

Abstract
The aberrant aggregation of α-synuclein in the brain is a hallmark of Parkinson’s disease (PD). In vivo soluble α-synuclein occurs as a monomer and several multimers, the latter of which may be important for the biological function of α-synuclein. Currently, there is a lack of reproducible methods to compare α-synuclein multimer abundance between complex biological samples. Here we developed a method, termed “multimer-PAGE,” that combines in-gel chemical cross-linking with several common electrophoretic techniques to measure the stoichiometry of soluble α-synuclein multimers in brain tissue lysates. Results show that soluble α-synuclein from the rat brain exists as several high molecular weight species of approximately 56 kDa (αS56), 80 kDa (αS80), and 100 kDa (αS100) that comigrate with endogenous lipids, detergents, and/or micelles during blue native gel electrophoresis (BN-PAGE). Co-extraction of endogenous lipids with α-synuclein was essential for the detection of soluble α-synuclein multimers. Homogenization of brain tissue in small buffer volumes (>50 mg tissue per 1 mL buffer) increased relative lipid extraction and subsequently resulted in abundant soluble multimer detection via multimer-PAGE. α-Synuclein multimers captured by directly cross-linking soluble lysates resembled those observed following multimer-PAGE. The ratio of multimer (αS80) to monomer (αS17) increased linearly with protein input into multimer-PAGE, suggesting to some extent, multimers were also formed during electrophoresis. Overall, soluble α-synuclein maintains lipid interactions following tissue disruption and readily forms multimers when this lipid–protein complex is preserved. Once the multimer-PAGE technique was validated, relative stoichiometric comparisons could be conducted simultaneously between 14 biological samples. Multimer-PAGE provides a simple inexpensive biochemical technique to study the molecular factors influencing α-synuclein multimerization.
Parkinson’s disease (PD) is a neurodegenerative disease characterized by the formation of proteinaceous inclusions, termed “Lewy bodies” that contain large amounts of α-synuclein, a small cytosolic protein for which the biological function is still unclear.1,2 In vivo α-synuclein occurs as a variety of multimers including a dimer, tetramer, and octomer which appear to be biologically important and possibly resistant to toxic aggregation.3−5 New therapeutic strategies for treating PD may involve the use of drugs to stabilize soluble multimeric α-synuclein to prevent the formation of higher order soluble toxic multimers.6
Soluble monomeric α-synuclein is an intrinsically disordered protein and behaves like a larger protein of ∼50–60 kDa when assessed using a number of techniques including size-exclusion chromatography and blue native gel electrophoresis (BN-PAGE).4 The unusual behavior of α-synuclein makes the separation of native multimers from the monomer, according to mass, particularly challenging. Chemical cross-linking can be used to preserve α-synuclein multimers allowing their separation from the monomer by traditional sodium dodecylsulfate polyacrylamide electrophoresis (SDS-PAGE).3,6,7 α-Synuclein multimerizes upon binding to phospholipid membranes, but it remains unclear whether free soluble α-synuclein can form stable soluble multimers. Upon binding to phospholipid membranes α-synuclein adopts defined secondary structure and subsequently multimerize.2,8 Although the interaction between α-synuclein with phospholipid membranes is intimately involved in the multimerization process and biological function of α-synuclein, the molecular details of this interaction remain unclear.
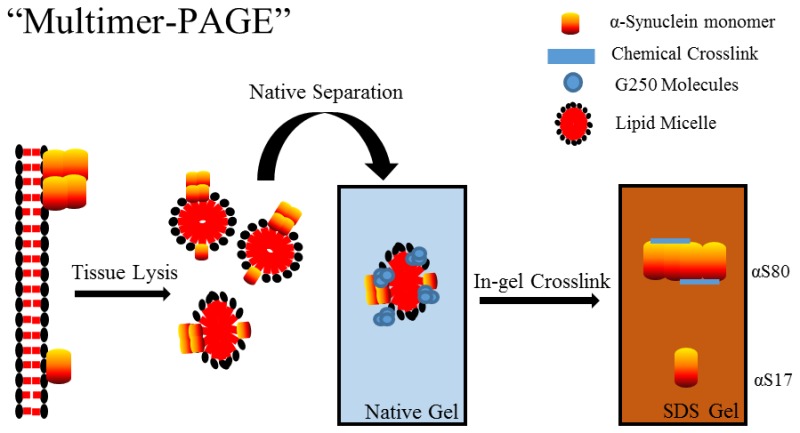
Here, we developed a new technique, termed “multimer-PAGE,” to quantify the multimerization of α-synuclein in complex biological samples. Using this technique it is possible to compare the ratio of α-synuclein multimers between 14 brain tissue samples without the need for specialized equipment.
Methods
Animals
Twenty adult male Sprague–Dawley (SD) rats (250–350g) were pair housed in polycarbonate cages at approximately 70 °F. Rats were euthanized via live decapitation. Following euthanasia, the brain was removed and dissected on ice. All collected brain tissue was placed on dry ice and frozen at −80 °C until assayed. All procedures were conducted in accordance with the Wayne State University institutional care and use committee approved protocol no. A3310-01.
Sample Preparation
All procedures were conducted on ice and in a cold room. Frozen brain tissue samples weighing approximately 25–200 mg were homogenized in 1 mL of 1× BN-PAGE sample buffer (Invitrogen, Waltham, MA) for native separation or phosphate buffered saline pH 7.4 (PBS) for in-solution cross-linking experiments. All solutions contained EDTA free protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). Tissue was disrupted using 30 strokes in a 7 mL dounce homogenizer. Following homogenization, samples were briefly vortexed and incubated on ice for 30 min. For some experiments, 0.5% m/vol NP40 (Boston Bioproducts, St. Ashland, MA) or 1% m/vol digitonin (Sigma) was added to the lysate prior to the 30 min incubation on ice. The sample was then centrifuged at 18 000g for 30 min. Following centrifugation the supernatant (S1) was carefully transferred to a prechilled 1.5 mL eppendorf tube. To determine protein concentration of the sample, 8 μL of the S1 was transferred to a separate eppendorf tube containing 2 μL of 10% m/vol SDS, mixed well, and incubated at room temperature for 10 min. This delipidated sample was then used to determine the protein content using a bicinchoninic acid assay (BCA assay, Thermofisher, Waltham, MA). Samples were diluted to a final concentration of 0.1–4 mg protein/mL, depending on the experiment. The insoluble pellet (P1) was resuspended in 1.5 mL of RIPA buffer (10 mM Tris-HCl pH 8.0, 1% vol/vol Triton X-100, 0.1% m/vol sodium dodecyl sulfate, and 140 mM NaCl), mixed thoroughly, and incubated on ice for 1 h. The P1 sample was then centrifuged at 18 000g for 30 min at 4 °C, and the supernatant retained (S2). The S2 was then processed for lipid and protein quantification. α-Synuclein was purified as previously described.9 Relative purity of α-synuclein was determined by SDS-PAGE and subsequent Coomassie Blue G-250 staining. The lyophilized purified α-synuclein was dissolved in 1× BN-PAGE sample buffer for native separation or PBS for in-sample cross-linking experiments to a final concentration of 5 mg protein/mL.
Lipid and Protein Isolation
Bulk lipid extraction was performed essentially as previously described.10 Briefly, 20 mL of a chloroform and methanol mixture (2:1) was added to 1 g of rat brain tissue. The tissue was homogenized via sonication for 2 min. The homogenate was vortexed well, 4 mL of PBS added, and the sample vortexed again. The sample was then allowed to sit for 5 min to allow the solution to separate into two phases. The organic phase containing the purified lipids at the bottom of the tube was carefully removed and dried under vacuum using a Centrivap DNA Concentrator (Labconco, Kansas City, MO). The purified lipids were then resuspended in 200 μL of 1× BN-PAGE sample buffer containing 10% vol/vol NP40, bath sonicated for 30 min, and centrifuged at 18 000g to remove any insoluble lipids not incorporated into the NP40 micelles. Isolated lipids were used within 24 h to avoid excessive lipid oxidation. Extracted brain lipids were used for generating a lipid standard for total lipid determination and to generate mixed micelles only (Figure 2A,D).
Figure 2.

Sample lipid content determines the migration of α-synuclein during BN-PAGE. (A) 200 mg rat brain tissue was homogenized in 1 mL of blue native page (BN-PAGE) sample buffer (50 mM BisTris, 6 N HCl, 50 mM NaCl, 10% vol/vol glycerol, 0.001% m/vol Ponceau S, pH ∼ 7.2) and incubated on ice for 30 min. The sample was centrifuged then at 18 000g for 30 min, S1 retained, and protein content determined using bicinchoninic acid (BCA) assay. Samples were then adjusted to a final concentration of 2 mg protein/mL. A volume of 150 μL of the samples were mixed with 600 μL of methanol and 150 μL of chloroform. A volume of 450 μL of ultrapure water was then added to the sample and mixed thoroughly. Following centrifugation at 18 000g for 5 min, the upper aqueous phase was discarded and the lower phase resuspended in 600 μL of methanol. The sample was centrifuged at 18 000g for 5 min. The liquid was discarded and precipitate (proteins) retained. Separately, bulk lipids were isolated by homogenizing 1 g of rat brain tissue in a mixture of chloroform and methanol (2:1). Following sonication for 2 min, the homogenate was vortexed well, 4 mL of PBS added, and vortexed again. The sample was then allowed to sit for 5 min to allow the solution to separate into two phases. The organic phase containing the purified lipids was removed and dried under vacuum. The lipid pellet was then completely dissolved in 200 μL of 10% NP40. The precipitated lipid free proteins were dissolved in BN-PAGE sample buffer, BN-PAGE sample buffer containing 0.5% w/v NP40, or BN-PAGE sample buffer containing 0.5% w/v NP40 mixed lipid micelles. Samples were mixed thoroughly, incubated on ice for 30 min, and mixed thoroughly again. All samples were then centrifuged at 18 000g and the resulting 10 μg of protein of the soluble fraction resolved by BN-PAGE. Top panel depicts immunobloting of samples with anti-α-synuclein antibody. Middle panel depicts the white light image of polyvinylidene fluoride (PVDF) membrane immediately following transfer of the BN-PAGE gel. Bottom panel depicts ponseau S staining of PVDF membrane following the removal of G250 from the membrane by washing in 100% methanol for ∼5 min. (B) 25, 50, 100, and 200 mg of rat brain tissue was homogenized in 1 mL of BN-PAGE sample buffer as described above and resolved via BN-PAGE. A total of 30 μg of total proteins were resolved in each lane without sample dilution (left column panels) and following dilution of samples to 2 mg protein/mL (right column panels). Top row depicts white light image of PVDF membrane immediately following transfer. Middle row depicts subsequent ponseau S staining of the PVDF membrane following removal of G-250. Bottom row depicts the immunoreactivity of α- synuclein. (C) Increasing concentrations of G-250 (0–2%) was added to rat brain tissue lyates from high concentration homogenization conditions (200 μg tissue/mL buffer). A total of 30 μg of protein was then resolved via BN-PAGE. Top panel depicts white light image of PVDF membrane. Middle row depicts ponseau S staining of PVDF membrane following removal of G-250. Bottom row dipicts the immunoreactivity of alpha-synuclein. (D) Lipid to protein content of insoluble fraction following tissue homogenization at different tissue to buffer ratios.
To extract lipid and proteins from individual lysates we first diluted the samples to 2 mg protein/mL using the appropriate buffer in a 1.5 mL eppendorf tube. The dilution was conducted to yield a final sample volume of 150 μL. Then 600 μL of methanol and 150 μL of chloroform were added to each sample. Samples were mixed well, 450 μL of dH20 added to each sample, and mixed well again. Samples were then centrifuged at 14 000g for 5 min. The upper aqueous phase was then carefully discarded, making sure not to disturb the precipitated proteins between the lower and upper phase. A volume of 450 μL of methanol was then added to each sample, vortexed, and centrifuged at 14 000g for 5 min. The methanol–chloroform mixture contained the lipids and precipitated proteins were found in the pellet. The extracted lipids from individual samples were used to quantify the amount of soluble lipids (Figure 2D). The precipitated proteins from individual samples were used for mixed micelles experiments (Figure 2A).
The precipitated proteins (300 μg) were dissolved in 100 μL of 1× BN-PAGE sample buffer containing either 0.5% NP40 or 0.5% NP40 brain lipid mixture. Samples were mixed thoroughly, allowed to sit on ice for 30 min, and mixed thoroughly again. All samples were then centrifuged 18 000g for 30 min to remove any remaining insoluble lipids or proteins. Protein content of each mixture was then determined using BCA assay.
BN-PAGE
Procedures were conducted as previously described.11 Briefly, linear gradient 2.8–13%T and nongradient 6%T gels were used for all BN-PAGE separations. All gradient gels were poured by hand using a linear gradient former built as previously described.12 A 2.8%T gel was used for the sample wells. The 0–0.5% m/v Coomassie Blue G-250 (Sigma-Aldrich, St. Louis, MO) was added to all samples immediately prior to electrophoresis and 4–50 μg of brain tissue lysate protein or 50–200 ng of purified α-synuclein were loaded into each sample well of the BN-PAGE gel. Either native page marker (Invitrogen) or 1 μg of bovine serum albumin (BSA, Santa Cruz Biotechnology, Dallas, TX) diluted in 1× BN-PAGE buffer was used as a protein standard. Electrophoresis was conducted using running buffer (100 mM bis-tris, 100 mM tricine) containing either 0.001% (for nondetergent samples) or 0.01% (for detergent samples) G-250 at 100 V until the samples entered the gel and then 180 V for approximately 3 h (until the dye front reached the end of the gel) at 4 °C. When detergent samples were resolved, the running buffer containing 0.01% G-250 was replaced by the running buffer containing 0.001% G-250, approximately halfway through the run. 6%T BN-PAGE gels were run under the same conditions but stopped once samples had migrated about 2 cm into the resolving gel. Once electrophoresis was complete the lanes of the gel were either excised for 2D electrophoresis or electroblotted onto methanol activated polyvinylidene fluoride (PVDF) membranes. Electroblotting of BN-PAGE gels was performed at 20 V for 3 h at 4 °C using the BN-PAGE running buffer. Following electroblotting, PVDF membranes were fixed for 20 min in 10% acetic acid, rinsed with ultrapure water, and then dried.
In-Gel Cross-Linking
Excised gel pieces were washed with PBS three times for 10 min at 4 °C to remove excess bis-tris and 6-aminocaproic acid because could compete with cross-linking reactions. Gel lanes were then incubated for 5–30 min at 4 °C in PBS containing either 1% paraformaldehyde (PFA), 1% glutaradehyde, 0.1–2 mM Dithiobis (succinimidylpropionate) (DSP, ThermoFisher Scientific, Waltham, MA), 0.1–2 mM 3,3′-dithiobis (sulfosuccinimidyl propionate) (DTSSP, Thermofisher Scientific), or 0.1–2 mM disuccinimidyl glutarate (DSG, ThermoFisher Scientific). DSP, DTSSP, and DSG must first be dissolved in DMSO to a concentration of 25 mM before adding dropwise to the PBS. Gels were then incubated in 375 mM Tris-HCl pH 8.8 buffer (when resolving by SDS-PAGE gradient gel) or 125 mM Tris-HCl pH 6.8 (when resolving by nongradient SDS-PAGE gel), both containing 2% SDS for 1 h at room temperature.
2D-SDS-PAGE
Excised gel strips were placed in-between Mini PROTEAN system glass plates with a 1 mm spacer (Biorad, Hercules, CA). Each gel strip was positioned with the lowest percentage gel facing down (to ensure proteins leave the gel strip during electrophoresis). When 6%T BN-PAGE gel was used the orientation of the gel strip was not crucial. Approximately 30 mm space is needed on the end of the gel strip for comb placement to form two wells. The glass plates containing the gel strip were then locked into the Bio-Rad gel stand and checked for leaks. Then a 3–16%T linear gradient or 12%T gel was poured between the glass plates leaving approximately 10 mm space under the excised gel piece for the sample gel. For 12%T gel, a 1 cm space was left for the stacking gel to ensure sufficient band focusing. Butanol was gently layered over the top of the gradient, and the gel was allowed to polymerize at room temperature for ∼1 h. Once the gel was polymerized, the butanol was removed and a sample gel of 3%T was poured around the gel strip. Tilting the gel stand slightly prevented bubbles from forming under the gel strip bubbles. The two-well comb was placed in between the glass plates and 30 min was allowed for polymerization. Alternatively, 2D-SDS-PAGE gels can also be precast (without the excised gel strip). However, this requires custom well comb, which can be fashioned out of polystyrene sheets purchased from most hobby stores. Reduction in band resolution, due to uneven migration of the sample out of excised gel, was observed when using precast gels and therefore the method should generally be avoided. Gels were then run at 125 V until the dye front reached the end and gels were then transferred to PVDF membrane in Towbin transfer buffer (25 mM Tris-HCl, 192 mM glycine, and 20% methanol) using the constant of 20 V for ∼16 h at 4 °C.
Immunodetection
Dried membranes were reactivated in 100% methanol for 1 min and then rinsed with ultrapure water. To confirm even transfer, all membranes were stained using Ponceau S (Sigma-Aldrich). Membranes were incubated in blocking buffer which consisted of 5% m/vol milk diluted in TBST (50 mM Tris-HCl, 150 mM NaCl, 0.05% Tween-80, pH 8.0) for 1 h at room temperature. Then membranes were incubated in blocking buffer containing either monoclonal (BD Biosciences) or polyclonal (Santa-Cruz) anti-α-synuclein antibodies diluted 1:1000, overnight at 4 °C. Parkin monoclonal and polyclonal antibodies were diluted 1:1 000 in blocking buffer and incubated with membranes overnight at 4 °C. Membranes were then washed 2 × 10 min with TBST before incubating in the appropriate HRP conjugated antibody diluted 1:5 000 in blocking buffer for 1 h at RT. Membranes were then washed 2 × 10 min with TBST, incubated 5 min in enhanced chemiluminescence substrate (ThermoFisher Scientific), and imaged using GE Imagequant LAS 4000 (GE Health Care, Little Chalfont, United Kingdom).
In-Sample Cross-Linking
Brain tissue lysates in PBS (see Sample Preparation) were diluted to concentrations ranging between 0.1 and 4 mg protein/mL. DSP or DSG (1 mM) were added to each sample and incubated at 4 °C for 10 min. The reaction was quenched with 50 mM Tris-HCl pH 8.0 for 15 min. The 1× SDS-PAGE sample buffer (50 mM Tris-HCl pH 6.8, 2% SDS, 10% glycerol, and 0.1% bromophenol blue) was then added to each sample, mixed well, and heated at 90 °C for 5 min. Protein (10 μg) from each sample was added to each well. DSP cross-linker arm was cleaved by adding 5 mM dithiothreitol (DTT) prior to heating the sample. Purified α-synuclein was dissolved in PBS to a concentration of 4 mg/mL and cross-linked with 2 mM DSP for 10 min at 4 °C. The reaction was quenched with 50 mM Tris-HCL pH 8.0 for 15 min.
Determination of Total Lipid Content
Total sample lipid content was determined essentially as previously described.13 Specifically, lipids isolated from the samples were dissolved in 100 μL of chloroform. The chloroform solution was then placed into a disposable 12 mm × 75 mm borosilicate glass tube (Corning Inc., Corning, NY) and allowed to dry completely. Standards were generated by dissolving a known mass of isolated rat brain lipids (see BN-PAGE) or canola oil (local grocery store) in chloroform. Standard solutions were then pipetted into six glass tubes to yield 5–400 μg of total lipid and dried completely. Then, 100 μL of concentrated sulfuric acid (Sigma-Aldrich) was placed in each tube and incubated at 90 °C for 20 min. The tubes are then cooled immediately using an ice water bath, and solutions are transferred to a clear polystyrene 96-well plate (Thermofisher). Light absorbance was measured at the wavelength 540 nm, then 50 μL of phosphor-vanillin solution (0.2 mg of vanillin dissolved in 1 mL of 17% phosphoric acid) was added, and the plate was incubated at room temperature for 10 min. The plate was then read at 540 nm again, and the background (i.e., first absorbance measurement) was subtracted from sulfo-phospho-vanillin absorbance (i.e., second absorbance measurement). The detection limit was determined to be approximately 5 μg of lipid.
Polyacrylamide Gel-Solutions
All gels were formed using two stock polyacrylamide solutions. The first 40%T acrylamide stock solution that can be purchased (Biorad) was used for all SDS-PAGE gels. The second 40%T acrylamide stock solution is made by mixing 48 g of acrylamide (Fisher Scientific) and 1.5 g of N,N-methylenebis(acrylamide) (Fisher Scientific) in 100 mL of ultrapure water (heating to 40 °C required to dissolve completely). This second acrylamide solution was used only for BN-PAGE gels. BN-PAGE gel buffers were prepared as previously described.14 Stacking gel buffer (125 mM Tris-HCl pH 6.8, 0.1% m/vol SDS) and resolving gel buffer (375 mM Tris-HCl pH 8.8, 0.1% m/vol SDS) were used for all SDS-PAGE gels. The 0.1% m/vol ammonium persulfate (Sigma) and 0.05% vol/vol N,N,N′,N′-tetramethylethylenediamine (Sigma) were used to the initiate polymerization of SDS-PAGE polyacrylamide gels.
Blot Quantification
Densitometry analysis was conducted on all images using Imagej software (National Institutes of Health). Linear regression analysis and t-tests were conducted using Graphpad Prism software (La Jolla, CA). All images are uncut (unless depicted by a black line) and representative of each experiment conducted. Several overexposed images are presented to display banding pattern, but these images were not used for any quantification.
Results/Discussion
Electrophoretic Mobility of α-Synuclein during BN-PAGE Is Dependent on Sample Preparation
To develop the multimer-PAGE protocol we first needed to describe, in detail, the migration pattern of α-synuclein during BN-PAGE (Figure 1). Extraction of proteins from rat brain tissue using 0.5% NP40, 1% digitonin, or in the absence of detergent under typical lysis concentrations (25 mg tissue/mL buffer) showed efficient tissue solubilization, an absence of abundant high molecular weight (>150 kDa) α-synuclein interactions and allowed for the separation of soluble α-synuclein via BN-PAGE (Figure 1A). Monoclonal and polyclonal antibodies against the C-terminus of α-synuclein produced similar band patterns for tissue lysates. α-Synulcein migrated to the gel position of approximately 50–146 kDa on the 2.8–13%T BN-PAGE gel. The addition of up to 50 μg lysate protein still produced one major band at 50 kDa. A mass shift to approximately ∼100 kDa was observed following the extraction with digitonin and NP40. The overall protein distribution on BN-PAGE appeared similar between extraction conditions, with the exception of an increased abundance of observable protein complexes with digitonin and NP40 (Figure 1A, Ponseau S stain). Together, these results show that under typical BN-PAGE conditions soluble α-synuclein from rat brain tissue was separated as a single species, predominantly migrating between the 50–146 kDa position on BN-PAGE, in agreement with several reports.4,15
Figure 1.

Electrophoretic mobility α-synuclein during BN-PAGE is determined by sample preparation. (A) Rat brain tissue lysates were resolved under typical blue native polyacrylamide gel electrophoresis (BN-PAGE) conditions. A total of 25 mg of rat brain tissue was homogenized in 1 mL of blue native page (BN-PAGE) sample buffer (50 mM BisTris, 6 N HCl, 50 mM NaCl, 10% vol/vol glycerol, 0.001% m/vol Ponceau S, pH ∼ 7.2). Then either 1% m/vol digitonin (Dig.) or 0.5% vol/vol NP40 was added to the sample and incubated at 4 °C for 30 min. Samples were then centrifuged at 18 000g for 30 min at 4 °C, and the supernatant (S1) was retained. Protein concentration of the S1 was determined using bicinchoninic acid (BCA) assay (ThermoFisher). In total, 10 μg and 50 μg of brain tissue lysate protein were resolved using BN-PAGE. Proteins were then immunoblotted onto polyvinylidene fluoride (PVDF) membranes and blue dye removed by washing the membrane with 100% methanol. PVDF membranes were then stained with ponseau S, imaged, and subsequently probed with both polyclonal and monoclonal anti-α-synuclein antibodies. (B) Rat brain tissue was then disrupted under high tissue to buffer ratio (m/vol) and separated by BN-PAGE. To do this, 200 mg rat brain tissue was homogenized, as described above, in 1 mL of BN-PAGE sample buffer in the absence of detergents. A total of 5–50 μg of total protein input of the undiluted S1 fraction was loaded into the first six wells of BN-PAGE gel. The S1 sample was then serial diluted from 0.8 to 4 mg protein/mL and 30 μg of protein loaded into the remaining wells (last six lanes). (C) Rat brain tissue lysates separated by BN-PAGE in two dimensions. A total of 200 mg of rat brain tissue was homogenized as described above. In total, 30 μg of the lysate was then resolved on a 2.8–13%T BN-PAGE gel. The gel lane was then excised and cast into a 2.8–13%T BN-PAGE gel perpendicular to the first dimension direction of migration. The sample was separated again via 2.8–13%T BN-PAGE. Top panel represents the immunoblot of first dimension BN-PAGE and the middle panel depicts immunoblot following the second dimension BN-PAGE. Bottom panel depicts ponseau S staining of the blotted proteins from the second dimension BN-PAGE. All membranes were probed with anti-α-synuclein antibodies and the appropriate HRP conjugated secondary antibodies. All separations shown were conducted using 2.8–13%T linear gradient BN-PAGE gels. Blue arrows indicate the direction of migration during BN-PAGE electrophoresis. All data representative of 3–10 experiments.
Previous reports demonstrate high lysis concentrations preserve α-synuclein multimers.7 Therefore, we then resolved high concentration rat brain tissue lysates (200 mg tissue/1 mL buffer) in the absence of detergents and observed several α-synuclein species migrating to 50 kDa, ∼100 kDa, 600 kDa, and 1048 kDa following BN-PAGE (Figure 1B). The mass shift of α-synuclein was sensitive to both total protein input, and to some extent, sample dilution. Under conditions of high sample concentration and/or high total protein input α-synuclein preferentially migrates to the 100 kDa BN-PAGE gel position. At the highest total protein input (50 μg protein) the predominant species of α-synuclein had an apparent mass of 100 kDa and two other massive species, at 600 kDa and 1048 kDa, could clearly be observed, albeit far less abundant than the 100 kDa position. The mass shift of α-synuclein during BN-PAGE was not completely lost following dilution of the sample, suggesting that the sample concentration at initial tissue lysis was also a crucial factor for the observed BN-PAGE mass shift. Interestingly, once the majority of α-synuclein had shifted to 100 kDa, several minor bands remained at the original position (more clearly seen in Figure 2B, bottom panel) suggesting the original band may be comprised of several species with distinct masses. Together, these results show that α-synuclein migrates to several positions on BN-PAGE depending on how the sample is prepared and that an unknown dilution sensitive factor was responsible for this apparent mass shift.
Next, we tested whether the observed α-synuclein mass-shift was a progressive process (i.e., a product of electrophoresis) by separating tissue lysates via BN-PAGE in two dimensions. Following the first dimension separation of 30 μg of brain lysate by BN-PAGE one main band at 50 kDa and a minor band just above ∼100 kDa, were observed (Figure 1C, top panel), as was shown previously in Figure 1B. When we subsequently separated the BN-PAGE gel lane by BN-PAGE perpendicular to the original migration direction, we again observed two distinct α-synuclein species at 50 kDa and ∼100 kDa (1C, middle panel), both at the original positions of the 1D-BN-PAGE. This suggests that the mass shift of α-synuclein during BN-PAGE was either due to a saturable process (i.e., nonprogressive) or the observed massive species existed in the original sample. The lack of a progressive mass shift during the second dimension BN-PAGE makes it highly unlikely that buffer components (i.e., coomassie blue g-250, Bis-Tris, aminocaporic acid, and tricine) were responsible for the observed mass shift.
Endogenous Lipids Determine α-Synuclein Migration Distance during BN-PAGE
An unknown biological factor or factors, lost upon dilution of the lysate and influenced the initial lysis concentration, were causing a mass shift of α-synuclein during BN-PAGE. Endogenous phospholipids are nearly insoluble amphiphiles that have the potential to form small soluble aggregates, termed micelles. Micelles form at threshold concentrations and are sensitive to dilution.16 Since α-synuclein is known to multimerize upon phospholipid membrane binding,5 endogenous lipid micelle-α-synuclein complex could be responsible for the α-synuclein mass shift during BN-PAGE. To test whether endogenous lipids were responsible for the apparent mass shift of α-synuclein, we removed lipids from the brain tissue lysates and then resolved the lipid free proteins by BN-PAGE. We found α-synuclein formed one distinct 50 kDa species in the absence of endogenous lipids (Figure 2A) similar to what was observed in low-lysis concentration detergent free samples (See Figure 1A). When 0.5% NP40 was added to lipid free proteins we observed a mass shift too approximately of ∼100 kDa with only a small amount of the 50 kDa α-synuclein species remaining (visualized a small spots toward the perimeter of the lane). The mass shift was similar to that observed with BN-PAGE of NP40 tissue extracts (Figure 1A). NP40/brain-lipid mixed micelles were then added to the lipid free proteins and another mass shift was observed, with α-synuclein now running at ∼50, ∼100, and ∼200 kDa, similar to that observed in the detergent free high concentration samples (refer to Figure 1B). Therefore, lipid and detergent micelles alone could produce a substantial apparent mass shift (+400% mass) during 1D-BNP. Interestingly, the micelles and endogenous lipids could be observed on the back of PVDF blots of the BN-PAGE following transfer, mostly stacked at the bottom of the membrane (Figure 2A, middle panel). Strikingly, the α-synuclein migration pattern was identical to the observed micelle migration pattern on the PVDF membrane. Ponceau S staining confirmed that the observed micelle positions were not highly proteinaceous (i.e., below ponseau S detection threshold, ∼10 μg of protein per lane). Taken together this data demonstrates that α-synuclein was migrating with endogenous lipids and/or detergent micelles during BN-PAGE, and this effect was likely responsible for mass shifts observed when tissue was disrupted at high concentrations.
When we resolved brain lipid/detergent mixed micelles by BN-PAGE, we noticed that they could be clearly visualized following transfer to PVDF membranes (Figure 2A, middle panel). Next we asked whether a similar pattern could be observed when tissue was lysed at high concentrations. Such a pattern would presumably confirm the hypothesis of endogenous lipid micelles being responsible for the observed mass shift during BN-PAGE. Results show that indeed the same type of pattern was observable most noticeably when tissue was lysed at high concentrations (Figure 2B, top panel). As with the lipid/detergent micelles, α-synuclein migration patterned mirrored the apparent endogenous lipid structures (Figure 2B, bottom panel) following BN-PAGE. Dilution of the samples did not abolish the visualization of the lipids but instead resulted in their shift to lower, discrete bands. Again, α-synuclein migration mirrored the position shift of the endogenous lipid micelles upon sample dilution. Gross protein distribution was relatively unaffected by the lipid structures migrating on BN-PAGE (Figure 2B, middle panel). Together, these observations demonstrate that α-synuclein was comigrating with soluble endogenous lipid structures.
Coomassie G-250 incorporates into detergent micelles,17 imparting a net negative charge, carrying them to the dye front during BN-PAGE, ultimately preventing distortion of protein separation.18 We wanted to determine whether G-250 could alter the distribution of the apparent endogenous lipid micelles and produce a corresponding mass shift in α-synuclein. When 0.01–1% G250 was added directly to samples from high concentration lysis conditions, the original lipid smear on the PVDF membrane was no longer observable (Figure 2C, top panel). Gross protein migration appeared unaffected by G-250 treatments (Figure 2C, middle panel), but α-synuclein migration became more focused to a position of 50 kDa as the G-250 concentration increased (Figure 2C, bottom panel). Therefore, excess G-250 added directly to the sample prevents the lipid mediated mass shift during BN-PAGE.
Lipids extracted under high concentration lysis conditions determined the electrophoretic mobility of α-synuclein during BN-PAGE; however, it was unclear whether high concentration tissue lysis resulted in increased lipid extraction from tissue. To directly address this, we measured the lipid and protein content in the remaining insoluble pellet (P1) at several lysis concentrations (Figure 2D). Results showed a sharp decrease in the lipid/protein ratio in the insoluble pellet when tissue was disrupted at high concentrations (i.e., 50–200 mg tissue/mL buffer). Interestingly, all lysis conditions above 50 mg tissue/mL buffer had similar lipid extraction efficacy. We attempted to directly measure the lipid content in S1 fractions, but the lipid concentration was near the assay’s threshold for detection (∼5 μg of lipid) and we were unable to obtain quantitative results. Even without the S1 lipid concentration it was clear that high lysis concentrations were increasing ratio of lipid to protein extracted from tissue.
Mass shift of a protein during BN-PAGE electrophoresis is commonly interpreted as a formation of protein complexes. However, as we have demonstrated here, the electrophoretic mobility of α-synuclein during BN-PAGE can vary drastically depending on sample preparation (specifically sample lipid content). A previous report demonstrated a similar effect for the mitochondrial ADP/ATP carrier protein AAC3.19 Therefore, BN-PAGE alone is insufficient to accurately detect and describe soluble α-synuclein multimers.
α-Synuclein Multimers Can Be Resolved via SDS-PAGE Following In-Gel Chemical Cross-Linking
Chemical cross-linking methods can be used to selectively capture protein–protein interactions. Once protein–protein interactions are captured the samples can be separated in the presence of SDS micelles via traditional SDS-PAGE. SDS solubilizes lipids20 and denatures proteins,21 thereby avoiding complications involved with BN-PAGE when separating lipid binding proteins, in this case α-synuclein. Therefore, following separation of tissue lysates via BN-PAGE, we incubated BN-PAGE gel lanes in several cross-linking solutions and then resolved them using 2D-SDS-PAGE (Figure 3A). Results showed that cross-linking with 0.1 mM DSP, 0.1 mM DTSSP (not shown), 1% glutaraldehyde, 0.1 mM DSG, but not 1% PFA produced several high molecular weight α-synuclein products in addition to the monomer detected following 2D-SDS-PAGE. We also observed two massive α-synuclein species of low abundance that were only captured and detectable with DSG or DSP. These elute from the BN-PAGE positions corresponding to the two high molecular weight species (∼600 kDa and ∼1048 kDa) clearly seen following BN-PAGE in Figure 1B. Both species were far less massive on the SDS-PAGE (∼80 kDa, and 170 kDa, respectively). Because DSP and DSG cross-linking is relatively specific for protein–protein interactions, the 600 kDa and 1048 kDa apparent masses on BN-PAGE were like due to processes other than true multimerization (e.g., lipid/detergent binding), as opposed to true multimers of 600 and 1048 kDa. PFA was ineffective at capturing α-synuclein multimers most likely because the spacer-arm length of PFA is relatively short (2 Å) while the effective cross-linkers had much longer spacer arm length (≥7 Å).7 Several other reports have also demonstrated the successful capture of α-synuclein multimers using cross-linkers with similar spacer-arm length.4−6
Figure 3.

In-gel chemical cross-linking reveals several soluble α-synuclein multimers following BN-PAGE. Rat brain tissue was homogenized in blue native page (BN-PAGE) sample buffer (50 mM Bis-Tris, 6N HCl, 50 mM NaCl, 10% vol/vol glycerol, 0.001% m/vol Ponceau S, pH ∼7.2) at a ratio of 200 mg of wet tissue to 1 mL of buffer. Following centrifugation at 18 000g for 30 min at 4 °C, the protein content of the S1 fraction was determined by bicinchoninic acid (BCA) assay. (A) 30 μg protein per lane was resolved via BN-PAGE. Individual lanes were then excised, washed with phosphate buffered saline pH 7.4 (PBS), and incubated in PBS solutions containing 0.1 mM dithiobis(succinimidyl propionate) (DSP), 0.1 mM disuccinimidyl glutarate (DSG), 1% glutaraldehyde (GLUT), or 1% paraformaldehyde (PFA) for 30 min at 4 °C. The excised gel pieces were then cast into SDS-PAGE and run perpendicular to the original migration direction. (B) Rat brain tissue was homogenized in PBS at a ratio of 200 mg wet tissue/1 mL buffer. Samples were centrifuged at 18 000g for 30 min at 4 °C. The sample was then diluted from 4 to 1 mg protein/mL. Following dilution, 1 mM DSG or 1 mM DSP was added to the samples and incubated for 10 min at 4 °C. The reaction was then quenched with 100 mM Tris- HCl pH 8.0 for 15 min at 4 °C. The 5× SDS-PAGE sample buffer was added to each sample, mixed well, and 10 μg of protein loaded into each lane of a 3–16%T SDS-PAGE gel. The 5 mM DTT was added to the 4 mg protein/mL DSP cross-linked sample to cleave the cross-linker (lane 1). (C) Increasing concentrations of total soluble protein were resolved via BN-PAGE (from the left; 4, 8, 12, 16, 20, 24, 28, 32, 36, and 40 μg of protein). BN-PAGE gel was cut above and below the 66 kDa molecular weight marker (red dotted box), washed with PBS, incubated with 0.1 mM DSP, and cast into a SDS-PAGE gel. Left panel depicts white light image of gel following BN-PAGE. Right panel depicts immunoblotting of the second dimension SDS-PAGE. All blots probed with rabbit polyclonal anti-α-synuclein antibody and appropriate horse radish peroxidase (HRP) conjugated secondary antibody. Images are representative of 3–5 separate experiments.
It remained unclear whether any α-synuclein multimers existed in solution prior to electrophoresis or were products of BN-PAGE. To address this, we directly cross-linked the soluble brain extracts using 1 mM DSP or DSG and resolved them via 4–16% SDS-PAGE (Figure 3B). Results show that α-synuclein multimers could be detected in lystates using both DSP and DSG cross-linkers. These multimers ranged from 34 kDa to >170 kDa and appeared to be formed predominantly by diffusion mediated cross-linking. Serial dilution of the sample prior to cross-linking prevented the capture of multimeric species, and for unclear reasons, also resulted in reduced immunodetection of the monomer (i.e., 17 kDa). Because directly cross-linking the sample did not produce α-synuclein multimers comparable to the in-gel crossslinking of BN-PAGE, the α-synuclein multimers observed following BN-PAGE were most likely, to some extent, products of electrophoresis.
Once we had determined that several multimers were found at the 50–100 kDa positions of the BN-PAGE gel, we then “scaled up” and optimized the procedure to make comparisons of many samples more reliable and less time-consuming. To do this we exploited the observation that under most sample conditions nearly all native α-synuclein migrated to the ∼50–100 kDa position on the BN-PAGE gel, and simply cut the gel just below the 66 kDa marker and just at the 146 kDa marker and ran the 2D-SDS-PAGE parallel to the BN-PAGE lanes (Figure 3C). The excised gel should encompass the 40–146 kDa positions, to ensure all α-synuclein is resolved by 2D-SDS-PAGE. This technique allowed the experimental conditions to be nearly identical between samples. Using this “scaled up” procedure would allow the cross-linked α-synuclein products to form more discrete bands when separated by 2D-SDS-PAGE, as opposed to smeared blebs observed with running the entire excised lanes, making them easier to visualize and compare. Several experimental conditions could now be run on a single gel making comparisons between the extracts simple and reproducible. Furthermore, several multimers at the gel position could be quantified simultaneously, as opposed to only the typically two α-synuclein species observed following BN-PAGE. For simplicity, we termed the entire process as “multimer-PAGE,” with the presented blots representing the blotted 2D-SDS-PAGE of cross-linked native complexes.
Optimization of In-Gel Chemical Cross-Linking
Diffusion based chemical cross-linking can randomly covalently bind proteins forming progressively massive products only limited by the diffusion of the protein in solution. Protocols using chemical cross-linking of proteins should be carefully validated to avoid diffusion based products, unless the product is desired, as was later exploited here to produce the α-synuclein multimer molecular weight “standard” (see Figure 4E). Generally, cross-linker concentration, time of cross-linking, and the molecular structure of the cross-linker are manipulated to determine true products (i.e., those that existed prior to cross-linking) vs diffusion based (i.e., those that formed during cross-linking). Here we tested two cross-linking molecules DSP and DSG, both of which have been used in tissue and solution to stabilize α-synuclein multimers.6 Cross-linking the excised BN-PAGE gel with as little as 0.1 mM DSP (Figure 4A) or DSG (Figure 4B) was sufficient to capture α-synuclein multimers. For reasons that are unclear, increasing DSG concentrations appeared to reduce the detection of both multimeric and monomeric α-synuclein. Because of the unusual behavior of DSG during in-gel cross-linking, we chose to use 0.1 mM DSP for the remaining studies. The main α-synuclein species observed following in-gel cross-linking was smeared between 34 and 130 kDa on the SDS-PAGE. Next we tested what cross-linker incubation time was optimal to capture the multimers (Figure 4C). Gels incubated with 0.1 mM DSP for 5 min preferentially capture αS80. The cross-linking time to capture multimers was so rapid it strongly suggested that diffusion based cross-linking was not a main driving factor of α-synuclein multimers detection following multimer-PAGE protocol. Cross-linking produced nearly equal multimer abundance at 30 and 45 min, and therefore 30 min was chosen for the remaining studies.
Figure 4.

Optimization of in-gel chemical cross-linking and multimer-PAGE gel format. 10, 20, and 40 μg of rat soluble brain tissue lysate were resolved by blue native polyacrylamide electrophoresis (BN-PAGE). 2.8–13% linear BN-PAGE gels were cut just above and below the 66 kDa molecular weight marker following electrophoresis. The excised gel pieces were washed in phosphate buffered saline pH 7.4 (PBS) for 30 min at 4 °C. Excised BN-PAGE gel pieces were incubated with cross-linking solution and cast directly to SDS-PAGE gels. The cross-linked proteins were resolved by SDS-PAGE perpendicular to the original direction of sample migration. SDS-PAGE gels were then immunoblotted. Excised gel pieces incubated with either (A) 0.1–2 mM dithiobis(succinimidyl propionate) (DSP) (B) or 0.1–2 mM disuccinimidyl glutarate (DSG). (C) Excised gel pieces incubated with 0.1 mM DSP for 5–45 min. (D) 30 μg of rat brain tissue lysate separated by BN-PAGE, blotted, and probed for parkin (Left panel). 10, 20, 30, and 40 μg of lysate protein resolved by BN-PAGE. BN-PAGE gel was excised between 40 and 242 kDa positions, incubated with 0.1 mM DSP for 30 min at 4 °C, and resolved via SDS-PAGE. Immunoblotting for parkin and α-synuclein shown (Right panels). (E) Purified α-synuclein was dissolved in PBS and diluted to a concentration of 5 mg protein/mL. The 2 mM DSP was added to the sample and incubated for 10 min at 4 °C. The reaction was quenched with 100 mM Tris-HCl pH 8.0. 200 ng of α-synuclein resolved on 12%T SDS-PAGE. (F) Increasing protein input resolved by 2.8–13%T BN-PAGE and 6%T BN-PAGE. Samples were allowed to migrate the length of the 2.8–13%T BN-PAGE and the 66 kDa gel position excised as before. In contrast, samples were allowed to migrate on approximately 2 cm into the resolving gel on the 6%T BN-PAGE, and this 2 cm gel piece excised. In-gel cross-linking was performed and both excised gel pieces resolved via SDS-PAGE. Bands are labeled with their apparent corresponding species according the standard (E). Dotted red lines indicate position of gel excision. All blots probed with rabbit polyclonal anti-α-synuclein antibody and appropriate horse radish peroxidase (HRP) conjugated secondary antibody. Images are representative of three separate experiments.
During validation of the multimer-PAGE protocol, we observed that optimal resolution of cross-linked products during SDS-PAGE requires that excised gel pieces be cast directly into the SDS-PAGE gel as described in the methods. Alternatively, when gel pieces were placed directly on top of a preformed SDS-PAGE gel (also mentioned in methods) lane distortion became evident (see Figure 4C) but did not appear to effect quantification. Therefore, this method can be used when sample processing speed is crucial, as it is more time intensive to cast excised gels directly into SDS-PAGE gels. We also observed that using a 12%T SDS-PAGE with a stacking layer ultimately provided superior band resolution to 3–16%T SDS-PAGE gels.
Next we tested the specificity of multimer-PAGE for α-synuclein. To do this we ran the multimer-PAGE protocol and probed for the E3 ligase parkin, a small globular soluble protein not known to interact with lipids or form progressively massive multimers. We found that parkin also migrates to approximately the same BN-PAGE gel position as α-synuclein (Figure 4D), making it a good internal control. Following multimer-PAGE we observed no detectable cross-linked parkin multimers following multimer-PAGE protocol. Instead we saw one species located at ∼43 kDa. This position is slightly below the 50 kDa position where parkin is normally found. The slight discrepancy in molecular weight of parkin is likely due to the stabilization of a compact tertiary structure of parkin by chemical cross-linking, which can increase proteins mobility on SDS-PAGE.22,23 The migration of parkin during multimer-PAGE was in stark contrast to α-synuclein, which under the same experimental conditions produced abundant multimers. This finding demonstrates that multimer-PAGE is relatively specific for α-synuclein and gross diffusion based protein cross-linking was not a key factor.
To determine stoichiometry of α-synuclein multimers following multimer-PAGE we generated a molecular weight “standard” by cross-linking concentrated purified α-synuclein (5 mg purified α-synuclein/1 mL) with 2 mM DSP and separated the resulting products by SDS-PAGE (Figure 4E). Results show five species at gel positions corresponding to the molecular weights of 17 (αS17), 35 (αS35), 56 (αS56), 80 (αS80), and 100 kDa (αS100). α-Synuclein was detected above 130 kDa position but was poorly resolved by SDS-PAGE and therefore was disregarded as a defined α-synuclein multimers species. The observed multimers were likely captured primarily via diffusion controlled cross-linking because there was an inverse relationship between multimers abundance and the observed molecular weight, which likely occurs if multimers were cross-linked in a stepwise fashion.
We next wanted to determine the optimal polyacrylamide gel format for multimer-PAGE (Figure 4F). We found that 6%T BN-PAGE gel followed by 12%T SDS-PAGE gel provided the greatest resolution of α-synuclein multimers with the least effort. We simply ran the 6%T BN-PAGE gel (with 2.8%T well gel) at 100 V until all soluble proteins had migrated ∼2 cm into the resolving gel. The gel was excised as before and cast into the 12%T SDS-PAGE. The new gel format had several key advantages: (1) avoided hand pouring linear gradient polyacrylamide gels which can be time-consuming and have poor reproducibility, (2) ensured all α-synuclein (and other proteins) in the sample was resolved by multimer-PAGE, (3) cross-linked products migrated out of a single 6%T BN-PAGE gel to the SDS-PAGE gel more consistently allowing for increased band resolution, (4) allowed for investigation of protein complexes other that α-synuclein without extensive validation of BN-PAGE migration (i.e., do not need to know a BN-PAGE migration pattern), and (5) faster BN-PAGE run times (∼30 min at 100 V).
Optimization of Brain Tissue Sample Preparation and Separation
Once in-gel cross-linking and electrophoresis conditions were optimized, we then tested the influence of sample preparation on multimer-PAGE output. First, we investigated whether lipid content of the sample would alter the observed multimer pattern. We had already observed that α-synuclein migrated with endogenous brain lipids during BN-PAGE (Figure 2A–C); however, when we assessed these samples using multimer-PAGE we found little evidence of any multimeric α-synuclein species in any of the samples (Figure 5A). Neither proteins alone nor proteins reconstituted into lipid/detergent micelles produced detectable multimers. This suggests that although α-synuclein was strongly interacting with lipid/detergent mixed micelles the binding to these micelles alone was insufficient to produce multimers. Furthermore, multimers observed following multimer-PAGE of tissue lysates were likely not due to protein–lipid cross-linking, as these products would then be predicted to be in abundance in the presence of excess brain lipids. However, phosphatidalserine and phosphatidylethanolamine both contain primary amines so protein–lipid cross-linked products cannot be conclusively ruled out.
Figure 5.

Sample preparation determines observed multimer abudance and pattern. Rat brain tissue was homogenized in blue native page (BN-PAGE) sample buffer (50 mM BisTris, 6 N HCL, 50 mM NaCl, 10% vol/vol glycerol, 0.001% m/vol Ponseau S, pH ∼7.2) at a ratio of 200 mg wet tissue/1 mL buffer. Following centrifugation at 18 000g for 30 min at 4 °C, the protein content of the S1 fraction was determined by the bicinchoninic acid (BCA) assay. (A) 300 μg proteins from the sample was precipitated and resusupended in either BN-PAGE sample buffer containing either 0.5% NP40 or 0.5% NP40 mixed lipid micelles. Protein (10 μg) was resolved by multimer-PAGE. (B) 50–200 ng of purified α-synuclein and 30 μg of brain tissue lysate were resolved via multimer-PAGE. (C) 0–1% G250 directly added to S1 samples. 30 μg of these samples was separated by multimer-PAGE. (D) Rat brain tissue was disrupted in BN-PAGE sample buffer at a tissue mass to buffer ratio of 25, 50, 100, and 200. A total of 30 μg of protein from each lysis condition was resolved via multimer-PAGE. SDS-PAGE of S1 and P1 found in the bottom two panels. (E) Rat brain tissue was homogenized in a tissue mass to a buffer ratio of 25 and 100. Increasing total protein input was then resolved via multimer-PAGE. (From the left: 5, 10, 20, 30, and 40 μg protein). All samples were centrifuged at 18 000g for 30 min prior to BN-PAGE. All blots probed with rabbit polyclonal anti-α-synuclein antibody and appropriate horse radish peroxidase (HRP) conjugated secondary antibody. Images are representative of 2–4 separate experiments.
Since recombining separated components (i.e., lipids and proteins) of complex biological samples did not produce detectable multimers by BN-PAGE, we wanted to determine whether α-synuclein alone could assemble into multimers during multimer-PAGE. To test this we resolved increasing α-synuclein input (50–200 ng) by multimer-PAGE and found multimers, to a limited extent, could be detected (Figure 5B). However, multimers from purified α-synuclein required excess protein input and were qualitatively distinct from those found in tissue lysates. High molecular weight α-synuclein multimers (i.e., αS56, αS80, and αS100) are preferentially detected in lysates while αS35 is the primary multimer detected when using purified α-synuclein. The multimer pattern when using purified α-synuclein was indicative of diffusion based cross-linking and distinct from multimers from lysates, as previously reported.7 Therefore, α-synuclein was insufficient to produce multimers observed in complex biological samples.
G-250 incorporates into micelles and can form micelle like structures in water, and therefore we wanted to know whether G-250 could affect multimer detection (Figure 5C). G250 added directly to the sample produced an increased multimer abundance only when G-250 concentrations were relatively high (0.5% and 1%). G-250 concentrations below 0.5% (first three lanes) were similar in multimer abundance and distribution. Because samples are not typically exposed to G-250 concentrations above 0.5% during multimer-PAGE protocol, this effect is likely minimal.
Previously we demonstrated that disruption of brain tissue at high concentrations increased relative lipid extraction and altered the migration of α-synuclein during BN-PAGE (see Figure 2D, B), so we explored whether the ratio of tissue to lysis buffer influenced the abundance of observed multimers. Results showed that when tissue was disrupted at increasing concentrations, multimers in the S1 fraction were in greater abundance but the pattern was relatively unchanged (Figure 5D). αS80 and αS100 appeared to be particularly abundant with αS56 being lower in abundance. Interestingly, the total amount of α-synuclein extracted (i.e., S1 fraction) per protein was similar when 50 mg tissue per mL buffer or greater was used for tissue lysis. Despite the similar extraction efficacy, multimers were clearly more abundant with increasing ratio of tissue to buffer. Both the 100 and 200 mg tissue per mL buffer showed nearly the same multimer abundance, and therefore the 100 mg tissue per mL buffer condition was selected for multimer quantification.
Few multimers were detected at typical tissue lysis concentration (i.e., 25 mg tissue/mL buffer) when 20 μg of total protein was input into multimer-PAGE (see Figure 5D). However, our previous data suggested that, to some extent, α-synuclein multimers were being formed during electrophoresis (see Figure 3C,B). Therefore, we next asked whether increasing sample input from low and high lysis concentrations would produce observable multimers. Results show that as sample input increased, multimers became more abundant in both the low and high concentration tissue lysis samples (Figure 5E). α-Synuclein multimers were detectable with 10 μg of protein in the high concentration lysis but required 15 μg following low concentration tissue lysis. Multimer-abundance increased disproportionally to monomer abundance following both lysis concentrations. This observation, as seen previously (Figure 3C), seems to support the idea that multimers were forming during multimer-PAGE and do not exist in the original sample. Therefore, although lysis at high concentration allows for ease of multimer detection, it does not seem absolutely necessary to produce multimers given sample input into the multimer-PAGE is sufficiently high.
Using Multimer-PAGE to Measure the Stoichiometry of α-Synuclein Multimers in Biological Samples
Next we wanted to determine a standardized method to analyze the data produced by multimer-PAGE. To do this we exploited the observation that the ratio of αS80 to αS17 increases linearly with increasing protein input into multimer-PAGE (see Figure 6C). This phenomenon allowed the assessment of multimerization while avoiding variability from factors such as relative α-synuclein abundance in the sample.
Figure 6.

Multimer-PAGE can be used to compare many complex biological samples. Brain tissue from 4 male Sprague–Dawley rats was weighed and homogenized in BN-PAGE sample buffer (100 mg tissue/mL buffer). Samples were centrifuged at 18 000g for 30 min at 4 °C. The supernatant was retained and 10, 20, and 30 μg of protein separated by multimer-PAGE. Multimer-PAGE of samples from the hypothalamus (A) and cortex (B). Top panels depict immunoblotting for α-synuclein. Bottom panels depict ponseau S stain of multimer-PAGE blots prior to immunoprobe. (C) The ratio of 17 kDa (αS17) to 80 kDa (αS80) α-synuclein was calculated using ImageJ software and values are plotted for each protein input. The R2 for each line is depicted on the graph. (D) Bar graph comparing the multimer curve slopes between the two brain regions. (“Rate of multimer formation” = slope of multimer curves. *students t test, t (6) = 11.29, p < 0.0001, n = 4).
We generated a multimer curve by separating 10, 20, and 30 μg protein of lysates from the rat hypothalamus (Figure 6A) and frontal cortex (Figure 6B) via multimer-PAGE. Care was taken to ensure tissue was disrupted in 1 mL of BN-PAGE sample buffer per 100 mg of tissue. Once the multimer curve was generated (Figure 6C) we could plot the ratio of αS80 to αS17 at each protein input. The corresponding slope of the multimer curve could then be calculated and compared. We refer to the calculated slope as the rate of multimer formation because the line likely represents the propensity of α-synuclein to form multimers during multimer-PAGE. We then compared multimer formation between two distict brain regions (Figure 6D). Results show, that within each brain region, animals had similar rate of multimer formation, resulting in very little variability. However, when we compared the two brain regions there was significant (Students t test, t (6) = 11.29, p < 0.0001, n = 4) reduction in the rate of multimer formation in the hypothalamus when compared to the frontal cortex. The significance the regional difference in multimer formation is not realized here, but the method appears to provide reproducible and accurate measurement of the α-synuclein multimer abundance between different biological samples.
Final Optimized Protocol for α-Synuclein Multimer-PAGE
The final optimized protocol for α-synuclein multimer-PAGE involves four key steps and takes approximately 1 day to complete (Figure 7). With this protocol we could routinely measure and compare several high molecular weight α-synuclein species across complex biological samples. Effort was taken to ensure the process was not overly complicated and could be conducted using common lab equipment. Although this protocol was optimized for α-synuclein detection, it should be emphasized that experimentation with protein separation, cross-linking, and sample handing conditions could potentially help answer questions about numerous other protein complexes. This technique likely has broad applicability to the study of the stoichiometry of numerous other protein-complexes. Although the current protocol is limited to measuring “low-n” multimers consisting of ∼10 or less α-synuclein molecules, the SDS-PAGE gel format could be easily adjusted to study ever more massive multimeric species.
Figure 7.

Final optimized protocol for α-synuclein multimer-PAGE. First, rat brain tissue was homogenized in blue native page electrophoresis (BN-PAGE) sample buffer (50 mM BisTris, 6 N HCl, 50 mM NaCl, 10% vol/vol glycerol, 0.001% m/vol Ponseau S, pH ∼7.2 ) at a specific buffer to tissue ratio (100 mg of wet tissue per 1 mL of buffer) (top panel). Homogenization was conducted on ice and consisted of 30 gentle strokes in a 7 mL glass dounce homogenizer. Samples were then centrifuged for 30 min at 18 000g, and the supernatant (S1) containing the soluble biological molecules was retained. Samples were then prepared to load into a BN-PAGE gel (see Step II). To do this, protein content of this sample was then determined using bicinchoninic acid (BCA) assay and the S1 diluted to 2 mg protein/mL using BN-PAGE sample buffer. In order to compare samples, 10, 20, and 30 μg of each sample was loaded into a 6%T BN-PAGE gel. A 100 V potential was applied to the gel until the sample had migrated to approximately 2 cm into the resolving layer of the 6%T BN-PAGE gel. The gel was then removed, well gel discarded, and the 2 cm section of gel containing the samples was excised. The excised gel piece was then washed in phosphate buffered saline pH 7.4 (PBS) for approximately 30 min at 4 °C (see step III). The PBS solution should be changed at least 3 times during the washing. Then the gel piece is placed in 10 mL of PBS and 40 μL of 25 mM dithiobis(succinimidyl propionate) (DSP) dissolved in DMSO added to the PBS. The gel is incubated in this cross-linking solution for 30 min at 4 °C, then the solution is discarded and replaced with quenching solution (125 mM Tris-HCL pH 6.8, 2% SDS) and incubated for 60 min at room temperature. The excised gel piece is then placed between glass plates and cast into 12%T SDS-PAGE gel. A constant voltage of 100 V is then applied to the gel using SDS-PAGE running buffer until coomassie dye has left the end of the gel. The 12%T SDS-PAGE gel was then electroblotted onto polyvinylidene fluoride (PVDF) membranes and probed with several α-synuclein antibodies (see Step IV). Densitometry analysis can then be conducted on the observed bands using imageJ software. The slope of the lines plotting multimer abundance and total protein input could be used to compare samples. (“ST” = well for molecular weight standard)
Conclusions
α-Synuclein formed several soluble multimers during BN-PAGE provided that lipid interactions were preserved during tissue lysis. Preservation of lipid interactions likely stabilized α-synuclein secondary structure8,24 subsequently “seeding” multimer formation3 and/or stabilizing existing multimers. We found that a practical method to preserve lipid interactions involved disrupting the tissue in small buffer volumes (i.e., 50–200 mg tissue/mL buffer) in the absence of detergents. The high ratio of tissue to buffer increased the extraction of lipids from tissue. Extracted lipids behaved like endogenous micelles. Although we did not provide a direct description of these micelles, they are logically consistent with the fact that free phospholipids are almost completely insoluble in water and the lipid structures migrated on BN-PAGE as complexes (i.e., above the dye front) and not monomers (i.e., with the dye front). The α-synuclein species observed following the multimer-PAGE, once optimized, were in general agreement with the previously reported α-synuclein multimers,2,5−7 and distinct from those produced by diffusion mediated protein cross-linking.
Future studies are needed to explore the applicability of the multimer-PAGE technique. Studies that may be fruitful should focus on the possible heterogeneity (e.g., differences in protein folding, post-translational modification etc.) of the observed α-synuclein multimers and how this may confer/abolish stability. Liquid chromatography–mass spectrometry (LC–MS) would be particularly useful in identifying possible heterogeneity post-translational modifications between multimers; although mass adjustments to account for the cross-linker would need to be applied, as has been done numerous reports.25 LC–MS may also allow for the identification of the specific lipid(s) interacting with α-synuclein.26 It is also important to further validate multimer-PAGE across several PD models to determine its usefulness in describing the pathology of the disease.
Multimer-PAGE provides several unique advantages over in-solution cross-linking or native-PAGE alone. Kinetics of the chemical cross-linking reaction could be controlled precisely making the comparison between many samples easily reproducible. The preseparation of proteins appeared to reduce diffusion based cross-linking, probably because the movement of complexes in the sample was restricted to the immediate vicinity of their position in the polyacrylamide gel, making chance interactions with other proteins unlikely. Multimer-PAGE seemed useful in detecting stable protein complexes other that α-synuclein multimers. In this regard, multimer-PAGE may be a particularly valuable supplemental technique for BN-PAGE, to verify true protein complexes, as protein mobility on BN-PAGE alone clearly is not sufficient.19 For example, multimer-PAGE detected parkin as a monomer (50 kDa) in contrast to a previous report that found parkin to be exclusively a tetramer, based on BN-PAGE experiments.27 Multimer-PAGE format also allows the simultaneous comparison of many biological samples, so that relative protein complex abundance can be compared on a single blot. Although not reported here, gels could also be incubated with solutions containing experimental molecules to assess their influence on complex stability.
Lipid–protein cross-linking could occur between α-synuclein and phosphatidalserine or phosphatidylethanolamine. This interpretation of the data has the advantage of explaining the seemingly anomalous behavior of α-synuclein following sample dilution prior to chemical cross-linking (seen in Figure 3B). DSP treatment enhances α-synuclein detection on PVDF membranes as described previously.28 The enhancement is lost upon sample dilution. Because we observed that α-synuclein association with endogenous lipids was sensitive to dilution, it could be that upon dilution α-synuclein disassociates from endogenous lipids, reverting to an unstable intrinsically dynamic protein in solution. Cross-linking of free α-synuclein would then likely form a more compact structure as adjacent lysine residues of α-synuclein cross-link to each other. In contrast, if lipid–protein cross-linking occurred between α-synuclein and several phospholipids, α-synuclein would not only have a larger molecular radius but would presumably be more hydrophobic, allowing for enhanced PVDF binding. Data provided here is insufficient to conclusively affirm or reject this interpretation and future studies should aim to address this potentially important molecular behavior of soluble α-synuclein.
Overall, multimer-page is a useful technique to study protein complexes in great detail. Multimer-PAGE was used successfully to describe and compare soluble α-synuclein multimers across complex biological samples. Future studies should focus on the applicability of this technique to studying PD pathogenesis.
Acknowledgments
We would like to thank the laboratory of Dr. Aloke Dutta for providing purified α-synuclein.
This work was funded by National Institutes of Health Grant DA034783.
The authors declare no competing financial interest.
References
- Bendor J. T.; Logan T. P.; Edwards R. H. Neuron 2013, 79, 1044–1066. 10.1016/j.neuron.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M.; Spillantini M. G.; Del Tredici K.; Braak H. Nat. Rev. Neurol. 2012, 9, 13–24. 10.1038/nrneurol.2012.242. [DOI] [PubMed] [Google Scholar]
- Burre J.; Sharma M.; Sudhof T. C. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, E4274–4283. 10.1073/pnas.1416598111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels T.; Choi J. G.; Selkoe D. J. Nature 2011, 477, 107–110. 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burre J.; Sharma M.; Sudhof T. C. J. Neurosci. 2015, 35, 5221–5232. 10.1523/JNEUROSCI.4650-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettmer U.; Newman A. J.; Soldner F.; Luth E. S.; Kim N. C.; von Saucken V. E.; Sanderson J. B.; Jaenisch R.; Bartels T.; Selkoe D. Nat. Commun. 2015, 6, 7314. 10.1038/ncomms8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettmer U.; Newman A. J.; Luth E. S.; Bartels T.; Selkoe D. J. Biol. Chem. 2013, 288, 6371–6385. 10.1074/jbc.M112.403311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N.; Lee H. J.; Li J.; Fink A. L.; Lee S. J. J. Biol. Chem. 2001, 276, 43495–43498. 10.1074/jbc.C100551200. [DOI] [PubMed] [Google Scholar]
- Huang C.; Ren G.; Zhou H.; Wang C. C. Protein Expression Purif. 2005, 42, 173–177. 10.1016/j.pep.2005.02.014. [DOI] [PubMed] [Google Scholar]
- Folch J.; Lees M.; Sloane Stanley G. H. J. Biol. Chem. 1957, 226, 497–509. [PubMed] [Google Scholar]
- Wittig I.; Braun H. P.; Schagger H. Nat. Protoc. 2006, 1, 418–428. 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- Shearer G. Jr. Anal. Biochem. 1994, 221, 397–400. 10.1006/abio.1994.1432. [DOI] [PubMed] [Google Scholar]
- McMahon A.; Lu H.; Butovich I. A. Lipids 2013, 48, 513–525. 10.1007/s11745-013-3755-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittig I.; Schagger H. Proteomics 2009, 9, 5214–5223. 10.1002/pmic.200900151. [DOI] [PubMed] [Google Scholar]
- Engelender S.; Kaminsky Z.; Guo X.; Sharp A. H.; Amaravi R. K.; Kleiderlein J. J.; Margolis R. L.; Troncoso J. C.; Lanahan A. A.; Worley P. F.; Dawson V. L.; Dawson T. M.; Ross C. A. Nat. Genet. 1999, 22, 110–114. 10.1038/8820. [DOI] [PubMed] [Google Scholar]
- Seddon A. M.; Curnow P.; Booth P. J. Biochim. Biophys. Acta, Biomembr. 2004, 1666, 105–117. 10.1016/j.bbamem.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Samsonoff C.; Daily J.; Almog R.; Berns D. S. J. Colloid Interface Sci. 1986, 109, 325–329. 10.1016/0021-9797(86)90310-3. [DOI] [Google Scholar]
- Wittig I.; Schagger H. Proteomics 2008, 8, 3974–3990. 10.1002/pmic.200800017. [DOI] [PubMed] [Google Scholar]
- Crichton P. G.; Harding M.; Ruprecht J. J.; Lee Y.; Kunji E. R. J. Biol. Chem. 2013, 288, 22163–22173. 10.1074/jbc.M113.484329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee P.; Joo J. B.; Buse J. T.; Dawson G. Chem. Phys. Lipids 1995, 77, 65–78. 10.1016/0009-3084(95)02455-R. [DOI] [PubMed] [Google Scholar]
- Bhuyan A. K. Biopolymers 2010, 93, 186–199. 10.1002/bip.21318. [DOI] [PubMed] [Google Scholar]
- Therien A. G.; Grant F. E.; Deber C. M. Nat. Struct. Biol. 2001, 8, 597–601. 10.1038/89631. [DOI] [PubMed] [Google Scholar]
- Rath A.; Glibowicka M.; Nadeau V. G.; Chen G.; Deber C. M. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 1760–1765. 10.1073/pnas.0813167106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson W. S.; Jonas A.; Clayton D. F.; George J. M. J. Biol. Chem. 1998, 273, 9443–9449. 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- Leitner A.; Walzthoeni T.; Kahraman A.; Herzog F.; Rinner O.; Beck M.; Aebersold R. Mol. Cell. Proteomics 2010, 9, 1634–1649. 10.1074/mcp.R000001-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubbens J.; Ruijter E.; de Fays L. E.; Damen J. M.; de Kruijff B.; Slijper M.; Rijkers D. T.; Liskamp R. M.; de Kroon A. I. Chem. Biol. (Oxford, U. K.) 2009, 16, 3–14. 10.1016/j.chembiol.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Van Humbeeck C.; Waelkens E.; Corti O.; Brice A.; Vandenberghe W. Eur. J. Neurosci. 2008, 27, 284–293. 10.1111/j.1460-9568.2007.06000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman A. J.; Selkoe D.; Dettmer U. PLoS One 2013, 8, e81314. 10.1371/journal.pone.0081314. [DOI] [PMC free article] [PubMed] [Google Scholar]