

Abstract
Proliferation is an important part of cancer development and progression. This is manifest by altered expression and/or activity of cell cycle related proteins. Constitutive activation of many signal transduction pathways also stimulates cell growth. Early steps in tumor development are associated with a fibrogenic response and the development of a hypoxic environment which favors the survival and proliferation of cancer stem cells. Part of the survival strategy of cancer stem cells may manifested by alterations in cell metabolism. Once tumors appear, growth and metastasis may be supported by overproduction of appropriate hormones (in hormonally dependent cancers), by promoting angiogenesis, by undergoing epithelial to mesenchymal transition, by triggering autophagy, and by taking cues from surrounding stromal cells. A number of natural compounds (e.g., curcumin, resveratrol, indole-3-carbinol, brassinin, sulforaphane, epigallocatechin-3-gallate, genistein, ellagitannins, lycopene and quercetin) have been found to inhibit one or more pathways that contribute to proliferation (e.g., hypoxia inducible factor 1, nuclear factor kappa B, phosphoinositide 3 kinase/Akt, insulin-like growth factor receptor 1, Wnt, cell cycle associated proteins, as well as androgen and estrogen receptor signaling). This data, in combination with bioinformatics analyses, will be very important for identifying signaling pathways and molecular targets that may provide early diagnostic markers and/or critical targets for the development of new drugs or drug combinations that block tumor formation and progression.
Keywords: proliferation, natural products, therapeutic targets, cancer stem cells, cancer hallmarks
1. The centrality of cell proliferation as a target in carcinogenesis
The cancer cell embodies characteristics that permit survival beyond its normal life span and to proliferate abnormally. Cancer therapy, involving cytotoxic drugs, kills cells that have a high basal level of proliferation and regeneration. While this type of therapy targets tumor cells, it affects rapidly proliferating, nontumor cells in the skin, hair, and epithelium of the gastrointestinal tract, accounting for the high level of toxicity associated with such treatments. Growth of normal tissue is tightly regulated while this regulation is lost in tumor cells. Lack of normal growth control is not only operative in early tumorigenesis but also during tumor metastasis. Thus, there is much to be learned from studies that address how and when abnormal growth begins, and then to use this knowledge to identify novel therapeutic targets and approaches that would more specifically treat cancer cells without damaging the normal host cells.
Carcinogenesis is a multistep process in which changes in tissue architecture and the formation of preneoplastic nodules precede the appearance of cancer. These alterations are associated with changes in cell phenotype that include epithelial to mesenchymal transition (EMT) and cell migration, resulting in local regions of hypoxia that promote the survival and growth of tissue stem cells [1-5], as well as angiogenesis [6-9] (Table 1). Autophagy also promotes the survival of preneoplastic and tumor cells under stressful conditions. While the growth and survival of normal cells are under partial control from growth factors and hormones, alterations in signaling pathways, resulting from mutations and/or epigenetic changes, renders cells resistant and independent of these pathways. Such changes promote survival and growth both by constitutively stimulating pathways that favor proliferation [10], and by inhibiting and/or overriding apoptotic pathways. Initially, altered signaling pathways, as well as changes in the metabolomics profile, epigenetically modify the patterns of gene expression in the cell, and as such are therapeutically reversible (Table 1). In contrast, tumor progression proceeds by “driver” mutations that are more difficult to target pharmacologically. Thus, elucidation of the underlying epigenetic mechanisms responsible for these alterations will provide meaningful targets for the development of novel therapeutics prior to or at the earliest stages of malignant transformation.
Table 1.
Factors in Cell Survival and Proliferation the Contribute to Carcinogenesis
| Factor | Contribution to carcinogenesis |
|---|---|
| EMT | Promotes stem cell growth, metastasis |
| Hypoxia | HIFs promote proliferation of CSCs and angiogenesis; alters metabolism; constitutive activation of signaling pathways |
| Autophagy | Promotes cell survival in response dysregulated signaling-mediated proliferation, enhanced glycolysis, and hypoxia |
| CSCs | Dysregulation in “stemness,” quiescence, self-renewal, the ability to produce differentiated progeny, resistance to apoptosis, and chemoresistance, resulting in altered cell fate and unregulated cell growth |
| Cell cycle proteins | Dysregulated expression of cell cycle proteins (Rb, CDKs, cdk inhibitors) promote uncontrolled cell proliferation |
| Signal transduction pathways | Constitutive activation of multiple signalling pathways promote uncontrolled proliferation (e.g., Wnt, Notch, IGF, PI3K/Akt, NF-κB, Hh) |
| Altered cell metabolism | Promotes altered survival and growth in the adverse conditions (e.g., hypoxia) in early stages of carcinogenesis (e.g., altered glycolysis and methionine metabolism) |
| Hormone signaling | Promote the growth of hormone responsive cancers through constitutive activation of estrogen and androgen signalling pathways |
| Tumor microenvironment | Stromal-tumor cell crosstalk promotes growth and metastasis of cancer stem cells |
To facilitate a better understanding of the early changes seen in carcinogenesis, this review presents discussion of the major pathways, disruption of which promote unregulated proliferation of cancer cells. This review focuses on changes in tissue architecture (EMT and migration), formation of preneoplastic nodules, development of hypoxia, survival and growth of cancer stem cells, autophagy and growth factor independent proliferation (Table 1). Each section attempts to identify the “best” molecular targets (e.g., receptors, signaling molecules, etc.) that might be exploited therapeutically. The “best” targets were chosen based upon their altered expression/function that promoted proliferation in many different human cancers. Additional questions include: Does loss of a target prevent tumor initiation or block tumor maintenance? What is the effect of global loss of a target in other tissues? Will there be off target effects due to additional functions and/or because of a high degree of homology with other proteins? Many of these targets are pleotropic, regulating different pathways and as such their targeting might abrogate additional required hallmarks. At the end of this review, there is a discussion of natural products that are likely to be effective against these molecular targets and pathways, which may be useful in delaying the onset of cancer and/or reversing cancer cell proliferation, with reduced associated toxicity. Many natural products have much lower toxicity than compounds or derivatives obtained from chemical libraries, suggesting that their further development could provide distinct advantages.
2. How does EMT contribute to tumor proliferation?
When EMT occurs in adult tissues in response to injury or during tumorigenesis, epithelial cells change morphological appearance, from an ordered structure with apical and basal polarity to a less ordered, migratory fibroblastic shape. The Snail family of transcription factors (Snail1/Snail and Snail2/Slug) is closely associated with EMT [11], because they suppress epithelial cadherin (E-cadherin) expression [12-14], which normally facilitates cell-cell interactions, providing polarity cues and preventing dissemination. Snail associated EMT is normally under stringent regulation [15-17], but when that is lost, cancer may appear [17-19]. Increased expression of Snail and Slug protects cells from death induced by the loss of survival factors or by apoptotic stimuli [15,20-24]. Elevated Snail1/2 results in increased protection from DNA damage [17,20,21], increased resistance to chemotherapeutic agents [25] and radiation therapy. Snail and Slug may also affect a cell's response to genotoxic stress, increasing DNA damage, which then may contribute to cancer development (Fig. 1).
Fig. 1.
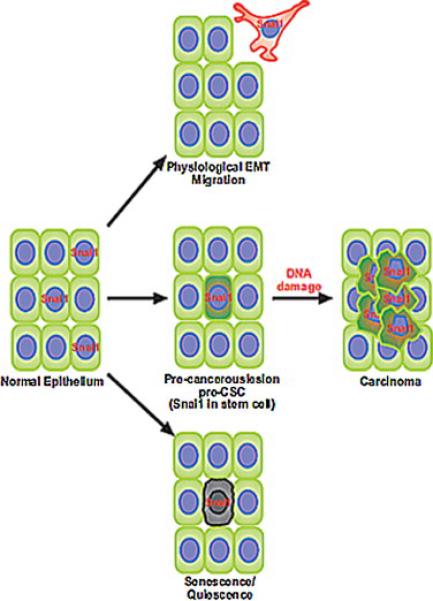
Senescence-resistant stem cells (SCs) are targets of Snail induced tumors. Tumor-associated Snail1/2 contribute to metastasis, but are also involved in early stages of cancer. In this model, cells expressing oncogenic Snail1/2 undergo EMT or senescence. However, SCs are resistant to this fate. Snail1/2 increases resistance to DNA damage, allowing those cells to accumulate mutations that fuel malignant transformation and uncontrolled cell growth.
Snail1 induced E-cadherin depletion is associated with the acquisition of invasive properties in several epithelial cell lines [12,26,27] and in tumors [18,19]. Snail expression also correlates with poor survival in human cancer [30-33]. Cells expressing Snail1 typically have an undifferentiated phenotype [18], suggesting a potential role in “stemness” and the genesis of cancer stem cells (CSC) (described below). CSCs are resistant to cell cycle arrest or senescence thereby accumulating oncogene induced DNA damage and mutations that guide malignant transformation. Cells undergoing EMT, and cells with properties of CSCs are resistant to typical cancer intervention strategies, tumor relapse following therapy, and metastasis. However, the expression of selected microRNAs (miRNA) that regulate gene expression in these cells can be altered by natural products, such as curcumin and epigallocatechin-3-gallate (EGCG) [34] (see below), suggesting a fresh therapeutic approach that needs to be developed in the future. EMT may occur prior to tumor appearance, resulting in aberrant tissue architecture and the development of hypoxia. In addition, CSCs drive tumor formation. These events further suggest the importance of these targets in cancer chemoprevention.
3. How does hypoxia contribute to tumor proliferation?
3.1 Hypoxia inducible factors [HIFs]
Cancer development results from the selection of cells with mutation(s) that provide survival and proliferative advantages. Normal barriers to proliferation are overcome as clones adapt to an ever changing hostile microenvironment, where low oxygen tension, low glucose levels, and an acidic extracellular pH (all of which increase genetic instability) are found. The hypoxia inducible factors, HIF-1 and HIF-2, are upregulated in response to these conditions. This could occur by constitutive activation of PI3K signaling or inactivating mutations in, for example, the von Hippel–Lindau tumor suppressor, VHL [35-37], which normally deacetylates HIF-1α, leading to HIF-1α polyubiquitination and proteasomal degradation [38]. HIFs trans activate genes mediating proliferation, angiogenesis, intermediate metabolism (glycolysis) and pH regulation, which promote tumor development [39].
HIF-1α stimulates production of growth factors, such as transforming growth factor β (TGF-β), insulin-like growth factor 2, interleukin-6 (IL-6), interleukin-8, macrophage migration inhibitory factor (MIF), and growth factor receptors, such as the epidermal growth factor receptor (EGFR), resulting in continuous proliferative signaling. In the hypoxic environment, constitutive activation of these signaling pathways (e.g., Ras [1] and PI3K [2]) stabilizes HIF-1 and may result in “oncogene addiction” that persists through the transition from adenoma to carcinoma. In the case of PI3K, constitutive activation may result from the appearance of mutations in tumor suppressor genes (e.g., the phosphatase and tensin homolog [PTEN]), from activating mutations in the PI3K complex itself, or from aberrant signaling in receptor tyrosine kinases [40]. Elevated PI3K stimulates the mechanistic target of rapamycin (mTOR) [35], and ATP production [41,42], both of which support cell proliferation. Strategies to block the proliferative effects of hypoxia include the design of small molecule HIF inhibitors, by enhanced degradation of HIF-1 via inhibition of heat shock protein 90 (Hsp90), or by inhibiting mTOR [43].
The Warburg effect describes the ability of tumor cells to switch from oxidative phosphorylation to glycolytic metabolism as their primary energy source. HIF-1 increases the expression of glycolytic enzymes and glucose transporters 1 and 3 [1], which facilitate glucose uptake necessitated by inefficient glycolysis. HIF-1 channels glucose towards glycolysis, and represses mitochondrial respiration, protecting cells from oxidative damage. Increased glycolytic metabolism promotes ATP production to sustain cell proliferation in the absence of oxygen. The development of glycolytic inhibitors has shown promising results in vitro [43]. Energy depletion and hypoxia also suppress mTOR signaling through activation of Ataxia telangiectasia mutated (ATM, involved in cell cycle arrest and DNA repair), saving on energy consuming protein synthesis and DNA damage responses. Thus, ATM or checkpoint 1 inhibitors may also abrogate metabolic adaptation of cells to hypoxia and subsequent survival [6,43].
HIF-1α also promotes autophagy, which is a mechanism whereby cells degrade macromolecules and organelles, and then reutilize the products for energy production and biosynthesis, thereby promoting cell survival. Thus, blocking autophagy via inhibition of IRE1 (a serine/threonine protein kinase/endoribonuclease that alters host cell gene expression under ER stress) may increase the sensitivity of cells to apoptosis in hypoxic environments [43]. Elevated HIF-1 levels may also increase de novo fatty acid synthesis [44,45] by upregulation of fatty acid synthase (FAS) transcription. This is mediated through sterol regulatory element binding protein 1 via Akt1 activation. Thus, inhibition of FAS or HIF-1 might block fatty acid synthesis mediated growth as well.
Despite the acidic pH due to accumulation of lactic acid during hypoxia, intracellular pH is maintained near neutral as a result of HIF-mediated up regulation/activation of membrane located transporters, exchangers, pumps and ectoenzymes. These include the amiloride sensitive Na+/H+ Exchanger, the H+/lactate cotransporter (monocarboxylate transporter, MCT4), and carbonic anhydrase (CA) IX and XII [6]. Although specific inhibitors of MCT4 are not available, disrupting pH homeostasis is justified by the antitumor and antimetastatic activity of CA inhibitors in xenografts [43]. Bioreductive agents may also be therapeutically useful as long as strategies are applied to increase their extravascular penetration [45]. Thus, while HIF-1/2 up regulation is a natural cellular response to hypoxia, this epigenetically fuels pathways that promote proliferation, creating an environment where mutation becomes more likely. Blocking hypoxia is attractive because it represents a predriver mutation state, where reversibility may be more feasible.
3.2 How does hypoxia promote growth in preneoplastic tissues?
Premalignant nodules are mostly devoid of blood vessels which limits the diffusion of substrates across the basement membrane from the local blood supply. Adaptation to these conditions is critical in the transition from a benign nodule to malignancy. As such, carcinoma in situ (CIS) becomes malignant following rupture of the basement membrane and invasion into the surrounding tissue, which may be facilitated by increased acid production [7]. Hypoxia may promote CIS progression by selecting for cells that are resistant to extracellular acidosis and those with upregulated glycolysis. Thus, the transition from preinvasive to invasive tumor may be closely linked to the CIS microenvironment [7,8].
In inflammation related carcinogenesis, altered tissue architecture due to necrosis and the development of hypoxia attracts inflammatory responses [46]. The latter usually includes tumor associated macrophages (TAM) that stimulate tumor proliferation (by promoting angiogenesis) and progression (by promoting invasion and metastasis) through the secretion of growth factors and cytokines [47,48]. HIF-1α is activated by these proinflammatory cytokines, which include tumor necrosis factor alpha (TNF-α) and interleukin-1β (IL-1β). Proinflammatory cyclooxygenase 2 (COX2) also mediates IL-1β-induced HIF-1α expression through production of prostaglandin E2 (PGE2). This leads to activation of the ras-mitogen activated protein kinase (MAPK) pathway, which maintains the prosurvival COX2/PGE2 pathway. Src is another key factor in hypoxia induced vascular endothelial growth factor (VEGF) and PGE2-mediated transactivation of EGFR. In addition, β-catenin-HIF-1 interaction results in the enhancement of HIF-1 transcriptional activity [8,49]. Importantly, HIF-1 is also activated in TAMs under hypoxic conditions [50,51], resulting in the stimulation of nuclear factor kappa B (NF-κB), and further inflammatory cytokine production, including sustained elevations in HIF-1. This feedback promotes tumor progression [52]. Thus, HIFs link hypoxia, chronic inflammation, and tumor progression by reprogramming tumor cells, macrophages and other cells during cancer development. Therefore, introducing natural compounds that target hypoxia in general, and NF-κB in particular, might delay or prevent the onset of dysplasia or/and neoplastic transformation, particularly in cell types where HIF promotes early steps of carcinogenesis. In this context, the antiinflammatory properties of many natural compounds may be able to attenuate the induction of HIF-1 (see below), thereby potentially preventing tumor development.
4. Autophagy and tumor cell proliferation
In normal cells, basal autophagy is a mechanism that maintains cellular homeostasis by removing protein aggregates and damaged organelles, whereas starvation induced autophagy prolongs cell survival by recycling amino acids and energy, which are both important for cellular fitness and preserving viability [53,54]. The basal level of autophagy increases in cancer cells to withstand stresses due to dysregulated signaling mediated proliferation [55], enhanced glycolysis [56], hypoxia [57], and to maintain cancer cells in a state of quiescence [58]. However, autophagy can promote tumor cell survival [59] or cell death [60] depending upon the tumor type, and thus, the implications of induced autophagy are not completely understood. It can be modulated therapeutically, either promoting survival or death [61,62].
4.1 Autophagy inducers
mTOR, which is part of a larger mechanistic target of rapamycin complex 1 (mTORC1), normally inhibits autophagy. When mTOR is inhibited by stress signals (e.g., HIF, dysregulated PI3K/Akt and elevated p53) [63,64], the Beclin 1/class III PI3K complex is activated [65], which promotes autophagy [66]. Alternatively, antiapoptotic B cell leukemia/lymphoma-2 (Bcl-2) family proteins, overexpressed in multiple tumor types, inhibit apoptosis and autophagy [67], suggesting that small molecule antagonists of Bcl-2 and related molecules (e.g., Bcl-2 larger isoform, Bcl-XL), known as BH3 mimetics (ABT-737/263, obatoclax), can competitively disrupt the Beclin 1-Bcl-2/Bcl-xL interaction to trigger autophagy [67] and apoptosis. A variety of mTORC1 inhibitors have been considered as antitumor agents that block proliferation. These include rapamycin, temsirolimus, deforolimus, metformin [68,69], the dual PI3K-mTOR inhibitors NVP-BEZ235 [70] and PI-103 [71], as well as combinations of these and other agents that induce autophagy [72,73] (Fig. 2). Interestingly, several polyphenolic compounds, such as resveratrol [74], curcumin [75], rottlerin, genistein, and quercetin [76], were reported to induce autophagy and cancer cell death, suggesting that these compounds may be clinically valuable in cancer treatment and/or chemoprevention.
Fig. 2.
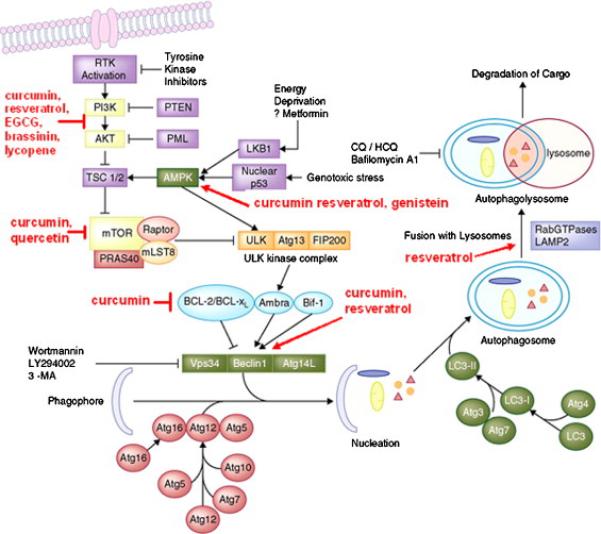
Major pathways of autophagy and natural compounds that inhibit these pathways. Autophagy inducers such as starvation (which may occur during hypoxic conditions) modulate the activity of the phagophore, consisting of the Atg1/unc-51-like kinase (ULK) complex, Beclin 1/PI3K complex, ubiquitin-like proteins (several Atg proteins), and proteins that mediate fusion between autophagosomes and lysosomes. Phagophore formation could be blocked with PI3K inhibitors. Autophagy induction involves budding of autophagosomes from the ER membranes, and inhibits interaction of TORC1 with the ULK1/2 complex. The latter regulates the activity of Beclin 1/class III PI3K complex. Beclin 1 interacts with factors that modulate its binding to Vps34, the catalytic unit of the PI3K, whose lipid kinase activity is essential for autophagy. This step could also be pharmacologically blocked. Fully mature autophagosomes can fuse with endosomes to form amphisomes. Autophagosomes or amphisomes fuse their external membranes with those from acidic lysosomes to acquire hydrolytic activity, degrade their cargo, and recycle essential biomolecules to the cytoplasm. Both fusion and degradation could also be inhibited by a variety of compounds, suggesting that autophagy would be a viable target in early stages of carcinogenesis [705].
4.2 Autophagy inhibitors
Knockdown of genes mediating autophagy [77,78] may contribute to tumor regression, as has been seen in human pancreatic cancer [79]. Human cancer cell lines harboring activating mutations in H-ras or K-ras have high basal levels of autophagy that promotes survival. Inhibition of autophagy in these lines reduces tumorigenicity [79,80]. Inhibition of autophagy also sensitizes tumor cells to alkylating agents and cetuximab [81]. In apoptosis defective leukemic and colon cancer cell lines, inhibition of autophagy sensitized resistant cells to TNF related apoptosis inducing ligand (TRAIL)-mediated apoptosis [82]. The natural compound, matrine, is a novel autophagy inhibitor that modulates the maturation of lysosomal proteases [82]. Combination therapies involving drugs that modulate autophagy are being classified as early or late stage inhibitors. Early inhibitors include 3-methyladenine, wortmannin and LY294022, which target the class III PI3K [83]. Late stage inhibitors include the antimalarial drugs bafilomycin A1 (which targets a vacuolar adenosine triphosphatase) [84], as well as monensin and chloroquine, both of which prevent the acidification of lysosomes [85]. Microtubule disrupting agents (e.g. taxanes, nocodizole, colchicine and vinca alkaloids) inhibit fusion of autophagosomes to lysosomes, thereby preventing steps in the formation of vacuoles that mediate autophagy. In addition, clomipramine (an anti-depressant) and lucanthone (an antischistome drug) block autophagosome degradation [86,87], suggesting new indications for existing drugs (Fig. 2). These observations imply that the modulation of autophagy may be an important therapeutic target in fighting cancer.
Similar to hypoxia, autophagy represents a viable way to treat cancer independent of targeting individual driver mutations. However, the main issue is that blocking or inducing autophagy appears to have opposite effects in different tumor types. Thus, markers to indicate which outcome would result must be identified before this line of targeting can be considered. Recently, a number of natural products were shown to be modulators of autophagy, such as bafilomycin A1 [88,89], feroniellin A [90] and oblongifolin C [91].
5. Survival and Growth of Cancer Stem Cells (CSCs)
5.1 Distinguishing features of adult and cancer stem cells
Stem cells (SCs) and CSCs share similar characteristics of “stemness,” quiescence, self renewal, the ability to produce differentialed progeny, resistance to apoptosis, and chemoresistance [92-103]. What distinguishes CSCs from adult SCs is the aberrant regulation of these processes in the former, resulting in altered cell fate and unregulated cell growth [94]. Aberrant Hedgehog (Hh), Notch and Wnt pathways, either through overexpression of wild type signaling molecules, or by activating mutations in these signaling pathways, contribute to the malignant conversion of adult stem cells to CSCs [103]. Further, the PTEN tumor suppressor maintains adult stem cells in quiescence, while in CSCs, PTEN is often mutated or deleted, resulting in increased expression of genes that promote the cell cycle and DNA replication.
The tumor mass contains a small proportion of CSCs that initiate/maintain malignant growth and differentiated progeny of these CSCs that do not [104,105]. Adult SCs divide asymmetrically, giving rise to a differentiated daughter cell and progenitor cell capable of a limited number of additional cell divisions [106]. In contrast, CSCs divide symmetrically into progenitor cells that possess an unlimited replicative potential that allows them to undergo an indefinite number of cell divisions. The latter may explain tumor relapse after initial therapy, where most of mature tumor cells are eliminated, while therapy resistant CSCs become reactivated and proliferate. Initial tumor responses might mean little if CSCs determine outcome [107], suggesting that CSCs are the cells that must be effectively targeted to achieve a definitive cure [107,108].
5.2 Stem cells and cancer initiation
Since the pathogenesis of cancer involves the appearance of driver mutations in long lived adult SC [109], only cells with self renewal capacity, especially in tissues with high cellular turnover (e.g., skin, intestine [109], breast [110-112] and hematopoetic cells [104,113]), should be most susceptible to malignant transformation. In chronic myelogenous leukemia (CML) [90], for example, the breakpoint cluster region/Abelson (Bcr-Abl) translocation appears at the beginning of the hematopoietic differentiation tree [114], implying an intimate relationship between SC, mutation, and tumor development. In some cancers, germline mutations in tissue SC (e.g., in colon cancer and medulloblastoma) also suggest a central role for these cells in tumor pathogenesis [115,116]. In other cancer types, including solid tumors [117,118], dedifferentiation and the reacquisition of the stem-like phenotype in mature cells may also be involved (e.g., observed in the pathogenesis of melanoma, breast and pancreatic cancers) [119-122]. This will influence the choice of cell target and the timing at which therapeutic intervention will have the greatest impact. However, phenotypic plasticity in tumors may preclude a simple approach to therapeutic intervention, since selected cell types in a tissue may have acquired the “stemness” phenotype in a given microenvironment.
Constitutively expressed oncogenes also contribute to cancer development, not just by inducing proliferation, but also because of their capacity to reprogram the epigenome of the target cell [123]. Reprogramming of differentiated cells can be achieved by the transient expression of the transcription factors octamer binding transcription factor 4 (Oct4), Kruppel-like factor 4, Nanog, and myc that “reset” the epigenetic status of cells and allow them to adopt a plethora of fates, including extended proliferation [123] (Fig. 3). If CSCs arise through a reprogramming like mechanism, then early intervention that target CSCs may be critical for the development and success of therapeutics.
Fig. 3.
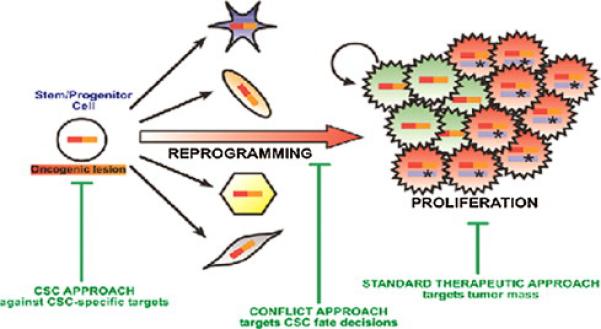
Cancer stem cells (CSCs) arise from tissue specific stem or progenitor cells that have undergone changes in gene expression (reprogramming) as a result of epigenetic mechanisms and/or oncogenic mutations. These CSCs undergo proliferation and differentiation into tumor cells. Standard therapeutic approaches target mostly the differentiated tumor cells, which reduce the bulk of the tumor, but CSCs are resistant to most therapies that are effective against the bulk of the tumor cells. In this model of carcinogenesis, it will be important to target key alterations in gene expression that drive reprogramming, be they natural compounds that epigenetically downregulate the expression of genes that contribute to reprogramming, and/or drugs that are effective against molecules that acquire driver mutations. Thus, blocking the reprogramming and proliferation of stem cells is likely to contribute importantly to cancer chemoprevention.
5.3 What regulates quiescence in stem cells?
If stem cell activation is important to the pathogenesis of cancer, maintaining stem cell quiescence and inhibiting their proliferation may have therapeutic value. This may prevent or delay the onset of primary tumors (e.g., in CML, melanoma, breast cancer, non small cell lung cancer, and osteosarcoma) [124-128], and help to prevent metastasis or relapse [129,130]. Micrometastases are quiescent for lengthy periods, and during this time, are resistant to most therapeutic approaches that target cell proliferation. This is why it is important to understand dormancy and growth regulation in stem cells.
Quiescence is most likely controlled by a combination of cell intrinsic and cell extrinsic (niche) interactions. The intracellular or “cell intrinsic” signals resemble normal processes that control cell cycle progression and survival. Thus, therapies that target G1 regulators (such as cyclin D), cyclin dependent kinase 4 (cdk4) or p27, or apoptotic regulators (such as Bcl-2), might be effective. Two canonical developmental pathways, Wnt/β-catenin and Hh, appear important in the self renewing potential of CSC [131,132]. Wnt and Hh are generally inactive in somatic adult cells, but are reactivated in adult SCs and CSC. The metastasis suppressor gene, mitogen activated protein kinase kinase 4, is part of a growing lists of genes that block proliferation through the activation of MAPK p38 [133-135], suggesting they may become therapeutic targets.
On the cell extrinsic side, the chemokine ligand 12 (CXC-12) and corresponding receptor (CXCR-4) interaction is required for breast, prostate, and multiple myeloma (MM) CSC colonization and subsequent quiescence [136]. In CML and MM, CSC can be mobilized from quiescence into the cell cycle by the addition of granulocyte colony stimulating factor (G-CSF), which degrades CXCL-12, or by an antagonist of CXCR-4 [137]. In the clinic, however, G-CSF has had mixed results, since it impacts both CSCs and adult SCs [137]. Thus, caution must be applied, as therapies should not affect or deplete adult SCs.
As the CSC fate decision is most likely controlled epigenetically, various transcription factors have been implicated in these processes. For example, CCAAT/enhancer binding protein alpha (C/EBPα) appears to regulate myeloid differentiation and self renewal of fetal liver hepatic SCs. Myeloid Elf-1-like factor is a transcriptional activator [138,139] that promotes the G1- to S-phase transition and enhances the movement of hepatic SCs out of a quiescent state (Go-phase) into the cell cycle [140,141]. The proangiogenic factor, angiopoietin-1, inactivates glycogen synthase kinase 3β (GSK3β) via phosphorylation, thereby releasing active β-catenin, which then migrates to the nucleus and upregulates the expression of genes that promote cell survival (by blocking apoptosis) and cell proliferation [142]. However, caution must be exercised in pursuing any of these as putative therapeutic targets, since the development of corresponding drugs would probably be associated with the appearance of considerable toxicity.
6. Targeting cell cycle proteins in sustained proliferative signaling
Cell cycle progression is controlled by cyclin-cdk complexes, that include cdk interacting protein p21CIP1, kinase inhibitory proteins (Kips) (p27KIP1, p57KIP2), and INhibitors of CDK4 (INK4s: p16INK4a, p15INK4b, p18INK4c, p19INK4d), which activate and inhibit these complexes, respectively [143]. The G1 phase of the cell cycle is the only time that a cell can respond to extracellular cues, and progression depends on the balance of proliferative and antiproliferative signals. In the presence of “go” signals, progression into S phase occurs; in the presence of “stop” signals, the cell arrests in G1. Thus, cancer can be thought of as a disease of the cell cycle: where a cancer cell ignores the “stop” signals and does not wait for the “go” signals. The result is excessive DNA replication, which increases the likelihood of replication induced mutations and telomere degeneration, further disabling other hallmark pathways.
6.1 Retinoblastoma (Rb) pathway
The Rb pathway (INK4-cyclin D-cdk4/6-Rb), which controls the G1-S phase transition, is universaly disrupted in human cancer. Cyclin D-cdk4/6 complexes initiate G1 progression by phosphorylating (inactivating) Rb, thus relieving transcriptional repression by the Rb-E2F complex (Fig. 4). Following Rb phosphorylation, E2F is released, inducing transcription of genes necessary for S-phase entry. Although Rb loss occurs in some tumor types, most cancers retain wild type Rb, and instead have mutated or activated cell cycle proteins that regulate Rb. In other tumor types, cell cycle proteins downstream of oncogenic pathways are frequently altered posttranslationally, demonstrating that these represent targets in cancer therapy [144].
Fig. 4.
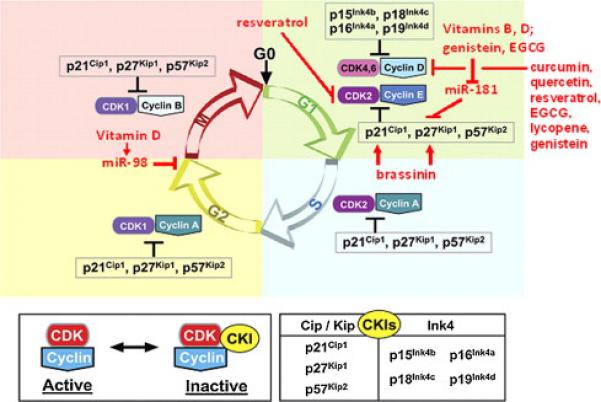
Selected natural products that block cell cycle progression. Receptor activation, via Raf, MEK, ERK, and AP1, increases cyclin D1 transcription. Cyclin D1 binds to cdk4 and the assembly factor, p27Kip1 to create an active ternary complex. This complex can be inactivated by association with Ink4A or loss of cyclin D1 via GSK-3β-mediated proteasomal degradation. Active cyclin D-cdk4-p27 complexes phosphorylate (inactivate) Rb, causing limited transcriptional activation of cyclin E. Increased cyclin E levels shifts the balance of inactive cyclin E-cdk2 complexes to active cyclin E-cdk2 complexes, which in turn phosphorylates its associated p27, targeting it for proteasomal degradation. p27-free cyclin E-cdk2 complexes now fully phosphorylate Rb, causing S phase gene transcription, and progression into S phase, where the cell cycle proceeds independently of extracellular signals. As shown in red, many natural compounds cause G1 arrest in several cancer cell culture models, due to effects on cyclin D1, p21, p27 or cyclin E. Some of these act via altered expresson of microRNAs. Modified from reference [650].
6.2 Cyclins D and E
The gene encoding cyclin D is the second most amplified locus in human cancer, and is directly downstream from many oncogenic pathways, suggesting it may be a good therapeutic target. In cancer, cyclin D-cdk4/6 activity may be increased by cdk4 and cdk6 amplification, mutation of cdk4 to an inhibitor resistant form, or loss of the INK4 inhibitors. Perhaps the best justification for targeting these molecules comes from clinical trials for the treatment of breast cancer, where palbociclib, a cdk4/6 specific inhibitor, delayed disease progression in human epidermal growth factor receptor 2 (HER2)+, estrogen receptor (ER)+ postmenopausal women. In addition, since cyclin D is transcriptionally linked to mitogenic signaling pathways, is expressed throughout the cell cycle, is degraded by GSK3β (which itself is a therapeutic target), and showed managable toxicity in clinical trials, suggests that cyclin D may be an important target for continued drug development [144].
Amplification of cyclin E or cdk2 is detected in some tumor types, but this is rare compared to cyclin D-cdk4/6 [145]. Cyclin E-cdk2 may be a good target, as its activation is a major consequence of Rb dependent phosphorylation. However, cdk2 activates origins of DNA replication, explaining its infrequent deregulation in tumors. Inactivation of the Rb checkpoint may also trigger cyclin E-cdk2 independent functions, such as the ability to overcome senescence. Moreover, several cdk2 inhibitors have failed in clinical trials for unknown reasons.
6.3 Cdk inhbitors
The cdk inhibitors of the INK4 class block cyclin D-cdk4/6 activity. They do not have additional targets, suggesting that therapeutic intervention could be highly specific. However, therapeutic restoration of INK4s presented problems, in that these loci are frequently deleted or mutated, which would preclude reactivation. For many cancers, frequent epigenetic inactivation of INK4 is due to extensive CpG methylation [146] raising the possibility that natural products capable of modulating DNA methylation such as EGCG, folate, and genistein are potential agents capable of reactivating INK4 genes [147].
The tumor suppressor, p27Kip1, inhibits cyclin E-cdk2, which would potentially block tumor growth. p27 levels are reduced posttranslationally with increasing tumor grade, resulting in increased cdk2 activity [148]. Re-expression of p27 could be achieved by interfering with protein turnover. The S-phase kinase associated protein 2 (Skp2) is the E3 ubiquitin ligase responsible for p27 degradation. Thus, Skp2 and p27 expression is inversely correlated [149,150]. Bortezomib-mediated proteosomal inhibition in multiple myeloma and mantle cell lymphoma [151,152] resulted in significant side effects, suggesting that more specific targets upstream from the proteasome might be less toxic. However, p27 inhibits cdk1 and proliferation of cdk2−/− mouse embryo fibroblasts, and also stabilizes the cyclin D-cdk4 complex, suggesting that targeting p27 may have global, deleterious effects.
7. Molecular pathways regulating tumor proliferation
Most clinically available targeted therapies focus on blocking the constitutive activation of signal transduction pathways (Bcr-Abl, EGFR, HER2, c-Met, and Raf). While these have initially been effective at blocking tumor proliferation, the emergence of resistant clones is a frequent clinical observation, suggesting that alternate therapeutic pathways should be investigated. Therefore, this part of the review will briefly introduce some of the major pathways that impact cell proliferation and fate, and that are targets for one or more natural compounds.
7.1 Wnt/β-catenin signaling
Wnt/β-catenin signaling is a developmental signaling pathway that regulates cell proliferation, differentiation, migration, polarity and asymmetric cell division [153,154]. It plays critical roles in embryonic stem cells [155], and can improve reprogramming of somatic cells towards induced pluripotent stem cells, highlighting the importance of this pathway for self renewal and pluripotency [156,157].
Aberrant Wnt/β-catenin signaling is implicated in numerous cancers (e.g., colorectal and breast cancers) [158-162]. Most involve stabilization of β-catenin by mutation which is often associated with tumor aggressiveness. Alternatively, aberrant Wnt signaling is due to either the inactivation of negative regulators of the Wnt signaling pathway, such as Frizzled related protein [161], or overexpression of positive regulators, such as disheveled [162]. In Wnt-1 transgenic mice, expanded mammary stem/progenitor cell populations are associated with the development of preneoplastic lesions or tumors [163,164]. Thus, constitutive activation of β-catenin appears to promote the survival and growth of stem cells in the early stages of tumor formation, suggesting it is an important target. Fortunately, there are many natural compounds that block Wnt signaling (see below).
7.2 Notch signaling
Notch is a family of mammalian transmembrane receptors (Notch 1-4) for membrane bound ligands (JAG1, JAG2, delta-like 1-4). Upon binding, Notch receptors undergo cleavage, releasing a Notch intracellular domain, which migrates to the nucleus, where it targets genes such as cyclin D [165], p21CIP1 [166], NF-κB [167], and c-myc [168-170]. Notch proteins contribute to angiogenesis, proliferation, differentiation, and apoptosis [171,172]. Notch signaling also contributes to cell fate in embryonic development, tissue homeostasis in adult tissues, and regulates stem cell maintenance and differentiation [173,174].
Notch signaling is detected in CSCs in breast cancer [175-177], embryonal brain tumors [178], gliomas [179], T cell leukemias, ovarian, cervical, colorectal, pancreatic, salivary gland, and lung carcinomas [93,94,178,180-182]. In breast cancer, constitutive activation of Notch prevented differentiation of mammary epithelial cells in vitro and resulted in the appearance of poorly differentiated adenocarcinomas [183-187]. Further, HER2 [188-190], Akt [191], signal tansducer and activator of transcription (STAT3) [101], NF-κB [192] were found to cross talk with Notch in breast CSCs [172], suggesting that Notch could impact breast cancer development and proliferation through these signals. In the hypoxic environment, HIFs activate Notch [3] and the expression of transcription factors such as Oct4 that control stem cell self renewal and pluripotency [4,193]. Thus, elevated Notch signaling permits CSCs to survive and proliferate in a hypoxic microenvironment. Natural compounds, such as resveratrol (see below) [194], down regulate transcription of the Notch and PI3K/Akt pathways and may prevent tumor appearance.
7.3 Insulin-like growth factor (IGF) signaling
The IGF-1 receptor/ligand system is implicated in self renewal/pluripotency in hematopoietic and embryonic stem cells and supports cell growth/survival by activation of PI3K/Akt and Ras/Raf/extracellular signal regulated kinase [195-197]. Recently, Nanog was shown to have a crucial role in maintaining the self renewal of CSCs through the IGF-1 signaling in hepatocellular carcinoma [198]. IGF-1 signaling could also crosstalk with other pathways, such as Notch, EGFR, leptin and promotes the transition of adult SC to CSCs [172,199,200].
7.4 PI3K/Akt/mTOR signaling
The PI3K/Akt/mTOR pathway plays a central role in growth, proliferation, motility, survival and angiogenesis in tumor cells [201,202]. mTOR is a ser/thr kinase that is a downstream target of PI3K/Akt in many types of cancer. Aberrant activation of mTOR by mutations or gene amplification [203], promotes cancer cell proliferation, EMT [204], and resistance to anticancer drugs [205,206].
PI3K/Akt/mTOR signaling plays a key role in CSC biology because this pathway is more sensitive to inhibition compared to healthy stem cells [191,207]. mTOR inhibition also suppresses EMT and CSC-like characteristics in colorectal cancer [208]. However, inhibition of mTOR is complex because several downstream targets in this pathway [e.g., mTORC1, S6 kinase 1 and eukaryotic translation initiation factor 4E binding protein 1 may be regulated in an mTOR independent manner [209-214]. Another challenge is to identify pharmacological profiles for mutations in these pathways. This could be aided using biomathematical algroithms like the COeXpression ExtrapolatioN (COXEN) model [215-217].
7.5 NF-κB signaling
NF-κB transcription factors regulate the expression of key genes for innate and adaptive immunity, cell proliferation and survival, and lymphoid organ development. NF-κB is activated in many cancers [218-220] by many divergent stimuli, including proinflammatory cytokines such as IL-1β, epidermal growth factor (EGF), T- and B-cell mitogens, bacteria and lipopolysaccharides, viruses, viral proteins, double stranded RNA, and physical and chemical stressors [220-222]. These events contribute to the link between inflammation and carcinogenesis. For example, NF-κB activation may be required for human ovarian CSC metastases [223], in human cervical CSC growth and migration [224], and in keeping differentiating glioblastoma CSCs from acquiring a mature post mitotic phenotype [225]. Mammary epithelial NF-κB also regulates the self-renewal of breast CSCs [226]. Thus, NF-κB is an important therapeutic target in carcinogenesis.
7.6 Hedgehog signaling
Hh signaling controls tissue polarity, patterning, and stem cells maintenance in a variety of tissues [227-230]. In vertebrates, three Hh ligands [Sonic Hedgehog (Shh), Desert Hedgehog, and Indian Hedgehog] bind to trans-membrane receptors [Patched (Ptch1 or Ptch2)]. Upon ligand binding, the complex containing Ptch and its inhibitor Smoothened (Smo) dissociates. Smo activates Gli transcription factor which translocates into the nucleus and initiates transcription of target genes that regulate the properties of stem cells [231,232].
Regulation of CSC proliferation in various human tumors including glioblastoma, breast cancer, pancreatic adenocarcinoma, MM and CML is through Hh signaling [97,233-241]. Use of the SMO antagonist cyclopamine or the Hh ligand neutralizing antibody 5E1 induced terminal differentiation and loss of clonogenic growth in gastric CSCs from primary tumors [242,243]. Mouse models of CML also suggest that Hh regulates the self renewal property of the tumor cells [240], providing an important preclinical model for intervention studies with natural compounds. Thus, Hh affects CSCs self renewal and differentiation [244]. IL-6 stimulated the growth of acute myeloid leukemia cells through Hh, and this effect was blocked by the natural compound resveratrol. Shh-Gli signaling controls the characteristics of pancreatic CSCs, and these are inhibited by the use of sulforaphane [132,245].
8. Is there a relationship between altered cellular metabolism and proliferation?
The contribution of altered cellular metabolism to cancer is exemplified by nonalcoholic fatty liver disease (NAFLD) [246], which includes alterations that range from triglyceride accumulation in hepatocytes (steatosis) to steatosis with inflammation (nonalcoholic steatohepatitis or NASH), with or without fibrosis [247,248]. NASH patients with liver fibrosis are at risk for the development of cirrhosis [249] and hepatocellular carcinoma (HCC). At the molecular level, altered methionine metabolism plays an essential role in the molecular bases of NAFLD related HCC.
Chronic liver disease among patients with cirrhosis is partially characterized by elevated serum levels of methionine [250,251]. The latter is associated with decreased methionine adenosyltransferase (MAT), and the product of its reaction, S-adenosylmethionine (SAM) [252,253]. MAT deficient mice developed chronic hepatic SAM deficiency, display increased proliferation, and spontaneously develop HCC [254]. SAM is a major methyl donor, where it mediates up to 85% of the methylation reactions in the liver, thereby promoting homeostasis [255]. Since SAM is also a precursor of the antioxidant glutathione (GSH), both are decreased in patients with cirrhosis. Treatment of these patients with SAM increased GSH levels and improved survival [255,256], suggesting the therapeutic use of SAM to treat liver diseases [257]. Conversely, mice deficient in the glycine N-methyltransferase gene, which encodes the enzyme responsible for SAM catabolism, developed elevated SAM, methionine, serum transaminase levels [258], hepatic steatosis, fibrosis and HCC [258-261]. Therefore, altered methionine metabolism resulted in increased proliferation through decreased levels of MAT and GSH.
In cancer, proliferating cells require rapid ATP generation, increased biosynthesis of macromolecules, and maintenance of an appropriate cellular redox status [262]. For example, the tumor suppressor, p53, stimulates glycolytic enzymes and the pentose phosphate pathway, which provide substrates for macromolecular synthesis. In addition, the M2 isoform of pyruvate kinase (PKM2), which converts phosphoenolpyruvate to pyruvate, attenuates glycolysis, thereby providing precursors for macromolecular synthesis and cell proliferation [263]. In these same reactions, PKM2 also promotes the development of nicotinamine adenine dinucleotide phosphate (NADPH) which provides reducing power for macromolecular synthesis as well as quenches free radicals. NADPH also contributes importantly to controlling the redox state of cells. At low levels, free radicals promote cell proliferation by activating signaling pathways [264,265]. At moderate levels, free radicals promote stress responsive genes (e.g., HIF-1α) and cell survival [266,267], while at high levels, free radicals cause macromolecular and organelle damage, triggering senescence or apoptosis, and in surviving cells, activating antioxidant pathways [268,269]. Thus, the control of free radical levels by cancer cells promotes proliferation but not the appearance of detrimental mutations. In this context, many natural polyphenols (see below) alter cancer cell metabolism by reducing intracellular free radicals to very low levels, thereby inhibiting the appearance of mutations and unwanted proliferation.
9. The role of estrogen and androgen receptors in cancer cell proliferation
Hormones are signaling molecules secreted by cells that modulate the function(s) of target tissues. This encompasses paracrine, autocrine, and intracrine hormonal actions. As one of the main functions of hormone stimulation is cell cycle regulation, it is not surprising that hormonal disregulation is involved in cancer progression. Hormone related cancers [270] make up almost 30% of all cancer cases, and include cancers of the breast, ovary, endometrium, prostate, and testis [271].
As cancer initiators, steroid hormones could cause irreversible damage to the genotype of the cell. For example, high doses and long term treatment with 17β-estradiol (E2) results in DNA damage among rodents [272,273]. However, it is unlikely that at physiological levels, estrogens and other hormones are carcinogens, and instead stimulate mitosis by shortening G1 and promoting entry into S phase [274]. For example, steroid hormones stimulate the proliferation of normal cells, increasing the chances of a cell acquiring DNA damage and oncogenic mutation as well as cells mutated by an initiator. Thus, deranged hormone signalling pathways can promote cancer. However, there is a lack of translational applications of this information, due to the complicated signals activated by hormones through their different receptors [275].
Two estrogen receptors (i.e., ERα and ERβ) and one androgen receptor (AR) mediate the mitogenic effects of estrogens and androgens, respectively. After ligand binding, the ligand-receptor complexes translocate to the nucleus where they recruit cofactor proteins and the basal transcription machinery onto estrogen or androgen responsive elements, respectively [274], impacting proliferation at the level of transcription.
Sex steroid hormones also signal through plasma membrane bound forms of AR and ER [276]. For some cancers, this occurs through activation of extracellular signal regulated kinase 1 in the MAPK family and via Akt in the PI3K pathway. These ERα-dependent pathways transduce proliferative, antiapoptotic and migration signals [277-279]. In this context, membranous ERα staining was observed in up to 1/3 of the cases in which the tumor was classified as ERα negative on the basis of ERα nuclear expression. Membrane ERα expressing breast cancers also show a strong positive correlation with phosphorylated Akt and HER2 overexpression [280], the latter of which is characteristic of ‘invasive carcinoma’ [281]. In addition, ERα plasma membrane localization and its interactions with IGFR1 and EGFR/ErbB2 may be one of the mechanisms underlying the development of drug resistance in breast cancer cells [282,283]. Androgen independent prostate cancer is also mediated by IGFR-1/IGF-1 [284] and elevated EGFR/ErbB-2 [285], combined with downstream Akt [286,287] and Janus kinase (JAK)/STAT [288] and MAPK signaling. These pathways activate AR, which translocates to the nucleus, where it alters host gene expression that promotes cell survival, proliferation, and metastasis. As indicated below, there are a number of natural compounds which target one or more of these pathways. While the success of surgical or medical castration has been demonstrated in androgen dependent tumors, the utility of natural compounds in andogen independent tumors will depend upon the mechanisms whereby andogen independence is achieved. This may include AR overexpression, AR mutations, altered recruitment of transcription cofactors, and/or sustained intratumoral synthesis of dihydrotestosterone, which binds to and activates the AR in the androgen dependent phase of prostate cancer [289].
Deregulation of ERα- and ERβ-mediated signal transduction, together with the deregulation of nuclear receptor activities, may explain the role of estrogen in promoting breast cancer [275]. In this respect, ERα positive breast tumors are treated with drugs that interfere with the availability of endogenous E2 (e.g., aromatase inhibitors) or ERα transcriptional activity (e.g., 4OH-tamoxifen) [290]. The same drugs could act on ERβ signaling, even if the expression of this receptor subtype is inversely correlated with the development of several cancers [291]. Thus, a selective agonist for ERβ could promote strong antiproliferative intracellular signals in breast, colon, and prostate tissues, where ERβ functions as a growth repressor and a dominant negative inhibitor of ERα-mediated proliferation [292,293]. In spite of these data, the role of the membrane initiated ER signaling in tumors has been underestimated. Nuclear localization of ERs [294,295] is now considered to have prognostic significance. Very few drugs for breast cancer have been shown to target ERs extranuclear mechanisms [283].
Anticancer drug development has been characterized by two different approaches: the chemical modification of preexisting therapeutics and the selection of new molecular targets. The latter approach better overcomes the limitations of available clinical treatments and could provide an opportunity to expand antihormonal treatments in new directions. A further promising strategy would rely on targeting ER membrane proliferative actions, but this remains to be explored.
10. The impact of stromal cells on tumor growth
Stromal components in tumor microenvironment contribute centrally to tumor progression and metastasis. Reciprocal interactions occur between neoplastic cells and stromal components leading to coevolution. In this context, either stromal cells support transformation of epithelial cells, or transformed tumor cells engage stromal cells, and the altered environment can influence the metastatic, dormancy related, and stem-like potential of tumor cells [296]. The stromal compartment of the tumor is complex, consisting of inflammatory/immune cells, endothelial cells (vascular), pericytes, fibroblasts, adipocytes and extracellular matrix components (e.g., collagen, fibronectin, laminin and proteoglycan complex). Tumor infiltrating inflammatory cells release EGF, VEGF, fibroblast growth factor-2 (FGF-2), chemokines, cytokines, and proinvasive matrix degrading enzymes to promote tumorigenesis [297-299]. Continued tumor growth and progression is mediated by the “angiogenic switch,” which occurs in response to VEGF and FGF-2 secreted from tumor cells, resulting in angiogenesis [300-302]. Adipocytes in the tumor microenvironment produce ‘adipokines’ [303,304] such as leptin, adiponectin, hepatocyte growth factor, collagen VI, IL-6 and TNF-α, which are important for tumor growth. Fibroblasts in the tumor microenvironment provide the structural framework of the stroma [305]. Fibroblasts remain quiescent, but they proliferate during wound healing, inflammation and cancer. Paracrine factors from tumor cells activate fibroblasts to become “cancer associated fibroblasts” (CAF) [306,307]. CAFs secrete factors that modulate tumor growth and modify the stroma to facilitate metastasis [308,309] and attenuate responses to anticancer therapies [310,311]. Thus, tumor stromal crosstalk is important when developing therapeutic options, since tumor centric approaches may not work in a stroma rich tumor microenvironment.
11. Natural and dietary substances that block cancer proliferation and augment anticancer therapy
More than half of current drugs originally came from natural products. Plant derived anticancer agents that block proliferation, resulting in cell cycle arrest and apoptosis, include vinblastine, etoposide, teniposide, homoharringtonine and camptothecin derivatives [312]. Epidemiological studies have shown that natural products and nutritional substances may be active in cancer chemoprevention. These have been most extensively described in colon, prostate and breast cancers.
Contrary to conventional chemotherapy, which exhibits cytotoxic effects against all dividing cells, targeted therapeutic drugs are active against proliferating cells involved in tumor progression. Targets of these therapeutic approaches include Bcr-Abl kinase (e.g., imatinib [313], nilotinib [314], and ponatinib [315]), EGFR (gefitinib [316] and erlotinib [317]), HER2/ErbB2 (lapatinib [318]), and c-Met (crizotinib [319]). Although intially effective, relapse is common, resulting in the appearance of drug resistance, the activation of alternative signaling pathways, and/or the generation of chemoresistant CSCs [320,321]. Given that cancer is multistep, targeting multiple pathways may yeild stronger antitumor activities. Accordingly, some of the leading natural compounds used in cancer therapy and in chemoprevention are presented below as examples of their potential utility in the pathogenesis of cancer.
11.1 Curcumin
Curcumin (diferuloylmethane), a yellow spice and phenolic compound derived from the plant Curcuma longa, is one of the most powerful and promising chemopreventive and anticancer agents [322] (Fig. 5). The consumption of a curcumin rich diet is inversely correlated with several human malignancies [323]. Curcumin blocks cancer cell proliferation by targeting Wnt (Table 2), NF-κB, STAT3, PI3K/Akt [324] (Fig. 6) and mTOR [325]. Curcumin also has fewer side effects than conventional therapy [326,327]. Curcumin indirectly affects tumor proliferation by altering the expression miRNAs, which regulate cellular signaling implicated in tumor cell proliferation. For example, the antiproliferative activity of curcumin against pancreatic cancer cells has been shown to be mediated by modulating the miR-200 family, which in turn regulates EMT [328]. Further, curcumin and its synthetic analog, diflourinated curcumin, downregulate miR-21 expression [329] and reduced the expression of Bcl-2 by upregulating miR-15a and miR-15b [330], indicating that curcumin impacts upon proliferation, in part, by epigenetic mechanisms. A major problem with curcumin is its low bioavailability, prompting the development and evaluation of structural analogs of curcumin, stabilization of curcumin with adjuvants (e.g., piperine), liposomal and nanoparticle associated curcumin, and the development of curcumin phospholipid complexes, aimed at increasing bioavailability while antitumor properties are maintained [331].
Fig. 5.
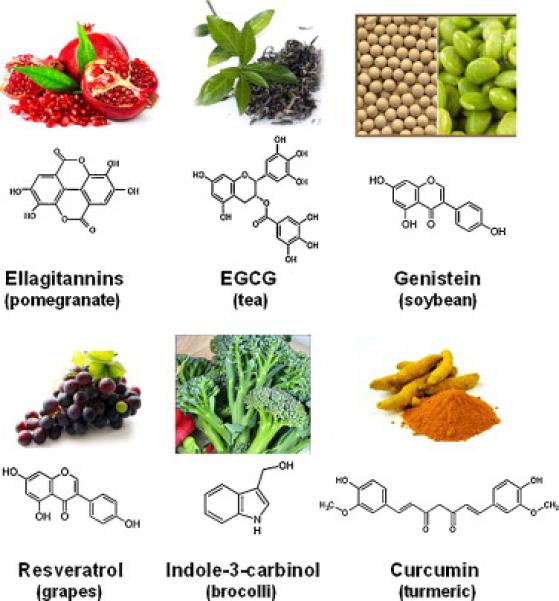
Examples of anti-proliferative compounds obtained from natural sources.
Table 2.
Summary of Selected Natural Compounds Active Against Wnt Signaling in Cell Proliferation
| Compound | Effects | Cancer Model | Concentration/dose | References |
|---|---|---|---|---|
| EGCG | reduced nuclear β-catenin; blocks β-catenin/TCF mediated transcription; increase Wnt inhibitor (WIF1) | Non small cell lung cancer (NSCLC) cells, breast cancer cells; familial adenomatous polyposis (APCMin/+) mice | NSCLC 0-50 μM; breast: 0-100 μM APC mice: high fat diet + 0.16% EGCG | [438-441] |
| Resveratrol | inhibits β-catenin migration to nucleus; disrupts β-catenin/TCF binding | colorectal cancer cells | 0-20 μM | [442,443] |
| Genistein | suppresses β-catenin/TCF mediated transcription; up-regulates GSK3β and E-cadherin; blocks Wnt signaling by down-regulation of miR-1260b; increase in DKK1 | gastric, renal, prostate, and colorectal cancer cells | gastric: 0-100 μM renal: 25 μM prostate: 25-50 μM colorectal: 75μM | [444-449] |
| Curcumin | decreases nuclear β-catenin; blocks β-catenin/TCF-mediated transcription; down-regulates Wnt4, GSK3β and Frizzled | breast and colorectal carcinoma cells; neuroblastoma cells | breast: 0-70 μM colorectal: 20 μM neuoblastoma: 0-20 μM | [450-453] |
| Vitamin A | inhibits Wnt/β-catenin signaling; vitamin receptor competes with β-catenin for TCF; blocks GSK3β | transformed human bronchial epithelial cells | 4 μM | [454] |
| Vitamin D | inhibits β-catenin signaling, induces E-cadherin | colorectal carcinoma cells | 0.1 μM | [455, 456] |
| Lycopene | Reduced nuclear β-catenin via attenuating GSK-3β phosphorylation | prostate cancer clinical trials | 5-50 mg/kg | [457] |
| 1p-XSC + docosahex-aenoic acid -3 fatty acid | target(s) unknown | colorectal cancer | 2.5 - 5 μM | [436] |
| 1p-XSC | pathway target(s) unknown | Apc mice a model for familial adenomatous polyposis | high fat diets containing 10-20 p.p.m. | [437] |
1,4-phenylene bis(methylene) selenocyanate
Fig. 6.
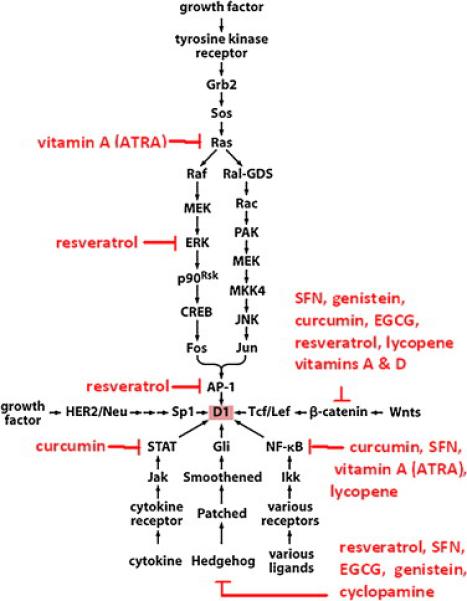
Impact of various natural compounds upon selected growth promoting signaling pathways. When proliferation is triggered by growth factor signaling, there are a number of natural compounds that could inhibit growth. For example, vitamin A, which promotes differentiation, downregulates ras signaling. Resveratrol could block downstream signaling components such as ERK, AP-1, and alternative pathways, such as Hedgehog. Other signaling pathways that promote growth, such as Wnt, cytokine triggered STAT signaling, and receptor mediated activation of NF-κB, could be blocked, in part, by a variety of natural compounds. This suggests that a combination of natural compounds could have a significant impact upon proliferation, even at early stages of carcinogenesis, by inhibiting normal signaling pathways.
11.2 Indole-3-carbinol, 3,3-diindolylmethane, sulforaphane and brassinin
Indole-3-carbinol, a natural hydrolysis product of glucobrassicin in cruciferous vegetables (Fig. 5), blocks tumor cell proliferation by modulating expression of the IGFR1, the insulin receptor substrate-1, and by triggering degradation of the ERα [332]. Such vegetables also contain sulforaphane, a naturally occurring organosulfur compound formed by the hydrolysis of glucosinolates. Sulforaphane may lower the risk of colon, prostate, and perhaps other cancers [333]. sulforaphane blocks proliferation, induces cell cycle arrest in vitro, and has anticancer activity in animal models [334]. Sulforaphane also suppresses NF-κB and the Wnt/β-catenin self renewal pathways in CSCs [335,336] (Fig. 6). Sulforaphane appears to synergize with sorafenib in shrinking pancreatic cancer by blocking proliferation, angiogenesis, and EMT [337]. Another indole compound derived from cruciferous vegetables, brassinin, arrests cancer cells in G1 via blocking PI3K signaling and upregulating p21 and p27 [338]. In comparison, 3,3-diindolylmethane increased the expression of miR-21 which reduced the expression of its target gene, cell division cycle 25 homolog A [339]. Moreover, 3,3-diindolylmethane has been shown to increase the level of the miR-200 family in pancreatic cancer cells, which impacts EMT [328].
11.3 Resveratrol
Resveratrol (3,4',5-trihydroxy-trans-stilbene) is a polyphenolic compound found in the skin of grapes, in red wine, peanuts and mulberries (Fig. 5). Resveratrol appears to have antiaging properties, cardiovascular protective and cancer prevention activities [340-342]. In cancer prevention, resveratrol blocks proliferation, promotes cell cycle arrest, and induces apoptosis by suppressing extracellular signal regulated kinase signaling, p53, Rb/E2F, cyclins and cdks. It sensitizes cells to extrinsic (Fas and TNF related apoptosis inducing ligand mediated) apoptosis by facilitating death receptor localization into membrane lipid rafts [243] and promotes intrinsic (mitochondrial) apoptosis, in part, by inhibiting survivin and Bcl-XL [344]. Resveratrol suppresses the activity of transcription factors involved in proliferation (e.g., NF-κB, activating protein 1 and the early growth response protein 1), MAPKs and tyrosine kinases (e.g., Src) [345,346] (Fig. 6). Resveratrol also inhibits the proliferation of prostate cancer cells by inhibiting AR transcriptional activity, stimulating PTEN expression, and by blocking Akt phosphorylation [347]. Resveratrol blocks the activity of β-catenin by preventing its accumulation in the nucleus as well as binding to transcription factor 3 [347] (Table 2). Resveratrol suppresses IGFR1 and Wnt pathways in colon cancer cells [348]. Thus, in vitro studies strongly support a role for resveratrol in mediating cell cycle arrest or triggering apoptosis.
Ingestion of resveratrol rich grape powder in humans suppressed expression of the Wnt target genes, cyclin D1 and axin in normal colonic mucosa, suggesting that Wnt pathway inhibition may contribute to resveratrol-mediated colon cancer prevention [349]. Constitutively activated Wnt is also proinflammatory [350]. Since many cancers are associated with prolonged inflammation and chronic tissue damage, resveratrol may also prevent tumor onset by attenuating the regenerative responses that accompany prolonged inflammation.
Resveratrol blocks PI3K and Akt signaling, which strongly promote growth [351], by downregulating cdk2, cyclin D1 and proliferative cell nuclear antigen. In ovarian cancer, resveratrol downregulates Akt and ERK signaling [352] (Fig. 6) and may inhibit prostate cancer growth via the Akt/miR-21 pathway [353]. Resveratrol also suppresses phosphorylation of the NF-κB inhibitor, IκB [354], thereby inhibiting NF-κB [355]. Resveratrol also inhibits Hh [356] and Jak2/STAT3 signaling [357], which contribute to cell proliferation and cancer progression (Fig. 6, Table 3), suggesting that it may be useful in vivo.
Table 3.
Summary of Selected Natural Compounds Active Against Hh Signaling in Cell Proliferation
| Compound | Effects | Cancer Model | References |
|---|---|---|---|
| Zerumbone | induces apoptosis | leukemia, breast cancer | [458,459] |
| Sulforaphane | inhibits Shh signaling by blocking Gli transcription, proliferation and induction of apoptosis. | pancreatic CSCs | [132,245] |
| Cyclopamine | targets Smo protein | targeted CSCs in murine medulloblastoma, pancreatic and breast cancers | [180,460. 461] |
| Curcumin | 95% inhibition of Gli1 mRNA; 80% down-regulates Gli reporter activity | medulloblastoma cells | [462] |
| Apigenin, Baicalein, EGCG, Curcumin, Genistein, Quercetin, Cyclopamine, Resveratrol | 95% inhibition of Gli1 mRNA; 80% down-regulates Gli reporter activity | prostate cancer cells | [463] |
| EGCG | 95% inhibition of Gli1 mRNA; 80% down-regulates Gli reporter activity | chondrosarcoma cells | [464] |
Resveratrol was shown to inhibit proliferation and NF-κB signaling in chemically induced rat liver carcinogenesis [358,359]. In addition, resveratrol affected the expression TGF-β and forkhead box protein C2 by regulating miR-520h [360], suggesting that resveratrol, like cucumin, mediates its effects via epigenetic mechanisms. Although most in vivo studies show that resveratrol has antitumor activity, its antiaging properties are paradoxical in that they promote cell survival [361,362]. These differences may depend upon bioavailability and serum concentrations and underscore the need to conduct carefully crafted clinical trials in the future. For example, the doses of natural compound(s) used for chemoprevention in “healthy” patients may be different than the generally higher pharmacologic doses given to patients already diagnosed with cancer, since in the former group, toxicity should be low, while in the latter group, the risk vs. benefit ratio would be more important.
11.4 Flavonoids
Flavonoids are polyphenolic herbal constituents with a wide range of antiallergic, antiinflammatory, antioxidant, antimicrobial and anticancer activities. Flavonoids inhibit hormone related cancers by modulating the activities of sex steroid hormone receptors [283,363,364]. Upon receptor binding, flavonoids reduce ERα association with the plasma membrane, impair ERα dependent proliferative signaling cascades, and promote apoptosis through the activation of ERα-mediated p38/MAPK [365-367]. This may explain the inverse correlation between the dietary consumption of flavonoids and the incidence of hormone related breast, prostate, testicular, and colorectal cancers [368,369]. While data support the potential medicinal use of flavonoids for the treatment of ER related cancers, their low bioavailability [370], combined with the length of time it takes to run human clinical tirals, has limited their use. Flavonoids were reported to exhibit a comparable activity to that of well known P-glycoprotein [P-gp] inhibitors (e.g., verapamil and cyclosporine), without toxicity to normal cells [371]. Thus, flavonoids have the potential of being useful as chemotherapeutic agents, through derivatives with increased bioavailability, as is the case with paclitaxel and vincristine in multidrug resistant tumor cells [372,373].
11.5 EGCG
Green tea is associated with decreased frequency of cancer development due to the presence of EGCG and other polyphenols (Fig. 5). EGCG suppresses AR expression and signaling. EGCG also blocks the nuclear translocation of NF-κB as a result of decreased inhibitor of NF-κB kinase activity, thereby blocking cancer cell proliferation. Green tea polyphenols also downregulate MAPK activity and VEGF production leading to a block in proliferation [374]. In addition, EGCG inhibits β-catenin nuclear accumulation and subsequent transcription of target genes (Fig. 6, Table 2). EGCG also exerts part of its anticancer activity through epigenetic mechanisms. For example, EGCG can reverse CpG island hypermethylation of various methylation silenced genes and reactivate their expression [375]. EGCG also upregulates such miRNAs as miR-16, let-7c, miR-18, miR-25, and miR-92 and downregulates miR-129, miR-196, miR-200, miR-342, and miR-526 [376]. Furtheremore, EGCG affects the expression of HIF-1α through the regulation of miR-210 [377]. EGCG protects against oxidative damage of DNA, proteins and lipids by acting as a chaperone, and by downregulating multiple signaling pathways (e.g., VEGFR1/R2, EGFR/HER2, PI3K/Akt, IGF/IGFR1, and MAPK) [378], and strongly inhibits the antiapoptotic proteins Bcl-XL and Bcl-2 [379]. However, EGCG promoted DNA damage in mouse leukaemic monocyte macrophage RAW 264.7 and human promyelocytic leukemia HL-60 cell lines in a dose dependent manner [380]. EGCG has also been associated with liver damage, perhaps because it triggers oxidative stress [381]. In light of these results, it is likely that the multiple properties and targets of EGCG and many other natural compounds (Fig. 6, Table 5) impact the outcome of treatment, depending upon dose, duration, and combinations with other therapeutic approaches. In this context, careful consideration must be given to the use of these compounds in the development of novel therapeutics.
Table 5.
Natural Compounds Effective Against Proliferation
| Natural product | Targets |
|---|---|
| Curcumin | NF-κB, PI3K/Akt, and STAT3 [324], mTOR [325]; HIF-1α [476] |
| Sulforaphane | NF-κB [335], Wnt (β-catenin) [336], Hh [132] |
| Resveratrol | NF-κB [354], AR [347], IGFR1 [348], Notch-1 [194], Cyclin/CDKs [351], Wnt (β-catenin) [349], STAT3 [357], PI3K/Akt [347], Hh [356] |
| Genistein | NF-κB [384], FOXO3 [386], Wnt (β-catenin) [392,393], ERβ [397] |
| EGCG | NF-κB, VEGFR1/R2, EGFR/HER2, PI3K/AKT, IGF/IGF1, MAPK, COX-2 [378] |
| Brassinin | PI3K/Akt [338] |
| Indole-3-carbinol | ERα, IGFR1, and IRS-1 [332] |
| Quercetin | ER [366], HER2 [477], mTOR [478] |
| Lycopene | NF-κB [479], IGFR1 [480], PI3K/Akt [481], Wnt (β-catenin) and AR [482], cyclins/CDKs [483] |
11.6 Genistein
Genistein is an isoflavone in soy that inhibits proliferation of breast cancer cells and has colon cancer prevention activity [382,383] (Fig. 5). Genistein blocks NF-κB [384], promotes apoptosis and alters polyamine metabolism [385]. It exerts antiproliferative activity by blocking EGF signaling through forkhead box O3 activity [386,387], has antitumor effects in a non small cell lung cancer cell line [388], regulates the expression of miRNA implicated in controlling proliferation [389], and exhibits additive effects when combined with trastuzumab and cetuximab in breast and oral squamous cell carcinoma cells, respectively [390,391]. Genistein suppresses prostate carcinogenesis in the transgenic adenocarcinoma of the mouse prostate model via inhibition of β-catenin signaling [392]. Treatment also reduced Wnt signaling in mammary epithelial cells [393] and in a colon cancer cell line [394] (Fig. 6, Table 2). Genistein inhibits β-catenin/TCF transcriptional activity, promotes GSK3-β activation (which phosphorylates and promotes degradation of β-catenin), and upregulates expression of E-cadherin (Fig. 6, Table 2). As a phytoestrogen, genistein acts through binding to the ER. ERα activation leads to cell proliferation [395], and ERβ activation promotes cellular differentiation [396]. ERβ signaling counteracts ERα related proliferation. Genistein preferentially activates ERβ-mediated gene transcription [397] which would inhibit proliferation (and tumorigenesis) and promote differentiation. In addition, genistein affects the expression of miRNAs [398], such as upregulating miR-200 [328]. However, its therapeutic actions in vivo have not been consistent, in that genistein exhibited a cancer promoting effect in some tumors [399], suggesting the need for careful selection of patients and safer planning in future clinical trials. This may be concentration dependent, because genistein inhibited cell proliferation at high concentrations and activated of estrogen signaling at low concentrations [331]. In order to better exploit the potential of genistein and limit off target effects, it has been coupled to a monoclonal antibody (B43) and then used for the treatment of patients with acute lymphocytic leukemia and Non-Hodgkin's lymphoma [400]. Genistein has also been coupled to recombinant EGF and then used to treat patients with EGFR+ breast cancer [401]. Additional human studies, where genistein is coupled to specific ligands, need to be conducted to see whether genistein's antitumor properties can be demonstated alone or with cytotoxic or radiation therapy.
11.7 Ellagitannins
Ellagitannins are bioactive polyphenols found in berries and pomegranates that have anticancer, antioxidant and antiinflammatory bioactivities (Fig. 5). Ellagitannins are not absorbed intact into the blood stream but are hydrolyzed to ellagic acid. They are also metabolized by gut flora into urolithins that are bioactive and inhibit prostate cancer cell proliferation by interfering with NF-κB activity [402]. Ellagitannin rich pomegranate extract inhibited proliferation of endothelial and prostate cancer cells, and blocked tumor associated angiogenesis [403]. Urolithin significantly inhibited testosterone induced MCF-7aro cell proliferation most likely by exhibiting antiaromatase activity [404]. In animal studies, ellagitannin rich pomegranate fraction has been shown to retard cell proliferation through suppression of β-catenin and NF-κB pathways in diethylnitrosamine induced hepatocarcinogenesis in rats [405,406]. In clinical studies, pomegranate juice led to a decrease in prostate specific antigen (PSA) levels after primary treatment with surgery or radiation [368]. Furthermore, ellagitannins were also active in regulating the expression of several miRNAs in HepG2 cells [407].
11.8 Lycopene
Lycopene is a lipid soluble carotenoid molecule found in high concentration in red fruit and vegetables. Lycopene has a significant antioxidative activity. Epidemiological studies have shown that consumption of lycopene is inversely related to human prostate cancer [408,409]. Lycopene blocked cell growth in breast, prostate and endometrial cancer cells by inhibiting NF-κB activity [410]. In colon cancer cells, lycopene inhibited Akt signaling. Lycopene treatment suppressed Akt activation, increased the phosphorylation (inactivation) of β-catenin, and stimulated expression of cdk inhibitor p27Kip1 [411]. In addition, lycopene inhibited IGF-1-mediated Akt and AR signaling in rat prostate cancer and reduced AR and β-catenin nuclear localization [412]. Independent evidence, however, failed to show that lycopene altered cell proliferation for a variety of cell types [413]. Given these circumstances, lycopene and many other natural products are currently available as herb and vitamin supplements that are not regulated by the Federal Drug Administration. Although these supplements have no serious side effects, future work will be needed to clarify the use of lycopene in cancer therapy.
11.9 Quercetin
Quercetin, a natural protective bioflavonoid, possesses diverse pharmacologic effects, such as antioxidant, antiinflammatory, antiproliferative, and antiangiogenic activities. Quercetin, at nontoxic concentrations, significantly inhibited Akt and mTOR. Moreover, quercetin exhibited antitumor activity that was manifested by a significant reduction of tumor size in a xenograft mouse model [414]. Quercetin inhibited P-gp function and consequently enhanced the bioavailability of chemotherapeutic agents [415]. Furthermore, tamoxifen underwent extensive hepatic metabolism as a substrate for the efflux of P-gp, breast cancer resistance protein, and multidrug resistance protein 2. As a dual inhibitor of the metabolizing enzyme cytochrome P450, family 3, subfamily A and the multidrug resistance transporter, quercetin increased the absorption and the bioavailability of tamoxifen [416].
11.10 Additional natural products
Additional natural products were also reported to affect tumor proliferation such as 13-cis-retinoic acid which significantly interferes with the activity of NF-κB, c-Fos, activated transcription factor-2, and cyclic adenosine monophosphate response element binding protein that directly or indirectly modulate tumor proliferation [417]. Other natural products, such as parthenolide, the active component in Feverfew (Tanacetum parthenium), also showed strong anticancer and antiinflammatory activities. Parthenolide exhibited strong NF-κB- and STAT-inhibition mediated transcriptional suppression of proapoptotic genes [418]. In addition to the antiproliferative activity of pure natural products, there are many promising medicinal plants and mushrooms [419]. For example, bitter melon (Momordica charantia), which is used as functional food to prevent and treat diabetes and associated complications, exhibited antitumor activity against a number of cancer cell lines without affecting normal cell growth [420]. Moreover, organic extracts of medicinal mushrooms, such as Ganoderma lucidum and Comatus caprinus, exhibited strong and promising antiproliferative activities against a variety of cancer cell lines [421-424]. To appreciate their full potential for future clinical use, isolation and elucidation of active chemical entities will be required, followed by preclinical and clinical evaluation.
11.11 Natural compounds in clinical trials
Although clinical data on natural products in cancer appears efficacious, most studies have not been conducted as randomized clinical trials. Appropriate clinical trials have begun with some natural compounds (Table 4), and these will be important for the development of stand alone or combination therapies. The goal of natural products or herbs is not to replace current cancer therapeutics but rather to augment their activity as adjuvants or to reduce side effects. Existing studies suggest synergistic interactions between cancer chemotherapeutics and natural products, especially when these two approaches act on cancer by different mechanisms. For example, lycopene supplements reduced tumor size and PSA levels in localized prostate cancer patients [425], which is consistent with the inhibition of AR nuclear translocation that was found by in vitro studies. Many of these bioactive natural products are consumed in diets, suggesting a need for developing “medicinal foods.” The latter will be based on phytochemicals directed against specific molecular targets that might be utilized as a diet based combinatorial approach in the prevention and treatment of cancer [426]. Table 5 presents a list of priority targets and corresponding natural products discussed herein. This could provide a foundation for the future research and development of these compounds or their derivatives that would be useful in cancer prevention and/or treatment. However, several “best” targets are not listed because so far there is no natural product that affects these molecules (e.g., Snail/Slug). In addition, several other phytochemicals have been evaluated over the past decade against a variety of different tumor types [331]. In some cases, plant extracts were used, even though the active phytochemical(s) were not known. Further, when purified compounds were used, they often potentially blocked the activities of multiple molecules or pathways, making it difficult to assess their relevant targets [331]. While the compounds in Table 5 have been selected for their activities against proliferation, many of them may also impact upon other hallmarks of cancer. For example, many of these chemicals have additional antiinflammatory and antioxidant effects, including their ability to scavenge free radicals. In generating a single “prototypical” approach for the treatment of preneoplastic and early tumor nodules with natural compounds, a combination of curcumin, genistein and resveratrol is suggested because they each have broad activities against many of the key signaling pathways and targets that drive proliferation. However, the concentration of these phytochemicals used experimentally in in vitro studies vastly exceeds the concentration detected in humans following vegetable and fruit intake. Thus, more stable, absorbable variants will have to be derived in order for these to be considered as chemopreventative or chemotherapeutic agents. The bioavailability, pharmacokinetics (especially when complex mixtures are being evaluated), isolation without copurifying contaminants (e.g., heavy metals), degradation profiles in human tissues, favorable drug-drug interactions in preclinical and clinical trials, and off target effects when used at doses that will probably exceed those found in the dietary sources will need to be determined. In addition, reproducible isolation of active compounds from natural sources is likely to be challenging. This is because activity may depend upon combinations of compounds that may be present in mixtures but separated during isolation, and because the amounts of these natural compounds produced by the plant or other host organism are likely to vary depending upon the environmental conditions in which these hosts grow. Thus, climate changes could greatly effect the reproducible isolation and characterization of compounds from natural sources. An alternative approach would involve chemical synthesis of active natural compounds, but the complex chemical structure of many compounds make them difficult or impossible to synthesize by conventional means. In this case, the key enzymes or metabolic pathways responsible for their synthesis could be isolated and transferred to easily manipulated bacteria or yeast to achieve synthesis of enough material for therapeutic applications. However, as the mechanism(s) whereby these natural products operate are elucidated, better complementation with other drugs (natural or synthetic) may provide opportunities to target proliferation at the appropriate time in the right cell type. Overcoming these limitations will provide an even broader base for their utility in cancer chemoprevention and in blocking tumor progression. Even if natural products are not suitable alone or in combination for cancer chemoprevention, they may be useful as an adjuvant to chemotherapy, radiation therapy, and/or small molecule inhibitors of key signaling molecules. This would prompt the design of clinical trials in which one or multiple natural products (or their derivatives) will be added to standard care therapy for individual tumor types, to see if they are active in reducing tumor burden and/or decreasing the frequency of relapse. Among tumor types where clear risk factors of cancer development are known, natural products could be evaluated to see whether they reduce the risk factors for tumor development.
Table 4.
Clinical Trials using Natural Products
| Target | Drug | Type of Cancer | Phase | Other agents | References |
|---|---|---|---|---|---|
| Notch | aMK0752 | pancreatic | I, II | gemcitabine/radiation | [465,466] |
| breast | I | ||||
| bRO4929097 | renal cell | II | [467] | ||
| colorectal | II | cetuximab, bevacizubmab/FOLFOX | |||
| vismodegib | |||||
| advanced breast | I | ||||
| advanced/meta-static sarcoma | |||||
| Hedgehog | cSaridegib.svg (IPI-926) | head and neck | I | cetuximab | [468,469] |
| colorectal | II | bevacizumab, chemotherapy | |||
| BCC | II | ||||
| solid tumors | I | ||||
| Hedgehog/Notch | bRO4929097 | breast | I | vismodegib | [467] |
| dBMS-833923 | BCC, solid tumors | I | dasatinib or bosutinib | [470,471] | |
| ePF-04449913 | hematologic | I | [472] | ||
| Wnt | Resveratrol | colon | I, II | [473,474] | |
| fPRI-724 | solid tumors | I | [475] | ||
3-((1r,4s)-4-(4-chlorophenylsulfonyl)-4-(2,5-difluorophenyl)cyclohexyl)-propanoic acid is a Ɣ-secretase inhibitor
2,2-dimethyl-N-((S)-6-oxo-6,7-dihydro-5H-dibenzo[b,d]azepin-7-yl)-N′-(2,2,3,3,3-pentafluoro-propyl)-malonamide is a Ɣ-secretase inhibitor
Saridegib is a cyclopamine (2-hydroxylpropyl)-β-cyclodextrin) which is a Smoothened (SMO) inhibitor.
BMS-833923 N-(2-methyl-5-((methylamino)methyl)phenyl)-4-((4-phenylquinazolin-2-yl) amino)benzamide is a Smoothened inhibitor
PF-04449913 (1-((2R,4R)-2-(1H-benzo[d]imidazol-2-yl)-1-methylpiperidin-4-yl)-3-(4-cyano-phenyl)urea is a SMO inhibitor.
PRI-724 specifically inhibits the recruiting of beta-catenin with its coactivator CBP (the binding protein of the cAMP response element-binding protein CREB)
12. Emerging genomic and bioinformatic tools to facilitate application of natural compounds to cancer
The “genomics revolution” is transforming the understanding of cellular processes involved in cancer such as cell proliferation, differentiation, and apoptosis. The Human Genome Project [427] provided the groundwork by generating human and model organism genome assemblies [428-431], new genome based technologies [432,433], and advanced computational infrastructure to handle newly generated massive datasets. New sequencing technologies are now outfitting researchers with unprecedented volumes of data to explore the etiology of cancer. For the first time, biologists are able to characterize how entire genomes, transcriptomes, proteomes, and epigenomes can respond to specific changes in genes and their cellular environments, including insights into underlying mechanisms of proliferation. By applying an integrative systems approach, cancer biologists are beginning to employ an enormously powerful perspective with important translational consequences. These tools will be critical in assessing the impact of natural compounds, alone or in combination, for cancer prevention and treatment.
Novel genomic data are accumulating at an ever increasing rate. Massive genome wide surveys of tumor and nontumor genomes from the Cancer Genome Project are providing new opportunities to track how cancers develop over time [434] and find common molecular mechanisms that could be therapeutically targeted. The ENCODE project has functionally annotated genomes with great precision [435]. Perhaps, the single most important feature of these data is that they are freely available via publicly accessible and expertly curated databases (e.g., NCBI's GenBank, Sanger Institute, University of California, Santa Cruz Genome Browser) allowing for greater opportunities to transform cancer research from single genomic phenomena to systems wide analyses (Fig. 7). As a result, bioinformatics has become a critical interdisciplinary tool enabling researchers to handle, analyze, mine, and explore high throughput data. Bioinformatics will become even more important as new genomic technologies increase the super exponential rate at which tumor based data of all sorts are generated.
Fig. 7.
Evolution of genomic resources aimed at identification of cancer targets. There are a growing number of accessible genomic resources that provide an empirical foundation to identify genome-wide targets of tumor cell proliferation.
In the postgenomics era, there is a transformational shift in the understanding of gene regulation, mutational changes, and epigenomic consequences in cell proliferation at the genomic level. While the study of cancer represents a difficult challenge due to the sheer diversity and complexity of its multiple genetic factors, the good news is that remarkable resources are rapidly accumulating in the public domain that will help to translate these recent advances in genomics and bioinformatics to the clinical setting through such approaches as the identification of biomarkers involved in tumor proliferation (Fig. 7). The challenge is to train researchers who can interpret the molecular data and are able to handle and integrate massive and diverse datasets. The ability to integrate these data types into comprehensive dynamic models using genetic pathways, protein-protein interaction networks, and genetic regulatory networks will ultimately connect changes at the molecular level to the phenotypic level, with potential clinical outcomes in arresting tumor progression.
13. Conclusions and prospects
Targeting mechanisms involved in uncontrolled cancer cell proliferation will increasingly become the standard of care for cancer patients, and this review has highlighted some the the “best” targets in several pathways that regulate proliferation. For example, in preneoplastic tissue the expression of Snail1/2 will need to be blocked in order to maintain E-cadherin expression and tissue integrity and to prevent EMT. Once E-cadherin is lost, and hypoxia develops, it will be important to block HIF-1 either directly or indirectly by utilizing inhibitors of Hsp90, topoisomerase, or mTOR. Targeting the generation and growth of CSCs will be a major challenge, due to the overlap of currently identified pathways between CSCs and SCs. Maintaining CSC dormancy may prevent reactivation of disease once the mature tumor cells have been eliminated by conventional therapies and remission has been achieved, but it is not clear how this can be done. Antagonists of Wnt, Hh and MAPK p38 signaling may be of some value in this, while increasing C/EBPα expression, which promotes differentiation, might push CSC into a therapy sensitive state. In addition to β-catenin, there is rationale to seek antagonists of Notch, HER2/ErbB2 and the IGF-1/IGFR1 signaling pathways, since these are central to cell fate decisions and are often reactivated in cancer. In the context of the cell cycle, it will be important to target cyclin D and cdk4/6. Among hormone sensitive cancers, ER and AR are prime targets for the continued development of antagonists. Thus, there are a number of putative targets in sustained proliferative signaling that may be considered for future drug development and evaluation.
Table 6 lists the target molecules and pathways that seem to be the most important in promoting sustained proliferation and enabling cells to acquire other hallmarks of cancer. As shown, there are 10 hallmarks listed, and an eleventh category, “tumor microenvironment,” which is not a hallmark but is being evaluated here as well. For example, HIF-1 signaling is an important therapeutic target for all the cancer hallmarks with the probable exception of evasion of growth suppression and the immune system evasion. Since HIF-1 promotes survival and growth under hypoxic conditions, it may be a therapeutic target in 8 out of the 10 cancer hallmarks where data is reported (which includes sustained proliferative signaling). Thus, it is likely that drugs that block HIF-1 signaling will be effective against multiple tumor types in their very early stages. Among other pathways, inhibiting PI3K/Akt signaling would block all of the hallmarks, while inhibiting Wnt signaling would block at least 6 of the 10 hallmarks (Table 6). Since blocking NF-κB or Wnt signaling promote or suppress replicative immortality, corresponding inhibitors will have to be evaluated more carefully for each tumor type. Likewise, drugs that would block development of some hallmarks actually promote the growth of androgen and estrogen dependent tumors (Table 6). When considering which hallmarks would make the best targets for therapeutic intervention, resistance to apoptosis, deregulated cell metabolism, evasion of growth suppression, the development of genomic instability, in addition to sustained proliferative signaling, would rank high. These hallmarks seem to be sensitive to inhibition from many signaling pathways, although which pathways are most important remains to be determined. Importantly, tissue/tumor interactions in the microenvironment would also be an excellent target for therapeutic intervention.
Table 6.
Priority targets for sustained proliferative signaling
| Targets for sustained proliferationa | (inhibits) HIF-1 signaling | (inhibits) NF-κB signaling | (inhibits) PI3K/Akt signaling | (inhibits) Wnt (β-catenin) | (inhibits) IGFR1 signaling | (attenuates) cell cycle (CDKs/cyclins) | (suppresses] AR signaling | (suppresses) ER signaling |
|---|---|---|---|---|---|---|---|---|
| Other cancer hallmarks | ||||||||
| Genomic instability | + [484,635]b |
+ [485] |
+ [486] |
0 | 0 | + [487] |
+ [488] |
+ [489] |
|
Tumor-promoting inflammation |
+ [490] |
+ [491,492] |
+ [493-495] |
+ [496-498] |
+ [499,500] |
+ [501,502] |
+/− [503,504] |
+/− [505.506] |
|
Evasion of anti- growth signaling |
0 | +/− [507] |
+ [508,509] |
+ [510.511] |
+ [512,513] |
+ [514,515] |
+ [516-518] |
+ [519,520] |
|
Resistance to apoptosis |
+ [521,522] |
+ [523,524] |
+ [525] |
+ [526] |
+ [527] |
+ [528] |
+ [529] |
+ [530] |
|
Replicative immortality |
+ [531] |
+/− [225,532,533] |
+ [534-536] |
+/− [537.538] |
+ [706,707] |
+ [539.540] |
+ [541,542] |
+ [543-546] |
|
Deregulated metabolism |
+ [45,547,548] |
+ [549] |
+ [548,560] |
+ [561] |
+ [562.563] |
+ [564,565] |
0 | + [566] |
|
Immune system evasion |
0 | + [567] |
+ [568] |
+ [569,570] |
0 | − [571,572] |
+ [573,574] |
+ [575,576] |
| Angiogenesis | + [727] |
+/− [355,577] |
+ [578,728,646] |
+/− [579,636] |
+ [580] |
+ [582] |
+ [583] |
+/− [584,585] |
|
Tissue invasion and metastasis |
+ [5,585] |
+ [586,587,708] |
+ [588] |
+/− [589] |
+ [563,590] |
− [591,592] |
+/− [593] |
− [594] |
| Other characteristics | + [595] |
+ [596] |
+ [597] |
+ [598,599] |
+ [600] |
+ [601,602] |
+ [603] |
+/− [601,604-606] |
|
Tissue interactions in the tumor microenvironment |
The cross validation above documents whether the therapeutic targets that are important to block sustained proliferative signaling also block additional hallmarks of cancer as indicated by the cited references. In identifying these putative therapeutic targets, + = putative target in sustained proliferative signaling that is shared with other hallmarks (complementary relationship); 0 = putative target not documented in other hallmarks (no relationship); +/− = putative target whose inhibition would promote other selected hallmarks (making it controversial as to whether the molecule/pathway should be targeted in cancer at all); − = putative target that promotes sustained proliferative signaling but inhibits other selected hallmarks (target inhibition has opposite effects, depending upon the hallmark; suggesting a contrary relationship). For example, HIF-1 is activated by mutation, confers resistance to apoptosis, supports replicative immortality, deregulates cell metabolism, stimulates angiogenesis, metastasis, and alters tumor microenvironment. All of these features promote tumorigenesis, suggesting it is a good target for therapeutic intervention in multiple steps of tumor pathogenesis.
The numbers in each box are the references that document whether the putative therapeutic target in each column is also a target for the other hallmarks.
To determine which natural compounds are likely to be most effective against multiple cancer hallmarks, a literature search was performed. Table 7 shows that curcumin, which targets HIF-1, NF-κB, and PI3K/Akt signaling (Table 5), is also active against all of the cancer hallmarks, as well as the tumor microenvironment. Genistein is effective against at least 6 of the 10 hallmarks while resveratrol is effective in 8 of 10. However, in some cases the, appropriate studies comparing a compound and a particular hallmark have not been performed, so that “no relationship” between these drugs and some of the hallmarks has been reported. In the case of genistein, it is not clear whether it promotes or inhibits the deregulation of cell metabolism, evasion of growth suppression, immune system evasion, and tumor promoting inflammation. For resveratrol, different reports document a positive or negative effect of this compound upon immune system evasion and angiogenesis. Importantly, all three of these compounds block the development of other hallmarks, which are critical steps in carcinogenesis. Importantly, all of these compounds also appear to be effective against the tumor microenvironment. Thus, combination therapy with curcumin, genistein and resveratrol may be effective against multiple targets (and cancer hallmarks) in many tumor types, suggesting that they would be very good choices for further clinical development and widespread application.
Table 7.
Therapeutic approaches targeting sustained proliferative signaling
| Phytochemical approaches | Curcumina | Resveratrol | Genistein |
|---|---|---|---|
| Other cancer hallmarksa | |||
| Genomic instability | + [611,617,651-652]b |
+ [673,674,709-711] |
+ [690,722] |
| Tumor-promoting Inflammation | + [323,607,609,653-655] |
+ [348,354,626,627,675-677] |
+/− [399,691-693,723-725] |
| Evasion of anti-growth signaling | + [619,643,656-658] |
+ [348,628,678.679] |
+/− [399,694-696] |
| Resistance to apoptosis | + [348,451,525,541,608,612,614,616,620-623,632,643,659] |
+ [194,345,348,625,628,637,680] |
+ [697] |
| Replicative immortality | + [631,660-662] |
+ [681,682] |
+ [630,698] |
| Deregulated metabolism | + [476,607,631,640,663,664] |
+ [6,28,74,76,348,627,683,684,712-716] |
+/− [76,649,699,700] |
| Immune system evasion | + [76,665] |
+/− [717-721] |
+/− [726] |
| Angiogenesis | + [476,610,615,633,666] |
+/− [624,681,685,714] |
+ [638,701] |
| Tissue invasion and metastasis | + [608,613,618,645,667-670] |
+ [353,629,639,641,686,687] |
+ [647,648,702] |
| Other characteristics | + [644,660,671,672] |
+ [688,689] |
+ [703,704] |
| Tissue interactions in the tumor microenvironment |
The cross validation above documents whether the compounds that are most likely to be effective against sustained proliferative signaling also block additional hallmarks of cancer as indicated by the cited references. In identifying natural compounds most useful for therapeutic intervention, + = compounds that are effective against sustained proliferative signaling and other hallmarks (complementary relationship); 0 = compound effective against sustained proliferative signaling that are not documented in other hallmarks (no relationship); +/− = compound effective against sustained proliferative signaling promotes but not consistently against other selected hallmarks (controversial as to whether the compound should be used against cancer at all); − = compound effective against sustained proliferative signaling shown to promote tumorigenesis by measuring other selected hallmarks (drug has opposite effects; suggesting a contrary relationship).
The numbers in each box are the references that document whether the putative compound in each column also targets other hallmarks.
Acknowledgements
Authors 1-13 contributed to the bulk of the work while the remaining authors contributed to cross-validation activities. Drs. Feitelson and Arzumanyan were supported by NIH (AI076535) and by Temple University. Dr. Rob J. Kulathinal was supported by the National Science Foundation, and by the American Cancer Society. Dr. Marino was supported by grant from University Roma Tre (CLA 2013) and by the Italian Association for Cancer Research (no. IG15221). Dr. Georgakilas was supported by the EU Marie Curie Reintegration Grant (MC-CIG-303514), Greek National funds through the Operational Program ‘Educational and Lifelong Learning of the National Strategic Reference Framework (NSRF)-Research Funding Program: THALES (MIS 379346) and COST Action CM1201 ‘Biomimetic Radical Chemistry.’ Dr. Amedei was supported by the Italian Ministry of University and University of Italy. Dr. Amin was supported by the Terry Fox Foundation (TF-36), UAEU Program for Advanced Research (UPAR25183), Al-Jalila Foundation (AJF201454) and Zayed Center for Health Sciences (ZCHS2014). Dr. Sanchez-Garcia was supported by FEDER, by MICINN (SAF2012-32810), by NIH (R01 CA109335-04A1), by Junta de Castilla y León (BIO/SA06/13), by the ARIMMORA project (FP7-ENV-2011, EU 7th Framework Program) and by the EuroSyStem and the DECIDE Network (EU FP7). Dr. Sharma was funded by NIH grants (R01CA131294, CA155686), the Avon Foundation and a Breast Cancer Research Foundation grant (90047965). Dr. Saxena was supported by a grant from NIH (K01DK077137 and R03DK089130). Dr. Singh was supported by the Fast Track Scheme for Young Scientists, Department of Science and Technology, India (SR/FT/LS-063/2008). Dr. Honoki was supported by a grant from the Ministry of Education, Culture, Sports, Science and Technology, Japan (no. 24590493). Dr. Ciriolo was supported by the Italian Association for Cancer Research (AIRC - grant #IG10636). Dr. Aquilano was supported by MIUR-PRIN (20125S38FA_002) and Ministero della Salute (GR-2011-02348047). Dr. Chen was funded from the Ovarian and Prostate Cancer Research Trust, UK. Dr. Mohammed is supported by the Purdue University Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Harris AL. Hypoxia a key regulatory factor in tumor growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 2.Qing G, Simon MC. Hypoxia inducible factor-2alpha: a critical mediator of aggressive tumor phenotypes. Curr Opin Genet Dev. 2009;19:60–66. doi: 10.1016/j.gde.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qiang L, Wu T, Zhang HW, Lu N, Hu R, Wang YJ, et al. HIF-1alpha is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ. 2012;19:284–294. doi: 10.1038/cdd.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iida H, Suzuki M, Goitsuka R, Ueno H. Hypoxia induces CD133 expression in human lung cancer cells by up-regulation of OCT3/4 and SOX2. Int J Oncol. 2012;40:71–79. doi: 10.3892/ijo.2011.1207. [DOI] [PubMed] [Google Scholar]
- 5.Bos R, van Diest PJ, van der Groep P, Shvarts A, Greijer AE, van der Wall E. Expression of hypoxia-inducible factor-1alpha and cell cycle proteins in invasive breast cancer are estrogen receptor related. Breast Cancer Res. 2004;6:R450–459. doi: 10.1186/bcr813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brahimi-Horn MC, Chiche J, Pouysségur J. Hypoxia and cancer. J Mol Med (Berl) 2007;85:1301–1307. doi: 10.1007/s00109-007-0281-3. [DOI] [PubMed] [Google Scholar]
- 7.Gillies RJ, Gatenby RA. Hypoxia and adaptive landscapes in the evolution of carcinogenesis. Cancer Metastasis Rev. 2007;26:311–317. doi: 10.1007/s10555-007-9065-z. [DOI] [PubMed] [Google Scholar]
- 8.Fang JS, Gillies RD, Gatenby RA. Adaptation to hypoxia and acidosis in carcinogenesis and tumor progression. Semin Cancer Biol. 2008;18:330–337. doi: 10.1016/j.semcancer.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hirota K, Semenza GL. Regulation of angiogenesis by hypoxia-inducible factor 1. Crit Rev Oncol Hematol. 2006;59:15–26. doi: 10.1016/j.critrevonc.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 10.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 11.Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132:3151–3161. doi: 10.1242/dev.01907. [DOI] [PubMed] [Google Scholar]
- 12.Batlle E, Sancho E, Francí C, Domínguez D, Monfar M, Baulida J, et al. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumor cells. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 13.Nieto MA. The Snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002;3:155–166. doi: 10.1038/nrm757. [DOI] [PubMed] [Google Scholar]
- 14.Yokoyama K, Kamata N, Fujimoto R, Tsutsumi S, Tomonari M, Taki M, et al. Increased invasion and matrix metalloproteinase-2 expression by Snail-induced mesenchymal transition in squamous cell carcinomas. Int J Oncol. 2003;22:891–898. [PubMed] [Google Scholar]
- 15.Perez-Losada J, Sánchez-Martín M, Rodríguez-García A, Sánchez ML, Orfao A, Flores T, et al. Zinc-finger transcription factor Slug contributes to the function of the stem cell factor c-kit signaling pathway. Blood. 2002;100:1274–1286. [PubMed] [Google Scholar]
- 16.Perez-Mancera PA, González-Herrero I, Pérez-Caro M, Gutiérrez-Cianca N, Flores T, Gutiérrez-Adán A, et al. SLUG in cancer development. Oncogene. 2005;24:3073–3082. doi: 10.1038/sj.onc.1208505. [DOI] [PubMed] [Google Scholar]
- 17.Perez-Mancera PA, Pérez-Caro M, González-Herrero I, Flores T, Orfao A, de Herreros AG, et al. Cancer development induced by graded expression of Snail in mice. Human Mol Genet. 2005;14:3449–3461. doi: 10.1093/hmg/ddi373. [DOI] [PubMed] [Google Scholar]
- 18.Franci C, Takkunen M, Dave N, Alameda F, Gómez S, Rodríguez R, et al. Expression of Snail protein in tumor-stroma interface. Oncogene. 2006;25:5134–5144. doi: 10.1038/sj.onc.1209519. [DOI] [PubMed] [Google Scholar]
- 19.Cobaleda C, Pérez-Caro M, Vicente-Dueñas C, Sánchez-García I. Function of the zinc-finger transcription factor SNAI2 in cancer and development. Annu Rev Genet. 2007;41:41–61. doi: 10.1146/annurev.genet.41.110306.130146. [DOI] [PubMed] [Google Scholar]
- 20.Escriva M, Peiró S, Herranz N, Villagrasa P, Dave N, Montserrat-Sentís B, et al. Repression of PTEN phosphatase by Snail1 transcriptional factor during gamma radiation-induced apoptosis. Mol Cell Biol. 2008;28:1528–1540. doi: 10.1128/MCB.02061-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kajita M, McClinic KN, Wade PA. Aberrant expression of the transcription factors Snail and Slug alters the response to genotoxic stress. Mol Cell Biol. 2004;24:7559–7566. doi: 10.1128/MCB.24.17.7559-7566.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y, Yue B, Yu X, Wang Z, Wang M. SLUG is activated by nuclear factor kappa B and confers human alveolar epithelial A549 cells resistance to tumor necrosis factor-alpha-induced apoptosis. World J Surg Oncol. 2013;11:12. doi: 10.1186/1477-7819-11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vega S, Morales AV, Ocaña OH, Valdés F, Fabregat I, Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004;18:1131–1143. doi: 10.1101/gad.294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bermejo-Rodriguez C, Pérez-Caro M, Pérez-Mancera PA, Sánchez-Beato M, Piris MA, Sánchez-García I. Mouse cDNA microarray analysis uncovers Slug targets in mouse embryonic fibroblasts. Genomics. 2006;87:113–118. doi: 10.1016/j.ygeno.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 25.Catalano A, Rodilossi S, Rippo MR, Caprari P, Procopio A. Induction of stem cell factor/c-Kit/slug signal transduction in multidrug-resistant malignant mesothelioma cells. J Biol Chem. 2004;279:46706–46714. doi: 10.1074/jbc.M406696200. [DOI] [PubMed] [Google Scholar]
- 26.Criswell L, Arteaga CL. Modulation of NFkappaB activity and E-cadherin by the type III transforming growth factor beta receptor regulates cell growth and motility. J Biol Chem. 2007;282:32491–32500. doi: 10.1074/jbc.M704434200. [DOI] [PubMed] [Google Scholar]
- 27.Leong KG, Niessen K, Kulic I, Raouf A, Eaves C, Pollet I, et al. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J Exp Med. 2007;204:2935–2948. doi: 10.1084/jem.20071082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 1998;392:190–193. doi: 10.1038/32433. [DOI] [PubMed] [Google Scholar]
- 29.Vleminck K, Vakaet L, Jr, Mareel M, Fiers W, van Roy F. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell. 1991;66:107–119. doi: 10.1016/0092-8674(91)90143-m. [DOI] [PubMed] [Google Scholar]
- 30.Blechschmidt K, Sassen S, Schmalfeldt B, Schuster T, Höfler H, Becker KF. The E-cadherin repressor Snail is associated with lower overall survival of ovarian cancer patients. Br J Cancer. 2008;98:489–495. doi: 10.1038/sj.bjc.6604115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mani SA, Yang J, Brooks M, Schwaninger G, Zhou A, Miura N, et al. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc Natl Acad Sci USA. 2007;104:10069–10074. doi: 10.1073/pnas.0703900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shih JY, Tsai MF, Chang TH, Chang YL, Yuan A, Yu CJ, et al. Transcription repressor Slug promotes carcinoma invasion and predicts outcome of patients with lung adenocarcinoma. Clin Cancer Res. 2005;11:8070–8078. doi: 10.1158/1078-0432.CCR-05-0687. [DOI] [PubMed] [Google Scholar]
- 33.Shioiri M, Shida T, Koda K, Oda K, Seike K, Nishimura M, et al. Slug expression is an independent prognostic parameter for poor survival in colorectal carcinoma patients. Br J Cancer. 2006;94:1816–1822. doi: 10.1038/sj.bjc.6603193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Z, Li Y, Ahmad A, Azmi AS, Kong D, Banerjee S, Sarkar FH. Targeting miRNAs involved in cancer stem cell and EMT regulation: An emerging concept in overcoming drug resistance. Drug Resist Updat. 2010;13:109–118. doi: 10.1016/j.drup.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 36.Kapitsinou PP, Haase VH. The VHL tumor suppressor and HIF: insights from genetic studies in mice. Cell Death Differ. 2008;15:650–659. doi: 10.1038/sj.cdd.4402313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaelin WG. The von Hippel-Lindau tumor suppressor protein: O2 sensing and cancer. Nature Rev Cancer. 2008;8:865–873. doi: 10.1038/nrc2502. [DOI] [PubMed] [Google Scholar]
- 38.Leiser SF, Kaeberlein M. A role for SIRT1 in the hypoxic response. Mol Cell. 2010;38:779–780. doi: 10.1016/j.molcel.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 39.Acker T, Plate KH. A role for hypoxia and hypoxia-inducible transcription factors in tumor physiology. J Mol Med (Berl) 2002;80:562–575. doi: 10.1007/s00109-002-0355-1. [DOI] [PubMed] [Google Scholar]
- 40.Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20:87–90. doi: 10.1016/j.gde.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–3899. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- 42.Fan Y, Dickman KG, Zong WX. Akt and c-Myc differentially activate cellular metabolic programs and prime cells to bioenergetic inhibition. J Biol Chem. 2010;285:7324–7333. doi: 10.1074/jbc.M109.035584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rapisarda A, Melillo G. Overcoming disappointing results with antiangiogenic therapy by targeting hypoxia. Nat Rev Clin Oncol. 2012;9:378–390. doi: 10.1038/nrclinonc.2012.64. [DOI] [PubMed] [Google Scholar]
- 44.Kim JW, Gao P, Dang CV. Effects of hypoxia on tumor metabolism. Cancer Metastasis Rev. 2007;26:291–298. doi: 10.1007/s10555-007-9060-4. [DOI] [PubMed] [Google Scholar]
- 45.Weljie AM, Jirik FR. Hypoxia-induced metabolic shifts in cancer cells: moving beyond the Warburg effect. Int J Biochem Cell Biol. 2011;43:981–989. doi: 10.1016/j.biocel.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 46.Shay JES, Simon MC. Hypoxia-inducible factors: Crosstalk between inflammation and metabolism. Semin Cell Develop Biol. 2012;23:389–394. doi: 10.1016/j.semcdb.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 47.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jedinak A, Dudhgaonkar S, Sliva D. Activated macrophages induce metastatic behavior of colon cancer cells. Immunobiology. 2010;215:242–249. doi: 10.1016/j.imbio.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 49.Greenhough A, Smartt HJ, Moore AE, Roberts HR, Williams AC, Paraskeva C, et al. Genetic aspects of inflammation and cancer. Biochem J. 2008;410:225–235. doi: 10.1042/BJ20071341. [DOI] [PubMed] [Google Scholar]
- 50.Imtiyaz HZ, Williams EP, Hickey MM, Patel SA, Durham AC, Yuan LJ, et al. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest. 2010;120:2699–2714. doi: 10.1172/JCI39506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ, et al. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol. 2000;157:411–421. doi: 10.1016/s0002-9440(10)64554-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. NFkappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eskelinen EL, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta. 2009;1793:664–673. doi: 10.1016/j.bbamcr.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 54.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 56.Hu Y, Lu W, Chen G, Wang P, Chen Z, Zhou Y, et al. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2011;22:399–416. doi: 10.1038/cr.2011.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brahimi-Horn M, C Bellot G, Pouysse'gur J. Hypoxia and energetic tumor metabolism. Curr Opin Genet Dev. 2011;21:67–72. doi: 10.1016/j.gde.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 58.Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, Khare S, et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J Clin Invest. 2008;118:3917–3929. doi: 10.1172/JCI35512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis inflammation and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rosenfeldt MT, Ryan KM. The multiple roles of autophagy in cancer. Carcinogenesis. 2011;32:955–963. doi: 10.1093/carcin/bgr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wilkinson S, Ryan KM. Autophagy: an adaptable modifier of tumorigenesis. Curr Opin Genet Dev. 2010;20:57–64. doi: 10.1016/j.gde.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 62.Levy JM, Thorburn A. Targeting autophagy during cancer therapy to improve clinical outcomes. Pharmacol Ther. 2011;131:130–141. doi: 10.1016/j.pharmthera.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, et al. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65:3336–3346. doi: 10.1158/0008-5472.CAN-04-3640. [DOI] [PubMed] [Google Scholar]
- 64.Maiuri MC, Malik SA, Morselli E, Kepp O, Criollo A, Mouchel PL, et al. Stimulation of autophagy by the p53 target gene Sestrin2. Cell Cycle. 2009;8:1571–1576. doi: 10.4161/cc.8.10.8498. [DOI] [PubMed] [Google Scholar]
- 65.He C, Levine B. The Beclin 1 interactome. Curr Opin Cell Biol. 2010;22:140–149. doi: 10.1016/j.ceb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pattingre S, Espert L, Biard-Piechaczyk M, Codogno P. Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie. 2008;90:313–323. doi: 10.1016/j.biochi.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 67.Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4:600–606. doi: 10.4161/auto.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Koehl GE, Spitzner M, Ousingsawat J, Schreiber R, Geissler EK, Kunzelmann K. Rapamycin inhibits oncogenic intestinal ion channels and neoplasia in APC(Min/þ) mice. Oncogene. 2010;29:1553–1560. doi: 10.1038/onc.2009.435. [DOI] [PubMed] [Google Scholar]
- 69.Memmott RM, Mercado JR, Maier CR, Kawabata S, Fox SD, Dennis PA. Metformin prevents tobacco carcinogen-induced lung tumorigenesis. Cancer Prev Res (Phila) 2010;3:1066–1076. doi: 10.1158/1940-6207.CAPR-10-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roper J, Richardson MP, Wang WV, Richard LG, Chen W, Coffee EM, et al. The dual PI3K/mTOR inhibitor NVP-BEZ235 induces tumor regression in a genetically engineered mouse model of PIK3CA wild-type colorectal cancer. PLoS One. 2011;6:e25132. doi: 10.1371/journal.pone.0025132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Janes MR, Limon JJ, So L, Chen J, Lim RJ, Chavez MA, et al. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med. 2010;16:205–213. doi: 10.1038/nm.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fan QW, Cheng C, Hackett C, Feldman M, Houseman BT, Nicolaides T, et al. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci Signal. 2010;3:ra81. doi: 10.1126/scisignal.2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cloonan SM, Williams DC. The antidepressants maprotiline and fluoxetine induce Type II autophagic cell death in drug-resistant Burkitt's lymphoma. Int J Cancer. 2010;128:1712–1723. doi: 10.1002/ijc.25477. [DOI] [PubMed] [Google Scholar]
- 74.Fu Y, Chang H, Peng X, Bai Q, Yi L, Zhou Y, et al. Resveratrol inhibits breast cancer stem-Like cells and induces autophagy via suppressing Wnt/β-catenin signaling pathway. PLoS One. 2014;9:e102535. doi: 10.1371/journal.pone.0102535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xiao K, Jiang J, Guan C, Dong C, Wang G, Bai L, et al. Curcumin induces autophagy via activating the AMPK signaling pathway in lung adenocarcinoma cells. J. Pharmacol Sci. 2013;123:102–109. doi: 10.1254/jphs.13085fp. [DOI] [PubMed] [Google Scholar]
- 76.Hasima N, Ozpolat B. Regulation of autophagy by polyphenolic compounds as a potential therapeutic strategy for cancer. Cell Death Dis. 2014;5:e1509. doi: 10.1038/cddis.2014.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15:5308–5316. doi: 10.1158/1078-0432.CCR-07-5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang C, Chung L, et al. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood. 2007;110:313–322. doi: 10.1182/blood-2006-10-050260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–729. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–470. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li X, Fan Z. The epidermal growth factor receptor antibody cetuximab induces autophagy in cancer cells by downregulating HIF-1alpha and Bcl-2 and activating the beclin 1/hVps34 complex. Cancer Res. 2010;70:5942–5952. doi: 10.1158/0008-5472.CAN-10-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Han J, Hou W, Goldstein LA, Lu C, Stolz DB, Yin XM, et al. Involvement of protective autophagy in TRAIL resistance of apoptosis-defective tumor cells. J Biol Chem. 2008;283:19665–19677. doi: 10.1074/jbc.M710169200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelárová H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem. 1997;243:240–246. doi: 10.1111/j.1432-1033.1997.0240a.x. [DOI] [PubMed] [Google Scholar]
- 84.Shacka JJ, Klocke BJ, Roth KA. Autophagy bafilomycin and cell death: the “a-B-cs” of plecomacrolide-induced neuroprotection. Autophagy. 2006;2:228–230. doi: 10.4161/auto.2703. [DOI] [PubMed] [Google Scholar]
- 85.Kimura T, Takabatake Y, Takahashi A, Isaka Y. Chloroquine in cancer therapy: A double-edged sword of autophagy. Cancer Res. 2013;73:3–7. doi: 10.1158/0008-5472.CAN-12-2464. [DOI] [PubMed] [Google Scholar]
- 86.Rossi M, Munarriz ER, Bartesaghi S, Milanese M, Dinsdale D, Guerra-Martin MA, et al. Desmethylclomipramine induces the accumulation of autophagy markers by blocking autophagic flux. J Cell Sci. 2009;122:3330–3339. doi: 10.1242/jcs.048181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Carew JS, Espitia CM, Esquivel JA, Mahalingam D, Kelly KR, Reddy G, et al. Lucanthone is a novel inhibitor of autophagy that induces cathepsin D-mediated apoptosis. J Biol Chem. 2011;286:6602–6613. doi: 10.1074/jbc.M110.151324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kawaguchi T, Miyazawa K, Moriya S, Ohtomo T, Che XF, Naito M, et al. Combined treatment with bortezomib plus bafilomycin A1 enhances the cytocidal effect and induces endoplasmic reticulum stress in U266 myeloma cells: crosstalk among proteasome, autophagy-lysosome and ER stress. Int J Oncol. 2011;38:643–654. doi: 10.3892/ijo.2010.882. [DOI] [PubMed] [Google Scholar]
- 89.Xie Z, Xie Y, Xu Y, Zhou H, Xu W, Dong Q. Bafilomycin A1 inhibits autophagy and induces apoptosis in MG63 osteosarcoma cells. Mol Med Rep. 2014;10:1103–1107. doi: 10.3892/mmr.2014.2281. [DOI] [PubMed] [Google Scholar]
- 90.Kaewpiboon C, Surapinit S, Malilas W, Moon J, Phuwapraisirisan P, Tip-Pyang S, et al. Feroniellin A-induced autophagy causes apoptosis in multidrug-resistant human A549 lung cancer cells. Int J Oncol. 2014;44:1233–1242. doi: 10.3892/ijo.2014.2297. [DOI] [PubMed] [Google Scholar]
- 91.Lao Y, Wan G, Liu Z, Wang X, Ruan P, Xu W, et al. The natural compound oblongifolin C inhibits autophagic flux and enhances antitumor efficacy of nutrient deprivation. Autophagy. 2014;10:736–749. doi: 10.4161/auto.28034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Trosko JE, Chang CC, Upham BL, Tai MH. Ignored hallmarks of carcinogenesis: stem cells and cell-cell communication. Ann NY Acad Sci. 2004;1028:192–201. doi: 10.1196/annals.1322.023. [DOI] [PubMed] [Google Scholar]
- 93.Mimeault M, Batra SK. Concise review: recent advances on the significance of stem cells in tissue regeneration and cancer therapies. Stem Cells. 2006;24:2319–2345. doi: 10.1634/stemcells.2006-0066. [DOI] [PubMed] [Google Scholar]
- 94.Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea - a paradigm shift. Cancer Res. 2006;66:1883–1890. doi: 10.1158/0008-5472.CAN-05-3153. [DOI] [PubMed] [Google Scholar]
- 95.Pece S. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140:62–73. doi: 10.1016/j.cell.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 96.Mimeault M, Batra SK. Interplay of distinct growth factors during epithelial mesenchymal transition of cancer progenitor cells and molecular targeting as novel cancer therapies. Ann Oncol. 2007;18:1605–1619. doi: 10.1093/annonc/mdm070. [DOI] [PubMed] [Google Scholar]
- 97.Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J, et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci USA. 2007;104:4048–4053. doi: 10.1073/pnas.0611682104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 99.Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66:7843–7848. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 100.Zeng X, Huang H, Tamai K, Zhang X, Harada Y, Yokota C, et al. Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, disheveled and axin functions. Development. 2008;135:367–375. doi: 10.1242/dev.013540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang Y, Deng J, et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem like cells is required for viability and maintenance. Proc Natl Acad Sci USA. 2007;104:16158–16163. doi: 10.1073/pnas.0702596104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fodde R, Brabletz T. Wnt/β-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol. 2007;19:150–158. doi: 10.1016/j.ceb.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 103.Hede K. PTEN takes center stage in cancer stem cell research, works as tumor suppressor. J Nat Cancer Inst. 2006;98:808–809. doi: 10.1093/jnci/djj270. [DOI] [PubMed] [Google Scholar]
- 104.Sánchez-García I, Vicente-Dueñas C, Cobaleda C. The theoretical basis of cancer-stem-cell-based therapeutics of cancer: can it be put into practice? BioEssays. 2007;29:1269–1280. doi: 10.1002/bies.20679. [DOI] [PubMed] [Google Scholar]
- 105.Hamburger A, Salmon SE. Primary bioassay of human myeloma stem cells. J Clin Invest. 1977;60:846–854. doi: 10.1172/JCI108839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Falzacappa V, Ronchini C, Reavie LB, Pelicci PG. Regulation of self-renewal in normal and cancer stem cells. FEBS J. 2012;279:3559–3572. doi: 10.1111/j.1742-4658.2012.08727.x. [DOI] [PubMed] [Google Scholar]
- 107.Sánchez-García I. The crossroads of oncogenesis and metastasis. New Engl J Med. 2009;360:297–299. doi: 10.1056/NEJMcibr0808031. [DOI] [PubMed] [Google Scholar]
- 108.Saito Y, Uchida N, Tanaka S, Suzuki N, Tomizawa-Murasawa M, Sone A, et al. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat Biotechnol. 2010;28:275–280. doi: 10.1038/nbt.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Westphalen CB, Asfaha S, Hayakawa Y, Takemoto Y, Lukin DJ, Nuber AH, et al. Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J Clin Invest. 2014;124:1283–1295. doi: 10.1172/JCI73434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Imaoka T, Hisatsune H, Sakanishi Y, Nishimura Y, Nishimura M, Shimada Y. Progesterone stimulates proliferation of a long-lived epithelial cell population in rat mammary gland. J Endocrinol Invest. 2012;35:828–834. doi: 10.3275/8189. [DOI] [PubMed] [Google Scholar]
- 111.Singh SK, Clarke ID, Hide T, Dirks PB. Cancer stem cells in nervous system tumors. Oncogene. 2004;23:7267–7273. doi: 10.1038/sj.onc.1207946. [DOI] [PubMed] [Google Scholar]
- 112.Jacques TS, Swales A, Brzozowski MJ, Henriquez NV, Linehan JM, Mirzadeh Z, et al. Combinations of genetic mutations in the adult neural stem cell compartment determine brain tumor phenotypes. EMBO J. 2010;29:222–235. doi: 10.1038/emboj.2009.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vicente-Duenas C, Perez-Caro M, Abollo-Jimenez F, Cobaleda C, Sanchez-Garcia I. Stem-cell driven cancer: “hands-off” regulation of cancer development. Cell Cycle. 2009;8:1314–1318. doi: 10.4161/cc.8.9.8217. [DOI] [PubMed] [Google Scholar]
- 114.Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer. 2007;7:441–453. doi: 10.1038/nrc2147. [DOI] [PubMed] [Google Scholar]
- 115.Fan X, Eberhart CG. Medulloblastoma stem cells. J Clin Oncol. 2008;26:2821–2127. doi: 10.1200/JCO.2007.15.2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Marsh Durban V, Jansen M, Davies EJ, Morsink FH, Offerhaus GJ, Clarke AR. Epithelial-specific loss of PTEN results in colorectal juvenile polyp formation and invasive cancer. Am J Pathol. 2014;184:86–91. doi: 10.1016/j.ajpath.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 117.Gort EH, van Haaften G, Verlaan I, Groot AJ, Plasterk RH, Shvarts A, et al. The TWIST1 oncogene is a direct target of hypoxia-inducible factor-2alpha. Oncogene. 2008;27:1501–1510. doi: 10.1038/sj.onc.1210795. [DOI] [PubMed] [Google Scholar]
- 118.Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, et al. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol. 2008;10:295–305. doi: 10.1038/ncb1691. [DOI] [PubMed] [Google Scholar]
- 119.Wilson A, Trumpp A. Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol. 2006;6:93–106. doi: 10.1038/nri1779. [DOI] [PubMed] [Google Scholar]
- 120.Kumar SM, Liu S, Lu H, Zhang H, Zhang PJ, Gimotty PA, et al. Acquired cancer stem cell phenotypes through Oct4-mediated dedifferentiation. Oncogene. 2012;31:4898–4911. doi: 10.1038/onc.2011.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nishi M, Sakai Y, Akutsu H, Nagashima Y, Quinn G, Masui S, et al. Induction of cells with cancer stem cell properties from nontumorigenic human mammary epithelial cells by defined reprogramming factors. Oncogene. 2014;33:643–652. doi: 10.1038/onc.2012.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ischenko I, Zhi J, Moll UM, Nemajerova A, Petrenko O. Direct reprogramming by oncogenic Ras and Myc. Proc Natl Acad Sci U S A. 2013;110:3937–3942. doi: 10.1073/pnas.1219592110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Vicente-Dueñas C, Romero-Camarero I, Cobaleda C, Sánchez-García I. Function of oncogenes in cancer development: a changing paradigm. EMBO J. 2013;32:1502–1513. doi: 10.1038/emboj.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tortorella SM, Hung A, Karagiannis T. The implication of cancer progenitor cells and the role of epigenetics in the development of novel therapeutic strategies for chronic myeloid leukemia. Antioxid Redox Signal. 2014 Nov 3; doi: 10.1089/ars.2014.6096. PubMed PMID: 25366930 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 125.Tomellini E, Touil Y, Lagadec C, Julien S, Ostyn P, Ziental-Gelus N, et al. NGF and proNGF simultaneously promote symmetric self-renewal, quiescence and EMT to enlarge the breast cancer stem cell compartment. Stem Cells. 2014 Oct 6; doi: 10.1002/stem.1849. PubMed PMID: 25286822 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 126.Tamari K, Hayashi K, Ishii H, Kano Y, Konno M, Kawamoto K, et al. Identification of chemoradiation-resistant osteosarcoma stem cells using an imaging system for proteasome activity. Int J Oncol. 2014;45:2349–2354. doi: 10.3892/ijo.2014.2671. [DOI] [PubMed] [Google Scholar]
- 127.Ostyn P, El Machhour R, Begard S, Kotecki N, Vandomme J, Flamenco P, et al. Transient TNF regulates the self-renewing capacity of stem-like label-retaining cells in sphere and skin equivalent models of melanoma. Cell Commun Signal. 2014;12(1):52. doi: 10.1186/s12964-014-0052-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zeuner A, Francescangeli F, Contavalli P, Zapparelli G, Apuzzo T, Eramo A, et al. Elimination of quiescent/slow-proliferating cancer stem cells by Bcl-XL inhibition in non-small cell lung cancer. Cell Death Differ. 2014;21:1877–1888. doi: 10.1038/cdd.2014.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Malanchi I, Santamaria-Martínez A, Susanto E, Peng H, Lehr HA, Delaloye JF, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2011;481:85–89. doi: 10.1038/nature10694. [DOI] [PubMed] [Google Scholar]
- 130.Liao WT, Ye YP, Deng YJ, Bian XW, Ding YQ. Metastatic cancer stem cells: from the concept to therapeutics. Am J Stem Cells. 2014;3:46–62. [PMC free article] [PubMed] [Google Scholar]
- 131.Crews L, Jamieson C. Selective elimination of leukemia stem cells: Hitting a moving target. Cancer Lett. 2013;338:15–22. doi: 10.1016/j.canlet.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 132.Rodova M, Fu J, Watkins D, Srivastava R, Shankar S. Sonic hedgehog signaling inhibition provides opportunities for targeted therapy by sulforaphane in regulating pancreatic cancer stem cell self-renewal. PLoS One. 2012;7:e46083. doi: 10.1371/journal.pone.0046083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Aguirre-Ghiso J, Bragado P, Sosa M. Metastasis awakening: targeting dormant cancer. Nat Med. 2013;19:276–277. doi: 10.1038/nm.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Krishnan V, Stadick N, Clark R, Bainer R, Veneris J, Khan S, et al. Using MKK4's metastasis suppressor function to identify and dissect cancer cell-microenvironment interactions during metastatic colonization. Cancer Metastasis Rev. 2013;31:605–613. doi: 10.1007/s10555-012-9371-y. [DOI] [PubMed] [Google Scholar]
- 135.Knopeke M, Ritschdorff E, Clark R, Vander Griend D, Khan S, Thobe M, et al. Building on the foundation of daring hypotheses: using the MKK4 metastasis suppressor to develop models of dormancy and metastatic colonization. FEBS Lett. 2011;585:3159–3165. doi: 10.1016/j.febslet.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 136.Trumpp A, Essers M, Wilson A. Awakening dormant haematopoietic stem cells. Nature Rev Immunol. 2010;10:201–209. doi: 10.1038/nri2726. [DOI] [PubMed] [Google Scholar]
- 137.Fang B, Mai L, Li N, Song Y, Chunhua Zhao R. Imatinib plus granulocyte colony-stimulating factor in chronic myeloid leukemia patients who have achieved partial or complete cytogenetic response while on Imatinib. Case Rep Oncol. 2011;4:192–197. doi: 10.1159/000327512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hedvat CV, Yao J, Sokolic RA, Nimer SD. Myeloid ELF1-like factor is a potent activator of interleukin-8 expression in hematopoietic cells. J Biol Chem. 2004;279:6395–6400. doi: 10.1074/jbc.M307524200. [DOI] [PubMed] [Google Scholar]
- 139.Lacorazza HD, Nimer SD. The emerging role of the myeloid Elf-1 like transcription factor in hematopoiesis. Blood Cells Mol Dis. 2003;31:342–350. doi: 10.1016/s1079-9796(03)00162-1. [DOI] [PubMed] [Google Scholar]
- 140.Lacorazza HD, Yamada T, Liu Y, Miyata Y, Sivina M, Nunes J, et al. The transcription factor MEF/ELF4 regulates the quiescence of primitive hematopoietic cells. Cancer Cell. 2006;9:175–187. doi: 10.1016/j.ccr.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 141.Liu Y, Elf SE, Miyata Y, Sashida G, Liu Y, Huang G, et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009;4(1):37–48. doi: 10.1016/j.stem.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhang J, Fukuhara S, Sako K, Takenouchi T, Kitani H, Kume T, et al. Angiopoietin-1/Tie2 signal augments basal Notch signal controlling vascular quiescence by inducing delta-like 4 expression through AKT-mediated activation of β-catenin. J Biol Chem. 2011;286:8055–8066. doi: 10.1074/jbc.M110.192641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 144.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11:558–572. doi: 10.1038/nrc3090. [DOI] [PubMed] [Google Scholar]
- 145.Hall M, Peters G. Genetic alterations of cyclins, cyclin-dependent kinases and cdk inhibitors in human cancer. Adv Cancer Res. 1996;68:67–108. doi: 10.1016/s0065-230x(08)60352-8. [DOI] [PubMed] [Google Scholar]
- 146.Chim CS, Wong KY, Loong F, Lam WW, Srivastava G. Frequent epigenetic inactivation of Rb1 in addition to p15 and p16 in mantle cell and follicular lymphoma. Hum Pathol. 2007;38:1849–1857. doi: 10.1016/j.humpath.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 147.Agbarya A, Ruimi N, Epelbaum R, Ben-Arye E, Mahajna J. Natural products as potential cancer therapy enhancers: A preclinical update. J SAGE Open Med. 2014;2:1–12. doi: 10.1177/2050312114546924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wander SA, Zhao D, Slingerland JM. p27: a barometer of signaling deregulation and potential predictor of response to targeted therapies. Clin Cancer Res. 2011;17:12–18. doi: 10.1158/1078-0432.CCR-10-0752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol. 2004;5:739–751. doi: 10.1038/nrm1471. [DOI] [PubMed] [Google Scholar]
- 150.Hershko DD. Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer. 2008;112:1415–1424. doi: 10.1002/cncr.23317. [DOI] [PubMed] [Google Scholar]
- 151.Bross PF, Kane R, Farrell AT, Abraham S, Benson K, Brower ME, et al. Approval summary for bortezomib for injection in the treatment of multiple myeloma. Clin Cancer Res. 2004;10:3954–3964. doi: 10.1158/1078-0432.CCR-03-0781. [DOI] [PubMed] [Google Scholar]
- 152.Kane RC, Dagher R, Farrell A, Ko CW, Sridhara R, Justice R, et al. Bortezomib for the treatment of mantle cell lymphoma. Clin Cancer Res. 2007;13:5291–5294. doi: 10.1158/1078-0432.CCR-07-0871. [DOI] [PubMed] [Google Scholar]
- 153.Kühl SJ, Kühl M. On the role of Wnt/β-catenin signaling in stem cells. Biochim Biophys Acta. 2013;1830:2297–3206. doi: 10.1016/j.bbagen.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 154.Curtin JC, Lorenzi MV. Drug discovery approaches to target Wnt signaling in cancer stem cells. Oncotarget. 2010;1:563–577. doi: 10.18632/oncotarget.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wray J, Hartmann C. WNTing embryonic stem cells. Trends Cell Biol. 2012;22:159–168. doi: 10.1016/j.tcb.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 156.Yuasa S, Fukuda K. Recent advances in cardiovascular regenerative medicine: the induced pluripotent stem cell era. Expert Rev Cardiovasc Ther. 2008;6:803–810. doi: 10.1586/14779072.6.6.803. [DOI] [PubMed] [Google Scholar]
- 157.Osakada F, Takahashi M. Neural induction and patterning in mammalian pluripotent stem cells. CNS Neurol Disord Drug Targets. 2011;10:419–432. doi: 10.2174/187152711795563958. [DOI] [PubMed] [Google Scholar]
- 158.Polakis P. Wnt signaling in cancer. Cold Spring Harb Perspect Biol. 2012;4:a008052. doi: 10.1101/cshperspect.a008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bishayee A. β-Catenin: a novel biomarker and therapeutic target in liver cancer. In: Georgakilas A, editor. Cancer Biomarkers. CRC Press; Boca Raton, FL: 2013. pp. 51–75. [Google Scholar]
- 160.Imbert A, Eelkema R, Jordan S, Feiner H, Cowin P. Delta N89 beta-catenin induces precocious development, differentiation, and neoplasia in mammary gland. J Cell Biol. 2001;153:555–568. doi: 10.1083/jcb.153.3.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ugolini F, Adelaide J, Charafe-Jauffret E, Nguyen C, Jacquemier J, Jordan B, et al. Differential expression assay of chromosome arm 8p genes identifies frizzled-related (FRP1/FRZB) and fibroblast growth factor receptor 1 (FGFR1) as candidate breast cancer genes. Oncogene. 1999;18:1903–1910. doi: 10.1038/sj.onc.1202739. [DOI] [PubMed] [Google Scholar]
- 162.Nagahata T, Shimada T, Harada A, Nagai H, Onda M, Yokoyama S, et al. Amplification, up-regulation and over-expression of DVL-1, the human counterpart of the Drosophila disheveled gene, in primary breast cancers. Cancer Sci. 2003;94:515–518. doi: 10.1111/j.1349-7006.2003.tb01475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML, et al. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84–88. doi: 10.1038/nature04372. [DOI] [PubMed] [Google Scholar]
- 164.Li Y, Welm B, Podsypanina K, Huang S, Chamorro M, Zhang X, et al. Evidence that transgenes encoding components of the Wnt signaling pathway preferentially induce mammary cancers from progenitor cells. Proc Natl Acad Sci USA. 2003;100:15853–15858. doi: 10.1073/pnas.2136825100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ronchini C, Capobianco AJ. Induction of cyclin D1 transcription and CDK2 activity by Notch(ic): implication for cell cycle disruption in transformation by Notch(ic). Mol Cell Biol. 2001;21:5925–5934. doi: 10.1128/MCB.21.17.5925-5934.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Rangarajan A, Talora C, Okuyama R, Nicolas M, Mammucari C, Oh H, et al. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J. 2001;20:3427–3436. doi: 10.1093/emboj/20.13.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Oswald F, Liptay S, Adler G, Schmid RM. NF-kappaB2 is a putative target gene of activated Notch-1 via RBP-Jkappa. Mol Cell Biol. 1998;18:2077–2088. doi: 10.1128/mcb.18.4.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci USA. 2006;103:18261–18266. doi: 10.1073/pnas.0606108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006;20:2096–2109. doi: 10.1101/gad.1450406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Oyama T, Harigaya K, Sasaki N, Okamura Y, Kokubo H, Hozumi K, et al. Mastermind-like 1 (MamL1) and mastermind-like 3 (MamL3) are essential for Notch signaling in vivo. Development. 2011;138:5235–5246. doi: 10.1242/dev.062802. [DOI] [PubMed] [Google Scholar]
- 171.Danovi SA. Angiogenesis: Turning it down a Notch. Nat Rev Cancer. 2008;8:572–573. [Google Scholar]
- 172.Guo S, Liu M, Gonzalez-Perez RR. Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochim Biophys Acta. 2011;1815:197–213. doi: 10.1016/j.bbcan.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Perdigoto CN, Bardin AJ. Sending the right signal: Notch and stem cells. Biochim Biophys Acta. 2013;1830:2307–2322. doi: 10.1016/j.bbagen.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 174.Chiba S. Notch signaling in stem cell systems. Stem Cells. 2006;24:2437–2447. doi: 10.1634/stemcells.2005-0661. [DOI] [PubMed] [Google Scholar]
- 175.Kakarala M, Wicha MS. Cancer stem cells: implications for cancer treatment and prevention. Cancer J. 2007;13:271–275. doi: 10.1097/PPO.0b013e318156da4e. [DOI] [PubMed] [Google Scholar]
- 176.Korkaya H, Wicha MS. Selective targeting of cancer stem cells: a new concept in cancer therapeutics. BioDrugs. 2007;21:299–310. doi: 10.2165/00063030-200721050-00002. [DOI] [PubMed] [Google Scholar]
- 177.Farnie G, Clarke RB. Mammary stem cells and breast cancer-role of Notch signalling. Stem Cell Rev. 2007;3:169–171. doi: 10.1007/s12015-007-0023-5. [DOI] [PubMed] [Google Scholar]
- 178.Fan X, Matsui W, Khaki L, Stearns D, Chun J, Li YM, et al. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006;66:7445–7452. doi: 10.1158/0008-5472.CAN-06-0858. [DOI] [PubMed] [Google Scholar]
- 179.Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N, et al. Notch pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells. 2010;28:5–16. doi: 10.1002/stem.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Liu S, Dontu G, Wicha MS, Patel S, Ahn N-s, Jackson KW, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66:6063–6071. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hassan KA, Wang L, Korkaya H, Chen G, Maillard I, Beer DG, et al. Notch pathway activity identifies cells with cancer stem cell-like properties and correlates with worse survival in lung adenocarcinoma. Clin Cancer Res. 2013;19:1972–1980. doi: 10.1158/1078-0432.CCR-12-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Abel EV, Kim EJ, Wu J, Hynes M, Bednar F, Proctor E, et al. The Notch pathway is important in maintaining the cancer stem cell population in pancreatic cancer. PLoS One. 2014;9:e91983. doi: 10.1371/journal.pone.0091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Pece S, Serresi M, Santolini E, Capra M, Hulleman E, Galimberti V, et al. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J Cell Biol. 2004;167:215–221. doi: 10.1083/jcb.200406140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Jhappan C, Gallahan D, Stahle C, Chu E, Smith GH, Merlino G, et al. Expression of an activated Notch-related int-3 transgene interferes with cell differentiation and induces neoplastic transformation in mammary and salivary glands. Genes Dev. 1992;6:345–355. doi: 10.1101/gad.6.3.345. [DOI] [PubMed] [Google Scholar]
- 185.Gallahan D, Jhappan C, Robinson G, Hennighausen L, Sharp R, Kordon E, et al. Expression of a truncated Int3 gene in developing secretory mammary epithelium specifically retards lobular differentiation resulting in tumorigenesis. Cancer Res. 1996;56:1775–1785. [PubMed] [Google Scholar]
- 186.Sansone P, Storci G, Giovannini C, Pandolfi S, Pianetti S, Taffurelli M, et al. p66Shc/Notch-3 interplay controls self-renewal and hypoxia survival in human stem/progenitor cells of the mammary gland expanded in vitro as mammospheres. Stem Cells. 2007;25:807–815. doi: 10.1634/stemcells.2006-0442. [DOI] [PubMed] [Google Scholar]
- 187.Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, Taffurelli M, et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest. 2007;117:3988–4002. doi: 10.1172/JCI32533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Osipo C, Patel P, Rizzo P, Clementz AG, Hao L, Golde TE, et al. ErbB-2 inhibition activates Notch-1 and sensitizes breast cancer cells to a Ɣ-secretase inhibitor. Oncogene. 2008;27:5019–5032. doi: 10.1038/onc.2008.149. [DOI] [PubMed] [Google Scholar]
- 189.Chen Y, Fischer WH, Gill GN. Regulation of the ERBB-2 promoter by RBPJκ and NOTCH. J Biol Chem. 1997;272:14110–14114. doi: 10.1074/jbc.272.22.14110. [DOI] [PubMed] [Google Scholar]
- 190.Korkaya H, Wicha MS. HER-2, notch, and breast cancer stem cells: targeting an axis of evil. Clin Cancer Res. 2009;15:1845–1847. doi: 10.1158/1078-0432.CCR-08-3087. [DOI] [PubMed] [Google Scholar]
- 191.Korkaya H, Paulson A, Charafe-Jauffret E, Ginestier C, Brown M, Dutcher J, et al. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol. 2009;7:e1000121. doi: 10.1371/journal.pbio.1000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Pratt MA, Tibbo E, Robertson SJ, Jansson D, Hurst K, Perez-Iratxeta C, et al. The canonical NF-kappaB pathway is required for formation of luminal mammary neoplasias and is activated in the mammary progenitor population. Oncogene. 2009;28:2710–2722. doi: 10.1038/onc.2009.131. [DOI] [PubMed] [Google Scholar]
- 193.Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Cecchinato V, Chiaramonte R, Nizzardo M, Cristofaro B, Basile A, Sherbet GV, et al. Resveratrol-induced apoptosis in human T-cell acute lymphoblastic leukaemia MOLT-4 cells. Biochem Pharmacol. 2007;74:1568–1574. doi: 10.1016/j.bcp.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 195.Seccareccia E, Brodt P. The role of the insulin-like growth factor-I receptor in malignancy: an update. Growth Horm IGF Res. 2012;22:193–199. doi: 10.1016/j.ghir.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 196.Grimberg A. Mechanisms by which IGF-I may promote cancer. Cancer Biol Ther. 2003;2:630–635. [PMC free article] [PubMed] [Google Scholar]
- 197.Chen L, Khillan JS. A novel signaling by vitamin A/retinol promotes self renewal of mouse embryonic stem cells by activating PI3K/Akt signaling pathway via insulin-like growth factor-1 receptor. Stem Cells. 2010;28:57–63. doi: 10.1002/stem.251. [DOI] [PubMed] [Google Scholar]
- 198.Shan J, Shen J, Liu L, Xia F, Xu C, Duan G, et al. Nanog regulates self-renewal of cancer stem cells through the insulin-like growth factor pathway in human hepatocellular carcinoma. Hepatology. 2012;56:1004–1014. doi: 10.1002/hep.25745. [DOI] [PubMed] [Google Scholar]
- 199.Guo S, Liu M, Wang G, Torroella-Kouri M, Gonzalez-Perez RR. Oncogenic role and therapeutic target of leptin signaling in breast cancer and cancer stem cells. Biochim Biophys Acta. 2012;1825:207–222. doi: 10.1016/j.bbcan.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Feng Y, Dai X, Li X, Wang H, Liu J, Zhang J, et al. EGF signalling pathway regulates colon cancer stem cell proliferation and apoptosis. Cell Prolif. 2012;45:413–419. doi: 10.1111/j.1365-2184.2012.00837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Steelman LS, Stadelman KM, Chappell WH, Horn S, Basecke J, Cervello M, et al. Akt as a therapeutic target in cancer. Expert Opin Ther Targets. 2008;12:1139–1165. doi: 10.1517/14728222.12.9.1139. [DOI] [PubMed] [Google Scholar]
- 202.Wickenden JA, Watson CJ. Key signalling nodes in mammary gland development and cancer. Signalling downstream of PI3 kinase in mammary epithelium: a play in 3 Akts. Breast Cancer Res. 2010;12:202–211. doi: 10.1186/bcr2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Montalto G, Cervello M, et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget. 2012;3:954–987. doi: 10.18632/oncotarget.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Sabbah M, Emami S, Redeuilh G, Julien S, Prevost G, Zimber A, et al. Molecular signature and therapeutic perspective of the epithelial-to-mesenchymal transitions in epithelial cancers. Drug Resist Updat. 2008;11:123–151. doi: 10.1016/j.drup.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 205.Ghayad SE, Cohen PA. Inhibitors of the PI3K/Akt/mTOR pathway: new hope for breast cancer patients. Recent Pat Anticancer Drug Discov. 2010;5:29–57. doi: 10.2174/157489210789702208. [DOI] [PubMed] [Google Scholar]
- 206.Hadad SM, Fleming S, Thompson AM. Targeting AMPK: a new therapeutic opportunity in breast cancer. Crit Rev Oncol Hematol. 2008;67:1–7. doi: 10.1016/j.critrevonc.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 207.Martelli AM, Evangelisti C, Follo MY, Ramazzotti G, Fini M, Giardino R, et al. Targeting the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin signaling network in cancer stem cells. Curr Med Chem. 2011;18:2715–2726. doi: 10.2174/092986711796011201. [DOI] [PubMed] [Google Scholar]
- 208.Wang Y, Liu Y, Lu J, Zhang P, Wang Y, Xu Y, et al. Rapamycin inhibits FBXW7 loss-induced epithelial-mesenchymal transition and cancer stem cell-like characteristics in colorectal cancer cells. Biochem Biophys Res Commun. 2013;434:352–356. doi: 10.1016/j.bbrc.2013.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Choo AY, Blenis J. Not all substrates are treated equally: implications for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle. 2009;8:567–572. doi: 10.4161/cc.8.4.7659. [DOI] [PubMed] [Google Scholar]
- 210.Zhang Y, Zheng XF. mTOR-independent 4E-BP1 phosphorylation is associated with cancer resistance to mTOR kinase inhibitors. Cell Cycle. 2012;11:594–603. doi: 10.4161/cc.11.3.19096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci USA. 2008;105:17414–17419. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Fox CJ, Hammerman PS, Cinalli RM, Master SR, Chodosh LA, Thompson CB. The serine/threonine kinase Pim-2 is a transcriptionally regulated apoptotic inhibitor. Genes Dev. 2003;17:1841–1854. doi: 10.1101/gad.1105003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Wang X, Yue P, Chan CB, Ye K, Ueda T, Watanabe-Fukunaga R, et al. Inhibition of mammalian target of rapamycin induces phosphatidylinositol 3-kinase-dependent and Mnk-mediated eukaryotic translation initiation factor 4E phosphorylation. Mol Cell Biol. 2007;27:7405–7413. doi: 10.1128/MCB.00760-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Ilic N, Utermark T, Widlund HR, Roberts TM. PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proc Natl Acad Sci USA. 2011;108:E699–708. doi: 10.1073/pnas.1108237108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Smith SC, Baras AS, Lee JK, Theodorescu D. The COXEN principle: translating signatures of in vitro chemosensitivity into tools for clinical outcome prediction and drug discovery in cancer. Cancer Res. 2010;70:1753–1758. doi: 10.1158/0008-5472.CAN-09-3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Gonzalez-Perez RR, Xu Y, Guo S, Watters A, Zhou W, Leibovich SJ. Leptin upregulates VEGF in breast cancer via canonic and non-canonical signalling pathways and NFkappaB/HIF-1alpha activation. Cell Signal. 2010;22:1350–1362. doi: 10.1016/j.cellsig.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 220.Prasad S, Ravindran J, Aggarwal BB. NF-kappaB and cancer: how intimate is this relationship. Mol Cell Biochem. 2010;336:25–37. doi: 10.1007/s11010-009-0267-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Takada Y, Ichikawa H, Badmaev V, Aggarwal BB. Acetyl-11-keto-beta-boswellic acid potentiates apoptosis, inhibits invasion, and abolishes osteoclastogenesis by suppressing NF-kappa B and NF-kappa B-regulated gene expression. J Immunol. 2006;176:3127–3140. doi: 10.4049/jimmunol.176.5.3127. [DOI] [PubMed] [Google Scholar]
- 222.Sethi G, Ahn KS, Sandur SK, Lin X, Chaturvedi MM, Aggarwal BB. Indirubin enhances tumor necrosis factor-induced apoptosis through modulation of nuclear factor-kappa B signaling pathway. J Biol Chem. 2006;281:23425–23435. doi: 10.1074/jbc.M602627200. [DOI] [PubMed] [Google Scholar]
- 223.Long H, Xie R, Xiang T, Zhao Z, Lin S, Liang Z, et al. Autocrine CCL5 signaling promotes invasion and migration of CD133+ ovarian cancer stem-like cells via NF-kappaB-mediated MMP-9 upregulation. Stem Cells. 2012;30:2309–2319. doi: 10.1002/stem.1194. [DOI] [PubMed] [Google Scholar]
- 224.Wang L, Guo H, Yang L, Dong L, Lin C, Zhang J, et al. Morusin inhibits human cervical cancer stem cell growth and migration through attenuation of NF-kappaB activity and apoptosis induction. Mol Cell Biochem. 2013;379:7–18. doi: 10.1007/s11010-013-1621-y. [DOI] [PubMed] [Google Scholar]
- 225.Nogueira L, Ruiz-Ontanon P, Vazquez-Barquero A, Lafarga M, Berciano MT, Aldaz B, et al. Blockade of the NFkappaB pathway drives differentiating glioblastoma-initiating cells into senescence both in vitro and in vivo. Oncogene. 2011;30:3537–3548. doi: 10.1038/onc.2011.74. [DOI] [PubMed] [Google Scholar]
- 226.Shostak K, Chariot A. NF-κB, stem cells and breast cancer: the links get stronger. Breast Cancer Res. 2011;13(4):214. doi: 10.1186/bcr2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.McMahon AP, Ingham PW, Tabin CJ. Developmental roles and clinical significance of Hedgehog signaling. Curr Top Dev Biol. 2003;53:1–114. doi: 10.1016/s0070-2153(03)53002-2. [DOI] [PubMed] [Google Scholar]
- 228.Ahn S, Joyner AL. In vivo analysis of quiescent adult neural stem cells responding to Sonic Hedgehog. Nature. 2005;437:894–897. doi: 10.1038/nature03994. [DOI] [PubMed] [Google Scholar]
- 229.Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog signaling within airway epithelial progenitors and in small-cell lung cancer. Nature. 2003;422:313–317. doi: 10.1038/nature01493. [DOI] [PubMed] [Google Scholar]
- 230.Beachy PA, Karhadkar SS, Berman DM. Tissue repair and stem cell renewal in carcinogenesis. Nature. 2004;432:324–331. doi: 10.1038/nature03100. [DOI] [PubMed] [Google Scholar]
- 231.Sasaki H, Nishizaki Y, Hui C, Nakafuku M, Kondoh H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development. 1999;126:3915–3924. doi: 10.1242/dev.126.17.3915. [DOI] [PubMed] [Google Scholar]
- 232.Ruiz I, Altaba A, Mas C, Stecca B. The Gli code: an information nexus regulating cell fate, stemness and cancer. Trends Cell Biol. 2007;17:438–447. doi: 10.1016/j.tcb.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Yang L, Xie G, Fan Q, Xie J. Activation of the Hedgehog signaling pathway in human cancer and the clinical implications. Oncogene. 2010;29:469–481. doi: 10.1038/onc.2009.392. [DOI] [PubMed] [Google Scholar]
- 234.Clement V, Sanchez P, de TN, Radovanovic I, Altaba A. Hedgehog-Gli1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007;17:165–172. doi: 10.1016/j.cub.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Bar EE, Chaudhry A, Lin A, Fan X, Schreck K, Matsui W, et al. Cyclopamine-mediated Hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. 2007;25:2524–2533. doi: 10.1634/stemcells.2007-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR, Manley P, et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell. 2008;14:238–249. doi: 10.1016/j.ccr.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 237.Chai F, Zhou J, Chen C, Xie S, Chen X, Su P, Shi J. The Hedgehog inhibitor cyclopamine antagonizes chemoresistance of breast cancer cells. Onco Targets Ther. 2013;6:1643–1647. doi: 10.2147/OTT.S51914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Feldmann G, Dhara S, Fendrich V, Bedja D, Beaty R, Mullendore M, et al. Blockade of Hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res. 2007;67:2187–2196. doi: 10.1158/0008-5472.CAN-06-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Tang SN, Fu J, Nall D, Rodova M, Shankar S, Srivastava RK. Inhibition of sonic Hedgehog pathway and pluripotency maintaining factors regulate human pancreatic cancer stem cell characteristics. Intl J Cancer. 2012;131:30–40. doi: 10.1002/ijc.26323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458:776–779. doi: 10.1038/nature07737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Liao HF, Su YC, Zheng ZY, Jhih Cai C, Hou MH, Chao KS, et al. Sonic Hedgehog signaling regulates Bcr-Abl expression in human chronic myeloid leukemia cells. Biomed Pharmacother. 2012;66:378–383. doi: 10.1016/j.biopha.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 242.Jeng KS, Sheen IS, Jengetal WJ. Blockade of the sonic hedgehog pathway effectively inhibits the growth of hepatoma in mice: an in vivo study. Oncology Lett. 2012;4:1158–1162. doi: 10.3892/ol.2012.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Song Z, Yue W, Wei B, Wang N, Li T, Guan L, et al. Sonic hedgehog pathway Is essential for maintenance of cancer stem-Like cells in human gastric cancer. PLoS One. 2011;6:e17687. doi: 10.1371/journal.pone.0017687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.LaBarge M. The difficulty of targeting cancer stem cell niches. Clin Cancer Res. 2010;16:3121–3129. doi: 10.1158/1078-0432.CCR-09-2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Li SH, Fu J, Watkins DN, Srivastava RK, Shankar S. Sulforaphane regulates self-renewal of pancreatic cancer stem cells through the modulation of Sonic Hedgehog-GLI pathway. Mol Cell Biochem. 2013;373:217–227. doi: 10.1007/s11010-012-1493-6. [DOI] [PubMed] [Google Scholar]
- 246.Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34:274–285. doi: 10.1111/j.1365-2036.2011.04724.x. [DOI] [PubMed] [Google Scholar]
- 247.Brunt EM. Pathology of nonalcoholic steatohepatitis. Hepatol Res. 2005;33:68–71. doi: 10.1016/j.hepres.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 248.Marra F, Gastaldelli A, Svegliati Baroni G, Tell G, Tiribelli C. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends Mol Med. 2008;14:72–81. doi: 10.1016/j.molmed.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 249.Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology. 2006;43:S99–112. doi: 10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- 250.Kinsell LW, Harper HA. Rate of disappearance from plasma of intravenously administered methionine in patients with liver damage. Science. 1947;106:589–590. doi: 10.1126/science.106.2763.589. [DOI] [PubMed] [Google Scholar]
- 251.Kinsell LW, Harper HA, Barton HC, Hutchin ME, Hess JR. Studies in methionine and sulfur metabolism. I. The fate of intravenously administered methionine in normal individuals and in patients with liver damage. J Clin Invest. 1948;27:677–688. doi: 10.1172/JCI102016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Duce AM, Ortíz P, Cabrero C, Mato JM. S-adenosyl-L-methionine synthetase and phospholipid methyltransferase are inhibited in human cirrhosis. Hepatology. 1988;8:65–68. doi: 10.1002/hep.1840080113. [DOI] [PubMed] [Google Scholar]
- 253.Avila MA, Berasain C, Torres L, Martín-Duce A, Corrales FJ, Yang H, et al. Reduced mRNA abundance of the main enzymes involved in methionine metabolism in human liver cirrhosis and hepatocellular carcinoma. J Hepatol. 2000;33:907–914. doi: 10.1016/s0168-8278(00)80122-1. [DOI] [PubMed] [Google Scholar]
- 254.Martínez-Chantar ML, Corrales FJ, Martínez-Cruz LA, García-Trevijano ER, Huang ZZ, Chen L, et al. Spontaneous oxidative stress and liver tumors in mice lacking methionine adenosyltransferase 1A. FASEB J. 2002;16:1292–1294. doi: 10.1096/fj.02-0078fje. [DOI] [PubMed] [Google Scholar]
- 255.Mato JM, Corrales FJ, Lu SC, Avila MA. S-Adenosylmethionine: a control switch that regulates liver function. FASEB J. 2002;16:15–26. doi: 10.1096/fj.01-0401rev. [DOI] [PubMed] [Google Scholar]
- 256.Mato JM, Cámara J, de Paz JF, Caballería L, Coll S, Caballero A, et al. S-Adenosylmethionine in alcoholic liver cirrhosis: a randomized, placebo-controlled, double-blind, multicenter clinical trial. J Hepatol. 1999;30:1081–1089. doi: 10.1016/s0168-8278(99)80263-3. [DOI] [PubMed] [Google Scholar]
- 257.Martínez-Chantar ML, Vázquez-Chantada M, Garnacho M, Latasa MU, Varela-Rey M, Dotor J, et al. S-adenosylmethionine regulates cytoplasmic HuR via AMP-activated kinase. Gastroenterology. 2006;131:223–232. doi: 10.1053/j.gastro.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 258.Luka Z, Cerone R, Phillips JA, 3rd, Mudd HS, Wagner C. Mutations in human glycine N-methyltransferase give insights into its role in methionine metabolism. Hum Genet. 2002;110:68–74. doi: 10.1007/s00439-001-0648-4. [DOI] [PubMed] [Google Scholar]
- 259.Mudd SH, Cerone R, Schiaffino MC, Fantasia AR, Minniti G, Caruso U, et al. Glycine N-methyltransferase deficiency: a novel inborn error causing persistent isolated hypermethioninaemia. J Inherit Metab Dis. 2001;24:448–464. doi: 10.1023/a:1010577512912. [DOI] [PubMed] [Google Scholar]
- 260.Luka Z, Capdevila A, Mato JM, Wagner C. A glycine N-methyltransferase knockout mouse model for humans with deficiency of this enzyme. Transgenic Res. 2006;15:393–397. doi: 10.1007/s11248-006-0008-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Martínez-Chantar ML, Vázquez-Chantada M, Ariz U, Martínez N, Varela M, Luka Z, et al. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology. 2008;47:1191–1199. doi: 10.1002/hep.22159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 263.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336–20342. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- 265.Cao J, Schulte J, Knight A, Leslie NR, Zagozdzon A, Bronson R, et al. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J. 2009;28:1505–1517. doi: 10.1038/emboj.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Gao P, Zhang H, Dinavahi R, Li F, Xiang Y, Raman V, et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007;12:230–238. doi: 10.1016/j.ccr.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Bell EL, Emerling BM, Chandel NS. Mitochondrial regulation of oxygen sensing. Mitochondrion. 2005;5:322–232. doi: 10.1016/j.mito.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 268.Takahashi A, et al. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nature Cell Biol. 2006;8:1291–1297. doi: 10.1038/ncb1491. [DOI] [PubMed] [Google Scholar]
- 269.Garrido C. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006;13:1423–433. doi: 10.1038/sj.cdd.4401950. [DOI] [PubMed] [Google Scholar]
- 270.Henderson BE, Feigelson HS. Hormonal carcinogenesis. Carcinogenesis. 2000;21:427–433. doi: 10.1093/carcin/21.3.427. [DOI] [PubMed] [Google Scholar]
- 271. http://www.cancerresearchuk.org/cancer-info/cancerstats/incidence/commoncancers/source1.
- 272.Han X, Liehr JG. 8-Hydroxylation of guanine bases in kidney and liver DNA of hamsters treated with estradiol: role of free radicals in estrogen-induced carcinogenesis. Cancer Res. 1994;54:5515–5517. [PubMed] [Google Scholar]
- 273.Mailander PC, Meza JL, Higginbotham S, Chakravarti D. Induction of A.T to G.C mutations by erroneous repair of depurinated DNA following estrogen treatment of the mammary gland of ACI rats. J Steroid Biochem Mol Biol. 2006;101:204–215. doi: 10.1016/j.jsbmb.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 274.Ascenzi P, Bocedi A, Marino M. Structure-function relationship of estrogen receptor α and β: impact on human health. Mol Aspects Med. 2006;27:299–402. doi: 10.1016/j.mam.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 275.Acconcia F, Marino M. The effects of 17β-estradiol in cancer are mediated by estrogen receptor signaling at the plasma membrane. Front Physiol. 2011;2:30–37. doi: 10.3389/fphys.2011.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Marino M, Ascenzi P. Membrane association of estrogen receptor α and β influences 17β-estradiol-mediated cancer cell proliferation. Steroids. 2008;73:853–858. doi: 10.1016/j.steroids.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 277.Marino M, Acconcia F, Trentalance A. Biphasic estradiol-induced AKT phosphorylation is modulated by PTEN via MAP kinase in HepG2 cells. Mol Biol Cell. 2003;14:2583–2591. doi: 10.1091/mbc.E02-09-0621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Song RX, Barnes CJ, Zhang Z, Bao Y, Kumar R, Santen RJ. The role of Shc and insulin-like growth factor 1 receptor in mediating the translocation of estrogen receptor α to the plasma membrane. Proc Natl Acad Sci USA. 2004;101:2076–2081. doi: 10.1073/pnas.0308334100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Acconcia F, Kumar R. Signaling regulation of genomic and nongenomic functions of estrogen receptors. Cancer Lett. 2006;238:1–14. doi: 10.1016/j.canlet.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 280.Kim R, Kaneko M, Arihiro K, Emi M, Tanabe K, Murakami S, et al. Extranuclear expression of hormone receptors in primary breast cancer. Ann Oncol. 2006;17:1213–1220. doi: 10.1093/annonc/mdl118. [DOI] [PubMed] [Google Scholar]
- 281.Mintz PJ, Habib NA, Jones LJ, Giamas G, Lewis JS, Bowen RL, et al. The phosphorylated membrane estrogen receptor and cytoplasmic signaling and apoptosis proteins in human breast cancer. Cancer. 2008;113:1489–1495. doi: 10.1002/cncr.23699. [DOI] [PubMed] [Google Scholar]
- 282.Métivier R, Stark A, Flouriot G, Hübner MR, Brand H, Penot G, et al. A dynamic structural model for estrogen receptor-α activation by ligands, emphasizing the role of interactions between distant A and E domains. Mol Cell. 2002;10:1019–1032. doi: 10.1016/s1097-2765(02)00746-3. [DOI] [PubMed] [Google Scholar]
- 283.Bolli A, Marino M. Current and future development of estrogen receptor ligands: applications in estrogen-related cancers. Recent Pat Endocr Metab Immune Drug Discov. 2011;5:210–229. doi: 10.2174/187221411797265881. [DOI] [PubMed] [Google Scholar]
- 284.Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, et al. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994;54:5474–5478. [PubMed] [Google Scholar]
- 285.Yeh S, Lin HK, Kang HY, Thin TH, Lin MF, Chang C. From HER2/Neu signal cascade to androgen receptor and its coactivators: a novel pathway by induction of androgen target genes through MAP kinase in prostate cancer cells. Proc Natl Acad Sci USA. 1999;96:5458–5463. doi: 10.1073/pnas.96.10.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Sun M, Yang L, Feldman RI, Sun XM, Bhalla KN, Jove R, et al. Activation of phosphatidylinositol 3-kinase/Akt pathway by androgen through interaction of p85alpha, androgen receptor, and Src. J Biol Chem. 2003;278:42992–30000. doi: 10.1074/jbc.M306295200. [DOI] [PubMed] [Google Scholar]
- 287.Cheng J, Watkins SC, Walker WH. Testosterone activates mitogen-activated protein kinase via Src kinase and the epidermal growth factor receptor in sertoli cells. Endocrinology. 2007;148:2066–2074. doi: 10.1210/en.2006-1465. [DOI] [PubMed] [Google Scholar]
- 288.Spiotto MT, Chung TD. STAT3 mediates IL-6-induced neuroendocrine differentiation in prostate cancer cells. Prostate. 2000;42:186–195. doi: 10.1002/(sici)1097-0045(20000215)42:3<186::aid-pros4>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 289.Chang KH, Ercole CE, Sharifi N. Androgen metabolism in prostate cancer: from molecular mechanisms to clinical consequences. Br J Cancer. 2014;111:1249–1254. doi: 10.1038/bjc.2014.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Keen JC, Davidson NE. The biology of breast carcinoma. Cancer. 2003;97:825–833. doi: 10.1002/cncr.11126. [DOI] [PubMed] [Google Scholar]
- 291.Galluzzo P, Caiazza F, Moreno S, Marino M. Role of ERβ palmitoylation in the inhibition of human colon cancer cell proliferation. Endocr Relat Cancer. 2007;14:153–167. doi: 10.1677/ERC-06-0020. [DOI] [PubMed] [Google Scholar]
- 292.Bardin A, Boulle N, Lazennec G, Vignon F, Pujol P. Loss of ERβ expression as a common step in estrogen-dependent tumor progression. Endocr Relat Cancer. 2004;11:537–551. doi: 10.1677/erc.1.00800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Warner M, Gustafsson J-Å. The role of estrogen receptor β (ERβ) in malignant diseases-a new potential target for antiproliferative drugs in prevention and treatment of cancer. Biochem Biophys Res Commun. 2010;396:63–66. doi: 10.1016/j.bbrc.2010.02.144. [DOI] [PubMed] [Google Scholar]
- 294.Kumar R. Another tie that binds the MTA family to breast cancer. Cell. 2003;113:142–143. doi: 10.1016/s0092-8674(03)00274-5. [DOI] [PubMed] [Google Scholar]
- 295.Barone M, Scavo MP, Papagni S, Piscitelli D, Guido R, Di Lena M, et al. ERβ expression in normal, adenomatous and carcinomatous tissues of patients with familial adenomatous polyposis. Scand J Gastroenterol. 2010;45:1320–1328. doi: 10.3109/00365521.2010.487915. [DOI] [PubMed] [Google Scholar]
- 296.Polyak K, Haviv I, Campbell IG. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009;25:30–38. doi: 10.1016/j.tig.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 297.Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumor angiogenesis. Nat Rev Cancer. 2008;8:618–631. doi: 10.1038/nrc2444. [DOI] [PubMed] [Google Scholar]
- 298.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.DeNardo DG, Andreu P, Coussens LM. Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Rev. 2010;29:309–316. doi: 10.1007/s10555-010-9223-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 301.Nagy JA, Chang SH, Shih SC, Dvorak AM, Dvorak HF. Heterogeneity of the tumor vasculature. Semin Thromb Hemost. 2010;36:321–331. doi: 10.1055/s-0030-1253454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Ruoslahti E. Specialization of tumor vasculature. Nat Rev Cancer. 2002;2:83–90. doi: 10.1038/nrc724. [DOI] [PubMed] [Google Scholar]
- 303.Rajala MW, Scherer PE. Minireview: The adipocyte at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology. 2003;144:3765–3673. doi: 10.1210/en.2003-0580. [DOI] [PubMed] [Google Scholar]
- 304.Tan J, Buache E, Chenard MP, Dali-Youcef N, Rio MC. Adipocyte is a non-trivial, dynamic partner of breast cancer cells. Int J Dev Biol. 2011;55:851–859. doi: 10.1387/ijdb.113365jt. [DOI] [PubMed] [Google Scholar]
- 305.Brouty-Boye D. Developmental biology of fibroblasts and neoplastic disease. Prog Mol Subcell Biol. 2005;40:55–77. doi: 10.1007/3-540-27671-8_3. [DOI] [PubMed] [Google Scholar]
- 306.De Wever O, Demetter P, Mareel M, Bracke M. Stromal myofibroblasts are drivers of invasive cancer growth. Int J Cancer. 2008;123:2229–2238. doi: 10.1002/ijc.23925. [DOI] [PubMed] [Google Scholar]
- 307.Rasanen K, Vaheri A. Activation of fibroblasts in cancer stroma. Exp Cell Res. 2010;316:2713–2722. doi: 10.1016/j.yexcr.2010.04.032. [DOI] [PubMed] [Google Scholar]
- 308.Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9:1392–1400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- 309.Kumar S, Weaver VM. Mechanics, malignancy, and metastasis: the force journey of a tumor cell. Cancer Metastasis Rev. 2009;28:113–127. doi: 10.1007/s10555-008-9173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Sethi T, Rintoul RC, Moore SM, MacKinnon AC, Salter D, Choo C, et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: a mechanism for small cell lung cancer growth and drug resistance in vivo. Nat Med. 1999;5:662–668. doi: 10.1038/9511. [DOI] [PubMed] [Google Scholar]
- 311.Berube M, Talbot M, Collin C, Paquet-Bouchard C, Germain L, Guerin SL, et al. Role of the extracellular matrix proteins in the resistance of SP6.5 uveal melanoma cells toward cisplatin. Int J Oncol. 2005;26:405–413. [PubMed] [Google Scholar]
- 312.Butler MS. Natural products to drugs: natural product-derived compounds in clinical trials. Nat Prod Rep. 2008;25:475–516. doi: 10.1039/b514294f. [DOI] [PubMed] [Google Scholar]
- 313.Buchdunger E, Zimmermann J, Mett H, Meyer T, Muller M, Regenass U, et al. Selective inhibition of the platelet-derived growth factor signal transduction pathway by a protein-tyrosine kinase inhibitor of the 2-phenylaminopyrimidine class. Proc Natl Acad Sci USA. 1995;92:2558–2562. doi: 10.1073/pnas.92.7.2558. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 314.Weisberg E, Catley L, Wright RD, Moreno D, Banerji L, Ray A, et al. Beneficial effects of combining nilotinib and imatinib in preclinical models of BCR-ABL+ leukemias. Blood. 2007;109:2112–2120. doi: 10.1182/blood-2006-06-026377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.O'Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16:401–412. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Pedersen MW, Pedersen N, Ottesen LH, Poulsen HS. Differential response to gefitinib of cells expressing normal EGFR and the mutant EGFRvIII. Br J Cancer. 2005;93:915–923. doi: 10.1038/sj.bjc.6602793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Moyer JD, Barbacci EG, Iwata KK, Arnold L, Boman B, Cunningham A, et al. Induction of apoptosis and cell cycle arrest by CP-358,774, an inhibitor of epidermal growth factor receptor tyrosine kinase. Cancer Res. 1997;57:4838–4848. [PubMed] [Google Scholar]
- 318.Rusnak DW, Lackey K, Affleck K, Wood ER, Alligood KJ, Rhodes N, et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Ther. 2001;1:85–94. [PubMed] [Google Scholar]
- 319.Zou HY, Li Q, Lee JH, Arango ME, McDonnell SR, Yamazaki S, et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007;67:4408–4417. doi: 10.1158/0008-5472.CAN-06-4443. [DOI] [PubMed] [Google Scholar]
- 320.Eramo A, Haas TL, De Maria R. Lung cancer stem cells: tools and targets to fight lung cancer. Oncogene. 2010;29:4625–4635. doi: 10.1038/onc.2010.207. [DOI] [PubMed] [Google Scholar]
- 321.Mani S, Guo W, Liao M, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Bar-Sela G, Epelbaum R, Schaffer M. Curcumin as an anti-cancer agent: review of the gap between basic and clinical applications. Curr Med Chem. 2010;17:190–197. doi: 10.2174/092986710790149738. [DOI] [PubMed] [Google Scholar]
- 323.Basnet P, Skalko-Basnet N. Curcumin: an anti-inflammatory molecule from a curry spice on the path to cancer treatment. Molecules. 2011;16:4567–4598. doi: 10.3390/molecules16064567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Jagtap S, Meganathan K, Wagh V, Winkler J, Hescheler J, Sachinidis A. Chemoprotective mechanism of the natural compounds, epigallocatechin-3-O-gallate, quercetin and curcumin against cancer and cardiovascular diseases. Curr Med Chem. 2009;16:1451–1462. doi: 10.2174/092986709787909578. [DOI] [PubMed] [Google Scholar]
- 325.Johnson SM, Gulhati P, Arrieta I, Wang X, Uchida T, Gao T, et al. Curcumin inhibits proliferation of colorectal carcinoma by modulating Akt/mTOR signaling. Anticancer Res. 2009;29:3185–3190. [PMC free article] [PubMed] [Google Scholar]
- 326.Kuhad A, Pilkhwal S, Sharma S, Tirkey N, Chopra K. Effect of curcumin on inflammation and oxidative stress in cisplatin-induced experimental nephrotoxicity. J Agric Food Chem. 2007;55:10150–10155. doi: 10.1021/jf0723965. [DOI] [PubMed] [Google Scholar]
- 327.Tirkey N, Kaur G, Vij G, Chopra K. Curcumin, a diferuloylmethane, attenuates cyclosporine-induced renal dysfunction and oxidative stress in rat kidneys. BMC Pharmacol. 2005;5:15–25. doi: 10.1186/1471-2210-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Li Y, VandenBoom TG, 2nd, Kong D, Wang Z, Ali S, Philip PA, et al. Upregulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009;69:6704–6712. doi: 10.1158/0008-5472.CAN-09-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Bao B, Ali S, Banerjee S, Wang Z, Logna F, Azmi AS, et al. Curcumin analogue CDF inhibits pancreatic tumor growth by switching on suppressor microRNAs and attenuating EZH2 expression. Cancer Res. 2012;72:335–345. doi: 10.1158/0008-5472.CAN-11-2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Yang J, Cao Y, Sun J, Zhang Y. Curcumin reduces the expression of Bcl-2 by upregulating miR-15a and miR-16 in MCF-7 cells. Med Oncol. 2010;27:1114–1118. doi: 10.1007/s12032-009-9344-3. [DOI] [PubMed] [Google Scholar]
- 331.Russo M, Spagnuolo C, Tedesco I, Russo GL. Phytochemicals in cancer prevention and therapy: truth or dare? Toxins (Basel) 2010;2:517–551. doi: 10.3390/toxins2040517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Marconett CN, Singhal AK, Sundar SN, Firestone GL. Indole-3-carbinol disrupts estrogen receptor-alpha dependent expression of insulin-like growth factor-1 receptor and insulin receptor substrate-1 and proliferation of human breast cancer cells. Mol Cell Endocrinol. 2012;363:74–84. doi: 10.1016/j.mce.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Clarke JD, Dashwood RH, Ho E. Multi-targeted prevention of cancer by sulforaphane. Cancer Lett. 2008;269:291–304. doi: 10.1016/j.canlet.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Qazi A, Pal J, Maitah M, Fulciniti M, Pelluru D, Nanjappa P, et al. Anticancer activity of a broccoli derivative, sulforaphane, in barrett adenocarcinoma: potential use in chemoprevention and as adjuvant in chemotherapy. Transl Oncol. 2010;3:389–399. doi: 10.1593/tlo.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Kwon JS, Joung H, Kim YS, Shim YS, Ahn Y, Jeong MH, et al. Sulforaphane inhibits restenosis by suppressing inflammation and the proliferation of vascular smooth muscle cells. Atherosclerosis. 2012;225:41–49. doi: 10.1016/j.atherosclerosis.2012.07.040. [DOI] [PubMed] [Google Scholar]
- 336.Li Y, Zhang T, Korkaya H, Liu S, Lee HF, Newman B, et al. Sulforaphane, a dietary component of broccoli/broccoli sprouts, inhibits breast cancer stem cells. Clin Cancer Res. 2010;16:2580–2590. doi: 10.1158/1078-0432.CCR-09-2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Rausch V, Liu L, Kallifatidis G, Baumann B, Mattern J, Gladkich J, et al. Synergistic activity of sorafenib and sulforaphane abolishes pancreatic cancer stem cell characteristics. Cancer Res. 2010;70:5004–5013. doi: 10.1158/0008-5472.CAN-10-0066. [DOI] [PubMed] [Google Scholar]
- 338.Izutani Y, Yogosawa S, Sowa Y, Sakai T. Brassinin induces G1 phase arrest through increase of p21 and p27 by inhibition of the phosphatidylinositol 3-kinase signaling pathway in human colon cancer cells. Int J Oncol. 2012;40:816–824. doi: 10.3892/ijo.2011.1246. [DOI] [PubMed] [Google Scholar]
- 339.Jin Y. 3,3′-Diindolylmethane inhibits breast cancer cell growth via miR-21-mediated Cdc25A degradation. Mol Cell Biochem. 2011;358:345–354. doi: 10.1007/s11010-011-0985-0. [DOI] [PubMed] [Google Scholar]
- 340.Bauer JA, Sinclair DA. Therapeutic potential of resveratrol: The in vivo evidence. Nat Rev Drug Discov. 2006;5:493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- 341.Schneider Y, Duranton B, Gosse F, Schleiffer R, Seiler N, Raul F. Resveratrol inhibits intestinal tumorigenesis and modulates host-defense-related gene expression in an animal model of human familial adenomatous polyposis. Nutr Cancer. 2001;39:102–107. doi: 10.1207/S15327914nc391_14. [DOI] [PubMed] [Google Scholar]
- 342.Bishayee A. Cancer prevention and treatment with resveratrol: from rodent studies to clinical trials. Cancer Prev Res (Phila) 2009;2:409–418. doi: 10.1158/1940-6207.CAPR-08-0160. [DOI] [PubMed] [Google Scholar]
- 343.Delmas D, Rebe C, Micheau O, Athias A, Gambert P, Grazide S, et al. Redistribution of CD95, DR4 and DR5 in rafts accounts for the synergistic toxicity of resveratrol and death receptor ligands in colon carcinoma cells. Oncogene. 2004;23:8979–8986. doi: 10.1038/sj.onc.1208086. [DOI] [PubMed] [Google Scholar]
- 344.Fulda S, Debatin KM. Resveratrol modulation of signal transduction in apoptosis and cell survival: A mini-review. Cancer Detect Prev. 2006;30:217–223. doi: 10.1016/j.cdp.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 345.Li Y, Liu J, Liu X, Xing K, Wang Y, Li F, et al. Resveratrol-induced cell inhibition of growth and apoptosis in MCF7 human breast cancer cells are associated with modulation of phosphorylated Akt and caspase-9. Appl Biochem Biotechnol. 2006;135:181–192. doi: 10.1385/abab:135:3:181. [DOI] [PubMed] [Google Scholar]
- 346.Signorelli P, Ghidoni R. Resveratrol as an anticancer nutrient: molecular basis, open questions and promises. J Nutr Biochem. 2005;16:449–466. doi: 10.1016/j.jnutbio.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 347.Wang Y, Romigh T, He X, Orloff MS, Silverman RH, Heston WD, et al. Resveratrol regulates the PTEN/AKT pathway through androgen receptor-dependent and -independent mechanisms in prostate cancer cell lines. Hum Mol Genet. 2010;19:4319–4329. doi: 10.1093/hmg/ddq354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Vanamala J, Reddivari L, Radhakrishnan S, Tarver C. Resveratrol suppresses IGF-1 induced human colon cancer cell proliferation and elevates apoptosis via suppression of IGF-1R/Wnt and activation of p53 signaling pathways. BMC Cancer. 2010;10:238–251. doi: 10.1186/1471-2407-10-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Nguyen AV, Martinez M, Stamos MJ, Moyer MP, Planutis K, Hope C, et al. Results of a phase I pilot clinical trial examining the effect of plant-derived resveratrol and grape powder on Wnt pathway target gene expression in colonic mucosa and colon cancer. Cancer Manag Res. 2009;1:25–37. [PMC free article] [PubMed] [Google Scholar]
- 350.Sen M, Ghosh G. Transcriptional outcome of Wnt-frizzled signal transduction in inflammation: evolving concepts. J Immunol. 2008;181:4441–4445. doi: 10.4049/jimmunol.181.7.4441. [DOI] [PubMed] [Google Scholar]
- 351.Parekh P, Motivale L, Naik N, Rao KV. Downregulation of cyclin D1 is associated with decreased levels of p38 MAP kinases, AKT/PKB and Pak1 during chemopreventive effects of resveratrol in liver cancer cells. Exp Toxicol Pathol. 2011;63:167–173. doi: 10.1016/j.etp.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 352.Vergara D, Simeone P, Toraldo D, Del Boccio P, Vergaro V, Leporatti S, et al. Resveratrol downregulates AKT/GSK and ERK signaling pathways in OVCAR-3 ovarian cancer cells. Mol Biosyst. 2012;8:1078–1087. doi: 10.1039/c2mb05486h. [DOI] [PubMed] [Google Scholar]
- 353.Sheth S, Jajoo S, Kaur T, Mukherjea D, Sheehan K, Rybak LP, et al. Resveratrol reduces prostate cancer growth and metastasis by inhibiting the AKT/miRNA21 pathway. PLoS One. 2012;7:e15655. doi: 10.1371/journal.pone.0051655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Kundu JK, Shin YK, Surh YJ. Resveratrol modulates phorbol ester-induced pro-inflammatory signal transduction pathways in mouse skin in vivo: NF-kappaB and AP-1 as prime targets. Biochem Pharmacol. 2006;72:1506–1515. doi: 10.1016/j.bcp.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 355.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 356.Mo W, Xu X, Wang F, Ke A, Wang X, Guo C. Resveratrol inhibits proliferation and induces apoptosis through the Hedgehog signaling pathway in pancreatic cancer cell. Pancreatology. 2011;11:601–609. doi: 10.1159/000333542. [DOI] [PubMed] [Google Scholar]
- 357.Quoc TL, Espinoza JL, Takami A, Nakao S. Resveratrol induces cell cycle arrest and apoptosis in malignant NK cells via JAK2/STAT3 pathway inhibition. PLoS One. 2013;8:e55183. doi: 10.1371/journal.pone.0055183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Bishayee A, Dhir N. Resveratrol-mediated chemoprevention of diethylnitrosamine-initiated hepatocarcinogenesis: inhibition of cell proliferation and induction of apoptosis. Chem Biol Interact. 2009;179:131–144. doi: 10.1016/j.cbi.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 359.Bishayee A, Waghray A, Barnes KF, Mbimba T, Bhatia D, Chatterjee M, et al. Suppression of the inflammatory cascade is implicated in resveratrol chemoprevention of experimental hepatocarcinogenesis. Pharm Res. 2010;27:1080–1091. doi: 10.1007/s11095-010-0144-4. [DOI] [PubMed] [Google Scholar]
- 360.Shah MS, Davidson LA, Chapkin RS. Mechanistic insights into the role of microRNAs in cancer: influence of nutrient crosstalk. Front Genet. 2012;3:305. doi: 10.3389/fgene.2012.00305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Knutson MD, Leeuwenburgh C. Resveratrol and novel potent activators of SIRT1: Effects on aging and age-related diseases. Nutr Rev. 2008;66:591–596. doi: 10.1111/j.1753-4887.2008.00109.x. [DOI] [PubMed] [Google Scholar]
- 362.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 363.Virgili F, Marino M. Regulation of cellular signals from nutritional molecules: a specific role for phytochemicals, beyond antioxidant activity. Free Radic Biol Med. 2008;45:1205–1216. doi: 10.1016/j.freeradbiomed.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 364.Whitehead SA, Rice S. Endocrine-disrupting chemicals as modulators of sex steroid synthesis. Best Pract Res Clin Endocrinol Metab. 2006;20:45–61. doi: 10.1016/j.beem.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 365.Bulzomi P, Bolli A, Galluzzo P, Leone S, Acconcia F, Marino M. Naringenin and 17β-estradiol coadministration prevents hormone-induced human cancer cell growth. IUBMB Life. 2010;62:51–60. doi: 10.1002/iub.279. [DOI] [PubMed] [Google Scholar]
- 366.Galluzzo P, Martini C, Bulzomi P, Leone S, Bolli A, Pallottini V, et al. Quercetin-induced apoptotic cascade in cancer cells: antioxidant versus estrogen receptor α-dependent mechanisms. Mol Nutr Food Res. 2009;53:699–708. doi: 10.1002/mnfr.200800239. [DOI] [PubMed] [Google Scholar]
- 367.Bulzomi P, Galluzzo P, Bolli A, Leone S, Acconcia F, Marino M. The pro-apoptotic effect of quercetin in cancer cell lines requires ERβ-dependent signals. J Cell Physiol. 2012;227:1891–1898. doi: 10.1002/jcp.22917. [DOI] [PubMed] [Google Scholar]
- 368.Amin A, Buratovich M. The anti-cancer charm of flavonoids: a cup-of-tea will do! Recent Pat Anticancer Drug Discov. 2007;2:109–117. doi: 10.2174/157489207780832414. [DOI] [PubMed] [Google Scholar]
- 369.Galluzzo P, Marino M. Nutritional flavonoids impact on nuclear and extranuclear estrogen receptor activities. Genes Nutr. 2006;1:161–176. doi: 10.1007/BF02829966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Wen J, Liu B, Yuan E, Ma Y, Zhu Y. Preparation and physicochemical properties of the complex of naringenin with hydroxypropyl-β-cyclodextrin. Molecules. 2010;15:4401–4407. doi: 10.3390/molecules15064401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Bansal T, Jaggi M, Khar RK, Talegaonkar S. Emerging significance of flavonoids as P-glycoprotein inhibitors in cancer chemotherapy. J Pharm Pharm Sci. 2009;12:46–78. doi: 10.18433/j3rc77. [DOI] [PubMed] [Google Scholar]
- 372.Choi JS, Jo BW, Kim YC. Enhanced paclitaxel bioavailability after oral administration of paclitaxel or prodrug to rats pretreated with quercetin. Eur J Pharm Biopharm. 2004;57:313–318. doi: 10.1016/j.ejpb.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 373.Ikegawa T, Ushigome F, Koyabu N, Morimoto S, Shoyama Y, Naito M, et al. Inhibition of P-glycoprotein by orange juice components, polymethoxyflavones in adriamycin-resistant human myelogenous leukemia (K562/ADM) cells. Cancer Lett. 2000;160:21–28. doi: 10.1016/s0304-3835(00)00549-8. [DOI] [PubMed] [Google Scholar]
- 374.Beltz LA, Bayer DK, Moss AL, Simet IM. Mechanisms of cancer prevention by green and black tea polyphenols. Anticancer Agents Med Chem. 2006;6:389–406. doi: 10.2174/187152006778226468. [DOI] [PubMed] [Google Scholar]
- 375.Fang MZ, Wang Y, Ai N, et al. Tea polyphenol epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003;63:7563–7570. [PubMed] [Google Scholar]
- 376.Tsang JS, Ebert MS, van Oudenaarden A. Genome-wide dissection of microRNA functions and cotargeting networks using gene set signatures. Mol Cell. 2010;38:140–153. doi: 10.1016/j.molcel.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Wang H, Bian S, Yang CS. Green tea polyphenol EGCG suppresses lung cancer cell growth through upregulating miR-210 expression caused by stabilizing HIF-1alpha. Carcinogenesis. 2011;32:1881–1889. doi: 10.1093/carcin/bgr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Khan N, Afaq F, Saleem M, Ahmad N, Mukhtar H. Targeting multiple signaling pathways by green tea polyphenol (−)-epigallocatechin-3-gallate. Cancer Res. 2006;66:2500–2505. doi: 10.1158/0008-5472.CAN-05-3636. [DOI] [PubMed] [Google Scholar]
- 379.Leone M, Zhai D, Sareth S, Kitada S, Reed JC, Pellecchia M. Cancer prevention by tea polyphenols is linked to their direct inhibition of antiapoptotic Bcl-2-family proteins. Cancer Res. 2003;63:8118–8121. [PubMed] [Google Scholar]
- 380.Kanadzu M, Lu Y, Morimoto K. Dual function of epigallocatechin gallate (EGCG) in healthy human lymphocytes. Cancer Lett. 2006;241:250–255. doi: 10.1016/j.canlet.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 381.Mazzanti G, Menniti-Ippolito F, Moro PA, Cassetti F, Raschetti R, Santuccio C, Mastrangelo S. Hepatotoxicity from green tea: A review of the literature and two unpublished cases. Eur J Clin Pharmacol. 2009;65:331–341. doi: 10.1007/s00228-008-0610-7. [DOI] [PubMed] [Google Scholar]
- 382.Barnes S, Peterson G, Coward L. Rationale for the use of genistein containing soy matrices in chemoprevention trials for breast and prostate cancer. J Cell Biochem. 1995;22:181–187. doi: 10.1002/jcb.240590823. [DOI] [PubMed] [Google Scholar]
- 383.Paggliaci MC, Smacchia M, Migliorati G, Grignani F, Riccardi C, Nicoletti I. Growth inhibitory effects of the natural phytoestrogen in MCF-7 human breast cancer cells. Eur J Cancer. 1994;30A:1675–1682. doi: 10.1016/0959-8049(94)00262-4. [DOI] [PubMed] [Google Scholar]
- 384.Li Y, Ahmed F, Ali S, Philip PA, Kucuk O, Sarkar FH. Inactivation of nuclear factor kappaB by soy isoflavone genistein contributes to increased apoptosis induced by chemotherapeutic agents in human cancer cells. Cancer Res. 2005;65:6934–6942. doi: 10.1158/0008-5472.CAN-04-4604. [DOI] [PubMed] [Google Scholar]
- 385.Linsalata M, Russo F, Notarnicola M, Guerra V, Cavallini A, Clemente C, et al. Effects of genistein on the polyamine metabolism and cell growth in DLD-1 human colon cancer cells. Nutr Cancer. 2005;52:84–93. doi: 10.1207/s15327914nc5201_11. [DOI] [PubMed] [Google Scholar]
- 386.Qi W, Weber CR, Wasland K, Savkovic SD. Genistein inhibits proliferation of colon cancer cells by attenuating a negative effect of epidermal growth factor on tumor suppressor FOXO3 activity. BMC Cancer. 2011;11:219–227. doi: 10.1186/1471-2407-11-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Pan H, Zhou W, He W, Liu X, Ding Q, Ling L, et al. Genistein inhibits MDA-MB-231 triple-negative breast cancer cell growth by inhibiting NF-kappaB activity via the Notch-1 pathway. Int J Mol Med. 2012;30:337–343. doi: 10.3892/ijmm.2012.990. [DOI] [PubMed] [Google Scholar]
- 388.Zhu H, Cheng H, Ren Y, Liu ZG, Zhang YF, De Luo B. Synergistic inhibitory effects by the combination of gefitinib and genistein on NSCLC with acquired drug-resistance in vitro and in vivo. Mol Biol Rep. 2012;39:4971–4979. doi: 10.1007/s11033-011-1293-1. [DOI] [PubMed] [Google Scholar]
- 389.Parker LP, Taylor DD, Kesterson J, Metzinger DS, Gercel-Taylor C. Modulation of microRNA associated with ovarian cancer cells by genistein. Eur J Gynaecol Oncol. 2009;30:616–621. [PubMed] [Google Scholar]
- 390.Lattrich C, Lubig J, Springwald A, Goerse R, Ortmann O, Treeck O. Additive effects of trastuzumab and genistein on human breast cancer cells. Anticancer Drugs. 2011;22:253–261. doi: 10.1097/CAD.0b013e3283427bb5. [DOI] [PubMed] [Google Scholar]
- 391.Park SJ, Kim MJ, Kim YK, Kim SM, Park JY, Myoung H. Combined cetuximab and genistein treatment shows additive anti-cancer effect on oral squamous cell carcinoma. Cancer Lett. 2010;292:54–63. doi: 10.1016/j.canlet.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 392.Shukla S, MacLennan GT, Flask CA, Fu P, Mishra A, Resnick MI, et al. Blockade of beta-catenin signaling by plant flavonoid apigenin suppresses prostate carcinogenesis in TRAMP mice. Cancer Res. 2007;67:6925–6935. doi: 10.1158/0008-5472.CAN-07-0717. [DOI] [PubMed] [Google Scholar]
- 393.Su Y, Simmen RC. Soy isoflavone genistein upregulates epithelial adhesion molecule E-cadherin expression and attenuates beta-catenin signaling in mammary epithelial cells. Carcinogenesis. 2009;30:331–339. doi: 10.1093/carcin/bgn279. [DOI] [PubMed] [Google Scholar]
- 394.Zhang Y, Chen H. Genistein attenuates WNT signaling by up-regulating sFRqP2 in a human colon cancer cell line. Exp Biol Med. 2011;236:714–722. doi: 10.1258/ebm.2011.010347. [DOI] [PubMed] [Google Scholar]
- 395.Monroe DG, Secreto FJ, Subramaniam M, Getz BJ, Khosla S, Spelsberg TC. Estrogen receptor alpha and beta heterodimers exert unique effects on estrogen and tamoxifen-dependent gene expression in human U2OS osteosarcoma cells. Mol Endocrinol. 2005;19:1555–1568. doi: 10.1210/me.2004-0381. [DOI] [PubMed] [Google Scholar]
- 396.Imamov O, Morani A, Shim GJ, Omoto Y, Thulin-Andersson C, Warner M, et al. Estrogen receptor beta regulates epithelial cellular differentiation in the mouse ventral prostate. Proc Natl Acad Sci USA. 2004;101:9375–9380. doi: 10.1073/pnas.0403041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Rietjens IMCM, Sotoca AM, Vervoort J, Louisse J. Mechanisms underlying the dualistic mode of action of major soy isoflavones in relation to cell proliferation and cancer risks. Mol Nutr Food Res. 2013;57:100–113. doi: 10.1002/mnfr.201200439. [DOI] [PubMed] [Google Scholar]
- 398.Parker LP, Taylor DD, Kesterson J, Metzinger DS, Gercel-Taylor C. Modulation of microRNA associated with ovarian cancer cells by genistein. Eur J Gynaecol Oncol. 2009;30:616–621. [PubMed] [Google Scholar]
- 399.Nakamura H, Wang Y, Kurita T, Adomat H, Cunha GR, Wang Y. Genistein increases epidermal growth factor receptor signaling and promotes tumor progression in advanced human prostate cancer. PLoS One. 2011;6:e20034. doi: 10.1371/journal.pone.0020034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Uckun FM, Evans WE, Forsyth CJ, Waddick KG, Ahlgren LT, Chelstrom LM, et al. Biotherapy of B-cell precursor leukemia by targeting genistein to CD19-associated tyrosine kinases. Science. 1995;267:886–891. doi: 10.1126/science.7531365. [DOI] [PubMed] [Google Scholar]
- 401.Uckun FM, Narla RK, Zeren T, Yanishevski Y, Myers DE, Waurzyniak B, et al. In vivo toxicity, pharmacokinetics, and anticancer activity of Genistein linked to recombinant human epidermal growth factor. Clin Cancer Res. 1998;4:1125–1134. [PubMed] [Google Scholar]
- 402.Heber D. Multitargeted therapy of cancer by ellagitannins. Cancer Lett. 2008;269:262–268. doi: 10.1016/j.canlet.2008.03.043. [DOI] [PubMed] [Google Scholar]
- 403.Sartippour MR, Seeram NP, Rao JY, Moro A, Harris DM, Henning SM, et al. Ellagitannin-rich pomegranate extract inhibits angiogenesis in prostate cancer in vitro and in vivo. Int J Oncol. 2008;32:475–480. [PubMed] [Google Scholar]
- 404.Adams LS, Zhang Y, Seeram NP, Heber D, Chen S. Pomegranate ellagitannin-derived compounds exhibit antiproliferative and antiaromatase activity in breast cancer cells in vitro. Cancer Prev Res (Phila) 2010;3:108–113. doi: 10.1158/1940-6207.CAPR-08-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Bishayee A, Thoppil RJ, Darvesh AS, Ohanyan V, Meszaros JG, Bhatia D. Pomegranate phytoconstituents blunt the inflammatory cascade in a chemically induced rodent model of hepatocellular carcinogenesis. J Nutr Biochem. 2013;24:178–187. doi: 10.1016/j.jnutbio.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 406.Bhatia D, Thoppil RJ, Mandal A, Samtani KA, Darvesh AS, Bishayee A. Pomegranate Bioactive Constituents Suppress Cell Proliferation and Induce Apoptosis in an Experimental Model of Hepatocellular Carcinoma: Role of Wnt/β-Catenin Signaling Pathway. Evid Based Complement Alternat Med. 2013;2013:371813. doi: 10.1155/2013/371813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Wen XY, Wu SY, Li ZQ, Liu ZQ, Zhang JJ, Wang GF, et al. Ellagitannin (BJA3121), an anti-proliferative natural polyphenol compound, can regulate the expression of MiRNAs in HepG2 cancer cells. Phytother Res. 2009;23:778–784. doi: 10.1002/ptr.2616. [DOI] [PubMed] [Google Scholar]
- 408.Mills PK, Beeson WL, Phillips RL, Fraser GE. Cohort study of diet, lifestyle, and prostate cancer in Adventist men. Cancer. 1989;64:598–604. doi: 10.1002/1097-0142(19890801)64:3<598::aid-cncr2820640306>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 409.Giovannucci E. Does prostate-specific antigen screening influence the results of studies of tomatoes, lycopene, and prostate cancer risk? J. Natl. Cancer Inst. 2007;99:1060–1062. doi: 10.1093/jnci/djm048. [DOI] [PubMed] [Google Scholar]
- 410.Hung CF, Huang TF, Chen BH, Shieh JM, Wu PH, Wu WB. Lycopene inhibits TNF-alpha-induced endothelial ICAM-1 expression and monocyte-endothelial adhesion. Eur J Pharmacol. 2008;586:275–282. doi: 10.1016/j.ejphar.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 411.Tang FY, Shih CJ, Cheng LH, Ho HJ, Chen HJ. Lycopene inhibits growth of human colon cancer cells via suppression of the Akt signaling pathway. Mol Nutr Food Res. 2008;52:646–654. doi: 10.1002/mnfr.200700272. [DOI] [PubMed] [Google Scholar]
- 412.Liu X, Allen JD, Arnold JT, Blackman MR. Lycopene inhibits IGF-I signal transduction and growth in normal prostate epithelial cells by decreasing DHT-modulated IGF-I production in co-cultured reactive stromal cells. Carcinogenesis. 2008;29:816–823. doi: 10.1093/carcin/bgn011. [DOI] [PubMed] [Google Scholar]
- 413.Burgess LC, Rice E, Fischer T, Seekins JR, Burgess TP, Sticka SJ, et al. Lycopene has limited effect on cell proliferation in only two of seven human cell lines (both cancerous and noncancerous) in an in vitro system with doses across the physiological range. Toxicol In Vitro. 2008;22:1297–1300. doi: 10.1016/j.tiv.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Pratheeshkumar P, Budhraja A, Son YO, Wang X, Zhang Z, et al. Quercetin inhibits angiogenesis mediated human prostate tumor growth by targeting VEGFR-2 regulated AKT/mTOR/P70S6K signaling pathways. PLoS One. 2012;7:e47516. doi: 10.1371/journal.pone.0047516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Choi JS, Jo BW, Kim YC. Enhanced paclitaxel bioavailability after oral administration of paclitaxel or prodrug to rats pretreated with quercetin. Eur J Pharm Biopharm. 2004;57:313–318. doi: 10.1016/j.ejpb.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 416.Shin SC, Choi JS, Li X. Enhanced bioavailability of tamoxifen after oral administration of tamoxifen with quercetin in rats. Int J Pharm. 2006;313:144–149. doi: 10.1016/j.ijpharm.2006.01.028. [DOI] [PubMed] [Google Scholar]
- 417.Guruvayoorappan C, Kuttan G. 13 cis-retinoic acid regulates cytokine production and inhibits angiogenesis by disrupting endothelial cell migration and tube formation. J Exp Ther Oncol. 2008;7:173–182. [PubMed] [Google Scholar]
- 418.Guo W, Kong E, Meydani M. Dietary polyphenols, inflammation, and cancer. Nutr Cancer. 2009;61:807–810. doi: 10.1080/01635580903285098. [DOI] [PubMed] [Google Scholar]
- 419.Zaidman BZ, Yassin M, Mahajna J, Wasser SP. Medicinal mushroom modulators of molecular targets as cancer therapeutics. Appl Microbiol Biotechnol. 2005;67:453–468. doi: 10.1007/s00253-004-1787-z. [DOI] [PubMed] [Google Scholar]
- 420.Nerurkar P, Ray RB. Bitter melon: antagonist to cancer. Pharm Res. 2010;27:1049–1053. doi: 10.1007/s11095-010-0057-2. [DOI] [PubMed] [Google Scholar]
- 421.Dotan N, Wasser SP, Mahajna J. Inhibition of the androgen receptor activity by Coprinus comatus substances. Nutr Cancer. 2011;63:1316–1327. doi: 10.1080/01635581.2011.607542. [DOI] [PubMed] [Google Scholar]
- 422.Dotan N, Wasser SP, Mahajna J. The culinary-medicinal mushroom Coprinus comatus as a natural antiandrogenic modulator. Integr Cancer Ther. 2011;10:148–159. doi: 10.1177/1534735410383169. [DOI] [PubMed] [Google Scholar]
- 423.Mahajna J, Dotan N, Zaidman BZ, Petrova RD, Wasser SP. Pharmacological values of medicinal mushrooms for prostate cancer therapy: the case of Ganoderma lucidum. Nutr Cancer. 2009;61:16–26. doi: 10.1080/01635580802379323. [DOI] [PubMed] [Google Scholar]
- 424.Zaidman BZ, Wasser SP, Nevo E, Mahajna J. Coprinus comatus and Ganoderma lucidum interfere with androgen receptor function in LNCaP prostate cancer cells. Mol Biol Rep. 2008;35:107–117. doi: 10.1007/s11033-007-9059-5. [DOI] [PubMed] [Google Scholar]
- 425.Kucuk O, Sarkar FH, Djuric Z, Sakr W, Pollak MN, Khachik F, et al. Effects of lycopene supplementation in patients with localized prostate cancer. Exp Biol Med (Maywood) 2002;227:881–885. doi: 10.1177/153537020222701007. [DOI] [PubMed] [Google Scholar]
- 426.Hsieh TC, Wu JM. Suppression of cell proliferation and gene expression by combinatorial synergy of EGCG, resveratrol and gamma-tocotrienol in estrogen receptor-positive MCF-7 breast cancer cells. Int J Oncol. 2008;33:851–859. [PubMed] [Google Scholar]
- 427.International Human Genome Sequencing Consortium Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 428.Adams MD, Celniker CE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- 429.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 430.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 431.Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 432.Mardis ER. Genome sequencing and cancer. Curr Opin Genet Dev. 2012;22:245–250. doi: 10.1016/j.gde.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Tran B, Dancey JE, Kamel-Reid S, McPherson JD, Bedard PL, Brown AM, et al. Cancer genomics: technology, discovery, and translation. J Clin Oncol. 2012;30:647–660. doi: 10.1200/JCO.2011.39.2316. [DOI] [PubMed] [Google Scholar]
- 434.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.The ENCODE Project Consortium Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Narayanan BA, Narayanan NK, Desai D, Pittman B, Reddy BS. Effects of a combination of docosahexaenoic acid and 1,4-phenylene bis(methylene) selenocyanate on cyclooxygenase 2, inducible nitric oxide synthase and beta-catenin pathways in colon cancer cells. Carcinogenesis. 2004;25:2443–2449. doi: 10.1093/carcin/bgh252. [DOI] [PubMed] [Google Scholar]
- 437.Rao CV, Cooma I, Rodriguez JG, Simi B, El-Bayoumy K, Reddy BS. Chemoprevention of familial adenomatous polyposis development In the APC(min) mouse model by 1,4-phenylene bis(methylene) selenocyanate. Carcinogenesis. 2000;21:617–621. doi: 10.1093/carcin/21.4.617. [DOI] [PubMed] [Google Scholar]
- 438.Gao Z, Xu Z, Hung MS, Lin YC, Wang T, Gong M, et al. Promoter demethylation of WIF-1 by epigallocatechin-3-gallate in lung cancer cells. Anticancer Res. 2009;29:2025–2030. [PubMed] [Google Scholar]
- 439.Kim J, Zhang X, Rieger-Christ KM, Summerhayes IC, Wazer DE, Paulson KE, et al. Suppression of Wnt signaling by the green tea compound (−)-epigallocatechin 3-gallate (EGCG) in invasive breast cancer cells. Requirement of the transcriptional repressor HBP1. J Biol Chem. 2006;281:10865–10875. doi: 10.1074/jbc.M513378200. [DOI] [PubMed] [Google Scholar]
- 440.Reguart N, He B, Taron M, You L, Jablons DM, Rosell R. The role of Wnt signaling in cancer and stem cells. Future Oncol. 2005;1:787–797. doi: 10.2217/14796694.1.6.787. [DOI] [PubMed] [Google Scholar]
- 441.Bose M, Hao X, Ju J, Husain A, Park S, Lambert JD, et al. Inhibition of tumorigenesis in Apc/Min mice by a combination of (−)-epigallocatechin-3-gallate and fish oil. J Agric Food Chem. 2007;55:7695–7700. doi: 10.1021/jf071004r. [DOI] [PubMed] [Google Scholar]
- 442.Hope C, Planutis K, Planutiene M, Moyer MP, Johal KS, Woo J, et al. Low concentrations of resveratrol inhibit Wnt signal throughput in colon-derived cells: implications for colon cancer prevention. Mol Nutr Food Res. 2008;52:S52–61. doi: 10.1002/mnfr.200700448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Chen HJ, Hsu LS, Shia YT, Lin MW, Lin CM. The β-catenin/TCF complex as a novel target of resveratrol in the Wnt/β-catenin signaling pathway. Biochem Pharmacol. 2012;84:1143–53. doi: 10.1016/j.bcp.2012.08.011. [DOI] [PubMed] [Google Scholar]
- 444.Park CH, Hahm ER, Lee JH, Jung KC, Yang CH. Inhibition of beta-catenin-mediated transactivation by flavanone in AGS gastric cancer cells. Biochem Biophys Res Commun. 2005;331:1222–1228. doi: 10.1016/j.bbrc.2005.03.242. [DOI] [PubMed] [Google Scholar]
- 445.Li Y, Wang Z, Kong D, Li R, Sarkar SH, Sarkar FH. Regulation of Akt/FOXO3a/GSK-3beta/AR signaling network by isoflavone in prostate cancer cells. J Biol Chem. 2008;283:27707–27716. doi: 10.1074/jbc.M802759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Zhang Y, Chen H. Genistein attenuates WNT signaling by up-regulating sFRP2 in a human colon cancer cell line. Exp Biol Med (Maywood) 2011;236:714–722. doi: 10.1258/ebm.2011.010347. [DOI] [PubMed] [Google Scholar]
- 447.Hirata H, Ueno K, Nakajima K, Tabatabai ZL, Hinoda Y, Ishii N, et al. Genistein downregulates onco-miR-1260b and inhibits Wnt-signalling in renal cancer cells. Br J Cancer. 2013;108:2070–2078. doi: 10.1038/bjc.2013.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Wang Z, Chen H. Genistein increases gene expression by demethylation of WNT5a promoter in colon cancer cell line SW1116. Anticancer Res. 2010;30:4537–4455. [PubMed] [Google Scholar]
- 449.Wang H, Li Q, Chen H. Genistein affects histone modifications on Dickkopf-related protein 1 (DKK1) gene in SW480 human colon cancer cell line. PLoS One. 2012;7:e40955. doi: 10.1371/journal.pone.0040955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Prasad CP, Rath G, Mathur S, Bhatnagar D, Ralhan R. Potent growth suppressive activity of curcumin in human breast cancer cells: Modulation of Wnt/beta-catenin signaling. Chem Biol Interact. 2009;181:263–271. doi: 10.1016/j.cbi.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 451.Jaiswal AS, Marlow BP, Gupta N, Narayan S. Beta-catenin-mediated transactivation and cell-cell adhesion pathways are important in curcumin (diferuylmethane)-induced growth arrest and apoptosis in colon cancer cells. Oncogene. 2002;21:8414–8427. doi: 10.1038/sj.onc.1205947. [DOI] [PubMed] [Google Scholar]
- 452.Yan C, Jamaluddin MS, Aggarwal B, Myers J, Boyd DD. Gene expression profiling identifies activating transcription factor 3 as a novel contributor to the proapoptotic effect of curcumin. Mol Cancer Ther. 2005;4:233–241. [PubMed] [Google Scholar]
- 453.Zhang X, Yin WK, Shi XD, Li Y. Curcumin activates Wnt/β-catenin signaling pathway through inhibiting the activity of GSK-3β in APPswe transfected SY5Y cells. Eur J Pharm Sci. 2011;42:540–546. doi: 10.1016/j.ejps.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 454.Ma Y, Feng Q, Sekula D, Diehl JA, Freemantle SJ, Dmitrovsky E. Retinoid targeting of different D-type cyclins through distinct chemopreventive mechanisms. Cancer Res. 2005;65:6476–6483. doi: 10.1158/0008-5472.CAN-05-0370. [DOI] [PubMed] [Google Scholar]
- 455.Palmer HG, Gonzalez-Sancho JM, Espada J, Berciano MT, Puig I, Baulida J, et al. Vitamin D3 promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J Cell Biol. 2001;154:369–387. doi: 10.1083/jcb.200102028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Larriba MJ, Valle N, Palmer HG, Ordóñez-Morán P, Alvarez-Díaz S, Becker KF, et al. The inhibition of Wnt/betacatenin signaling by 1alpha,25-dihydroxyvitamin D3 is abrogated by Snail1 in human colon cancer cells. Endocr Relat Cancer. 2007;14:141–151. doi: 10.1677/ERC-06-0028. [DOI] [PubMed] [Google Scholar]
- 457.Sarkar FH, Li Y, Wang Z, Kong D. The role of nutraceuticals in the regulation of Wnt and Hedgehog signaling in cancer. Cancer Metastasis Rev. 2010;29:383–394. doi: 10.1007/s10555-010-9233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Abdelwahab SI, Abdul AB, Mohan S, Taha MM, Syam S, Ibrahim MY, et al. Zerumbone induces apoptosis in T-acute lymphoblastic leukemia cells. Leuk Res. 2011;35:268–271. doi: 10.1016/j.leukres.2010.07.025. [DOI] [PubMed] [Google Scholar]
- 459.Sehrawat A, Arlotti JA, Murakami A, Singh SV. Zerumbone causes Bax- and Bak-mediated apoptosis in human breast cancer cells and inhibits orthotopic xenograft growth in vivo. Breast Cancer Res Treat. 2012;136(2):429–441. doi: 10.1007/s10549-012-2280-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Berman DM, Karhadkar SS, Hallahan AR, Pritchard JI, Eberhart CG, Watkins DN, et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science. 2002;297:1559–1561. doi: 10.1126/science.1073733. [DOI] [PubMed] [Google Scholar]
- 461.Mueller MT, Hermann PC, Witthauer J, Rubio-Viqueira B, Leicht SF, Huber S, et al. Combined targeted treatment to eliminate tumorigenic cancer stem cells in human pancreatic cancer. Gastroenterology. 2009;137(3):1102–1113. doi: 10.1053/j.gastro.2009.05.053. [DOI] [PubMed] [Google Scholar]
- 462.Elamin MH, Shinwari Z, Hendrayani SF, Al-Hindi H, Al-Shail E, Khafaga Y, et al. Curcumin inhibits the sonic hedgehog signaling pathway and triggers apoptosis in medulloblastoma cells. Mol Carcinogen. 2010;49:302–314. doi: 10.1002/mc.20604. [DOI] [PubMed] [Google Scholar]
- 463.Slusarz A, Shenouda NS, Sakla MS, Drenkhahn SK, Narula AS, MacDonald RS, et al. Common botanical compounds inhibit the hedgehog signaling pathway in prostate cancer. Cancer Res. 2010;70:3382–3390. doi: 10.1158/0008-5472.CAN-09-3012. [DOI] [PubMed] [Google Scholar]
- 464.Tang GQ, Yan TQ, Guo W, Ren TT, Peng CL, Zhao H, et al. (−)-Epigallocatechin3-gallate induces apoptosis and suppresses proliferation by inhibiting the human Indian Hedgehog pathway in human chondrosarcoma cells. J Cancer Res Clin Oncol. 2010;136:1179–1185. doi: 10.1007/s00432-010-0765-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Krop I, Demuth T, Guthrie T, Wen PY, Mason WP, Chinnaiyan P, et al. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J Clin Oncol. 2012;30:2307–2313. doi: 10.1200/JCO.2011.39.1540. [DOI] [PubMed] [Google Scholar]
- 466.Whitehead J, Thygesen H, Jaki T, Davies S, Halford S, Turner H, Cook N, Jodrell D. A novel Phase I/IIa design for early phase oncology studies and its application in the evaluation of MK-0752 in pancreatic cancer. Stat Med. 2012;31:1931–1943. doi: 10.1002/sim.5331. [DOI] [PubMed] [Google Scholar]
- 467.Nair JS, Sheikh T, Ho AL, Schwartz GK. PTEN regulates sensitivity of melanoma cells to RO4929097, the γ-secretase inhibitor. Anticancer Res. 2013;33:1307–1316. [PubMed] [Google Scholar]
- 468.Ma H, Li HQ, Zhang X. Cyclopamine, a naturally occurring alkaloid, and its analogues may find wide applications in cancer therapy. Curr Top Med Chem. 2013;13:2208–2215. doi: 10.2174/15680266113139990153. [DOI] [PubMed] [Google Scholar]
- 469.Cheng H, Merika E, Syrigos KN, Saif MW. Novel agents for the treatment of pancreatic adenocarcinoma.. JOP; Highlights from the “2011 ASCO Annual Meeting”.; Chicago, IL, USA. June 3-7, 2011; 2011. pp. 334–338. [PubMed] [Google Scholar]
- 470 370w.Brechbiel J, Miller-Moslin K, Adjei AA. Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treat Rev. 2014;40:750–759. doi: 10.1016/j.ctrv.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 471.Ali FR, Lear JT. Systemic treatments for basal cell carcinoma (BCC): the advent of dermato-oncology in BCC. Br J Dermatol. 2013;169:53–57. doi: 10.1111/bjd.12311. [DOI] [PubMed] [Google Scholar]
- 472.Sandhiya S, Melvin G, Kumar SS, Dkhar SA. The dawn of hedgehog inhibitors: Vismodegib. J Pharmacol Pharmacother. 2013;4:4–7. doi: 10.4103/0976-500X.107628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Temraz S, Mukherji D, Shamseddine A. Potential targets for colorectal cancer prevention. Int J Mol Sci. 2013;14:17279–17303. doi: 10.3390/ijms140917279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Juan ME, Alfaras I, Planas JM. Colorectal cancer chemoprevention by trans-resveratrol. Pharmacol Res. 2012;65:584–591. doi: 10.1016/j.phrs.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 475.Lenz HJ, Kahn M. Safely targeting cancer stem cells via selective catenin coactivator antagonism. Cancer Sci. 2014;105:1087–92. doi: 10.1111/cas.12471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Shan B, Schaaf C, Schmidt A, Lucia K, Buchfelder M, Losa M, et al. Curcumin suppresses HIF1A synthesis and VEGFA release in pituitary adenomas. J Endocrinol. 2012;214(3):389–398. doi: 10.1530/JOE-12-0207. [DOI] [PubMed] [Google Scholar]
- 477.Huynh H, Nguyen TT, Chan E, Tran E. Inhibition of ErbB-2 and ErbB-3 expression by quercetin prevents transforming growth factor alpha (TGF-alpha)- and epidermal growth factor (EGF)-induced human PC-3 prostate cancer cell proliferation. Int J Oncol. 2003;23:821–829. [PubMed] [Google Scholar]
- 478.Pratheeshkumar P, Budhraja A, Son YO, Wang X, Zhang Z, Ding S, et al. Quercetin inhibits angiogenesis mediated human prostate tumor growth by targeting VEGFR-2 regulated AKT/mTOR/P70S6K signaling pathways. PLoS One. 2012;7:e47516. doi: 10.1371/journal.pone.0047516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Huang CS, Fan YE, Lin CY, Hu ML. Lycopene inhibits matrix metalloproteinase-9 expression and down-regulates the binding activity of nuclear factor-kappa B and stimulatory protein-1. J Nutr Biochem. 2007;18:449–456. doi: 10.1016/j.jnutbio.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 480.Liu X, Allen JD, Arnold JT, Blackman MR. Lycopene inhibits IGF-I signal transduction and growth in normal prostate epithelial cells by decreasing DHT-modulated IGF-I production in co-cultured reactive stromal cells. Carcinogenesis. 2008;29:816–823. doi: 10.1093/carcin/bgn011. [DOI] [PubMed] [Google Scholar]
- 481.Tang FY, Shih CJ, Cheng LH, Ho HJ, Chen HJ. Lycopene inhibits growth of human colon cancer cells via suppression of the Akt signaling pathway. Mol Nutr Food Res. 2008;52:646–654. doi: 10.1002/mnfr.200700272. [DOI] [PubMed] [Google Scholar]
- 482.Wertz K. Lycopene effects contributing to prostate health. Nutr Cancer. 2009;61:775–783. doi: 10.1080/01635580903285023. [DOI] [PubMed] [Google Scholar]
- 483.Palozza P, Sheriff A, Serini S, Boninsegna A, Maggiano N, Ranelletti FO, et al. Lycopene induces apoptosis in immortalized fibroblasts exposed to tobacco smoke condensate through arresting cell cycle and down-regulating cyclin D1, pAKT and pBad. Apoptosis. 2005;10:1445–1456. doi: 10.1007/s10495-005-1393-2. [DOI] [PubMed] [Google Scholar]
- 484.Luoto KR, Kumareswaran R, Bristow RG. Tumor hypoxia as a driving force in genetic instability. Genome Integr. 2013;4(1):5. doi: 10.1186/2041-9414-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Xia Y, Shen S, Verma IM. NF-κB, an active player in human cancers. Cancer Immunol Res. 2014;2(9):823–830. doi: 10.1158/2326-6066.CIR-14-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Xu N, Lao Y, Zhang Y, Gillespie DA. Akt: a double-edged sword in cell proliferation and genome stability. J Oncol. 2012;2012:951724. doi: 10.1155/2012/951724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Moore JD. In the wrong place at the wrong time: does cyclin mislocalization drive oncogenic transformation? Nat Rev Cancer. 2013;13(3):201–208. doi: 10.1038/nrc3468. [DOI] [PubMed] [Google Scholar]
- 488.Nyquist MD, Dehm SM. Interplay between genomic alterations and androgen receptor signaling during prostate cancer development and progression. Horm Cancer. 2013;4(2):61–69. doi: 10.1007/s12672-013-0131-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Kabil A, Silva E, Kortenkamp A. Estrogens and genomic instability in human breast cancer cells--involvement of Src/Raf/Erk signaling in micronucleus formation by estrogenic chemicals. Carcinogenesis. 2008t;29:1862–1868. doi: 10.1093/carcin/bgn138. [DOI] [PubMed] [Google Scholar]
- 490.Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF Transcription Factors, Inflammation, and Immunity. Immunity. 2014;41:518–528. doi: 10.1016/j.immuni.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Vander Broek R, Snow GE, Chen Z, Van Waes C. Chemoprevention of head and neck squamous cell carcinoma through inhibition of NF-κB signaling. Oral Oncol. 2013;S1368-8375(13):00714–00718. doi: 10.1016/j.oraloncology.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. 2009;1(5):a000141. doi: 10.1101/cshperspect.a000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Dituri F, Mazzocca A, Giannelli G, Antonaci S. PI3K functions in cancer progression, anticancer immunity and immune evasion by tumors. Clin Dev Immunol. 2011;2011:947858. doi: 10.1155/2011/947858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Smith DA, Kiba A, Zong Y, Witte ON. Interleukin-6 and oncostatin-M synergize with the PI3K/AKT pathway to promote aggressive prostate malignancy in mouse and human tissues. Mol Cancer Res. 2013;11:1159–1165. doi: 10.1158/1541-7786.MCR-13-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Liu Y, Xu Y, Sun J, Ma A, Zhang F, Xia S, et al. AKT hyperactivation confers a Th1 phenotype in thymic Treg cells deficient in TGF-β receptor II signaling. Eur J Immunol. 2014;44:521–532. doi: 10.1002/eji.201243291. [DOI] [PubMed] [Google Scholar]
- 496.Han J, Soletti RC, Sadarangani A, Sridevi P, Ramirez ME, Eckmann L, et al. Nuclear expression of β-catenin promotes RB stability and resistance to TNF-induced apoptosis in colon cancer cells. Mol Cancer Res. 2013;11:207–218. doi: 10.1158/1541-7786.MCR-12-0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Keerthivasan S, Aghajani K, Dose M, Molinero L, Khan MW, Venkateswaran V, et al. β-Catenin promotes colitis and colon cancer through imprinting of proinflammatory properties in T cells. Sci Transl Med. 2014;6(225):225ra28. doi: 10.1126/scitranslmed.3007607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Salim T, Sand-Dejmek J, Sjölander A. The inflammatory mediator leukotriene D4 induces subcellular β-catenin translocation and migration of colon cancer cells. Exp Cell Res. 2014;321:255–266. doi: 10.1016/j.yexcr.2013.10.021. [DOI] [PubMed] [Google Scholar]
- 499.Durfort T, Tkach M, Meschaninova MI, Rivas MA, Elizalde PV, Venyaminova AG, et al. Small interfering RNA targeted to IGF-IR delays tumor growth and induces proinflammatory cytokines in a mouse breast cancer model. PLoS One. 2012;7:e29213. doi: 10.1371/journal.pone.0029213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Li S, Wang N, Brodt P. Metastatic cells can escape the proapoptotic effects of TNF-α through increased autocrine IL-6/STAT3 signaling. Cancer Res. 2012;72:865–875. doi: 10.1158/0008-5472.CAN-11-1357. [DOI] [PubMed] [Google Scholar]
- 501.Leitch AE, Haslett C, Rossi AG. Cyclin-dependent kinase inhibitor drugs as potential novel anti-inflammatory and pro-resolution agents. Br J Pharmacol. 2009;158:1004–1016. doi: 10.1111/j.1476-5381.2009.00402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Farahi N, Uller L, Juss JK, Langton AJ, Cowburn AS, Gibson A, et al. Effects of the cyclin-dependent kinase inhibitor R-roscovitine on eosinophil survival and clearance. Clin Exp Allergy. 2011;41:673–687. doi: 10.1111/j.1365-2222.2010.03680.x. [DOI] [PubMed] [Google Scholar]
- 503.Kaikkonen S, Paakinaho V, Sutinen P, Levonen AL, Palvimo JJ. Prostaglandin 15d-PGJ(2) inhibits androgen receptor signaling in prostate cancer cells. Mol Endocrinol. 2013;27:212–223. doi: 10.1210/me.2012-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Vera-Badillo FE, Templeton AJ, de Gouveia P, Diaz-Padilla I, Bedard PL, Al-Mubarak M, et al. Androgen receptor expression and outcomes in early breast cancer: a systematic review and meta-analysis. J Natl Cancer Inst. 2014;106:djt319. doi: 10.1093/jnci/djt319. [DOI] [PubMed] [Google Scholar]
- 505.Baumgarten SC, Frasor J. Minireview: Inflammation: an instigator of more aggressive estrogen receptor (ER) positive breast cancers. Mol Endocrinol. 2012;26(3):360–371. doi: 10.1210/me.2011-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Dey P, Ström A, Gustafsson JA. Estrogen receptor β upregulates FOXO3a and causes induction of apoptosis through PUMA in prostate cancer. Oncogene. 2013 doi: 10.1038/onc.2013.384. doi: 10.1038/onc.2013.384. [DOI] [PubMed] [Google Scholar]
- 507.Perkins ND. NF-kappaB: tumor promoter or suppressor? Trends Cell Biol. 2004;14:64–69. doi: 10.1016/j.tcb.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 508.Georgescu MM. PTEN tumor suppressor network in PI3K-Akt pathway control. Genes Cancer. 2010;1:1170–1177. doi: 10.1177/1947601911407325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Kennedy SG, Wagner AJ, Conzen SD, Jordán J, Bellacosa A, Tsichlis PN, et al. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 1997;11:701–713. doi: 10.1101/gad.11.6.701. [DOI] [PubMed] [Google Scholar]
- 510.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 511.Pai R, Dunlap D, Qing J, Mohtashemi I, Hotzel K, French DM. Inhibition of fibroblast growth factor 19 reduces tumor growth by modulating beta-catenin signaling. Cancer Res. 2008;68:5086–5095. doi: 10.1158/0008-5472.CAN-07-2325. [DOI] [PubMed] [Google Scholar]
- 512.Larsson O, Girnita A, Girnita L. Role of insulin-like growth factor 1 receptor signalling in cancer. Br J Cancer. 2005;92:2097–2101. doi: 10.1038/sj.bjc.6602627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Kuijjer ML, Peterse EF, van den Akker BE, Briaire-de Bruijn IH, Serra M, Meza-Zepeda LA, et al. IR/IGF1R signaling as potential target for treatment of high-grade osteosarcoma. BMC Cancer. 2013;13:245. doi: 10.1186/1471-2407-13-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Agarwal C, Dhanalakshmi S, Singh RP, Agarwal R. Inositol hexaphosphate inhibits growth and induces G1 arrest and apoptotic death of androgen-dependent human prostate carcinoma LNCaP cells. Neoplasia. 2004;6:646–59. doi: 10.1593/neo.04232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Deshpande A, Sicinski P, Hinds PW. Cyclins and cdks in development and cancer: a perspective. Oncogene. 2005;24:2909–2915. doi: 10.1038/sj.onc.1208618. [DOI] [PubMed] [Google Scholar]
- 516.Niu Y, Altuwaijri S, Lai KP, Wu CT, Ricke WA, Messing EM, Yao J, Yeh S, Chang C. Androgen receptor is a tumor suppressor and proliferator in prostate cancer. Proc Natl Acad Sci U S A. 2008;105:12182–12187. doi: 10.1073/pnas.0804700105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Shiota M, Takeuchi A, Yokomizo A, Kashiwagi E, Tatsugami K, Kuroiwa K, et al. Androgen receptor signaling regulates cell growth and vulnerability to doxorubicin in bladder cancer. J Urol. 2012;188:276–286. doi: 10.1016/j.juro.2012.02.2554. [DOI] [PubMed] [Google Scholar]
- 518.Léotoing L, Manin M, Monté D, Baron S, Communal Y, Lours C, et al. Crosstalk between androgen receptor and epidermal growth factor receptor-signalling pathways: a molecular switch for epithelial cell differentiation. J Mol Endocrinol. 2007;39:151–162. doi: 10.1677/JME-07-0021. [DOI] [PubMed] [Google Scholar]
- 519.Chen C, Baumann WT, Clarke R, Tyson JJ. Modeling the estrogen receptor to growth factor receptor signaling switch in human breast cancer cells. FEBS Lett. 2013;587:3327–3334. doi: 10.1016/j.febslet.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Schiff R, Massarweh S, Shou J, Osborne CK. Breast cancer endocrine resistance: how growth factor signaling and estrogen receptor coregulators modulate response. Clin Cancer Res. 2003;9:447S–454S. [PubMed] [Google Scholar]
- 521.Gabellini C, De Luca T, Trisciuoglio D, Desideri M, Di Martile M, Passeri D, et al. BH4 domain of bcl-2 protein is required for its proangiogenic function under hypoxic condition. Carcinogenesis. 2013;34:2558–2567. doi: 10.1093/carcin/bgt242. [DOI] [PubMed] [Google Scholar]
- 522.Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumor angiogenesis. Nature. 1998;394:485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- 523.Caamaño J, Hunter CA. NF-kappaB family of transcription factors: central regulators of innate and adaptive immune functions. Clin Microbiol Rev. 2002;15:414–429. doi: 10.1128/CMR.15.3.414-429.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Wang H, Cho CH. Effect of NF-κB signaling on apoptosis in chronic inflammation-associated carcinogenesis. Curr Cancer Drug Targets. 2010;10:593–539. doi: 10.2174/156800910791859425. [DOI] [PubMed] [Google Scholar]
- 525.Cassinelli G, Zuco V, Gatti L, Lanzi C, Zaffaroni N, Colombo D, et al. Targeting the Akt kinase to modulate survival, invasiveness and drug resistance of cancer cells. Curr Med Chem. 2013;20:1923–1945. doi: 10.2174/09298673113209990106. [DOI] [PubMed] [Google Scholar]
- 526.Zimmerman ZF, Kulikauskas RM, Bomsztyk K, Moon RT, Chien AJ. Activation of Wnt/β-catenin signaling increases apoptosis in melanoma cells treated with trail. PLoS One. 2013;8:e69593. doi: 10.1371/journal.pone.0069593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Negi A, Ramarao P, Kumar R. Recent advancements in small molecule inhibitors of insulin-like growth factor-1 receptor (IGF-1R) tyrosine kinase as anticancer agents. Mini Rev Med Chem. 2013;13:653–681. doi: 10.2174/1389557511313050004. [DOI] [PubMed] [Google Scholar]
- 528.Kang J, Sergio CM, Sutherland RL, Musgrove EA. Targeting cyclin-dependent kinase 1 (CDK1) but not CDK4/6 or CDK2 is selectively lethal to MYC-dependent human breast cancer cells. BMC Cancer. 2014;14:32. doi: 10.1186/1471-2407-14-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Eder IE, Egger M, Neuwirt H, Seifarth C, Maddalo D, Desiniotis A, et al. Enhanced inhibition of prostate tumor growth by dual targeting the androgen receptor and the regulatory subunit type iα of protein kinase a in vivo. Int J Mol Sci. 2013;14:11942–11962. doi: 10.3390/ijms140611942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530.Wang L, Gallo KA, Conrad SE. Targeting mixed lineage kinases in ER-positive breast cancer cells leads to G2/M cell cycle arrest and apoptosis. Oncotarget. 2013;4:1158–1171. doi: 10.18632/oncotarget.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Heeg S, Hirt N, Queisser A, Schmieg H, Thaler M, Kunert H, et al. EGFR overexpression induces activation of telomerase via PI3K/AKT-mediated phosphorylation and transcriptional regulation through Hif1-alpha in a cellular model of oral-esophageal carcinogenesis. Cancer Sci. 2011;102:351–360. doi: 10.1111/j.1349-7006.2010.01796.x. [DOI] [PubMed] [Google Scholar]
- 532.Akiyama M, Hideshima T, Hayashi T, Tai YT, Mitsiades CS, Mitsiades N, et al. Nuclear factor-kappaB p65 mediates tumor necrosis factor alpha-induced nuclear translocation of telomerase reverse transcriptase protein. Cancer Res. 2003;63:18–21. [PubMed] [Google Scholar]
- 533.Mowla SN, Perkins ND, Jat PS. Friend or foe: emerging role of nuclear factor kappa-light-chain-enhancer of activated B cells in cell senescence. Onco Targets Ther. 2013;6:1221–1229. doi: 10.2147/OTT.S36160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Sasaki T, Kuniyasu H, Luo Y, Kitayoshi M, Tanabe E, Kato D, et al. AKT activation and telomerase reverse transcriptase expression are concurrently associated with prognosis of gastric cancer. Pathobiology. 2014;81:36–41. doi: 10.1159/000351721. [DOI] [PubMed] [Google Scholar]
- 535.Kim HD, Jang CY, Choe JM, Sohn J, Kim J. Phenylbutyric acid induces the cellular senescence through an Akt/p21(WAF1) signaling pathway. Biochem Biophys Res Commun. 2012;422:213–218. doi: 10.1016/j.bbrc.2012.04.086. [DOI] [PubMed] [Google Scholar]
- 536.Axanova LS, Chen YQ, McCoy T, Sui G, Cramer SD. 1,25-dihydroxyvitamin D(3) and PI3K/AKT inhibitors synergistically inhibit growth and induce senescence in prostate cancer cells. Prostate. 2010;70:1658–1671. doi: 10.1002/pros.21201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 537.Jaitner S, Reiche JA, Schäffauer AJ, Hiendlmeyer E, Herbst H, Brabletz T, et al. Human telomerase reverse transcriptase (hTERT) is a target gene of β-catenin in human colorectal tumors. Cell Cycle. 2012;11:3331–3338. doi: 10.4161/cc.21790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.Listerman I, Gazzaniga FS, Blackburn EH. An investigation of the effects of the core protein telomerase reverse transcriptase on Wnt signaling in breast cancer cells. Mol Cell Biol. 2014;34:280–289. doi: 10.1128/MCB.00844-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Leontieva OV, Blagosklonny MV. CDK4/6-inhibiting drug substitutes for p21 and p16 in senescence: duration of cell cycle arrest and MTOR activity determine geroconversion. Cell Cycle. 2013;12:3063–3069. doi: 10.4161/cc.26130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Ohtani N, Yamakoshi K, Takahashi A, Hara E. The p16INK4a-RB pathway: molecular link between cellular senescence and tumor suppression. J Med Invest. 2004;51:146–153. doi: 10.2152/jmi.51.146. [DOI] [PubMed] [Google Scholar]
- 541.Pernicová Z, Slabáková E, Kharaishvili G, Bouchal J, Král M, Kunická Z, et al. Androgen depletion induces senescence in prostate cancer cells through down-regulation of Skp2. Neoplasia. 2011;13:526–536. doi: 10.1593/neo.11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542.Liu S, Qi Y, Ge Y, Duplessis T, Rowan BG, Ip C, et al. Telomerase as an important target of androgen signaling blockade for prostate cancer treatment. Mol Cancer Ther. 2010;9:2016–2025. doi: 10.1158/1535-7163.MCT-09-0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543.Marconett CN, Sundar SN, Tseng M, Tin AS, Tran KQ, Mahuron KM, et al. Indole-3-carbinol downregulation of telomerase gene expression requires the inhibition of estrogen receptor-alpha and Sp1 transcription factor interactions within the hTERT promoter and mediates the G1 cell cycle arrest of human breast cancer cells. Carcinogenesis. 2011;32:1315–1323. doi: 10.1093/carcin/bgr116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Zhou C, Steplowski TA, Dickens HK, Malloy KM, Gehrig PA, Boggess JF, et al. Estrogen induction of telomerase activity through regulation of the mitogen-activated protein kinase (MAPK) dependent pathway in human endometrial cancer cells. PLoS One. 2013;8:e55730. doi: 10.1371/journal.pone.0055730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Boggess JF, Zhou C, Bae-Jump VL, Gehrig PA, Whang YE. Estrogen-receptor-dependent regulation of telomerase activity in human endometrial cancer cell lines. Gynecol Oncol. 2006;103:417–24. doi: 10.1016/j.ygyno.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 546.Kimura A, Ohmichi M, Kawagoe J, Kyo S, Mabuchi S, Takahashi T, et al. Induction of hTERT expression and phosphorylation by estrogen via Akt cascade in human ovarian cancer cell lines. Oncogene. 2004;23:4505–4515. doi: 10.1038/sj.onc.1207582. [DOI] [PubMed] [Google Scholar]
- 547.Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23:537–548. doi: 10.1101/gad.1756509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548.Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell. 2008;13:472–482. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 549.Martinez-Outschoorn UE, Curry JM, Ko YH, Lin Z, Tuluc M, Cognetti D, et al. Oncogenes and inflammation rewire host energy metabolism in the tumor microenvironment: AS and NFκB target stromal MCT4. Cell Cycle. 2013;12:2580–2597. doi: 10.4161/cc.25510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Du J, Li Q, Tang F, Puchowitz MA, Fujioka H, Dunwoodie SL, et al. Cited2 is required for the maintenance of glycolytic metabolism in adult hematopoietic stem cells. Stem Cells Dev. 2014;23:83–94. doi: 10.1089/scd.2013.0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561.Liu JJ, Dai XJ, Xu Y, Liu PQ, Zhang Y, Liu XD, et al. Inhibition of lymphoma cell proliferation by peroxisomal proliferator-activated receptor-γ ligands via Wnt signaling pathway. Cell Biochem Biophys. 2012;62:19–27. doi: 10.1007/s12013-011-9253-x. [DOI] [PubMed] [Google Scholar]
- 562.Fukushima T, Nakamura Y, Yamanaka D, Shibano T, Chida K, Minami S, et al. Phosphatidylinositol 3-kinase (PI3K) activity bound to insulin-like growth factor-I (IGF-I) receptor, which is continuously sustained by IGF-I stimulation, is required for IGF-I-induced cell proliferation. J Biol Chem. 2012;287:29713–29721. doi: 10.1074/jbc.M112.393074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563.Zumsteg A, Caviezel C, Pisarsky L, Strittmatter K, García-Echeverría C, Hofmann F, et al. Repression of malignant tumor progression upon pharmacologic IGF1R blockade in a mouse model of insulinoma. Mol Cancer Res. 2012;10:800–809. doi: 10.1158/1541-7786.MCR-11-0522. [DOI] [PubMed] [Google Scholar]
- 564.Yalcin A, Clem BF, Simmons A, Lane A, Nelson K, Clem AL, et al. Nuclear targeting of 6-phosphofructo-2-kinase (PFKFB3) increases proliferation via cyclin-dependent kinases. J Biol Chem. 2009;284:24223–24232. doi: 10.1074/jbc.M109.016816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565.Salpeter SJ, Klochendler A, Weinberg-Corem N, Porat S, Granot Z, Shapiro AM, et al. Glucose regulates cyclin D2 expression in quiescent and replicating pancreatic β-cells through glycolysis and calcium channels. Endocrinology. 2011;152:2589–2598. doi: 10.1210/en.2010-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566.Cai Q, Lin T, Kamarajugadda S, Lu J. Regulation of glycolysis and the Warburg effect by estrogen-related receptors. Oncogene. 2013;32:2079–2086. doi: 10.1038/onc.2012.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567.Mineharu Y, Muhammad AK, Yagiz K, Candolfi M, Kroeger KM, Xiong W, et al. Gene therapy-mediated reprogramming tumor infiltrating T cells using IL-2 and inhibiting NF-κB signaling improves the efficacy of immunotherapy in a brain cancer model. Neurotherapeutics. 2012;9:827–843. doi: 10.1007/s13311-012-0144-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Heavey S, O'Byrne KJ, Gately K. Strategies for co-targeting the PI3K/AKT/mTOR pathway in NSCLC. Cancer Treat Rev. 2014;40:445–456. doi: 10.1016/j.ctrv.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 569.Yaguchi T, Goto Y, Kido K, Mochimaru H, Sakurai T, Tsukamoto N, et al. Immune suppression and resistance mediated by constitutive activation of Wnt/β-catenin signaling in human melanoma cells. J Immunol. 2012;189:2110–2117. doi: 10.4049/jimmunol.1102282. [DOI] [PubMed] [Google Scholar]
- 570.Oishi N, Wang XW. Novel therapeutic strategies for targeting liver cancer stem cells. Int J Biol Sci. 2011;7:517–535. doi: 10.7150/ijbs.7.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571.Baychelier F, Vieillard V. The modulation of the cell-cycle: a sentinel to alert the NK cells of dangers. Front Immunol. 2013;4:325. doi: 10.3389/fimmu.2013.00325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572.Nishinakagawa T, Fujii S, Nozaki T, Maeda T, Machida K, Enjoji M, et al. Analysis of cell cycle arrest and apoptosis induced by RCAS1. Int J Mol Med. 2010;25:717–722. doi: 10.3892/ijmm_00000396. [DOI] [PubMed] [Google Scholar]
- 573.Thakur A, Vaishampayan U, Lum LG. Immunotherapy and immune evasion in prostate cancer. Cancers (Basel) 2013;5:569–590. doi: 10.3390/cancers5020569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574.Wong CP, Bray TM, Ho E. Induction of proinflammatory response in prostate cancer epithelial cells by activated macrophages. Cancer Lett. 2009;276:38–46. doi: 10.1016/j.canlet.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Curran EM, Judy BM, Duru NA, Wang HQ, Vergara LA, Lubahn DB, et al. Estrogenic regulation of host immunity against an estrogen receptor-negative human breast cancer. Clin Cancer Res. 2006;12:5641–5647. doi: 10.1158/1078-0432.CCR-05-1117. [DOI] [PubMed] [Google Scholar]
- 576.Maeyama Y, Otsu M, Kubo S, Yamano T, Iimura Y, Onodera M, et al. Intracellular estrogen receptor-binding fragment-associated antigen 9 exerts in vivo tumor-promoting effects via its coiled-coil region. Int J Oncol. 2011;39:41–49. doi: 10.3892/ijo.2011.1026. [DOI] [PubMed] [Google Scholar]
- 577.Tabruyn SP, Griffioen AW. A new role for NF-kappaB in angiogenesis inhibition. Cell Death Differ. 2007;14:1393–1397. doi: 10.1038/sj.cdd.4402156. [DOI] [PubMed] [Google Scholar]
- 578.Zhong XS, Zheng JZ, Reed E, Jiang BH. SU5416 inhibited VEGF and HIF-1alpha expression through the PI3K/AKT/p70S6K1 signaling pathway. Biochem Biophys Res Commun. 2004;324:471–80. doi: 10.1016/j.bbrc.2004.09.082. [DOI] [PubMed] [Google Scholar]
- 579.Dejana E. The role of wnt signaling in physiological and pathological angiogenesis. Circ Res. 2010;107:943–952. doi: 10.1161/CIRCRESAHA.110.223750. [DOI] [PubMed] [Google Scholar]
- 580.Reinmuth N, Liu W, Fan F, Jung YD, Ahmad SA, Stoeltzing O, et al. Blockade of insulin-like growth factor I receptor function inhibits growth and angiogenesis of colon cancer. Clin Cancer Res. 2002;8:3259–3269. [PubMed] [Google Scholar]
- 581.Jacks T, Weinberg RA. The expanding role of cell cycle regulators. Science. 1998;280:1035–1036. doi: 10.1126/science.280.5366.1035. [DOI] [PubMed] [Google Scholar]
- 582.Yoshida S, Aihara K, Ikeda Y, Sumitomo-Ueda Y, Uemoto R, Ishikawa K, et al. Androgen receptor promotes sex-independent angiogenesis in response to ischemia and is required for activation of vascular endothelial growth factor receptor signaling. Circulation. 2013;128:60–71. doi: 10.1161/CIRCULATIONAHA.113.001533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583.Péqueux C, Raymond-Letron I, Blacher S, Boudou F, Adlanmerini M, Fouque MJ, et al. Stromal estrogen receptor-α promotes tumor growth by normalizing an increased angiogenesis. Cancer Res. 2012;72:3010–3019. doi: 10.1158/0008-5472.CAN-11-3768. [DOI] [PubMed] [Google Scholar]
- 584.Hartman J, Lindberg K, Morani A, Inzunza J, Ström A, Gustafsson JA. Estrogen receptor beta inhibits angiogenesis and growth of T47D breast cancer xenografts. Cancer Res. 2006;66:11207–11213. doi: 10.1158/0008-5472.CAN-06-0017. [DOI] [PubMed] [Google Scholar]
- 585.Liao D, Corle C, Seagroves TN, Johnson RS. Hypoxia-inducible factor-1alpha is a key regulator of metastasis in a transgenic model of cancer initiation and progression. Cancer Res. 2007;67:563–572. doi: 10.1158/0008-5472.CAN-06-2701. [DOI] [PubMed] [Google Scholar]
- 586.Nakshatri H, Bhat-Nakshatri P, Martin DA, Goulet RJ, Jr, Sledge GW., Jr Constitutive activation of NF-kappaB during progression of breast cancer to hormone-independent growth. Mol Cell Biol. 1997;17:3629–3639. doi: 10.1128/mcb.17.7.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 587.Yan M, Xu Q, Zhang P, Zhou XJ, Zhang ZY, Chen WT. Correlation of NF-kappaB signal pathway with tumor metastasis of human head and neck squamous cell carcinoma. BMC Cancer. 2010;10:437. doi: 10.1186/1471-2407-10-437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588.Meng Q, Xia C, Fang J, Rojanasakul Y, Jiang BH. Role of PI3K and AKT specific isoforms in ovarian cancer cell migration, invasion and proliferation through the p70S6K1 pathway. Cell Signal. 2006;18:2262–2271. doi: 10.1016/j.cellsig.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 589.Bilir B, Kucuk O, Moreno CS. Wnt signaling blockage inhibits cell proliferation and migration, and induces apoptosis in triple-negative breast cancer cells. J Transl Med. 2013;11:280. doi: 10.1186/1479-5876-11-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590.Sachdev D, Zhang X, Matise I, Gaillard-Kelly M, Yee D. The type I insulin-like growth factor receptor regulates cancer metastasis independently of primary tumor growth by promoting invasion and survival. Oncogene. 2010;29:251–262. doi: 10.1038/onc.2009.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591.Velasco-Velázquez MA, Li Z, Casimiro M, Loro E, Homsi N, Pestell RG. Examiningthe role of cyclin D1 in breast cancer. Future Oncol. 2011;7:753–765. doi: 10.2217/fon.11.56. [DOI] [PubMed] [Google Scholar]
- 592.Tobin NP, Sims AH, Lundgren KL, Lehn S, Landberg G. Cyclin D1, Id1 and EMT in breast cancer. BMC Cancer. 2011;11:417. doi: 10.1186/1471-2407-11-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Chang C, Lee SO, Yeh S, Chang TM. Androgen receptor (AR) differential roles in hormone-related tumors including prostate, bladder, kidney, lung, breast and liver. Oncogene. 2014;33(25):3225–3234. doi: 10.1038/onc.2013.274. [DOI] [PubMed] [Google Scholar]
- 594.Platet N, Cathiard AM, Gleizes M, Garcia M. Estrogens and their receptors in breast cancer progression: a dual role in cancer proliferation and invasion. Crit Rev Oncol Hematol. 2004;51:55–67. doi: 10.1016/j.critrevonc.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 595.Bendinelli P, Maroni P, Matteucci E, Luzzati A, Perrucchini G, Desiderio MA. Hypoxia inducible factor-1 is activated by transcriptional co-activator with PDZ-binding motif (TAZ) versus WWdomain-containing oxidoreductase (WWOX) in hypoxic microenvironment of bone metastasis from breast cancer. Eur J Cancer. 2013;49:2608–2618. doi: 10.1016/j.ejca.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 596.He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas-Ahner J, et al. NF-κB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest. 2013;123:4821–4835. doi: 10.1172/JCI68523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597.Rosich L, Saborit-Villarroya I, López-Guerra M, Xargay-Torrent S, Montraveta A, Aymerich M, et al. The phosphatidylinositol-3-kinase inhibitor NVP-BKM120 overcomes resistance signals derived from microenvironment by regulating the Akt/FoxO3a/Bim axis in chronic lymphocytic leukemia cells. Haematologica. 2013;98:1739–1747. doi: 10.3324/haematol.2013.088849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598.Macheda ML, Stacker SA. Importance of Wnt signaling in the tumor stroma microenvironment. Curr Cancer Drug Targets. 2008;8:454–465. doi: 10.2174/156800908785699324. [DOI] [PubMed] [Google Scholar]
- 599.Ren Z, Chen X, Cui G, Yin S, Chen L, Jiang J, et al. Nanosecond pulsed electric field inhibits cancer growth followed by alteration in expressions of NF-κB and Wnt/β-catenin signaling molecules. PLoS One. 2013;8:e74322. doi: 10.1371/journal.pone.0074322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600.Peretz S, Kim C, Rockwell S, Baserga R, Glazer PM. IGF1 receptor expression protects against microenvironmental stress found in the solid tumor. Radiat Res. 2002;158:174–180. doi: 10.1667/0033-7587(2002)158[0174:irepam]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 601.Nwabo Kamdje AH, Seke Etet PF, Vecchio L, Muller JM, Krampera M, Lukong KE. Signaling pathways in breast cancer: Therapeutic targeting of the microenvironment. Cell Signal. 2014;26:2843–2856. doi: 10.1016/j.cellsig.2014.07.034. [DOI] [PubMed] [Google Scholar]
- 602.Diaz-Moralli S, Tarrado-Castellarnau M, Miranda A, Cascante M. Targeting cell cycle regulation in cancer therapy. Pharmacol Ther. 2013;138:255–271. doi: 10.1016/j.pharmthera.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 603.Zhu ML, Kyprianou N. Role of androgens and the androgen receptor in epithelial-mesenchymal transition and invasion of prostate cancer cells. FASEB J. 2010;24:769–777. doi: 10.1096/fj.09-136994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604.Yamaguchi Y, Hayashi S. Estrogen-related cancer microenvironment of breast carcinoma. Endocr J. 2009;56:1–7. doi: 10.1507/endocrj.k08e-099. [DOI] [PubMed] [Google Scholar]
- 605.Frasor J, Weaver A, Pradhan M, Dai Y, Miller LD, Lin CY, et al. Positive cross-talk between estrogen receptor and NF-kappaB in breast cancer. Cancer Res. 2009;69:8918–8925. doi: 10.1158/0008-5472.CAN-09-2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606.Galien R, Garcia T. Estrogen receptor impairs interleukin-6 expression by preventing protein binding on the NF-kappaB site. Nucleic Acids Res. 1997;25:2424–2429. doi: 10.1093/nar/25.12.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607.Shehzad A, Ha T, Subhan F, Lee YS. New mechanisms and the anti-inflammatory role of curcumin in obesity and obesity-related metabolic diseases. Eur J Nutr. 2011;50:151–161. doi: 10.1007/s00394-011-0188-1. [DOI] [PubMed] [Google Scholar]
- 608.Aggarwal S, Ichikawa H, Takada Y, Sandur SK, Shishodia S, Aggarwal BB. Curcumin (diferuloylmethane) down-regulates expression of cell proliferation and antiapoptotic and metastatic gene products through suppression of IkappaBalpha kinase and Akt activation. Mol Pharmacol. 2006;69:195–206. doi: 10.1124/mol.105.017400. [DOI] [PubMed] [Google Scholar]
- 609.Jobin C, Bradham CA, Russo MP, Juma B, Narula AS, Brenner DA, et al. Curcumin blocks cytokine-mediated NF-kappa B activation and proinflammatory gene expression by inhibiting inhibitory factor I-kappa B kinase activity. J Immunol. 1999;163:3474–3483. [PubMed] [Google Scholar]
- 610.Bae MK, Kim SH, Jeong JW, Lee YM, Kim HS, Kim SR, et al. Curcumin inhibits hypoxia-induced angiogenesis via down-regulation of HIF-1. Oncol Rep. 2006;15:1557–1562. [PubMed] [Google Scholar]
- 611.Wolanin K, Magalska A, Mosieniak G, Klinger R, McKenna S, Vejda S, et al. Curcumin affects components of the chromosomal passenger complex and induces mitotic catastrophe in apoptosis-resistant Bcr-Abl-expressing cells. Mol Cancer Res. 2006;4:457–469. doi: 10.1158/1541-7786.MCR-05-0172. [DOI] [PubMed] [Google Scholar]
- 612.Zheng M, Ekmekcioglu S, Walch ET, Tang CH, Grimm EA. Inhibition of nuclear factor-kappaB and nitric oxide by curcumin induces G2/M cell cycle arrest and apoptosis in human melanoma cells. Melanoma Res. 2004;14:165–171. doi: 10.1097/01.cmr.0000129374.76399.19. [DOI] [PubMed] [Google Scholar]
- 613.Aggarwal BB, Shishodia S, Takada Y, Banerjee S, Newman RA, Bueso-Ramos CE, et al. Curcumin suppresses the paclitaxel-induced nuclear factor-kappaB pathway in breast cancer cells and inhibits lung metastasis of human breast cancer in nude mice. Clin Cancer Res. 2005;11:7490–7498. doi: 10.1158/1078-0432.CCR-05-1192. [DOI] [PubMed] [Google Scholar]
- 614.Dorai T, Gehani N, Katz A. Therapeutic potential of curcumin in human prostate cancer-I. curcumin induces apoptosis in both androgen-dependent and androgen independent prostate cancer cells. Prostate Cancer Prostatic Dis. 2000;3:84–93. doi: 10.1038/sj.pcan.4500399. [DOI] [PubMed] [Google Scholar]
- 615.Park MJ, Kim EH, Park IC, Lee HC, Woo SH, Lee JY, et al. Curcumin inhibits cell cycle progression of immortalized human umbilical vein endothelial (ECV304) cells by up-regulating cyclin-dependent kinase inhibitor, p21WAF1/CIP1, p27KIP1 and p53. Int J Oncol. 2002;21(2):379–83. [PubMed] [Google Scholar]
- 616.Squires MS, Hudson EA, Howells L, Sale S, Houghton CE, Jones JL, et al. Relevance of mitogen activated protein kinase (MAPK) and phosphotidylinositol-3-kinase/protein kinase B (PI3K/PKB) pathways to induction of apoptosis by curcumin in breast cells. Biochem Pharmacol. 2003;65(3):361–76. doi: 10.1016/s0006-2952(02)01517-4. [DOI] [PubMed] [Google Scholar]
- 617.Thomas SL, Zhong D, Zhou W, Malik S, Liotta D, Snyder JP, et al. EF24, a novel curcumin analog, disrupts the microtubule cytoskeleton and inhibits HIF-1. Cell Cycle. 2008;7:2409–2417. doi: 10.4161/cc.6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 618.Su CC, Chen GW, Lin JG, Wu LT, Chung JG. Curcumin inhibits cell migration of human colon cancer colo 205 cells through the inhibition of nuclear factor kappa B /p65 and down-regulates cyclooxygenase-2 and matrix metalloproteinase-2 expressions. Anticancer Res. 2006;26:1281–1288. [PubMed] [Google Scholar]
- 619.Li M, Zhang Z, Hill DL, Wang H, Zhang R. Curcumin, a dietary component, has anticancer, chemosensitization, and radiosensitization effects by down-regulating the MDM2 oncogene through the PI3K/mTOR/ETS2 pathway. Cancer Res. 2007;67:1988–1996. doi: 10.1158/0008-5472.CAN-06-3066. [DOI] [PubMed] [Google Scholar]
- 620.Bhuiyan MM, Li Y, Banerjee S, Ahmed F, Wang Z, Ali S, Sarkar FH. Down-regulation of androgen receptor by 3,3'-diindolylmethane contributes to inhibition of cell proliferation and induction of apoptosis in both hormone-sensitive LNCaP and insensitive C4-2B prostate cancer cells. Cancer Res. 2006;66:10064–10072. doi: 10.1158/0008-5472.CAN-06-2011. [DOI] [PubMed] [Google Scholar]
- 621.Ye MX, Zhao YL, Li Y, Miao Q, Li ZK, Ren XL, et al. Curcumin reverses cis-platin resistance and promotes human lung adenocarcinoma A549/DDP cell apoptosis through HIF-1α and caspase-3 mechanisms. Phytomedicine. 2012;19:779–787. doi: 10.1016/j.phymed.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 622.Xia Y, Jin L, Zhang B, Xue H, Li Q, Xu Y. The potentiation of curcumin on insulin-like growth factor-1 action in MCF-7 human breast carcinoma cells. Life Sci. 2007;80(23):2161–2169. doi: 10.1016/j.lfs.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 623.Patel BB, Sengupta R, Qazi S, Vachhani H, Yu Y, Rishi AK, Majumdar AP. Curcumin enhances the effects of 5-fluorouracil and oxaliplatin in mediating growth inhibition of colon cancer cells by modulating EGFR and IGF-1R. Int J Cancer. 2008;122:267–273. doi: 10.1002/ijc.23097. [DOI] [PubMed] [Google Scholar]
- 624.Hu Y, Sun CY, Huang J, Hong L, Zhang L, Chu ZB. Antimyeloma effects of resveratrol through inhibition of angiogenesis. Chin Med J (Engl) 2007;120:1672–7. [PubMed] [Google Scholar]
- 625.Chen Y, Tseng SH, Lai HS, Chen WJ. Resveratrol-induced cellular apoptosis and cell cycle arrest in neuroblastoma cells and antitumor effects on neuroblastoma in mice. Surgery. 2004;136:57–66. doi: 10.1016/j.surg.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 626.Holmes-McNary M, Baldwin AS., Jr Chemopreventive properties of trans-resveratrol are associated with inhibition of activation of the IkappaB kinase. Cancer Res. 2000;60(13):3477–3483. [PubMed] [Google Scholar]
- 627.Manna SK, Mukhopadhyay A, Aggarwal BB. Resveratrol suppresses TNF-induced activation of nuclear transcription factors NF-kappa B, activator protein-1, and apoptosis: potential role of reactive oxygen intermediates and lipid peroxidation. J Immunol. 2000;164(12):6509–6519. doi: 10.4049/jimmunol.164.12.6509. [DOI] [PubMed] [Google Scholar]
- 628.Benitez DA, Pozo-Guisado E, Alvarez-Barrientos A, Fernandez-Salguero PM, Castellón EA. Mechanisms involved in resveratrol-induced apoptosis and cell cycle arrest in prostate cancer-derived cell lines. J Androl. 2007;28:282–293. doi: 10.2164/jandrol.106.000968. [DOI] [PubMed] [Google Scholar]
- 629.Ji Q, Liu X, Fu X, Zhang L, Sui H, Zhou L, et al. Resveratrol inhibits invasion and metastasis of colorectal cancer cells via MALAT1 mediated Wnt/β-catenin signal pathway. PLoS One. 2013;8:e78700. doi: 10.1371/journal.pone.0078700. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 630.Jagadeesh S, Kyo S, Banerjee PP. Genistein represses telomerase activity via both transcriptional and post-translational mechanisms in human prostate cancer cells. Cancer Res. 2006;66:2107–2115. doi: 10.1158/0008-5472.CAN-05-2494. [DOI] [PubMed] [Google Scholar]
- 631.Aoki H, Takada Y, Kondo S, Sawaya R, Aggarwal BB, Kondo Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: role of Akt and extracellular signal-regulated kinase signaling pathways. Mol Pharmacol. 2007;72:29–39. doi: 10.1124/mol.106.033167. [DOI] [PubMed] [Google Scholar]
- 632.Li L, Aggarwal BB, Shishodia S, Abbruzzese J, Kurzrock R. Nuclear factor-kappaB and IkappaB kinase are constitutively active in human pancreatic cells, and their down-regulation by curcumin (diferuloylmethane) is associated with the suppression of proliferation and the induction of apoptosis. Cancer. 2004;101:2351–2362. doi: 10.1002/cncr.20605. [DOI] [PubMed] [Google Scholar]
- 633.Lin YG, Kunnumakkara AB, Nair A, Merritt WM, Han LY, Armaiz-Pena GN, et al. Curcumin inhibits tumor growth and angiogenesis in ovarian carcinoma by targeting the nuclear factor-kappaB pathway. Clin Cancer Res. 2007;13:3423–3430. doi: 10.1158/1078-0432.CCR-06-3072. [DOI] [PubMed] [Google Scholar]
- 634.Nwachukwu JC, Srinivasan S, Bruno NE, Parent AA, Hughes TS, Pollock JA, et al. Resveratrol modulates the inflammatory response via an estrogen receptor-signal integration network. Elife. 2014;3:e02057. doi: 10.7554/eLife.02057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 635.Ding G, Liu HD, Liang HX, Ni RF, Ding ZY, Ni GY, Hua HW, Xu WG. HIF1-regulated ATRIP expression is required for hypoxia induced ATR activation. FEBS Lett. 2013;587:930–935. doi: 10.1016/j.febslet.2013.02.020. [DOI] [PubMed] [Google Scholar]
- 636.Yeh CT, Yao CJ, Yan JL, Chuang SE, Lee LM, Chen CM, et al. Apoptotic cell death and inhibition of Wnt/β-catenin signaling pathway in human colon cancer cells by an active fraction (HS7) from Taiwanofungus camphoratus. Evid Based Complement Alternat Med. 2011;2011:750230. doi: 10.1155/2011/750230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 637.Johnson GE, Ivanov VN, Hei TK. Radiosensitization of melanoma cells through combined inhibition of protein regulators of cell survival. Apoptosis. 2008;13(6):790–802. doi: 10.1007/s10495-008-0212-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 638.Büchler P, Reber HA, Büchler MW, Friess H, Lavey RS, Hines OJ. Antiangiogenic activity of genistein in pancreatic carcinoma cells is mediated by the inhibition of hypoxia-inducible factor-1 and the down-regulation of VEGF gene expression. Cancer. 2004;100:201–210. doi: 10.1002/cncr.11873. [DOI] [PubMed] [Google Scholar]
- 639.Wu H, Liang X, Fang Y, Qin X, Zhang Y, Liu J. Resveratrol inhibits hypoxia-induced metastasis potential enhancement by restricting hypoxia-induced factor-1 alpha expression in colon carcinoma cells. Biomed Pharmacother. 2008;62:613–621. doi: 10.1016/j.biopha.2008.06.036. [DOI] [PubMed] [Google Scholar]
- 640.Shinojima N, Yokoyama T, Kondo Y, Kondo S. Roles of the Akt/mTOR/p70S6K and ERK1/2 signaling pathways in curcumin-induced autophagy. Autophagy. 2007;3:635–637. doi: 10.4161/auto.4916. [DOI] [PubMed] [Google Scholar]
- 641.Tang FY, Su YC, Chen NC, Hsieh HS, Chen KS. Resveratrol inhibits migration and invasion of human breast-cancer cells. Mol Nutr Food Res. 2008;52:683–691. doi: 10.1002/mnfr.200700325. [DOI] [PubMed] [Google Scholar]
- 642.Azios NG, Krishnamoorthy L, Harris M, Cubano LA, Cammer M, Dharmawardhane SF. Estrogen and resveratrol regulate Rac and Cdc42 signaling to the actin cytoskeleton of metastatic breast cancer cells. Neoplasia. 2007;9:147–158. doi: 10.1593/neo.06778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643.Srivastava RK, Chen Q, Siddiqui I, Sarva K, Shankar S. Linkage of curcumin-induced cell cycle arrest and apoptosis by cyclin-dependent kinase inhibitor p21(/WAF1/CIP1). Cell Cycle. 2007;6:2953–2961. doi: 10.4161/cc.6.23.4951. [DOI] [PubMed] [Google Scholar]
- 644.Chen A, Zheng S. Curcumin inhibits connective tissue growth factor gene expression in activated hepatic stellate cells in vitro by blocking NF-kappaB and ERK signalling. Br J Pharmacol. 2008;153:557–567. doi: 10.1038/sj.bjp.0707542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 645.Leow PC, Tian Q, Ong ZY, Yang Z, Ee PL. Antitumor activity of natural compounds, curcumin and PKF118-310, as Wnt/β-catenin antagonists against human osteosarcoma cells. Invest New Drugs. 2010;28:766–782. doi: 10.1007/s10637-009-9311-z. [DOI] [PubMed] [Google Scholar]
- 646.Zhang C, Yang N, Yang CH, Ding HS, Luo C, Zhang Y, et al. S9, a novel anticancer agent, exerts its anti-proliferative activity by interfering with both PI3K-Akt-mTOR signaling and microtubule cytoskeleton. PLoS One. 2009;4:e4881. doi: 10.1371/journal.pone.0004881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 647.Nakamura A, Aizawa J, Sakayama K, Kidani T, Takata T, Norimatsu Y, et al. Genistein inhibits cell invasion and motility by inducing cell differentiation in murine osteosarcoma cell line LM8. BMC Cell Biol. 2012;13:24. doi: 10.1186/1471-2121-13-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 648.Wang SD, Chen BC, Kao ST, Liu CJ, Yeh CC. Genistein inhibits tumor invasion by suppressing multiple signal transduction pathways in human hepatocellular carcinoma cells. BMC Complement Altern Med. 2014;14:26. doi: 10.1186/1472-6882-14-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 649.Nakamura Y, Yogosawa S, Izutani Y, Watanabe H, Otsuji E, Sakai T. A combination of indol-3-carbinol and genistein synergistically induces apoptosis in human colon cancer HT-29 cells by inhibiting Akt phosphorylation and progression of autophagy. Mol Cancer. 2009;8:100. doi: 10.1186/1476-4598-8-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 650.Fuster JJ, Fernández P, González-Navarro H, Silvestre C, Nabah YN, Andrés V. Control of cell proliferation in atherosclerosis: insights from animal models and human studies. Cardiovasc Res. 2010;86:254–264. doi: 10.1093/cvr/cvp363. [DOI] [PubMed] [Google Scholar]
- 651.Holy JM. Curcumin disrupts mitotic spindle structure and induces micronucleation in MCF-7 breast cancer cells. Mutat Res. 2002;518:71–84. doi: 10.1016/s1383-5718(02)00076-1. [DOI] [PubMed] [Google Scholar]
- 652.Shakibaei M, Mobasheri A, Lueders C, Busch F, Shayan P, Goel A. Curcumin enhances the effect of chemotherapy against colorectal cancer cells by inhibition of NF-κB and Src protein kinase signaling pathways. PLoS One. 2013;8:e57218. doi: 10.1371/journal.pone.0057218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 653.Kawamori T, Lubet R, Steele VE, Kelloff GJ, Kaskey RB, Rao CV, et al. Chemopreventive effect of curcumin, a naturally occurring anti-inflammatory agent, during the promotion/progression stages of colon cancer. Cancer Res. 1999;59:597–601. [PubMed] [Google Scholar]
- 654.Xu Y, Zhang J, Han J, Pan X, Cao Y, Guo H, et al. Curcumin inhibits tumor proliferation induced by neutrophil elastase through the upregulation of α1-antitrypsin in lung cancer. Mol Oncol. 2012;6:405–17. doi: 10.1016/j.molonc.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 655.Killian PH, Kronski E, Michalik KM, Barbieri O, Astigiano S, Sommerhoff CP, et al. Curcumin inhibits prostate cancer metastasis in vivo by targeting the inflammatory cytokines CXCL1 and -2. Carcinogenesis. 2012;33:2507–19. doi: 10.1093/carcin/bgs312. [DOI] [PubMed] [Google Scholar]
- 656.Nonn L, Duong D, Peehl DM. Chemopreventive anti-inflammatory activities of curcumin and other phytochemicals mediated by MAP kinase phosphatase-5 in prostate cells. Carcinogenesis. 2007;28:1188–96. doi: 10.1093/carcin/bgl241. [DOI] [PubMed] [Google Scholar]
- 657.Wilken R, Veena MS, Wang MB, Srivatsan ES. Curcumin: A review of anti-cancer properties and therapeutic activity in head and neck squamous cell carcinoma. Mol Cancer. 2011;10:12. doi: 10.1186/1476-4598-10-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 658.Cai YY, Lin WP, Li AP, Xu JY. Combined effects of curcumin and triptolide on an ovarian cancer cell line. Asian Pac J Cancer Prev. 2013;14:4267–71. doi: 10.7314/apjcp.2013.14.7.4267. [DOI] [PubMed] [Google Scholar]
- 659.Yu LL, Wu JG, Dai N, Yu HG, Si JM. Curcumin reverses chemoresistance of human gastric cancer cells by downregulating the NF-κB transcription factor. Oncol Rep. 2011;26:1197–203. doi: 10.3892/or.2011.1410. [DOI] [PubMed] [Google Scholar]
- 660.Hendrayani SF, Al-Khalaf HH, Aboussekhra A. Curcumin triggers p16-dependent senescence in active breast cancer-associated fibroblasts and suppresses their paracrine procarcinogenic effects. Neoplasia. 2013;15:631–40. doi: 10.1593/neo.13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 661.Mosieniak G, Adamowicz M, Alster O, Jaskowiak H, Szczepankiewicz AA, Wilczynski GM, Ciechomska IA, Sikora E. Curcumin induces permanent growth arrest of human colon cancer cells: link between senescence and autophagy. Mech Ageing Dev. 2012;133:444–55. doi: 10.1016/j.mad.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 662.Malhotra A, Nair P, Dhawan DK. Premature mitochondrial senescence and related ultrastructural changes during lung carcinogenesis modulation by curcumin and resveratrol. Ultrastruct Pathol. 2012;36:179–84. doi: 10.3109/01913123.2011.652765. [DOI] [PubMed] [Google Scholar]
- 663.Divya CS, Pillai MR. Antitumor action of curcumin in human papillomavirus associated cells involves downregulation of viral oncogenes, prevention of NFκB and AP-1 translocation, and modulation of apoptosis. Mol Carcinog. 2006;45:320–32. doi: 10.1002/mc.20170. [DOI] [PubMed] [Google Scholar]
- 664.Panchal HD, Vranizan K, Lee CY, Ho J, Ngai J, Timiras PS. Early anti-oxidative and anti-proliferative curcumin effects on neuroglioma cells suggest therapeutic targets. Neurochem Res. 2008;33:1701–10. doi: 10.1007/s11064-008-9608-x. [DOI] [PubMed] [Google Scholar]
- 665.Bhattacharyya S, Md Sakib Hossain D, Mohanty S, Sankar Sen G, Chattopadhyay S, Banerjee S, et al. Curcumin reverses T cell-mediated adaptive immune dysfunctions in tumor-bearing hosts. Cell Mol Immunol. 2010;7:306–15. doi: 10.1038/cmi.2010.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 666.Arbiser JL, Klauber N, Rohan R, van Leeuwen R, Huang MT, Fisher C, et al. Curcumin is an in vivo inhibitor of angiogenesis. Mol Med. 1998;4:376–83. [PMC free article] [PubMed] [Google Scholar]
- 667.Chen JW, Tang YL, Liu H, Zhu ZY, Lü D, Geng N, et al. [Anti-proliferative and anti-metastatic effects of curcumin on oral cancer cells]. Hua Xi Kou Qiang Yi Xue Za Zhi. 2011;29:83–6. [PubMed] [Google Scholar]
- 668.Kunnumakkara AB, Anand P, Aggarwal BB. Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett. 2008;269:199–225. doi: 10.1016/j.canlet.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 669.Guo LD, Chen XJ, Hu YH, Yu ZJ, Wang D, Liu JZ. Curcumin inhibits proliferation and induces apoptosis of human colorectal cancer cells by activating the mitochondria apoptotic pathway. Phytother Res. 2013;27:422–30. doi: 10.1002/ptr.4731. [DOI] [PubMed] [Google Scholar]
- 670.Prakobwong S, Gupta SC, Kim JH, Sung B, Pinlaor P, Hiraku Y, et al. Curcumin suppresses proliferation and induces apoptosis in human biliary cancer cells through modulation of multiple cell signaling pathways. Carcinogenesis. 2011;32:1372–80. doi: 10.1093/carcin/bgr032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 671.Park W, Amin AR, Chen ZG, Shin DM. New perspectives of curcumin in cancer prevention. Cancer Prev Res (Phila) 2013;6:387–400. doi: 10.1158/1940-6207.CAPR-12-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 672.Jin QH, Shen HX, Wang H, Shou QY, Liu Q. Curcumin improves expression of SCF/c-kit through attenuating oxidative stress and NF-κB activation in gastric tissues of diabetic gastroparesis rats. Diabetol Metab Syndr. 2013;5:12. doi: 10.1186/1758-5996-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 673.Denissova NG, Nasello CM, Yeung PL, Tischfield JA, Brenneman MA. Resveratrol protects mouse embryonic stem cells from ionizing radiation by accelerating recovery from DNA strand breakage. Carcinogenesis. 2012;33:149–55. doi: 10.1093/carcin/bgr236. [DOI] [PubMed] [Google Scholar]
- 674.Lin HY, Shih A, Davis FB, Tang HY, Martino LJ, Bennett JA, et al. Resveratrol induced serine phosphorylation of p53 causes apoptosis in a mutant p53 prostate cancer cell line. J Urol. 2002;168:748–55. [PubMed] [Google Scholar]
- 675.Narayanan BA. Chemopreventive agents alters global gene expression pattern: predicting their mode of action and targets. Curr Cancer Drug Targets. 2006;6:711–27. doi: 10.2174/156800906779010218. [DOI] [PubMed] [Google Scholar]
- 676.Talero E, Ávila-Roman J, Motilva V. Chemoprevention with phytonutrients and microalgae products in chronic inflammation and colon cancer. Curr Pharm Des. 2012;18:3939–65. doi: 10.2174/138161212802083725. [DOI] [PubMed] [Google Scholar]
- 677.Subbaramaiah K, Sue E, Bhardwaj P, Du B, Hudis CA, Giri D, et al. Dietary polyphenols suppress elevated levels of proinflammatory mediators and aromatase in the mammary gland of obese mice. Cancer Prev Res (Phila) 2013;6:886–97. doi: 10.1158/1940-6207.CAPR-13-0140. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 678.He X, Wang Y, Zhu J, Orloff M, Eng C. Resveratrol enhances the anti-tumor activity of the mTOR inhibitor rapamycin in multiple breast cancer cell lines mainly by suppressing rapamycin-induced AKT signaling. Cancer Lett. 2011;301:168–76. doi: 10.1016/j.canlet.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 679.Kim KH, Back JH, Zhu Y, Arbesman J, Athar M, Kopelovich L, et al. Resveratrol targets transforming growth factor-β2 signaling to block UV-induced tumor progression. J Invest Dermatol. 2011;131:195–202. doi: 10.1038/jid.2010.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 680.Wesolowska O, Wisniewski J, Bielawska-Pohl A, Paprocka M, Duarte N, Ferreira MJ, et al. Stilbenes as multidrug resistance modulators and apoptosis inducers in human adenocarcinoma cells. Anticancer Res. 2010;30:4587–93. [PubMed] [Google Scholar]
- 681.Fuggetta MP, Lanzilli G, Tricarico M, Cottarelli A, Falchetti R, Ravagnan G, et al. Effect of resveratrol on proliferation and telomerase activity of human colon cancer cells in vitro. J Exp Clin Cancer Res. 2006;25:189–93. [PubMed] [Google Scholar]
- 682.Lanzilli G, Fuggetta MP, Tricarico M, Cottarelli A, Serafino A, Falchetti R, Ravagnan G, Turriziani M, Adamo R, Franzese O, Bonmassar E. Resveratrol down-regulates the growth and telomerase activity of breast cancer cells in vitro. Int J Oncol. 2006;28:641–8. [PubMed] [Google Scholar]
- 683.Kang NH, Hwang KA, Lee HR, Choi DW, Choi KC. Resveratrol regulates the cell viability promoted by 17β-estradiol or bisphenol A via down-regulation of the cross-talk between estrogen receptor α and insulin growth factor-1 receptor in BG-1 ovarian cancer cells. Food Chem Toxicol. 2013;59:373–9. doi: 10.1016/j.fct.2013.06.029. [DOI] [PubMed] [Google Scholar]
- 684.Wolter F, Akoglu B, Clausnitzer A, Stein J. Downregulation of the cyclin D1/Cdk4 complex occurs during resveratrol-induced cell cycle arrest in colon cancer cell lines. J Nutr. 2001;131:2197–203. doi: 10.1093/jn/131.8.2197. [DOI] [PubMed] [Google Scholar]
- 685.Tseng SH, Lin SM, Chen JC, Su YH, Huang HY, Chen CK, et al. Resveratrol suppresses the angiogenesis and tumor growth of gliomas in rats. Clin Cancer Res. 2004;10:2190–202. doi: 10.1158/1078-0432.ccr-03-0105. [DOI] [PubMed] [Google Scholar]
- 686.Vanamala J, Radhakrishnan S, Reddivari L, Bhat VB, Ptitsyn A. Resveratrol suppresses human colon cancer cell proliferation and induces apoptosis via targeting the pentose phosphate and the talin-FAK signaling pathways-A proteomic approach. Proteome Sci. 2011;9:49. doi: 10.1186/1477-5956-9-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 687.Kozuki Y, Miura Y, Yagasaki K. Resveratrol suppresses hepatoma cell invasion independently of its anti-proliferative action. Cancer Lett. 2001;167:151–6. doi: 10.1016/s0304-3835(01)00476-1. [DOI] [PubMed] [Google Scholar]
- 688.Shamim U, Hanif S, Albanyan A, Beck FW, Bao B, Wang Z, et al. Resveratrol-induced apoptosis is enhanced in low pH environments associated with cancer. J Cell Physiol. 2012;227:1493–500. doi: 10.1002/jcp.22865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 689.Sun C, Hu Y, Liu X, Wu T, Wang Y, He W, et al. Resveratrol downregulates the constitutional activation of nuclear factor-kappaB in multiple myeloma cells, leading to suppression of proliferation and invasion, arrest of cell cycle, and induction of apoptosis. Cancer Genet Cytogenet. 2006;165:9–19. doi: 10.1016/j.cancergencyto.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 690.Shao ZM, Alpaugh ML, Fontana JA, Barsky SH. Genistein inhibits proliferation similarly in estrogen receptor-positive and negative human breast carcinoma cell lines characterized by P21WAF1/CIP1 induction, G2/M arrest, and apoptosis. J Cell Biochem. 1998;69:44–54. [PubMed] [Google Scholar]
- 691.Zhang Y, Li Q, Chen H. DNA methylation and histone modifications of Wnt genes by genistein during colon cancer development. Carcinogenesis. 2013;34:1756–63. doi: 10.1093/carcin/bgt129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 692.Zhang Y, Li Q, Zhou D, Chen H. Genistein, a soya isoflavone, prevents azoxymethane-induced up-regulation of WNT/β-catenin signalling and reduces colon pre-neoplasia in rats. Br J Nutr. 2013;109:33–42. doi: 10.1017/S0007114512000876. [DOI] [PubMed] [Google Scholar]
- 693.Chen FP, Chien MH. Phytoestrogens induce apoptosis through a mitochondria/caspase pathway in human breast cancer cells. Climacteric. 2014;17:385–92. doi: 10.3109/13697137.2013.869671. [DOI] [PubMed] [Google Scholar]
- 694.Wang J, Eltoum IE, Lamartiniere CA. Genistein alters growth factor signaling in transgenic prostate model (TRAMP). Mol Cell Endocrinol. 2004;219:171–80. doi: 10.1016/j.mce.2003.12.018. [DOI] [PubMed] [Google Scholar]
- 695.Myoung H, Hong SP, Yun PY, Lee JH, Kim MJ. Anti-cancer effect of genistein in oral squamous cell carcinoma with respect to angiogenesis and in vitro invasion. Cancer Sci. 2003;94:215–20. doi: 10.1111/j.1349-7006.2003.tb01422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 696.Shim HY, Park JH, Paik HD, Nah SY, Kim DS, Han YS. Genistein-induced apoptosis of human breast cancer MCF-7 cells involves calpain-caspase and apoptosis signaling kinase 1-p38 mitogen-activated protein kinase activation cascades. Anticancer Drugs. 2007;18:649–57. doi: 10.1097/CAD.0b013e3280825573. [DOI] [PubMed] [Google Scholar]
- 697.Choi EJ, Jung JY, Kim GH. Genistein inhibits the proliferation and differentiation of MCF-7 and 3T3-L1 cells via the regulation of ERα expression and induction of apoptosis. Exp Ther Med. 2014;8:454–8. doi: 10.3892/etm.2014.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 698.Khaw AK, Yong JW, Kalthur G, Hande MP. Genistein induces growth arrest and suppresses telomerase activity in brain tumor cells. Genes Chromosomes Cancer. 2012;51:961–74. doi: 10.1002/gcc.21979. [DOI] [PubMed] [Google Scholar]
- 699.Fiorentini D, Hakim G, Bonsi L, Bagnara GP, Maraldi T, Landi L. Acute regulation of glucose transport in a human megakaryocytic cell line: difference between growth factors and H(2)O(2). Free Radic Biol Med. 2001;31:923–31. doi: 10.1016/s0891-5849(01)00678-5. [DOI] [PubMed] [Google Scholar]
- 700.Pons DG, Nadal-Serrano M, Blanquer-Rossello MM, Sastre-Serra J, Oliver J, Roca P. Genistein modulates proliferation and mitochondrial functionality in breast cancer cells depending on ERalpha/ERbeta ratio. J Cell Biochem. 2014;115:949–58. doi: 10.1002/jcb.24737. [DOI] [PubMed] [Google Scholar]
- 701.Fotsis T, Pepper M, Adlercreutz H, Fleischmann G, Hase T, Montesano R, et al. Genistein, a dietary-derived inhibitor of in vitro angiogenesis. Proc Natl Acad Sci U S A. 1993;90:2690–4. doi: 10.1073/pnas.90.7.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 702.Gupta SC, Kim JH, Prasad S, Aggarwal BB. Regulation of survival, proliferation, invasion, angiogenesis, and metastasis of tumor cells through modulation of inflammatory pathways by nutraceuticals. Cancer Metastasis Rev. 2010;29:405–34. doi: 10.1007/s10555-010-9235-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 703.Chen SS, Michael A, Butler-Manuel SA. Advances in the treatment of ovarian cancer: a potential role of antiinflammatory phytochemicals. Discov Med. 2012;13:7–17. [PubMed] [Google Scholar]
- 704.Dai W, Wang F, He L, Lin C, Wu S, Chen P, et al. Genistein inhibits hepatocellular carcinoma cell migration by reversing the epithelial-mesenchymal transition: Partial mediation by the transcription factor NFAT(1.). Mol Carcinog. 2013 doi: 10.1002/mc.22100. Doi 10.1002/mc.22100 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 705.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10:1533–41. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 706.Wetterau LA, Francis MJ, Ma L, Cohen P. Insulin-like growth factor I stimulates telomerase activity in prostate cancer cells. J Clin Endocrinol Metab. 2003;88:3354–9. doi: 10.1210/jc.2002-021326. [DOI] [PubMed] [Google Scholar]
- 707.Mendivil A, Zhou C, Cantrell LA, Gehrig PA, Malloy KM, Blok LJ, et al. AMG 479, a novel IGF-1-R antibody, inhibits endometrial cancer cell proliferation through disruption of the PI3K/Akt and MAPK pathways. Reprod Sci. 2011;18:832–41. doi: 10.1177/1933719111398501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 708.Huber MA, Azoitei N, Baumann B, Grünert S, Sommer A, Pehamberger H, et al. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114:569–81. doi: 10.1172/JCI21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 709.Saud SM, Li W, Morris NL, Matter MS, Colburn NH, Kim YS, Young MR. Resveratrol prevents tumorigenesis in mouse model of Kras activated sporadic colorectal cancer by suppressing oncogenic Kras expression. Carcinogenesis. 2014;35:2778–86. doi: 10.1093/carcin/bgu209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 710.Lissa D, Senovilla L, Rello-Varona S, Vitale I, Michaud M, Pietrocola F, et al. Resveratrol and aspirin eliminate tetraploid cells for anticancer chemoprevention. Proc Natl Acad Sci USA. 2014;111:3020–5. doi: 10.1073/pnas.1318440111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 711.Tyagi A, Gu M, Takahata T, Frederick B, Agarwal C, Siriwardana S, et al. Resveratrol selectively induces DNA Damage, independent of Smad4 expression, in its efficacy against human head and neck squamous cell carcinoma. Clin Cancer Res. 2011;17:5402–11. doi: 10.1158/1078-0432.CCR-11-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 712.Khuda-Bukhsh AR, Das S, Saha SK. Molecular approaches toward targeted cancer prevention with some food plants and their products: inflammatory and other signal pathways. Nutr Cancer. 2014;66:194–205. doi: 10.1080/01635581.2014.864420. [DOI] [PubMed] [Google Scholar]
- 713.Ren Z, Wang L, Cui J, Huoc Z, Xue J, Cui H, Mao Q, Yang R. Resveratrol inhibits NF-kB signaling through suppression of p65 and IkappaB kinase activities. Pharmazie. 2013;68:689–94. [PubMed] [Google Scholar]
- 714.Fouad MA, Agha AM, Merzabani MM, Shouman SA. Resveratrol inhibits proliferation, angiogenesis and induces apoptosis in colon cancer cells: calorie restriction is the force to the cytotoxicity. Hum Exp Toxicol. 2013;32:1067–80. doi: 10.1177/0960327113475679. [DOI] [PubMed] [Google Scholar]
- 715.Gomez LS, Zancan P, Marcondes MC, Ramos-Santos L, Meyer-Fernandes JR, Sola-Penna M, et al. Resveratrol decreases breast cancer cell viability and glucose metabolism by inhibiting 6-phosphofructo-1-kinase. Biochimie. 2013;95:1336–43. doi: 10.1016/j.biochi.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 716.Lee SC, Chan JY, Pervaiz S. Spontaneous and 5-fluorouracil-induced centrosome amplification lowers the threshold to resveratrol-evoked apoptosis in colon cancer cells. Cancer Lett. 2010;288:36–41. doi: 10.1016/j.canlet.2009.06.020. [DOI] [PubMed] [Google Scholar]
- 717.Lee-Chang C, Bodogai M, Martin-Montalvo A, Wejksza K, Sanghvi M, Moaddel R, et al. Inhibition of breast cancer metastasis by resveratrol-mediated inactivation of tumor-evoked regulatory B cells. J Immunol. 2013;191:4141–51. doi: 10.4049/jimmunol.1300606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 718.Buttari B, Profumo E, Facchiano F, Ozturk EI, Segoni L, Saso L, et al. Resveratrol prevents dendritic cell maturation in response to advanced glycation end products. Oxid Med Cell Longev. 2013;2013:574029–041. doi: 10.1155/2013/574029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 719.Sayeed A, Luciani-Torres G, Meng Z, Bennington JL, Moore DH, Dairkee SH. Aberrant regulation of the BST2 (Tetherin) promoter enhances cell proliferation and apoptosis evasion in high grade breast cancer cells. PLoS One. 2013;8:e67191. doi: 10.1371/journal.pone.0067191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 720.Iwasaki K, Ray PD, Huang BW, Sakamoto K, Kobayashi T, Tsuji Y. Role of AMP-activated protein kinase in ferritin H gene expression by resveratrol in human T cells. Biochemistry. 2013;52:5075–83. doi: 10.1021/bi400399f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 721.Noh KT, Chae SH, Chun SH, Jung ID, Kang HK, Park YM. Resveratrol suppresses tumor progression via the regulation of indoleamine 2,3-dioxygenase. Biochem Biophys Res Commun. 2013;431:348–53. doi: 10.1016/j.bbrc.2012.12.093. [DOI] [PubMed] [Google Scholar]
- 722.Charles C, Nachtergael A, Ouedraogo M, Belayew A, Duez P. Effects of chemopreventive natural products on non-homologous end-joining DNA double-strand break repair. Mutat Res Genet Toxicol Environ Mutagen. 2014;768:33–41. doi: 10.1016/j.mrgentox.2014.04.014. [DOI] [PubMed] [Google Scholar]
- 723.Sotoca AM, Ratman D, van der Saag P, Ström A, Gustafsson JA, Vervoort J, et al. Phytoestrogen-mediated inhibition of proliferation of the human T47D breast cancer cells depends on the ERalpha/ERbeta ratio. J Steroid Biochem Mol Biol. 2008;112:171–8. doi: 10.1016/j.jsbmb.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 724.Bao B, Azmi AS, Ali S, Ahmad A, Li Y, Banerjee S, et al. The biological kinship of hypoxia with CSC and EMT and their relationship with deregulated expression of miRNAs and tumor aggressiveness. Biochim Biophys Acta. 2012;1826:272–96. doi: 10.1016/j.bbcan.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 725.Nadal-Serrano M, Pons DG, Sastre-Serra J, Blanquer-Rosselló Mdel M, Roca P, et al. Genistein modulates oxidative stress in breast cancer cell lines according to ERα/ERβ ratio: effects on mitochondrial functionality, sirtuins, uncoupling protein 2 and antioxidant enzymes. Int J Biochem Cell Biol. 2013;45:2045–51. doi: 10.1016/j.biocel.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 726.Breen MJ, Moran DM, Liu W, Huang X, Vary CP, Bergan RC. Endoglin-mediated suppression of prostate cancer invasion is regulated by activin and bone morphogenetic protein type II receptors. PLoS One. 2013;8:e72407. doi: 10.1371/journal.pone.0072407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 727.Bryant CS, Munkarah AR, Kumar S, Batchu RB, Shah JP, Berman J, et al. Reduction of hypoxia-induced angiogenesis in ovarian cancer cells by inhibition of HIF-1 alpha gene expression. Arch Gynecol Obstet. 2010;282:677–83. doi: 10.1007/s00404-010-1381-9. [DOI] [PubMed] [Google Scholar]
- 728.Fang J, Ding M, Yang L, Liu LZ, Jiang BH. PI3K/PTEN/AKT signaling regulates prostate tumor angiogenesis. Cell Signal. 2007;19:2487–97. doi: 10.1016/j.cellsig.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]