
Keywords: Gold nanorod, femtosecond laser, nanoparticle assembly, dimer, welded nanoparticles
Research on gold nanoparticles (AuNPs) is of increasing interest in connection with their unique optical properties, which originate from the interaction of light with free conduction electrons. This phenomenon is known as localized surface plasmon resonance (LSPR)1,2 and has shown great potential for applications in sensing,3,4 photovoltaics, and photocatalysis,5,6 as well as in waveguides and metamaterials,7,8 to cite a few examples. In this context, many of such applications are based on the collective optical properties of AuNPs that are in close proximity of each other.2 When the distance between two AuNPs is sufficiently short, new hybridized plasmon modes appear through LSPR coupling,9 in such a way that the intensity of the resulting interaction is controlled by the interparticle distance.10−12 In the case of anisotropic AuNPs, such as gold nanorods (AuNRs), dimer structures display hybridized bonding and antibonding resonance modes, which are highly sensitive to the relative orientation of the nanocrystals and the separation distance.11,13 For tip-to-tip AuNR assemblies, the bonding longitudinal mode registers a significant redshift and an increase of the effective polarizability with respect to the monomer (single AuNR) state, as the distance is reduced.12 Moreover, the magnitude of the near field enhancement at the interparticle gap depends directly on the distance between the nanocrystals, being greater as the gap gets smaller.2,11,12 Therefore, AuNP ensembles act as nanolenses that are able to confine light at subwavelenght dimensions, giving rise to electromagnetic field enhancements that are several orders of magnitude larger than those of the incident field at the interparticle gap space, known as hot spots.14
Regardless of nanocrystal morphology, control over the directed assembly of AuNPs is a crucial factor to build up plasmonic nanomaterials with tailored functionalities.15 For instance, a number of organizations based on molecular concepts,16 such as spherical clusters,17 linear and branched chains,18,19 polymers,20 and supercrystals,21 can be produced depending on the AuNPs surface functionalization. These attempts to assemble AuNPs in a controlled fashion generally include the use of supramolecular complexes,20 surfactants,21 (bio)macromolecules,22 amino acids,23 and thiol-functionalized ligands.13,24 In the case of AuNRs, the inherent structural anisotropy gives rise to a different reactivity of the tips as compared to the lateral facets of the nanocrystals, thus allowing their preferential functionalization.25 Notably, most assemblies are controlled by means of chemical parameters (stabilizer, linker and nanoparticle concentrations, solvent, evaporation rates and temperature),16−24 and/or photochemical conditions (light activation of the reactants).26,27 In this respect, we questioned ourselves whether electromagnetic field enhancements at hot spots, which are directly associated with a high temperature increase,28 could be used to control the assembly of AuNPs via a photothermal process.
The strong interaction of light, particularly of laser radiation with AuNPs, can result in irreversible morphological changes of the nanocrystals.1 Ultrafast femtosecond (fs) irradiation can induce melting of AuNPs, whereas nanosecond (ns) laser light produces both photothermal melting as well as fragmentation.29,31 All these effects have been ascribed to the relaxation dynamics of the localized surface plasmon electron oscillations in resonance with the laser wavelength. In the case of AuNP irradiation with a fs laser, a thermal equilibration process takes place after irradiation, whereas for the ns laser the electrons continue absorbing photons when the nanocrystal lattice is still “hot”, ultimately resulting in AuNPs fragmentation.30,32 In all these investigations, reshaping and fragmentation of single AuNPs of various sizes and geometries have been described using relatively high fluence laser irradiation.29−32 Under such conditions, Baumberg and co-workers employed a fs laser to efficiently weld spherical AuNP assemblies through the generation of hot spots at interparticle gaps.33 Inspired by these examples, we decided to exploit not only the AuNP melting capability of fs lasers but also the possibility of using lower fluence fs laser irradiation to control the assembly of AuNPs. This kind of control over the nanostructures’ morphology by means of laser pulses may be exploited in the scalable manufacturing of electronic and optoelectronic devices.16
For this purpose, we reasoned that irradiation with 800 nm Ti:sapphire low fluence 50 fs laser pulses during tip-to-tip assembly by monomer addition of AuNRs with initial longitudinal LSPR bands well below 800 nm for the monomers can selectively inhibit the formation of longer oligomers (larger clusters) with longitudinal LSPRs in resonance with the laser wavelength. The inhibition process is proposed to occur by photothermal decomposition of interparticle molecular linkers. This hypothesis relies on the relatively strong hot spots generated at the interparticle gaps, where a dithiolated molecule (1,8-octanedithiol) is preferentially located as the molecular linker during the assembly.13,25 We selected the AuNR aspect ratio to ensure that the respective maxima of the LSPRs for the monomer and dimer species are located at 600 and ca. 700 nm, respectively, that is, far away from the wavelength of the fs pulses, which are in resonance with the trimer structure (ca. 800 nm). Hence, field enhancement is negligible for the shorter species but not for the trimer, selectively disrupting formation of the latter due to temperature increase at the interparticle gaps. This leads to a significant increase of the population of dimers beyond what is attainable through the nonirradiated reaction. Finally, we explored the possibility of welding intermediate species, such as dimers and trimers, during the AuNRs assembly by increasing the fluence of the fs laser pulses. Those welded nanostructures form high-aspect ratio nanorods with a great potential for applications in electronics and plasmonics, which are hard to produce by chemical or physical means.16 Moreover, welding can be attained at relatively low fluences due to the extreme field enhancement in the interparticle gaps.
Finite-difference time-domain (FDTD) calculations using the free software MEEP (see the Supporting Information for details, Figure S1)34 were performed for guidance in predicting the ideal conditions for the experiments (Figure 1). An interparticle separation of 1.5 nm was considered for the calculation, which is in agreement with the length of the linking molecule assuming a fully outstretched conformation of the alkyl chain. We found that for AuNRs with an aspect ratio of 1.8 (see Supporting Information, Figure S3), the longitudinal LSPR of the trimer was in resonance with the laser wavelength whereas shorter species were not affected. Because the dithiols may allow some degree of flexibility at the linking point, we also analyzed the dependence of the LSPR with the angle between the AuNRs in the dimer. Our results show that for a wide angular range (∼60–180°) the LSPR position is barely affected,11,35 whereas the plasmon intensity varies significantly with the angle (Figure 1a) being maximum at 180°. We additionally studied the field enhancement (|E|/|E0|) in the surroundings of the AuNRs (Figure 1b–e) for the particular case of parallel polarization. Maximum field enhancements of 15, 180, and 530 were obtained for single AuNRs, dimers, and trimers, respectively (Figure 1b).
Figure 1.

(a) Optical extinction spectra of single AuNRs, rod dimers at various angles (0, 90, 120, and 180°) and linear trimers, calculated using FDTD. (b) Longitudinal field enhancement profiles for single AuNRs (black line), AuNR dimers (red line) and AuNR trimers (green line). Local field enhancement contour plots in the middle plane for (c) single AuNRs, (d) AuNR dimers, and (e) AuNR trimers.
It can be readily seen that in particular for the trimer the field enhancement is so large that even for illumination with moderate intensity the energy concentrated in the gap may be sufficient to decompose the linking molecules and even to melt the AuNR tips at higher laser intensities. For instance, AuNRs illuminated with fs laser pulses have been reported to reach gold melting temperatures (1337 K for the bulk material) for irradiation fluences above 500 μJ/cm2 per pulse.28 Illumination with a fluence of 100 μJ/cm2 per pulse would be translated into temperature increments of ca. 550 K for single AuNRs, and very likely far larger values around the interparticle gaps for dimers and trimers,33 due to the enhanced electric field in these regions. It is therefore clear that the largest fluence (650 μJ/cm2) used in our work should be high enough to melt AuNRs, thus favoring the formation of welded structures during tip-to-tip assembly. On the other hand, we expect that intermediate fluences (130 μJ/cm2) may lead to a temperature increase high enough to decompose the organic linker but not to melt AuNRs species, and that the temperature produced at lower fluence (13 μJ/cm2) is so small that the AuNR assembly is barely affected by such fs laser pulses.
The use of dithiols as molecular linkers required, as a first step, the transfer of AuNRs into ethanol, which is a more suitable solvent for the organic linkers. Solvent transfer was achieved by addition of a high molecular weight branched polyethylenimine polymer (see Supporting Information, Figure S4), which provided sufficient stability to AuNRs in ethanol via steric hindrance effects.36 Optimized linker concentrations were then used to control both initiation and assembly rates (see Supporting Information, Figure S5). Termination of the reaction was achieved by blocking the free thiols of the linker by means of a “click” thiol-maleimide reaction.37 The redshift of the longitudinal LSPR band resulting from tip-to-tip AuNR assembly was monitored to follow the reaction process (Figure 2a), and the resulting products were analyzed by transmission electron microscopy (TEM) (Figure 2b,c). Initiation occurs along with a decrease of the LSPR band intensity at 600 nm and formation of a new intense band centered at ca. 700 nm, likely due to the longitudinal LSPR of dimers. After 8 min, the LSPR bands corresponding to AuNR monomers and dimers showed similar intensities, while after 10 min the reaction was stopped when the LSPR of trimers at ca. 800 nm reached an analogous intensity.
Figure 2.

(a) Extinction spectra during AuNRs (∼109 M) tip-to-tip assembly in ethanol, using 1,8-octanedithiol (0.5 mM) as linker, acquired at time intervals of 90 s. Typical TEM micrographs of AuNRs (b) before and (c) after assembly (10 min of reaction). (d–f) Extinction spectra after the assembly of AuNRs exposed for 10 min to 800 nm 50 fs laser pulses: (d) equal frequency (1 kHz) at different pulse fluences, 13, 130, and 650 μJ/cm2; (e) equal fluence (650 μJ/cm2) at different frequencies, 10 Hz, 200 Hz, and 1 kHz; and (f) the same average power fluence (130 mW/cm2) attained with different pulse fluences and frequencies, 130 μJ/cm2 at 1 kHz and 650 μJ/cm2 at 200 Hz.
A systematic analysis of the influence of 50 fs (fwhm) laser pulses on AuNR assembly was carried out by varying: (i) pulse fluence (13, 130, and 650 μJ/cm2) at a pulse frequency of 1 kHz (Figure 2d); (ii) pulse frequency (10 Hz, 200 Hz, and 1 kHz) at a pulse fluence of 650 μJ/cm2 (Figure 2e); and (iii) both laser fluence and pulse frequency (130 μJ/cm2 at 1 kHz, and 650 μJ/cm2 at 200 Hz) at an equal average power fluence of 130 mW/cm2 (Figure 2f). When the pulse frequency was maintained constant at 1 kHz, no significant differences were observed on the assembly process at 13 μJ/cm2, resulting in a final extinction spectrum similar to that of the reaction in the absence of laser irradiation. By contrast, at 650 μJ/cm2 the final extinction spectrum showed a new broad band ranging from 900 to 1400 nm, which may be attributed to the welding of AuNRs, which then effectively behave as longer rods.33 Moreover, the peak at 800 nm is considerably damped after irradiation with an intermediate pulse fluence of 130 μJ/cm2, indicating the coexistence of AuNR monomers and dimers as the main reaction products. Therefore, at this irradiation regime the concentration of trimers is significantly reduced with respect to the nonirradiated case.
Interestingly, irradiation with 650 μJ/cm2 while varying the pulse frequency from 10 Hz to 1 kHz led to similar results (Figure 2e) in which welded plasmonic AuNRs register LSPR maxima in the NIR between 900 and 1400 nm. This indicates that the pulse frequency is a rather irrelevant parameter, probably because relaxation dynamics of LSPR oscillations and pulse irradiation occur at very different time scales. In other words, when AuNR trimers are exposed to such ultrafast laser pulses, free electrons absorb the energy of incident photons, thereby increasing their kinetic energy. Highly energetic electrons with an initial energy distribution out of equilibrium are then relaxed through electron–electron scattering on the order of 10–50 fs.38,39 Within these time scales, no energy exchange occurs between electrons and phonons. The AuNRs lattice temperature starts increasing as a result of electron–phonon scattering, reaching thermal equilibrium between the electrons and the lattice within a time scale of tens of ps, depending on the initial rise in electron temperature. As the AuNR temperature increases, energy exchange between the particle and its surrounding medium occurs through phonon–phonon coupling. Finally, thermal equilibrium between the AuNR and the aqueous solution is achieved within 100 ps to 1 ns, depending on particle size and laser pulse intensity.40,41 Interestingly, an experiment in which both the pulse fluence and frequency were modified while keeping constant the average power fluence (130 mW/cm2) showed significant differences in the final UV–vis-NIR spectrum, thus revealing that the average power fluence is not a significant parameter regarding reaction control. Hence, this series of experiments showed that the most important parameter toward controlling the reaction kinetics of AuNRs is pulse fluence.
We investigated the resulting assembled products by using transmission electron microscopy (TEM) on AuNR oligomers, both assembled (Figure 3a) and welded (Figure 3b), through their tips. Figure 3c shows the distribution of AuNR oligomers upon irradiation for 10 min at 130 and 650 μJ/cm2 (1 kHz), as compared to the nonirradiated process (see Supporting Information, Figure S6). An analysis of the number of nanocrystals (either linked or welded) per ensemble showed that although the proportion of long chains (>6 AuNRs) is ∼50% at 650 μJ/cm2, this value decreased to ∼10% at 130 μJ/cm2, which is significantly lower than the ∼25% observed in absence of irradiation. Interestingly, the yield of dimers at 130 μJ/cm2 doubled those obtained both without irradiation and in the presence of high pulse energies, meaning that the intermediate dimer assembly can be trapped during the assembly via fs laser irradiation at the selected pulse fluence. The proportion of monomers is similar (∼50%) in the absence of irradiation and at 130 μJ/cm2, which indicates that both monomers and dimers are formed under the photothermal decomposition of the linker molecule. Although the fraction of dimers is limited to 25% under irradiation at 130 μJ/cm2, it should be noted that this value corresponds to the number of assembled dimers, which means that the number of AuNRs with a tip-to-tip dimer configuration is the double of such fraction. We attribute this limit to the natural polydispersity of gold nanorods obtained from the seed-mediated growth method, which increases upon gold nanorod tip-to-tip assembly: (i) dimers with large aspect ratios (with LSPRs close to 800 nm) may be disrupted by the laser pulses increasing the population of monomers, which is in contrast to the trend of the reaction; and (ii) trimers with large aspect ratios (with LSPRs above 800 nm) can elude the effect of the laser pulses, forming larger oligomers.
Figure 3.

Representative TEM micrographs of AuNRs (∼109 M) assemblies after 10 min in ethanol, using 1,8-octanedithiol (0.5 mM) as molecular linker: (a) 130 μJ/cm2 per pulse at 1 kHz and (b) 650 μJ/cm2 per pulse at 1 kHz. (c) Statistical distribution of AuNRs in assembled species after 10 min of reaction without laser irradiation (black), 130 μJ/cm2 per pulse at 1 kHz irradiation (red) and 650 μJ/cm2 per pulse at 1 kHz irradiation (blue). (d) Angular distribution of the AuNRs dimers obtained after irradiation with 130 mJ/cm2 laser pulses irradiation. TEM micrographs of a dimer (e) and a tetramer (g) obtained by irradiation at 130 μJ/cm2 per pulse. (f,h) Magnifications of the interparticle region within (e,g), respectively. (i) HAADF-STEM image of a trimer with the CBED patterns shown as insets. (j,k) High-resolution HAADF-STEM images of the connection points between the AuNRs in the trimer.
High-resolution high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) and TEM analysis confirmed that at pulse fluences of 130 μJ/cm2, AuNRs were assembled tip-to-tip, mainly into dimers (Figure 3a,e) with and angular distribution centered at 60–120°, and interparticle gaps of 1.0 ± 0.5 nm (Figure 3f), which is in good agreement with the length of 1,8-octanedithiol in a fully outstretched conformation of the alkyl chain. Additionally, other types of larger ensembles are also obtained in low yield (Figure 3a,g) without significant differences of the interparticle distances (Figure 3h). In Figure 3i, an HAADF-STEM image of a trimer is presented with the convergent beam electron diffraction (CBED) patterns from the AuNRs shown as insets. From the latter, it can be concluded that the three nanocrystals are not oriented along the same crystallographic orientation, an observation that suggests the absence of structural connection between the AuNRs. In Figure 3j,k, high resolution HAADF-STEM images of the connection points between the AuNRs are presented and the presence of a layer of ligands can be observed (low contrast regions between the rods). By contrast, irradiation under pulse fluences of 650 μJ/cm2 led to the formation of welded oligomers (Figure 3b). High-resolution HAADF-STEM images of a dimer and a trimer are shown in Figure 4a,b, respectively, for the case of welded AuNRs. Figure 4c,d show high-resolution HAADF-STEM images of the connection between two AuNRs in a dimer and a trimer, respectively. A grain boundary is always present at the connection point of two rods. From the CBED patterns, shown as insets in Figure 4b for the case of the trimer, it is clear that the three nanocrystals are in almost the same crystallographic orientation, but yield a slight misorientation with respect to each other. It should be noted, however, that in other cases no common crystallographic orientations were found to be apparent for welded AuNRs. This rather surprising effect may be due to the extreme concentration of energy on this region and/or to the fact that the energy deposition occurs in such short times that the welding is essentially a nonthermal process.33 Of course, we also should keep in mind that the HAADF-STEM images are 2D projections of 3D objects and therefore electron tomography experiments might be able to yield a further understanding concerning the connection mechanism. As such experiments should be carried out with atomic resolution, they are outside the scope of this work.
Figure 4.

HAADF-STEM images of a dimer (a) and a trimer (b) of AuNRs obtained at 650 μJ/cm2. The insets in figure (b) show the respective CBED patterns of the AuNRs in the trimer conformation. (c,d) High-resolution HAADF-STEM images of the connection points between the rods within (a,b), respectively.
To gain insight into the way fs laser affects AuNRs tip-to-tip assembly, we repeated a number of experiments acquiring in situ extinction spectra (see Supporting Information, Figure S2) at time intervals of 2 s (Figure 5a–c). The relative concentration of the main species was determined by fitting the experimental spectra with those calculated using FDTD (Figure 1a and Supporting Information). Unfortunately, the possible configurations for the AuNRs oligomers are rather numerous, far more than can be simulated, which introduces an error in the concentrations predicted by the model. We assume that the AuNRs are preferentially bonded by the tips, according to TEM analysis,13,25 which considerably reduces the possible number of configurations. As discussed above, the angle between AuNRs does not change the position of the LSPR but only its intensity.35 Because the only configuration without tip-to-tip links that was found in meaningful amounts during TEM analysis was the side-by-side dimer (see Supporting Information, Figure S7), we also included the corresponding extinction spectrum in the fit. We additionally considered the spectra for the main species (single AuNRs, dimers, and trimers), whereas all larger-chain oligomers were approximated by a Gaussian curve. Despite its simplicity, the model is able to yield reasonable results for the relative concentrations of the main species (see Supporting Information, Figure S8).
Figure 5.

Evolution of AuNRs tip-to-tip assembly for different irradiation conditions. (a–c) Extinction spectra at 20 s intervals for 10 min (a) without laser irradiation, (b) 130 μJ/cm2 per pulse at 1 kHz, and (c) 650 μJ/cm2 per pulse at 1 kHz. Arrows point to the spectral region at the LSPR maxima for the monomer (orange), dimer (red), and trimer (black). Concentration of side-by-side (SBS, green lines) dimers yielded by the fit is very close to zero. (d–f) Concentration of single AuNRs, dimers, and trimers obtained from the fits for (d) no laser and pulse fluences of (e) 130 μJ/cm2 and (f) 650 μJ/cm2.
Concentration of the main species as a function of time are shown in Figure 5d–f for the nonirradiated reaction, as well as those for medium and large fluences. Data for the low fluence was omitted because they were virtually identical to those of the nonirradiated reaction. The rate of AuNR assembly is slightly decreased by laser irradiation with respect to the nonirradiated reaction, which is apparent for a fluence of 130 μJ/cm2. This suggests that fs irradiation not only affects trimers but also a small fraction of dimers due to the natural polydispersity of AuNRs in aspect ratio, resulting in an increase of monomer concentration at early reaction stages. Our model predicts an inversion of the concentrations of dimers and trimers due to fs irradiation (Figure 5d,e), which is in good agreement with the proportion of oligomers determined by TEM (Figure 3c). We additionally found a significant increase of the rate at which monomers vanish at the early stages of reaction at the fluence of 650 μJ/cm2, suggesting that an alternative reaction pathway based on the activation of chemical reactants by light26,27 takes place for larger fluences. It should be noted that by the end of the reaction there is still a significant remaining concentration of monomers (∼1010 particles/cm3) because the reaction rate stagnates from this point onward. This is probably due to the polydispersity of AuNRs from which dimers with large aspect ratios are affected by the laser pulses, increasing the population of nonreactive monomers.
In summary, we have shown that femtosecond laser irradiation is a powerful tool to control the assembly of AuNRs. Laser pulse fluence was found to be the most important parameter affecting tip-to-tip assembly kinetics. This effect was demonstrated using AuNRs that were tailored in such a way that the longitudinal LSPR of the rod trimers is in resonance with the wavelength of a Ti:sapphire fs laser (800 nm). We were thus able to use fs pulses with a fluence of ∼100 μJ/cm2 to considerably reduce the formation of trimers and longer oligomers, thereby increasing the relative population of AuNR dimers. We also found that if the fluence is increased beyond 500 μJ/cm2, the increase of temperature at the interparticle gaps is large enough to melt AuNR tips, thus producing new species: welded AuNRs with LSPR bands at the NIR. Preliminary results in this direction indicate that it can be used to produce nanostructures with tailored LSPR modes in the IR region. Although we have only illustrated the use of laser irradiation for controlling the assembly kinetics of gold nanorods in high yields and short reaction times, this technique can be generalized to a variety of shapes, limited only by the wavelength of available fs lasers. Therefore, we show that under irradiation of femtosecond laser pulses with wavelengths in resonance with the LSPR of larger tip-to-tip AuNR oligomers, plasmonic polymers with relatively high monodispersities may be obtained.
Acknowledgments
This work has been funded by the Spanish MINECO (MAT2012-38541, MAT2013-46101-R, MAT2014-59678-R and CTQ2012-37404-C02-01). A.G.-M. and G.G.-R., respectively, acknowledge receipt of Ramón y Cajal and FPI Fellowships from the Spanish MINECO. O.P.-R. is grateful with Moncloa Campus of International Excellence (UCM-UPM) for the PICATA postdoctoral fellowship. The facilities provided by the Center for Ultrafast Lasers at Complutense University of Madrid are gratefully acknowledged. S.B. acknowledges funding from the European Research Council under the Seventh Framework Program (FP7), ERC Grant 335078 COLOURATOMS.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nanolett.5b03844.
MEEP calculations, synthetic details and characterization techniques, femtosecond laser control of the AuNRs assembly and welding, and calculation of the concentration of the AuNR ensembles. (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Link S.; El-Sayed M. A. Annu. Rev. Phys. Chem. 2003, 54 (1), 331–366. 10.1146/annurev.physchem.54.011002.103759. [DOI] [PubMed] [Google Scholar]
- Myroshnychenko V.; Rodríguez-Fernández J.; Pastoriza-Santos I.; Funston A. M.; Novo C.; Mulvaney P.; Liz-Marzán L. M.; García de Abajo F. J. Chem. Soc. Rev. 2008, 37 (9), 1792–1805. 10.1039/b711486a. [DOI] [PubMed] [Google Scholar]
- Anker J. N.; Hall W. P.; Lyandres O.; Shah N. C.; Zhao J.; Van Duyne R. P. Nat. Mater. 2008, 7 (6), 442–453. 10.1038/nmat2162. [DOI] [PubMed] [Google Scholar]
- Willets K. A.; Van Duyne R. P. Annu. Rev. Phys. Chem. 2007, 58 (1), 267–297. 10.1146/annurev.physchem.58.032806.104607. [DOI] [PubMed] [Google Scholar]
- Brongersma M. L.; Halas N. J.; Nordlander P. Nat. Nanotechnol. 2015, 10 (1), 25–34. 10.1038/nnano.2014.311. [DOI] [PubMed] [Google Scholar]
- Mukherjee S.; Libisch F.; Large N.; Neumann O.; Brown L. V.; Cheng J.; Lassiter J. B.; Carter E. A.; Nordlander P.; Halas N. J. Nano Lett. 2013, 13 (1), 240–247. 10.1021/nl303940z. [DOI] [PubMed] [Google Scholar]
- Gramotnev D. K.; Bozhevolnyi S. I. Nat. Photonics 2010, 4 (2), 83–91. 10.1038/nphoton.2009.282. [DOI] [Google Scholar]
- Guerrero-Martínez A.; Auguié B.; Alonso-Gómez J. L.; Džolić Z.; Gómez-Graña S.; Žinić M.; Cid M. M.; Liz-Marzán L. M. Angew. Chem., Int. Ed. 2011, 50 (24), 5499–5503. 10.1002/anie.201007536. [DOI] [PubMed] [Google Scholar]
- Nordlander P.; Oubre C.; Prodan E.; Li K.; Stockman M. I. Nano Lett. 2004, 4 (5), 899–903. 10.1021/nl049681c. [DOI] [Google Scholar]
- Huang J.-S.; Kern J.; Geisler P.; Weinmann P.; Kamp M.; Forchel A.; Biagioni P.; Hecht B. Nano Lett. 2010, 10 (6), 2105–2110. 10.1021/nl100614p. [DOI] [PubMed] [Google Scholar]
- Funston A. M.; Novo C.; Davis T. J.; Mulvaney P. Nano Lett. 2009, 9 (4), 1651–1658. 10.1021/nl900034v. [DOI] [PubMed] [Google Scholar]
- Jain P. K.; Huang W.; El-Sayed M. A. Nano Lett. 2007, 7 (7), 2080–2088. 10.1021/nl071008a. [DOI] [PubMed] [Google Scholar]
- Pramod P.; Thomas K. G. Adv. Mater. 2008, 20 (22), 4300–4305. 10.1002/adma.200703057. [DOI] [Google Scholar]
- Alvarez-Puebla R.; Liz-Marzán L. M.; García de Abajo F. J. J. Phys. Chem. Lett. 2010, 1 (16), 2428–2434. 10.1021/jz100820m. [DOI] [Google Scholar]
- Grzelczak M.; Vermant J.; Furst E. M.; Liz-Marzán L. M. ACS Nano 2010, 4 (7), 3591–3605. 10.1021/nn100869j. [DOI] [PubMed] [Google Scholar]
- Guerrero-Martínez A.; Grzelczak M.; Liz-Marzán L. M. ACS Nano 2012, 6 (5), 3655–3662. 10.1021/nn301390s. [DOI] [PubMed] [Google Scholar]
- Sánchez-Iglesias A.; Grzelczak M.; Altantzis T.; Goris B.; Pérez-Juste J.; Bals S.; Van Tendeloo G.; Donaldson S. H.; Chmelka B. F.; Israelachvili J. N.; Liz-Marzán L. M. ACS Nano 2012, 6 (12), 11059–11065. 10.1021/nn3047605. [DOI] [PubMed] [Google Scholar]
- Tang Z.; Kotov N. A. Adv. Mater. 2005, 17 (8), 951–962. 10.1002/adma.200401593. [DOI] [Google Scholar]
- Zhao Y.; Xu L.; Liz-Marzán L. M.; Kuang H.; Ma W.; Asenjo-García A.; García de Abajo F. J.; Kotov N. A.; Wang L.; Xu C. J. Phys. Chem. Lett. 2013, 4 (4), 641–647. 10.1021/jz400045s. [DOI] [PubMed] [Google Scholar]
- Nie Z.; Fava D.; Rubinstein M.; Kumacheva E. J. Am. Chem. Soc. 2008, 130 (11), 3683–3689. 10.1021/ja711150k. [DOI] [PubMed] [Google Scholar]
- Gómez-Graña S.; Pérez-Juste J.; Alvarez-Puebla R. A.; Guerrero-Martínez A.; Liz-Marzán L. M. Adv. Opt. Mater. 2013, 1 (7), 477–481. 10.1002/adom.201300162. [DOI] [Google Scholar]
- Lukach A.; Liu K.; Therien-Aubin H.; Kumacheva E. J. Am. Chem. Soc. 2012, 134 (45), 18853–18859. 10.1021/ja309475e. [DOI] [PubMed] [Google Scholar]
- Zhu Z.; Liu W.; Li Z.; Han B.; Zhou Y.; Gao Y.; Tang Z. ACS Nano 2012, 6 (3), 2326–2332. 10.1021/nn2044802. [DOI] [PubMed] [Google Scholar]
- Shibu Joseph S. T.; Ipe B. I.; Pramod P.; Thomas K. G. J. Phys. Chem. B 2006, 110 (1), 150–157. 10.1021/jp0544179. [DOI] [PubMed] [Google Scholar]
- Caswell K. K.; Wilson J. N.; Bunz U. H. F.; Murphy C. J. J. Am. Chem. Soc. 2003, 125 (46), 13914–13915. 10.1021/ja037969i. [DOI] [PubMed] [Google Scholar]
- Klajn R.; Bishop K. J. M.; Grzybowski B. A. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (25), 10305–10309. 10.1073/pnas.0611371104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzelczak M.; Liz-Marzán L. M. Chem. Soc. Rev. 2014, 43 (7), 2089–2097. 10.1039/C3CS60256G. [DOI] [PubMed] [Google Scholar]
- Ekici O.; Harrison R. K.; Durr N. J.; Eversole D. S.; Lee M.; Ben-Yakar A. J. Phys. D: Appl. Phys. 2008, 41 (18), 185501. 10.1088/0022-3727/41/18/185501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link S.; Burda C.; Nikoobakht B.; El-Sayed M. A. J. Phys. Chem. B 2000, 104 (26), 6152–6163. 10.1021/jp000679t. [DOI] [Google Scholar]
- Link S.; El-Sayed M. A. J. Phys. Chem. B 1999, 103 (40), 8410–8426. 10.1021/jp9917648. [DOI] [Google Scholar]
- Aguirre C. M.; Moran C. E.; Young J. F.; Halas N. J. J. Phys. Chem. B 2004, 108 (22), 7040–7045. 10.1021/jp036222b. [DOI] [Google Scholar]
- Link S.; Wang Z. L.; El-Sayed M. A. J. Phys. Chem. B 2000, 104 (33), 7867–7870. 10.1021/jp0011701. [DOI] [Google Scholar]
- Herrmann L. O.; Valev V. K.; Tserkezis C.; Barnard J. S.; Kasera S.; Scherman O. A.; Aizpurua J.; Baumberg J. J. Nat. Commun. 2014, 5, 4568. 10.1038/ncomms5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskooi A. F.; Roundy D.; Ibanescu M.; Bermel P.; Joannopoulos J. D.; Johnson S. G. Comput. Phys. Commun. 2010, 181 (3), 687–702. 10.1016/j.cpc.2009.11.008. [DOI] [Google Scholar]
- Wu J.; Lu X.; Zhu Q.; Zhao J.; Shen Q.; Zhan L.; Ni W. Angle-Resolved Plasmonic Properties of Single Gold Nanorod Dimers. Nano-Micro Lett. 2014, 6 (4), 372–380. 10.1007/s40820-014-0011-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pong B.-K.; Lee J.-Y.; Trout B. L. Langmuir 2005, 21 (25), 11599–11603. 10.1021/la052116k. [DOI] [PubMed] [Google Scholar]
- Northrop B. H.; Frayne S. H.; Choudhary U. Polym. Chem. 2015, 6 (18), 3415–3430. 10.1039/C5PY00168D. [DOI] [Google Scholar]
- Sundaram S. K.; Mazur E. Nat. Mater. 2002, 1 (4), 217–224. 10.1038/nmat767. [DOI] [PubMed] [Google Scholar]
- Plech A.; Kotaidis V.; Grésillon S.; Dahmen C.; von Plessen G. Phys. Rev. B: Condens. Matter Mater. Phys. 2004, 70, 195423. 10.1103/PhysRevB.70.195423. [DOI] [Google Scholar]
- Qin Z.; Bischof J. C. Chem. Soc. Rev. 2012, 41 (3), 1191–1217. 10.1039/C1CS15184C. [DOI] [PubMed] [Google Scholar]
- Baffou G.; Quidant R.; García de Abajo F. J. ACS Nano 2010, 4 (2), 709–716. 10.1021/nn901144d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.