

Abstract
The prevalence and incidence of stroke rises with life expectancy. However, except for the use of recombinant tissue-type plasminogen activator, the translation of new therapies for acute stroke from animal models into humans has been relatively unsuccessful. Oxidative DNA and protein damage following stroke is typically associated with cell death. Cause-effect relationships between reactive oxygen species and epigenetic modifications have been established in aging, cancer, acute pancreatitis, and fatty liver disease. In addition, epigenetic regulatory mechanisms during stroke recovery have been reviewed, with focuses mainly on neural apoptosis, necrosis, and neuroplasticity. However, oxidative stress-induced epigenetic regulation in vascular neural networks following stroke has not been sufficiently explored. Improved understanding of the epigenetic regulatory network upon oxidative stress may provide effective antioxidant approaches for treating stroke. In this review, we summarize the epigenetic events, including DNA methylation, histone modification, and microRNAs, that result from oxidative stress following experimental stroke in animal and cell models, and the ways in which epigenetic changes and their crosstalk influence the redox state in neurons, glia, and vascular endothelial cells, helping us to understand the foregone and vicious epigenetic regulation of oxidative stress in the vascular neural network following stroke.
Keywords: Stroke, brain, epigenetics, oxidative stress
Oxidative stress is crucial in the pathogenesis of neurodegenerative disorders [1], and the relevant mechanisms of oxidative neuronal death especially mitochondrial dysfunction [2] in ischemic stroke have been extensively studied. More importantly, a recent study showed that androgens were neuroprotective when oxidative stress levels were minimal, but exacerbated oxidative stress damage in immortalized rat dopaminergic neuronal cells when oxidative stress levels were elevated [3]. This indicated that oxidative stress was not only strongly implicated in the progression of cell death, but could also define the neuroprotective or neurotoxic properties of other drugs following stroke [3].
Oxidative stress represents an imbalance between the elevated production of reactive oxygen species (ROS)/reactive nitrogen species (RNS) and missing antioxidants, leading to oxidative modifications of proteins, lipids, and DNA [4-9]. Epigenetics have been reported to be profoundly involved in oxidative stress responses, and cause-effect relationships have been established between ROS and epigenetic modifications in aging, cancer, acute pancreatitis, and non-alcoholic fatty liver disease [10-15]. Moreover, epigenetic regulatory mechanisms, especially the role of histone deacetylase inhibition during stroke recovery, have been summarized in several reviews focusing mainly on apoptosis, necrosis, and neuroplasticity [16-26]. However, the role of oxidative stress-induced epigenetic regulation in vascular neural networks following stroke has not been sufficiently explored.
Epigenetic mechanisms, referring to heritable changes in gene expression without changing the DNA sequence, mainly include DNA methylation, histone modification, and microRNAs (miRNAs), which specifically modulate the expression levels of single genes and functional gene networks [27-28]. In this review, we emphasize the epigenetic events resulting from oxidative stress in experimental stroke models in vivo and in vitro, and how epigenetic changes influence the redox state in vascular neural networks, with implications for the discovery of more sensitive and specific therapeutic targets based on a combination of antioxidant and epigenetic regulatory strategies for stroke.
Oxidative stress regulate DNA methylation in stroke
CpG island methylation is a well-characterized epigenetic change that generally regulates global and specific gene expression by transcription inhibition [29]. That is mediated by DNA methyltransferases (DNMTs), which are abundant in the brain, includes Dnmt 3a and Dnmt 3b (de novo methylation), as well as Dnmt 1 (maintains methylation) [30]. In addition, gene silencing mediated by DNA methylation also contains the interactions of protein-DNA and protein-protein, with the primary recruitment of methyl-CpG-binding-domain (MBD) family includes MBD1-4 and MeCP2 (methyl-CpG-binding protein 2) and subsequent combination of histone-modifying enzymes, that together raises chromatin condensation and deactivation [31]. In this section, we summarize the changes in global and gene-specific methylation following ischemic stroke in vivo and oxidative stress in vitro, and especially the related epigenetic mechanisms induced by ROS/RNS and hyperhomocysteinemia (HHcy).
ROS/RNS induced DNA methylation following stroke
The generation of ROS/RNS (e.g. H2O2, nitric oxide) directly modify cytosine chemically, with oxidative conversion of 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC), inhibiting the binding of Dnmt1 and MBP to DNA, therefore changes its methylation pattern [32-34]. 5hmC level is increased in blood cell DNA from patients with acute ischemic stroke [35]. Moreover, peroxides also cause nucleobases modification like 5-chlorocytosine, which mimics 5-mC and induce improper Dnmt1 methylation within CpG sequences, inducing gene silence [36, 37]. These evidences provide a mechanistic link between oxidative stress and epigenetic changes via chemical DNA modifications and altering DNA-protein interactions.
It’s noted that global DNA methylation in neural cells was changed by ischemia or oxidative stress, and DNMT inhibitors could alleviate ischemia or oxidative stress-induced neural injury. In vivo, the global DNA methylation was significantly increased in infarcted tissue in model of cerebral ischemia [38,39]. And DNMT inhibitors conducted neuroprotection, treatment with the broad-spectrum DNMT inhibitor 5-aza-2′-deoxycytidine (5-aza-dC) and zebularine [38], as well as reduced levels of Dnmt1 in postmitotic neurons in transgenic mice [39], could alleviate cerebral ischemic injury. Except for DNMTs, the expression of MBD-family proteins is altered orderly within the hippocampus: MBD3 expression was significantly reduced 3 h after ischemia, while MBD2 expression was increased by 6 h after ischemia, and MBD1 and MeCP2 levels were both elevated by 24 h after ischemia [40]. In vitro, treatment with H2O2 for 1 h increased the global DNA methylation level in SH-SY5Y human neuroblastoma cells, however, long-term treatment (72 h) had the opposite effect, along with decreased expressions of Dnmt 1, Dnmt 3a and Dnmt 3b [41]. Furthermore, H2O2 treatment in SH-SY5Y cells for 1 h increased the DNA-binding activities of nuclear factor (NF)-κB and SP1/3, while 5-aza-dC pretreatment resulted in increased NF-κB DNA-binding activity [41]. In addition, DNMT inhibitors 5-azacytidine and 5-aza-dC reduced photodynamic-treatment, a therapy based on photosensitizer-mediated oxidative cytotoxicity, induced necrosis of glial cells [42]. Conversely, knockdown of a critical enzyme for DNA demethylation, ten-eleven translocation methylcytosine dioxygenase (Tet1), notably increased the H2O2-induced apoptosis of cerebellar granule cells [43].
Gene-specific hypermethylation also plays a vital role in the vulnerability to ischemic stroke. For instance, oxygen glucose deprivation (OGD) induced raised methylation in thrombospondin 1 (THBS1) promoter and consequently decreased gene expression in murine cerebral endothelial cells, while reoxygenation led to the opposite effect [44]. Additionally, increased methylation at promoter of angiotensin II type 2 receptor (AT2R) and resultant gene depression in the developing brain augmented the vulnerability of brain hypoxic-ischemic injury in the neonate, which is reversed by 5-aza-dC [45]. Besides, gestational hypoxia causes epigenetic repression of glucocorticoid receptor (GR) gene expression in the developing brain, resulted from the increased DNA methylation, decreased binding of transcription factors early growth response protein (EGR1) and Sp1 to GR gene promoters, therefore, enhances brain vulnerability to hypoxic-ischemic injury in neonatal rats [46]. In addition, clinical study showed that obesity and ischemic stroke modulate the methylation levels of KCNQ1 (potassium channel, voltage gated KQT-like subfamily Q, member 1) and WT1 (Wilms tumor 1) in white blood cells [47], and weight loss intervention program changed the methylation patterns of two stroke-related genes KCNQ1 and WT1 (Wilms tumor 1) in obese stroke patients [48]. Moreover, DNMTs not only catalyze DNA methylation, but also is involved in the removal of amino groups [49]. Taken together, the global and gene-specific methylation following stroke is multifunctional, and influences the susceptibility to brain lesion. Although the protection against cerebral ischemia of DNMT inhibitors which is aiming at the global DNA hypermethylation is promising, and functional study of gene-specific methylation following stroke is increasing, the oxidative-stress-related epigenetic mechanisms remain understood.
HHcy induced DNA methylation in stroke
Methylenetetrahydrofolate reductase (MTHFR) is important in DNA methylation, involving in the formation of methyl group donor S-adenosylmethionine. MTHFR deficiency-induced hyperhomocysteinemia (HHcy) could result in protein and lipoperoxidation, and further endothelial and neuronal degeneration and carotid artery plaques formation that may lead to an increased risk of ischemic stroke [50,51], and the genetic and epigenetic mechanisms are both involved [52-54]. Animals administered a folate/methyl-deficient diet showed global hypermethylation in the brain [55]. And clinical study showed that CpG A is a potential epigenetic marker in mediating serum folate and vitamin B12 to contribute to ischemic stroke [56]. Understanding HHcy-related epigenetics during brain ischemia may help in the discovery of sensitive biomarkers and in developing new therapeutic approaches for stroke.
N-methyl D-aspartate receptors (NMDAR)-mediated oxidative stress is the key pathway to the HHcy-related epigenetic regulation of vascular neural network following stroke. Endothelial cells and neurons contain NMDAR that have a high-affinity binding for Hcy, binding to the glutamate site of NMDAR1 initiates nitric oxide synthase (NOS) and generation of superoxide anions [57-60]. Hcy treatment was shown to increase the levels of DNMT1 and DNMT3a, but decrease DNMT3b levels in mouse brain endothelial cells, inducing mitochondrial toxicity and endothelial dysfunction [61]. In addition, HHcy inhibited the growth of arterial endothelial cells through altering promoter DNA methylation and inducing transcriptional repression of fibroblast growth factor-2 involving G-protein pathway [62]. Similar to endothelial cells, epigenetic alterations including increased levels of SAM and increased DNMT3a and DNMT3b activities were induced by HHcy in vascular smooth muscle cells, resulting in DNA hypermethylation and consequent gene silencing [63]. In contrast to vessel, primary cultures of astrocytes exposed to HHcy leaded to reduce total DNMT activity and a remarkable reduction in protein levels of DNMT 3b, accompanied by global DNA hypomethylation notably [64]. The discrepant pattern of global methylation and DNMTs activity surveyed in these investigations may suggest that the effects of HHcy on genomic DNA methylation are cell specific.
In addition to this nuclear mechanism, mitochondrial epigenetics (mito-epigenetics) are also involved in HHcy-related bone remodeling and skeletal muscle weakness [65, 66]. Mitochondrial DNA methylation has been demonstrated in humans and other mammalians and DNMT activity was present in mitochondria, and mitochondrial DNA was less methylated than nuclear DNA [67]. Recent studies of epigenetics along with the discovery of histone-like proteins in mitochondria indicate exciting new areas for mito-epigenetics. Because mitochondria is pivotal following ischemia-reperfusion injury through generating excessive ROS and hence damaging neural cells, Hcy-related mito-epigenetics during brain ischemia represents a promising area for stroke therapy (Fig. 1).
Figure 1. Schematic representation of the proposed mechanisms of hyperhomocysteinemia (HHcy) triggering epigenetic modifications following stroke.
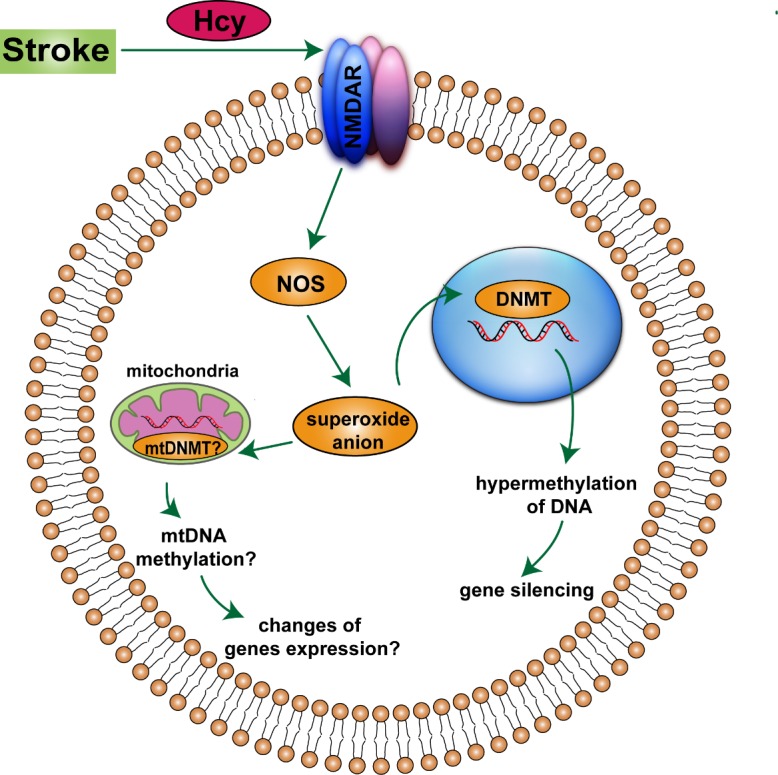
Hcy, homocysteine. NMDAR, N-methyl D-aspartate receptors. NOS, nitric oxide synthase. DNMT, DNA methyltransferase. mtDNMT, mitochondrial DNA methyltransferase.
Histone modification induced by oxidative stress following stroke
Histones wrap around DNA to form the nucleosome, and a variety of histone-modifying enzymes change the DNA conformation, leading to repositioning of nucleosomes, activating or preventing transcription [68]. The modifications are largely reversible and allow dynamic gene expression changes in response to the cellular environment. And modulation of histone-modifying enzymes has produced interesting results in stroke models, which has been reviewed recently [16-26], and we therefore focus on oxidative stress-induced histone modifications in stroke, and the influence of these modifications on gene transcription in the brain.
Histone acetylation modulate oxidative stress following stroke
Histone acetylation/deacetylation is linked to transcriptional activation/depression, and is modified by histone acetyltransferases (HATs) and histone deacetylases (HDACs) respectively [69-72]. Given the clinic application of HDAC inhibitors (HDACi) in cancer treatment [73-80], and the promising result of HDACi in preclinical and clinical studies of stroke [81-86], the reuse of HDACi in stroke clinic should be expedited. Moreover, HDACi increase the acetylation of many non-histone proteins including hormone receptors, chaperone proteins, and transcription factors, thus modifying their activity or function [74]. In this part, we summarize the specific changes in HDAC proteins following stroke, and the function of pan-HDAC inhibition and distinctive HDAC isoforms involved in oxidative stress in cerebral ischemia.
Gene-expression mapping of the HDAC isoforms (classes I, II, and IV) under normal condition and following stroke has demonstrated their distinct regional, cellular and subcellular localizations and discrete substrates [87-89]. Under normal condition, HDACs are located mainly in neurons and mature oligodendrocytes; following stroke, the expression feature of the HDACs was changed [88, 89]. In vivo, HDAC1-2 was decreased in the ischemic core area, but increased in neurons of the penumbra in the subventricular zone and cortex, and in glial cells in the subcortical white matter following 45 min MCAO. HDAC1 was bright and encircled in the capillaries of contralateral tissue. HDAC2 is strongly expressed in the astrocyte end-feet in the hippocampus and cortex [88]. In vitro, HDAC1-3 expression levels were upregulated after 60 min of OGD in all glial cell nuclei and astrocyte processes, with HDAC3 being the most strongly upregulated [88]. Another study identified HDAC3 and HDAC6 as probable regulators of neurotoxicity in ischemic stroke, implying that therapeutic approaches aiming at specific HDAC subtype may be considered [89]. Given the toxicity of pan-HDACi towards a host of CNS cell types and the opposing effects of HDACi on unique cell types [90], identification of HDAC isoforms involved in stroke and of those responsible for specifically mediating the beneficial function of pan-HDAC inhibition is needed to conquer this barrier.
HDACs participate in the progress of oxidative stress following stroke by altering the functions of histone or non-histone proteins through posttranslational deacetylation. Pre-treatment with the HDACi trichostatin A (TSA) was demonstrated to protect against OGD in primary cortical neurons by enhancing histone acetylation in the promoter region of gelsolin, a pivotal mediator of actin-filament assembly-disassembly, in dose- and time-dependent manners [91]. The transcription factors Sp1 and Nrf-2 participate in the antioxidant-responsive function of HDACi. TSA augmented transcription factor Sp1 acetylation, and associated loss of DNA binding and its downstream gene expression of Catalase, MnSOD and p21 waf1/cip1, mitigating the glutamate analog homocysteate-induced oxidative neuronal death in vitro and 3-nitroproprionic acid-caused oxidative neuronal death in vivo [92]. Nrf2 is a critical mediator of antioxidant-responsive genes in stroke, and pharmacologic inhibition of HDAC could not protect Nrf2-deficient mice against cerebral ischemia. Moreover, HDACi reduced expression of the Keap1, induced Keap1/Nrf2 dissociation and Nrf2 nuclear translocation, upregulating proteins downstream of Nrf2, including heme oxygenase 1 (HO-1), glutamate-cysteine ligase catalytic subunit (GCLC), and NAD(P)H:quinone oxidoreductase 1 (NQO1) in neuronal cultures and brain tissue [93]. Resveratrol leads to Nrf-2 protein acetylation thereby providing cell protection against cerebral ischemia through modulation of sirtuin activity, a nicotinamide adenosine dinucleotide-dependent histone deacetylase [94]. These results suggested that Nrf2 activation might be a vital mechanism by which HDACi provides neuroprotection. In addition, the HDACi valproic acid and TSA can inhibit photodynamic-therapy-induced necrosis and apoptosis of satellite glial cells [42]. HDACi can protect against oxidative neuronal death induced by peroxide addition or glutathione depletion [95]. Several hydroxamate-based HDACi can protect neurons from oxidative stress via an HDAC-independent mechanism, involving the in situ formation of hydroxamate-iron complexes that catalyze the decomposition of H2O2 in a manner reminiscent of catalase [96].
Importantly, HDAC subtypes play different roles in oxidative stress following stroke. Ischemia/reperfusion (I/R) reduced phosphorylation at Ser 394 of HDAC2, and weakened the HDAC2-FOXO3a reciprocity in mouse brain tissue. Moreover, H2O2 also reduced the HDAC2-FOXO3a interaction in cerebellar granule neurons, resulting in elevated histone H4K16 acetylation in the promoter region of p21 and upregulated its expression. This study revealed novel epigenetic regulation of FOXO3a-mediated gene expression during oxidative stress-induced neuronal cell death, which could be developed therapeutically [97]. In addition, H2O2 treatment induced translocation of HDAC4 from the cytoplasm into the nucleus in cultured cortical neurons, where it interacted physically with peroxisome proliferator-activated receptor-γ and repressed its transcriptional activity and inhibited its pro-survival activity, thus regulating neuronal death [98]. HDAC5 and HDAC4 were markedly reduced in both cerebral ischemia/reperfusion injury and OGD model, and NADPH oxidase-reduced HDAC4 and HDAC5 accelerates cerebral ischemia injury via increasing the expression and release of high mobility group box-1 protein (HMGB1) [99]. A selective and robust increase of HDAC6 expression associated with homocysteic acid-induced oxidative neuronal injury was demonstrated by real-time polymerase chain reaction, and inhibition of HDAC6 can promote the neuronal survival [100]. In accordance with its cytoplasmic localization, the function of HDAC6 inhibition appears to be transcription-independent. Particularly, the selective inhibition of HDAC6 avoids cell death associated with pan-HDAC inhibition, defining HDAC6 as a latent non-toxic therapeutic target for alleviating CNS injury against oxidative stress-induced neurodegeneration.
Despite numerous studies, the mechanisms responsible for the protection of HDACi remain to be adequately illuminated, while the specific subtypes of HDACs associated with ischemic stroke remain unclear. Investigating the effects of these types of modulation on oxidative stress-induced inhibition of synaptic plasticity in relation to stroke recovery will provide important mechanistic insights. Given that some small-molecule HDACi are currently in use in patients or clinical trials, HDACi represent promising treatment approach for stroke patients.
Histone methylation and demethylation under oxidative stress in stroke
Histone methylation, modified by histone methyltransferases (HMTs), was always considered to be a permanent epigenetic modification [101-103], but the discovery of histone demethylases (HDMs) has changed this perception [104-106]. Although increasing numbers of HMTs and HDMs have been identified, their functions in the experimental stroke remain inadequately understood. In contrast to the neuroprotective effects of DNA methylation and histone acetylation inhibition, the role of histone methylation in transcriptional response following stroke remains intricate. In this section, we summarize the changes and functions of HMTs such as SUV39H1 and G9a, and HDMs such as JmjC-domain-containing histone demethylases (JHDMs) and lysine-specific demethylase 1 (LSD1) following stroke respectively, and the mechanism involved in relation to oxidative stress.
H3K4 HMT activity was recently shown to be decreased in astrocytes from middle-aged female rats compared to adult females of stroke [85]. And enhanced cell survival following ischemia in adult females was correlated with enriched H3K4me3 at the miR-17-20 cluster and VEGFa and subsequent greater VEGF protein expression and miR-20 mRNA expression [85]. However, no methylation difference was detected at the H3K36 position in neonatal hypoxic-ischemic brain injury [107]. A new study showed that inhibition of the repressive H3K9 HMTs SUV39H1 and G9a using either RNA interference or the specific blocker chaetocin improved neuronal survival in an OGD model, partly mediated by the increased H3K9ac in promoter regions of brain-derived neurotrophic factor and its active transcription [108].
The JHDMs and LSD1 are needed to demethylate histone H3 at Lys4 or Lys 9, two specific tags for epigenetic transcriptional activation. It was proved that JmjC domain uses Fe(II) and α-ketoglutarate as cofactors in an oxidative demethylation reaction via hydro-xymethyl lysine, indicated by that excessive Fe(II) or ascorbate can rescue H2O2-mediated impairment of histone demethylase activity [109]. The members of this family functions in brain development, however, their effect in stroke remains to be illuminated. In addition, the expression of LSD1 changes temporally and spatially following brain ischemia and reperfusion injury. The numbers of LSD1-positive cells in the DG and CA1 regions significantly increased as soon as 1 h post-ischemia, peaked at 6 h and day 3 respectively, suggesting that LSD1 may be involved in neural regeneration following stroke [110]. And it was proved that LSD1 plays an important role in silencing neuronal-specific genes in non-neuronal cells [111], also promoted long-term memory [112]. Importantly, LSD1 is a flavin-dependent amine oxidase, which could stimulate androgen-receptor-dependent transcription coverting oxygen to H2O2 [113, 114]. Given the involvement of JHDMs and LSD1 in oxidative response, future studies are needed to explore their significance in regulation of oxidative stress following stroke.
Interestingly, LSD1 acts as a component of various transcriptional co-repressor complexes rather than a free-functioning enzyme in vivo [115-119]. LSD1 participates in HDAC1/2-mediated deacetylation of H3K9Ac which is thought to precede the binding of CoREST, followed by LSD1-mediated H3K4me1/2 demethylation [120]. In addition, HDAC4 plays a central role in the rapid modification of histone methylation in response to variations in cardiac load in patients with heart failure [121]. The existence of crosstalk between histone modifications suggest that further investigations is required to clarify the function of LSD1 inhibition on histone methylation and the link between methylation changes and acetylation. However, the role of LSD1 remains largely unexplored. In addition, the potentially reversible modes of LSD1 inhibition that may alter LSD1 through mechanisms other than competitive inhibition of substrates are required to be explored, and specific molecular targets of LSD1 upon oxidative stress and how they are linked to the regulation of transcription are also needed to be identified.
Histone phosphorylation-mediated neural necrosis upon oxidative stress in stroke
In addition to the methylation and acetylation of histone lysine residues, serine and threonine phosphorylation also occur during cerebral ischemia [122-124]. Phosphorylated Histone 2AX (γ-H2AX) occurs under oxidative stress and accumulates with progressive injury following stroke. The overactive glutamate receptor (GluR) after ischemia increases oxidative stress and evoked γ-H2AX in neurons, which was alleviated by pretreatment with the antioxidant. The generation of γ-H2AX following GluR activation corresponded to the increases observed following exposure to H2O2. These data suggest that insults not necessarily resulting in neuronal death may induce the DNA-damage-evoked chromatin modification, γ-H2AX, implicating histone alterations in determining neuronal vulnerability following neurological insults [122]. Glutamate-induced calcium influx in neurons activates the ERK1/2 and its downstream of MSK1/2/JIL-1, which increases phosphorylation of histone H3 at serine 28 (pH3S28), displace polycomb repressive complex 1 from chromatin, then activates Trithorax, leading to increased H3K4me3, resulting in cell necrosis by an unknown mechanism [123]. Oxidative stress induced enhanced pH3 at serine 10 (pH3S10) in mouse brain endothelial cells after OGD, resulting in cell death and pH3 interacted with IKKa in the nucleus. And IKKa siRNA treatment significantly reduced cell death and pH3 level after OGD, suggesting the crucial function of pIKKa and subsequent phosphorylation of histone H3 in response to oxidative stress in cell death after cerebral ischemia [124]. Together, these results indicated that phosphorylation of histone-mediated chromatin-modifying cascade was involved in neuronal necrosis following cerebral ischemia and oxidative stress. However, the mechanism of phosphorylation histone related with oxidative stress following stroke is rarely studied and need further elucidation.
MiRNA-mediated regulation of oxidative stress following stroke
MiRNAs are approximately 22 nucleotides small RNA molecules, which negatively regulate the expression of the target genes post-transcriptionally. MiRNAs are involved in stroke risk factors including atherosclerosis, hypertension, atrial fibrillation, diabetes, and dyslipidemia. The function of miRNAs in the pathophysiology of stroke become one subject of recent researches [125], including miRNAs regulating apoptosis and autophagy of neurons, astrocytes, and cerebral vascular endothelial cells after stroke [126-130], though studies of their mechanisms in relation to oxidative stress are limited. In this section, we examine the relationships between miRNAs, oxidative stress, and epigenetic machinery.
The effect of oxidative stress on miRNA expression profile in mouse primary hippocampal neurons was studied. MiR-708 and miR-135b were significantly increased upon H2O2 stimulation, and their targets were related with DNA recombination, protein autophosphorylation, protein ubiquitination, and neurons development [131]. Cell death was reduced when miR-181a levels were reduced, and increased when miR-181a levels increased in N2a cells upon serum deprivation (SD) and oxidative stress; protection was associated with increased Bcl-2 protein [132]. Our group showed that miR-424 reduced oxidative stress in the cortex and protected against transient cerebral ischemia-reperfusion injury. MiR-424 treatment abrogated H2O2-induced lactate dehydrogenase leakage and increased manganese superoxide dismutase activity in neuronal cultures, and its protective effects against oxidative stress were reversed by Nrf-2 knockdown and superoxide dismutase (SOD) inhibition [133]. We also demonstrated that miR-23a-3p dose-dependently reduced H2O2-induced generation of nitric oxide and 3-nitrotyrosine, thus reversing the decreased activities of total SOD and manganese SOD in N2a neuroblastoma cells. Furthermore, miR-23a-3p suppressed oxidative stress and relieved cerebral ischemia-reperfusion injury [134]. In addition, vagus nerve stimulation was neuroprotective against cerebral ischemia-reperfusion injury and modulates redox status through activating neuronal and astrocyte α7n acetylcholine receptor and possibly associated with increased miR-210 expression [135].
Oxidative stress-induced endothelial dysfunction plays a pivotal role in ischemia-reperfusion injury. Recent evidence indicates that endothelial progenitor-cell-derived microvesicles can promote angiogenesis of endothelial cells. A hypoxia/reoxygenation model of human brain microvascular endothelial cells was produced by 6 h hypoxia and 24 h reoxygenation. Functionally, serum deprivation had beneficial effects on hypoxia/reoxygenation-exposed endothelial cells, whereas serum-deprived medium containing tumor necrosis factor-α (apoptotic stress) had detrimental effects. These results suggest that the serum-deprived and apoptotic-stress endothelial progenitor-cell-derived microvesicles were functionally different in terms of apoptosis and dysfunction via their RNAs, such as miR126 associated with ROS production and the phosphoinositide 3-kinase/endothelial NOS/nitric oxide pathway [136]. MiR-204 augmenting the susceptibility of RSC96 Schwann cells to H2O2-induced apoptosis through down-regulating the expression of neuritin, which act as a neurotrophin and play crucial role in plasticity and repair following nervous system injury. Therefore, low-level expression of miR-204 may create a suitable microenvironment for the nerves repair by lightening the transcriptional inhibition of neuritin transcription [137]. MiRNAs are currently undergoing evaluation for feasible clinical use as biomarkers for neurological diseases [138].
In another study, exosomes were used to deliver therapeutic mRNA/protein to treat cancer [139]. However, the delivery of therapeutic miRNAs for stroke treatment remains largely unexplored.
Conclusions
Clinical and preclinical investigations suggest a critical relationship between oxidative stress and epigenetic mechanisms following stroke. Peroxide could induce global and gene-specific DNA methylation, and influences the susceptibility to brain ischemia. HDAC subtypes play different roles in oxidative stress following stroke by altering the functions of histone or non-histone proteins. In contrast, the role of histone methylation in transcriptional response following stroke remains intricate, but JHDMs and LSD1 are important in oxidative response, future studies are needed to explore their significance in stroke. Histone phosphorylation mainly mediated neural necrosis upon oxidative stress in stroke. MiRNAs are changed upon oxidative stress in the vascular neural networks and may connect the ischemic brain with other organs. However, although compelling, these findings raise new questions. The issues of whether epigenetic remodeling promotes susceptibility to oxidative stress insults following ischemia-reperfusion, and if epigenetic modifications occur in response to oxidative stress insults following ischemia-reperfusion remain to be answered. Given the redox state following stroke could influence the effect of neuroprotective drugs, the investigations of the function of oxidative stress on epigenetic modifications are critical for formulating effective individualized therapeutic approaches for stroke patients.
Acknowledgments
This work was supported by Projects of Beijing Nova Program (Z151100000315065) and Chinese Natural Science Foundation grants (81471340, 81571280, and 81325007).
Footnotes
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- [1].Melo A, Monteiro L, Lima RM, de Oliveira DM, de Cerqueira MD, El-Bachá RS (2011). Oxidative stress in neurodegenerative diseases: mechanisms and therapeutic perspectives. Oxid Med Cell Longev, 2011: 467180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Baxter P, Chen Y, Xu Y, Swanson RA (2014). Mitochondrial dysfunction induced by nuclear poly (ADP-ribose) polymerase-1: a treatable cause of cell death in stroke. Transl Stroke Res, 5: 136-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Holmes S, Abbassi B, Su C, Singh M, Cunningham RL (2013). Oxidative stress defines the neuroprotective or neurotoxic properties of androgens in immortalized female rat dopaminergic neuronal cells. Endocrinology, 154: 4281-4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lambeth JD, Neish AS (2014). Nox enzymes and new thinking on reactive oxygen: a double-edged sword revisited. Annu Rev Pathol, 9: 119-145. [DOI] [PubMed] [Google Scholar]
- [5].Keyer K, Gort AS, Imlay JA (1995). Superoxide and the production of oxidative DNA damage. J Bacteriol, 177: 6782-6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Thomas SR, Witting PK, Drummond GR (2008). Redox control of endothelial function and dysfunction: molecular mechanisms and therapeutic opportunities. Antioxid Redox Signal, 10: 1713-1766. [DOI] [PubMed] [Google Scholar]
- [7].Nishikawa T, Edelstein D, Du XL, Yamagishi SI, Matsumura T, Kaneda Y, et al. (2000). Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature, 404: 787-790. [DOI] [PubMed] [Google Scholar]
- [8].Hall ED, Braughler JM (1993). Free radicals in CNS injury. Res Publ Assoc Res Nerv Ment Dis, 71: 81-105. [PubMed] [Google Scholar]
- [9].Zaleska MM, Floyd RA (1985). Regional lipid peroxidation in rat brain in vitro: possible role of endogenous iron. Neurochem Res, 10: 397-410. [DOI] [PubMed] [Google Scholar]
- [10].Wu Q, Ni X (2015). ROS-mediated DNA methylation pattern alterations in carcinogenesis. Curr Drug Targets, 16: 13-19. [DOI] [PubMed] [Google Scholar]
- [11].Afanas’ev I (2013). New nucleophilic mechanisms of ros-dependent epigenetic modifications: comparison of aging and cancer. Aging Dis, 5: 52-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cencioni C, Spallotta F, Martelli F, Valente S, Mai A, Zeiher AM, et al. (2013). Oxidative stress and epigenetic regulation in ageing and age-related diseases. Int J Mol Sci, 14: 17643-17663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Monks TJ, Xie R, Tikoo K, Lau SS (2006). Ros-induced histone modifications and their role in cell survival and cell death. Drug Metab Rev, 38: 755-767. [DOI] [PubMed] [Google Scholar]
- [14].Escobar J, Pereda J, López-Rodas G, Sastre J (2012). Redox signaling and histone acetylation in acute pancreatitis. Free Radic Biol Med, 52: 819-837. [DOI] [PubMed] [Google Scholar]
- [15].Podrini C, Borghesan M, Greco A, Pazienza V, Mazzoccoli G, Vinciguerra M (2013). Redox homeostasis and epigenetics in non-alcoholic fatty liver disease (NAFLD). Curr Pharm Des, 19: 2737-2746. [DOI] [PubMed] [Google Scholar]
- [16].Qureshi IA, Mehler MF (2010). Emerging role of epigenetics in stroke: part 1: DNA methylation and chromatin modifications. Arch Neurol, 67: 1316-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Qureshi IA, Mehler MF (2010). The emerging role of epigenetics in stroke: II. RNA regulatory circuitry. Arch Neurol, 67: 1435-1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Qureshi IA, Mehler MF (2010).The emerging role of epigenetics in stroke: III. Neural stem cell biology and regenerative medicine. Arch Neurol, 68: 294-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kalani A, Kamat PK, Tyagi SC, Tyagi N (2013). Synergy of homocysteine, microRNA, and epigenetics: a novel therapeutic approach for stroke. Mol Neurobiol, 48: 157-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hwang JY, Aromolaran KA, Zukin RS (2013). Epigenetic mechanisms in stroke and epilepsy. Neuropsychopharmacology, 38: 167-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Schweizer S, Meisel A, Märschenz S (2013). Epigenetic mechanisms in cerebral ischemia. J Cereb Blood Flow Metab, 33: 1335-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Elder J, Cortes M, Rykman A, Hill J, Karuppaqounder S, Edwards D, Ratan RR (2013). The epigenetics of stroke recovery and rehabilitation: from polycomb to histonedeacetylases. Neurotherapeutics, 10: 808-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Thompson JW, Dave KR, Young JI, Perez-Pinzon MA (2013). Ischemic preconditioning alters the epigenetic profile of the brain from ischemic intolerance to ischemic tolerance. Neurotherapeutics, 10: 789-797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Krupinski J, Slevin M (2013). Emerging molecular targets for brain repair after stroke. Stroke Res Treat, 2013: 473416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ma Q, Zhang L (2015). Epigenetic programming of hypoxic-ischemic encephalopathy in response to fetal hypoxia. Prog Neurobiol 124, 28-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Felling RJ, Song H (2015). Epigenetic mechanisms of neuroplasticity and the implications for stroke recovery. Exp Neurol, 268: 37-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Moore LD, Le T, Fan G (2013). DNA Methylation and Its Basic Function. Neuropsychopharmacology, 38: 23-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kebede AF, Schneider R, Daujat S (2015). Novel types and sites of histone modifications emerge as players in the transcriptional regulation contest. FEBS J, 282: 1658-1674. [DOI] [PubMed] [Google Scholar]
- [29].Reinhart B, Chaillet JR (2005). Genomic imprinting: cis-acting sequences and regional control. Int Rev Cytol, 243: 173-213. [DOI] [PubMed] [Google Scholar]
- [30].Cirio MC, Ratnam S, Ding F, Reinhart B, Navara C, Chaillet JR (2008). Preimplantation expression of the somatic form of Dnmt1 suggests a role in the inheritance of genomic imprints. BMC Dev Biol, 8: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Du Q, Luu PL, Stirzaker C, Clark SJ (2015). Methyl-CpG-binding domain proteins: readers of the epigenome. Epigenomics, 2015 Apr 30: 1-23. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- [32].Chia N, Wang L, Lu X, Senut MC, Brenner C, Ruden DM (2011). Hypothesis: environmental regulation of 5-hydroxymethylcytosine by oxidative stress. Epigenetics, 6: 853-856. [DOI] [PubMed] [Google Scholar]
- [33].Valinluck V, Tsai HH, Rogstad DK, Burdzy A, Bird A, Sowers LC (2004). Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res, 32: 4100-4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Valinluck V, Sowers LC (2007). Endogenous Cytosine Damage Products Alter the Site Selectivity of Human DNA Maintenance Methyltransferase DNMT1. Cancer Res, 67: 946-950. [DOI] [PubMed] [Google Scholar]
- [35].Miao Z, He Y, Xin N, Sun M, Chen L, Lin L, et al. (2015). Altering 5-hydroxymethylcytosine modification impacts ischemic brain injury. Hum Mol Genet, 24:5855-5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Valinluck V, Sowers LC (2007). Inflammation-mediated cytosine damage: a mechanistic link between inflammation and the epigenetic alterations in human cancers. Cancer Res, 67: 5583-5586. [DOI] [PubMed] [Google Scholar]
- [37].Lao VV, Herring JL, Kim CH, Darwanto A, Soto U, Sowers LC (2009). Incorporation of 5-chlorocytosine into mammalian DNA results in heritable gene silencing and altered cytosine methylation patterns. Carcinogenesis, 30: 886-893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Endres M, Meisel A, Biniszkiewicz D, Namura S, Prass K, Ruscher K, et al. (2000). DNA methyltransferase contributes to delayed ischemic brain injury. J Neurosci, 20: 3175-3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Endres M, Fan G, Meisel A, Dirnagl U, Jaenisch R (2001). Effects of cerebral ischemia in mice lacking DNA methyltransferase 1 in post-mitotic neurons. Neuroreport, 12: 3763-3766. [DOI] [PubMed] [Google Scholar]
- [40].Jung BP, Zhang G, Ho W, Francis J, Eubanks J H (2002). Transient forebrain ischemia alters the mRNA expression of methyl DNA-binding factors in the adult rat hippocampus. Neuroscience, 115: 515-524. [DOI] [PubMed] [Google Scholar]
- [41].Gu X, Sun J, Li S, Wu X, Li L (2013). Oxidative stress induces DNA demethylation and histone acetylation in SH-SY5Y cells: potential epigenetic mechanisms in gene transcription in Aβ production. Neurobiol Aging, 34: 1069-1079. [DOI] [PubMed] [Google Scholar]
- [42].Sharifulina SA, Komandirov MA, Uzdensky AB (2014). Epigenetic regulation of death of crayfish glial cells but not neurons induced by photodynamic impact. Brain Res Bull, 102: 15-21. [DOI] [PubMed] [Google Scholar]
- [43].Xin YJ, Yuan B, Yu B, Wang YQ, Wu JJ, Zhou WH, et al. (2015). Tet1-mediated DNA demethylation regulates neuronal cell death induced by oxidative stress. Sci Rep, 5: 7645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hu CJ, Chen SD, Yang DI, Lin TN, Chen CM, Huang TH, et al. (2006). Promoter region methylation and reduced expression of thrombospondin-1 after oxygen-glucose deprivation in murine cerebral endothelial cells. J Cereb Blood Flow Metab, 26: 1519-1526. [DOI] [PubMed] [Google Scholar]
- [45].Li Y, Xiao D, Yang S, Zhang L (2013). Promoter methylation represses AT2R gene and increases brain hypoxic-ischemic injury in neonatal rats. Neurobiol Dis, 60: 32-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gonzalez-Rodriguez PJ, Xiong F, Li Y, Zhou J, Zhang L (2014). Fetal hypoxia increases vulnerability of hypoxic-ischemic brain injury in neonatal rats: role of glucocorticoid receptors. Neurobiol Dis, 65:172-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gómez-Úriz AM, Milagro FI, Mansego ML, Cordero P, Abete I, De Arce A, et al. (2015). Obesity and ischemic stroke modulate the methylation levels of KCNQ1 in white blood cells. Hum Mol Genet, 24:1432-1440. [DOI] [PubMed] [Google Scholar]
- [48].Abete I, Gomez-Uriz AM, Mansego ML, De Arce A, Goyenechea E, Blazquez V, et al. (2015). Epigenetic Changes in the Methylation Patterns of KCNQ1 and WT1 after a Weight Loss Intervention Program in Obese Stroke Patients. Curr Neurovasc Res, 12:321-333. [DOI] [PubMed] [Google Scholar]
- [49].Van der Wijst MG, Venkiteswaran M, Chen H, Xu GL, Plösch T, Rots MG (2015). Local chromatin microenvironment determines DNMT activity: from DNA methyltransferase to DNA demethylase or DNA dehydroxymethylase. Epigenetics, 10: 671-676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Casas JP, Hingorani AD, Bautista LE, Sharma P (2004). Meta-analysis of genetic studies in ischemic stroke: thirty-two genes involving approximately 18,000 cases and 58,000 controls. Arch Neurol, 61: 1652-1661. [DOI] [PubMed] [Google Scholar]
- [51].Kelly PJ, Rosand J, Kistler JP, Shih VE, Silvera S, Plomaritoqlou A, et al. (2002). Homocysteine, MTHFR 677C-->T polymorphism, and risk of ischemic stroke: results of a meta-analysis. Neurology, 59: 529-536. [DOI] [PubMed] [Google Scholar]
- [52].Austin RC, Lentz SR, Werstuck GH (2004). Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ, 11: S56-64. [DOI] [PubMed] [Google Scholar]
- [53].Jeon SB, Kang DW, Kim JS, Kwon SU (2014). Homocysteine, small-vessel disease, and atherosclerosis: an MRI study of 825 stroke patients. Neurology, 83: 695-701. [DOI] [PubMed] [Google Scholar]
- [54].Lehotsky J, Petras M, Kovalska M, Tothova B, Drgova A, Kaplan P (2015). Mechanisms Involved in the Ischemic Tolerance in Brain: Effect of the Homocysteine. Cell Mol Neurobiol, 35: 7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bagnyukova TV, Powell CL, Pavliv O, Tryndyak VP, Pogribny IP (2008). Induction of oxidative stress and DNA damage in rat brain by a folate/methyl-deficient diet. Brain Res, 1237: 44-51. [DOI] [PubMed] [Google Scholar]
- [56].Wei LK, Sutherland H, Au A, Camilleri E, Haupt LM, Gan SH, et al. (2015). A potential epigenetic marker mediating serum folate and vitamin B12 levels contributes to the risk of ischemic stroke. Biomed Res Int, 2015: 167976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Qureshi I, Chen H, Brown AT, Fitzgerald R, Zhang X, Breckenridge J, et al. (2005). Homocysteine-induced vascular dysregulation is mediated by the NMDA receptor. Vasc Med, 10: 215-223. [DOI] [PubMed] [Google Scholar]
- [58].Jara-Prado A, Ortega-Vazquez A, Martinez-Ruano L, Rios C, Santamaria A (2003). Homocysteine-induced brain lipid peroxidation: effects of NMDA receptor blockade, antioxidant treatment, and nitric oxide synthase inhibition. Neurotox Res, 5: 237-243. [DOI] [PubMed] [Google Scholar]
- [59].Moshal KS, Singh M, Sen U, Rosenberger DS, Henderson B, Tyagi N, et al. (2006). Homocysteine-mediated activation and mitochondrial translocation of calpain regulates MMP-9 in MVEC. Am J Physiol Heart Circ Physiol, 291: H2825-H2835. [DOI] [PubMed] [Google Scholar]
- [60].Doronzo G, Russo I, Del Mese P, Viretto M, Mattiello L, Trovati M, et al. (2010). Role of NMDA receptor in homocysteine-induced activation of mitogen-activated protein kinase and phosphatidyl inositol 3-kinase pathways in cultured human vascular smooth muscle cells. Thromb Res, 125: e23-32. [DOI] [PubMed] [Google Scholar]
- [61].Kamat P K, Kalani A, Tyagi SC, Tyagi N (2015). Hydrogen Sulfide Epigenetically Attenuates Homocysteine-Induced Mitochondrial Toxicity Mediated through NMDA Receptor in Mouse Brain Endothelial (bEnd3) Cells. J Cell Physiol, 230: 378-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chang PY, Lu SC, Lee CM, Chen YJ, Dugan TA, Huang WH, et al. (2008). Homocysteine inhibits arterial endothelial cell growth through transcriptional downregulation of fibroblast growth factor-2 involving G protein and DNA methylation. Circ Res 102, 933-941. [DOI] [PubMed] [Google Scholar]
- [63].Yideng J, Jianzhong Z, Ying H, Juan S, Jinge Z, Shenglan W, et al. (2007). Homocysteine-mediated expression of SAHH, DNMTs, MBD2, and DNA hypomethylation potential pathogenic mechanism in VSMCs. DNA Cell Biol, 26: 603-611. [DOI] [PubMed] [Google Scholar]
- [64].Jin Y, Amaral A, McCann A, Brennan L (2011). Homocysteine levels impact directly on epigenetic reprogramming in astrocytes. Neurochem Int, 58: 833-838. [DOI] [PubMed] [Google Scholar]
- [65].Kalani A, Kamat PK, Voor MJ, Tyagi SC and Tyagi N (2014). Mitochondrial epigenetics in bone remodeling during hyperhomocysteinemia. Mol Cell Biochem, 395: 89-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Veeranki S, Winchester LJ, Tyagi SC (2015). Hyperhomocysteinemia associated skeletal muscle weakness involves mitochondrial dysfunction and epigenetic modifications. Biochim Biophys Acta, 1852: 732-741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Manev H, Dzitoyeva S (2013). Progress in mitochondrial epigenetics. Biomol Concepts, 4: 381-389. [DOI] [PubMed] [Google Scholar]
- [68].Kouzarides T. Chromatin modifications and their function (2007). Cell, 128: 693-705 [DOI] [PubMed] [Google Scholar]
- [69].Gong F, Miller KM (2013). Mammalian DNA repair: HATs and HDACs make their mark through histone acetylation. Mutat Res, 750: 23-30. [DOI] [PubMed] [Google Scholar]
- [70].Pham TX, Lee J (2012). Dietary regulation of histone acetylases and deacetylases for the prevention of metabolic diseases. Nutrients, 4: 1868-1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Zhou Y, Peng J, Jiang S (2014). Role of histone acetyltransferases and histone deacetylases in adipocyte differentiation and adipogenesis. Eur J Cell Biol, 93: 170-177. [DOI] [PubMed] [Google Scholar]
- [72].Haery L, Thompson RC, Gilmore TD (2015). Histone acetyltransferases and histone deacetylases in B- and T-cell development, physiology and malignancy. Genes Cancer, 6: 184-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kazantsev AG, Thompson LM (2008). Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov, 7: 854-868. [DOI] [PubMed] [Google Scholar]
- [74].Zwergel C, Valente S, Jacob C, Mai A (2015). Emerging approaches for histone deacetylase inhibitor drug discovery. Expert Opin Drug Discov, 10: 599-613. [DOI] [PubMed] [Google Scholar]
- [75].Rasheed WK, Johnstone RW, Prince HM (2007). Histone deacetylase inhibitors in cancer therapy. Expert Opin Investig Drugs, 16: 659-678. [DOI] [PubMed] [Google Scholar]
- [76].Lu X, Wang L, Yu C, Yu D, Yu G (2015). Histone Acetylation Modifiers in the Pathogenesis of Alzheimer’s Disease. Front Cell Neurosci, 9: 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Geyer HL, Mesa RA (2015). Emerging drugs for the treatment of myelofibrosis. Expert Opin Emerg Drugs, 2015. July 8:1-16. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- [78].Mimura N, Hideshima T, Anderson KC (2015). Novel therapeutic strategies for multiple myeloma. Exp Hematol, 43: 732-741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Neele AE, Van den Bossche J, Hoeksema MA, de Winther MP (2015). Epigenetic pathways in macrophages emerge as novel targets in atherosclerosis. Eur J Pharmacol, 763:79-89. [DOI] [PubMed] [Google Scholar]
- [80].Aune SE, Herr DJ, Kutz CJ, Menick DR (2015). Histone Deacetylases Exert Class-Specific Roles in Conditioning the Brain and Heart Against Acute Ischemic Injury. Front Neurol, 6: 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].LangleyB, Brochier C, Rivieccio MA (2009). Targeting histone deacetylases as a multifaceted approach to treat the diverse outcomes of stroke. Stroke, 40: 2899-905. [DOI] [PubMed] [Google Scholar]
- [82].Fessler EB, Chibane FL, Wang Z, Chuang DM (2013). Potential roles of HDAC inhibitors in mitigating ischemia-induced brain damage and facilitating endogenous regeneration and recovery. Curr Pharm Des, 19: 5105-5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Baltan S, Morrison RS, Murphy SP (2013). Novel protective effects of histone deacetylase inhibition on stroke and white matter ischemic injury. Neurotherapeutics, 10: 798-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Wang ZY, Qin W Yi F (2015). Targeting histone deacetylases: perspectives for epigenetic-based therapy in cardio-cerebrovascular disease. J Geriatr Cardiol, 12: 153-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Chisholm NC, Henderson ML, Selvamani A, Park MJ, Dindot S, Miranda RC, et al. (2015). Histone methylation patterns in astrocytes are influenced by age following ischemia. Epigenetics, 10: 142-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Dregan A, Charlton J, Wolfe CD, Gulliford MC, Markus HS (2014). Is sodium valproate, an HDAC inhibitor, associated with reduced risk of stroke and myocardial infarction? A nested case-control study. Pharmacoepidemiol Drug Saf, 23: 759-767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Broide RS, Redwine JM, Aftahi N, Young W, Bloom FE, Winrow CJ (2007). Distribution of histone deacetylases 1-11 in the rat brain. J Mol Neurosci, 31: 47-58. [DOI] [PubMed] [Google Scholar]
- [88].Baltan S, Bachleda A, Morrison RS and Murphy SP (2011). Expression of histone deacetylases in cellular compartments of the mouse brain and the effects of ischemia. Transl Stroke Res, 2: 411-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Chen YT, Zang XF, Pan J, Zhu XL, Chen F, Chen ZB, et al. (2012). Expression patterns of histone deacetylases in experimental stroke and potential targets for neuroprotection. Clin Exp Pharmacol Physiol, 39: 751-758. [DOI] [PubMed] [Google Scholar]
- [90].Dietz KC, Casaccia P. HDAC inhibitors and neurodegeneration: at the edge between protection and damage (2010). Pharmacol Res, 62: 11-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Meisel A, Harms C, Yildirim F, Bösel J, Kronenberg G, Harms U, et al. (2006). Inhibition of histone deacetylation protects wild-type but not gelsolin-deficient neurons from oxygen/glucose deprivation. J Neurochem, 98: 1019-1031. [DOI] [PubMed] [Google Scholar]
- [92].Ryu H, Lee J, Olofsson BA, Mwidau A, Dedeoglu A, Escudero M, et al. (2014). Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Brain Res Bull, 102: 15-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Wang B, Zhu X, Kim Y, Li J, Huang S, Saleem S, et al. (2012). Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radic Biol Med, 52: 928-936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Raval AP1, Dave KR, Pérez-Pinzón MA (2006). Resveratrol mimics ischemic preconditioning in the brain. J Cereb Blood Flow Metab, 26: 1141-1147. [DOI] [PubMed] [Google Scholar]
- [95].Langley B, D’Annibale MA, Suh K, Ayoub I, Tolhurst A, Bastan B, et al. (2008). Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(waf1/cip1) in cell cycle-independent neuroprotection. J Neurosci 28, 163-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Olson DE, Sleiman SF, Bourassa MW, Wagner FF, Gale JP, Zhang YL, et al. (2015). Hydroxamate-Based Histone Deacetylase Inhibitors Can Protect Neurons from Oxidative Stress via a Histone Deacetylase-Independent Catalase-Like Mechanism. Chem Biol, 22: 439-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Peng S, Zhao S, Yan F, Cheng J, Huang L, Chen H, et al. (2015). HDAC2 Selectively Regulates FOXO3a-Mediated Gene Transcription during Oxidative Stress-Induced Neuronal Cell Death. J Neurosci, 35: 1250-1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Yang Y, Qin X, Liu S, Li J, Zhu X, Gao T, et al. (2011). Peroxisome proliferator-activated receptor γ is inhibited by histone deacetylase 4 in cortical neurons under oxidative stress. J Neurochem, 118: 429-439. [DOI] [PubMed] [Google Scholar]
- [99].He M, Zhang B, Wei X, Wang Z, Fan B, Du P, et al. (2013). HDAC4/5-HMGB1 signalling mediated by NADPH oxidase activity contributes to cerebral ischaemia/reperfusion injury. J Cell Mol Med, 17: 531-542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Rivieccio MA, Brochier C, Willis DE, Walker BA, D’Annibale MA, McLaughlin K, et al. (2009). HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc Natl Acad Sci USA, 106: 19599-19604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Shi Y, Whetstine JR (2007). Dynamic regulation of histone lysine methylation by demethylases. Mol Cell, 25: 1-14. [DOI] [PubMed] [Google Scholar]
- [102].Martin C, Zhang Y (2005). The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol, 6: 838-849. [DOI] [PubMed] [Google Scholar]
- [103].Jenuwein T, Allis CD (2001). Translating the histone code. Science, 293: 1074-1080. [DOI] [PubMed] [Google Scholar]
- [104].Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell, 119: 941-953. [DOI] [PubMed] [Google Scholar]
- [105].Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, et al. (2006). Histone demethylation by a family of JmjC domain-containing proteins. Nature, 439: 811-816. [DOI] [PubMed] [Google Scholar]
- [106].Bannister AJ, Kouzarides T (2005). Reversing histone methylation. Nature, 436: 1103-1106. [DOI] [PubMed] [Google Scholar]
- [107].Kumral A, Tuzun F, Tugyan K, Ozbal S, Yılmaz O, Yesilirmak CD, et al. (2013). Role of epigenetic regulatory mechanisms in neonatal hypoxic-ischemic brain injury. Early Hum Dev, 89: 165-173. [DOI] [PubMed] [Google Scholar]
- [108].Schweizer S, Harms C, Lerch H, Flynn J, Hecht J, Yildirim F, et al. (2015). Inhibition of histone methyltransferases SUV39H1 and G9a leads to neuroprotection in an invitro model of cerebral ischemia. J Cereb Blood Flow Metab, 35:1640-1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Niu Y, DesMarais TL, Tong Z, Yao Y, Costa M (2015). Oxidative stress alters global histone modification and DNA methylation. Free Radic Biol Med, 82:22-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Zhang YZ, Zhang QH, Ye H, Zhang Y, Luo YM, Ji XM, et al. (2010). Distribution of lysine-specific demethylase 1 in the brain of rat and its response in transient global cerebralischemia. Neurosci Res, 68: 66-72. [DOI] [PubMed] [Google Scholar]
- [111].Ceballos-Chávez M, Rivero S, García-Gutiérrez P, Rodríguez-Paredes M, García-Domínguez M, Bhattacharya S, et al. (2012). Control of neuronal differentiation by sumoylation of BRAF35, a subunit of the LSD1-CoREST histone demethylase complex. Proc Natl Acad Sci U S A, 21:8085-8090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Neelamegam R, Ricq EL, Malvaez M, Patnaik D, Norton S, Carlin SM, et al. (2012). Brain-Penetrant LSD1 Inhibitors Can Block Memory Consolidation. ACS Chem Neurosci, 3: 120-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Forneris F, Binda C, Vanoni MA, Battaglioli E, Mattevi A (2005). Human histone demethylase LSD1 reads the histone code. J Biol Chem, 50:41360-41365. [DOI] [PubMed] [Google Scholar]
- [114].Forneris F, Binda C, Battaglioli E, Mattevi A (2008). LSD1: oxidative chemistry for multifaceted functions in chromatin regulation. Trends Biochem Sci, 4:181-189. [DOI] [PubMed] [Google Scholar]
- [115].Lee MG, Wynder C, Cooch N, Shiekhattar R (2005). An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature, 437: 432-435. [DOI] [PubMed] [Google Scholar]
- [116].Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y (2005). Regulation of LSD1 histone demethylase activity by its associated factors. Mol. Cell, 19: 857-864. [DOI] [PubMed] [Google Scholar]
- [117].Humphrey GW, Wang Y, Russanova VR, Hirai T, Qin J, Nakatani Y, et al. (2001). Stable histone deacetylase complexes distinguished by the presence of SANT domain proteins CoREST/kiaa0071 and Mta-L1. J Biol Chem, 276: 6817-6824. [DOI] [PubMed] [Google Scholar]
- [118].Shi Y, Sawada J, Sui G, Affar el B, Whetstine JR, Lan F, et al. (2003). Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature, 422: 735-738. [DOI] [PubMed] [Google Scholar]
- [119].You A, Tong JK, Grozinger CM, Schreiber SL (2001). CoREST is an integral component of the CoREST-human histone deacetylase complex. Proc Natl Acad Sci USA, 98: 1454-1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Lan F, Nottke AC, Shi Y (2008). Mechanisms involved in the regulation of histone lysine demethylases. Curr Opin Chem Biol, 20: 316-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Hohl M, Wagner M, Reil JC, Müller SA, Tauchnitz M, Zimmer AM, et al. (2013). HDAC4 controls histone methylation in response to elevated cardiac load. J Clin Invest, 123: 1359-1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Crowe SL, Movsesyan VA, Jorgensen TJ, Kondratyev A (2006). Rapid phosphorylation of histone H2A.X following ionotropic glutamate receptor activation. Eur J Neurosci, 23: 2351-2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Liu K, Ding L, Li Y, Yang H, Zhao C, Lei Y, et al. (2014). Neuronal necrosis is regulated by a conserved chromatin-modifying cascade. Proc Natl Acad Sci USA, 111: 13960-13965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Song YS, Kim MS, Kim HA, Jung BI, Yang J, Narasimhan P, et al. (2010). Oxidative stress increases phosphorylation of IjB kinase-a by enhancing NF-jB-inducing kinase after transient focal cerebral ischemia. J Cereb Blood Flow Metab, 30: 1265-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Koutsis G, Siasos G, Spengos K (2013). The emerging role of microRNA in stroke. Curr Top Med Chem, 13: 1573-1588. [DOI] [PubMed] [Google Scholar]
- [126].Huang W, Liu X, Cao J, Meng F, Li M, Chen B, et al. (2015). miR-134 regulates ischemia/reperfusion injury-induced neuronal cell death by regulating CREB signaling. J Mol Neurosci, 55: 821-829. [DOI] [PubMed] [Google Scholar]
- [127].Chang CY, Lui TN, Lin JW, Lin YL, Hsing CH, Wang JJ, et al. (2014). Roles of microRNA-1 in hypoxia-induced apoptotic insults to neuronal cells. Arch Toxicol, 2014. September 20 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- [128].Ziu M, Fletcher L, Rana S, Jimenez DF, Digicaylioglu M (2011). Temporal differences in microRNA expression patterns in astrocytes and neurons after ischemic injury. PLoS One, 6: e14724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Yin KJ, Deng Z, Hamblin M, Xiang Y, Huang H, Zhang J, et al. (2010). Peroxisome proliferator-activated receptor delta regulation of miR-15a in ischemia-induced cerebral vascular endothelial injury. J Neurosci, 30: 6398-6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Wang P, Liang J, Li Y, Li J, Yang X, Zhang X, et al. (2014). Down-regulation of miRNA-30a alleviates cerebral ischemic injury through enhancing beclin 1-mediated autophagy. Neurochem Res, 39: 1279-1291. [DOI] [PubMed] [Google Scholar]
- [131].Xu S, Zhang R, Niu J, Cui D, Xie B, Zhang B, et al. (2012). Oxidative Stress Mediated-Alterations of the MicroRNA Expression Profile in Mouse Hippocampal Neurons. Int J Mol Sci, 13: 16945-16960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Moon JM, Xu L, Giffard RG (2013). Inhibition of microRNA-181 reduces forebrain ischemia-induced neuronal loss. J Cereb Blood Flow Metab, 33: 1976-1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Liu P, Zhao H, Wang R, Wang P, Tao Z, Gao L, et al. (2015). MicroRNA-424 Protects Against Focal Cerebral Ischemia and Reperfusion Injury in Mice by Suppressing Oxidative Stress. Stroke, 46: 513-519. [DOI] [PubMed] [Google Scholar]
- [134].Zhao H, Tao Z, Wang R, Liu P, Yan F, Li J, et al. (2014). MicroRNA-23a-3p attenuates oxidative stress injury in a mouse model of focal cerebral ischemia-reperfusion. Brain Res, 1592: 65-72. [DOI] [PubMed] [Google Scholar]
- [135].Jiang Y, Li L, Tan X, Liu B, Zhang Y, Li C (2015). MiR-210 mediates vagus nerve stimulation-induced antioxidant stress and anti-apoptosis reactions following cerebral ischemia/reperfusion injury in rats. J Neurochem, 134: 173-181. [DOI] [PubMed] [Google Scholar]
- [136].Wang J, Chen S, Ma X, Cheng C, Xiao X, Chen J, et al. (2013). Effects of Endothelial Progenitor Cell-Derived Microvesicles on Hypoxia/Reoxygenation-Induced Endothelial Dysfunction and Apoptosis. Oxid Med Cell Longev, 2013: 572729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Gao R, Wang L, Sun J, Nie K, Jian H, Gao L, et al. (2014). MiR-204 promotes apoptosis in oxidative stress-induced rat Schwann cells by suppressing neuritin expression. FEBS Lett, 588: 3225-3232. [DOI] [PubMed] [Google Scholar]
- [138].De Planell-Saguer M, Rodicio MC (2011). Analytical aspects of microRNA in diagnostics: a review. Anal Chim Acta, 699: 134-152. [DOI] [PubMed] [Google Scholar]
- [139].Mizrak A, Bolukbasi MF, Ozdener GB, Brenner GJ, Madlener S, Erkan EP, et al. (2013). Genetically engineered microvesicles carrying suicide mRNA/protein inhibit schwannoma tumor growth. Mol Ther, 21: 101-108. [DOI] [PMC free article] [PubMed] [Google Scholar]