

Abstract
BCL2L1 and MCL1 are key anti‐apoptotic genes, and critical for cancer progression. The prognostic values of BCL2L1 and MCL1 copy‐number variations (CNVs) in non‐small‐cell lung cancer (NSCLC) remain largely unknown. Somatic CNVs in BCL2L1 and MCL1 genes were tested in tumor tissues from 516 NSCLC patients in southern China; afterward, survival analyses were conducted with overall survival (OS) as outcome. Additionally, the associations between CNVs and mRNA expression levels were explored using data from 986 NSCLC patients in the Cancer Genome Atlas project. It was found that amplifications of BCL2L1 and MCL1 were associated with unfavorable OS of NSCLC, with adjusted hazards ratio of 1.62 (95% confident interval [CI] = 1.10–2.40; P = 0.015) and 1.39 (95% CI = 1.05–1.84; P = 0.020), respectively. Amplifications of MCL1, but not BCL2L1, were related with higher mRNA expression levels of corresponding gene, compared with non‐amplifications (P = 0.005). Interestingly, after incorporating with MCL1 CNV status, clinical variables (age, sex, TNM stage, and surgical approach) showed an improved discriminatory ability to classify OS (area under curve increased from 72.2% to 74.1%; P = 0.042, DeLong's test). Overall, MCL1 CNV might be a prognostic biomarker for NSCLC, and additional investigations are needed to validate our findings.
Keywords: BCL2L1, copy‐number variation, MCL1, non‐small‐cell lung cancer, prognosis
Introduction
An estimated 1.8 million new lung cancer cases occurred in 2012, accounting for about 13% of total cancer diagnoses 1. Lung cancer was the most frequently diagnosed cancer and the leading cause of cancer death among males in 2012. Among females, lung cancer was the leading cause of cancer death in more developed countries, and the second leading cause of cancer death in less developed countries 1. In 2013, lung cancer was the fourth most common cause of death for men and women in China overall 2, and lung cancer is the number one cause of death among people with malignant tumors in China 3. Non‐small‐cell lung cancer (NSCLC) is the most common type of lung cancer. Although treatment (such as surgery, radiotherapy, and/or chemotherapy) has improved the rate of survival of patients with NSCLC in recent years, the long‐term survival rate still has room to improve. TNM classification is the basis for prognostic management of NSCLC; however, it does not provide sufficient information about biological tumor progression 4. There is still demand for revealing biomarkers for patients’ survival. Recent genomic studies revealed potential therapeutic targets for lung cancer, including ROS1 rearrangements, MET amplification, RET fusions, and activating mutations in BRAF, HER2, and KRAS in frequencies exceeding 1%5. Nevertheless, it is possible that some other unknown genetic factors may also modulate survival outcomes of NSCLC patients.
Studies suggest that Bcl2 family not only plays an important role in resisting cell death but also has function in cell‐cycle control 6. At the same time, it is proposed that the anti‐apoptotic Bcl‐2 family members may participate in the inhibition of autophagy, which is believed to be a nonapoptotic form of programmed cell death 7. Altered expression of members in this family leads to aberrant cell proliferation and malignant growth 8, 9. Study also supports the idea that Bcl2 family proteins represent important therapeutic targets 10. BCL2L1, also known as BCL‐X, is mapped to chromosome 20. Amplifications of 20q have been previously noted in various cancers 11, 12. BCL2L1 proteins regulate outer mitochondrial membrane channel opening, which in turn regulates mitochondrial membrane potential, and thus controls the release of cytochrome and the production of reactive oxygen species, both of which are potent inducers of programmed cell death 13. It is shown that BCL2L1, but not BCL2, could suppress mitophagy mediated by FUNDC1 through its BH3 domain, eventually mediating mitochondrial quality control that is essential for cell survival 14. BCL2L1 DNA copy number is increased in colorectal cancers compared to adenomas 15. Myeloid cell leukemia sequence 1 (MCL1), is located in 1q21.2. MCL1 functions as an anti‐apoptotic molecule and is capable of blocking apoptosis induced by various apoptotic stimuli, including etoposide, staurosporine, UV irradiation, calcium ionophore A23187, and c‐Myc overexpression 16. High levels of MCL1 expression were found in many different cancer types 10, 17. MCL1 copy number gain is a frequent event in several cancers, like mantle cell lymphoma, lung cancer, and breast cancer 18, 19. What is more, overexpression of MCL1 has also been reported to be correlated with poor survival and resistance to chemotherapeutic agents 10.
Above all, we can hypothesize that copy‐number variations (CNVs) in BCL2L1 and MCL1 may be associated with cancer prognosis. To date and to the best of our knowledge, there have been rare studies addressing the roles of CNVs of BCL2L1 and MCL1 in NSCLC outcomes. In this study, we detected BCL2L1, MCL1 CNVs in DNA from NSCLC tumors tissues in a southern Chinese population; additionally, we conducted survival analyses to analyze prognostic values of BCL2L1, MCL1 CNVs on overall survival (OS).
Materials and Methods
Ethics statement
The study was approved by the institutional review boards of Tongji Medical College. All patients were informed about the aims of specimen collection and gave signed written consent in accordance with the ethical guidelines of Tongji Medical College.
Study subjects
All the patients were histopathologically confirmed, without preoperative chemotherapy or radiotherapy, and underwent lung cancer surgery in the Affiliated Tongji Hospital of Huazhong University of Science and Technology from October 2006 to June 2012. Only patients with fully characterized somatic CNV status and intact survival information were included in this study, and there was no selection bias (Table S1). Eventually, 516 patients were recruited, among which 390 patients provided formalin‐fixed paraffin‐embedded (FFPE) tissues; the other 126 tumor specimens were obtained during surgery, then snap frozen in liquid nitrogen, and finally stored at −80°C until usage. The tumor specimens selected for DNA isolation were verified to consist of a minimum of 80% tumor cells in each case on hematoxylin and eosin‐stained tissue sections. All tumor specimens were histologically reviewed.
Data were collected on demographic characteristics, smoking status, alcohol intake, and family history of cancer. The definition of “ever smokers”, “never smokers”, “ever drinkers” and “never drinkers” could be found in Appended Method. Age at cancer diagnosis and additional clinical characteristics were extracted from medical records; TNM stages were defined according to American Joint Committee on Cancer Staging Manual, 7th edition 20.
Follow‐up was designed to carry out every 3 months for the first year; every 6 months for the next 2 years, and every 12 months from the fourth year after surgery. Information on event (death) and event time was obtained by trained nurses and medical students via telephone interview. For this study, the last follow‐up was performed on 31 July 2014.
DNA extraction and quality control
Genomic DNA was isolated from frozen and FFPE samples using the TIANamp Genomic DNA Kit DP304 (Tiangen, Beijing, China) and TIANamp FFPE DNA Kit DP331 (Tiangen, Beijing,China), respectively.
PCR‐based approaches to analyze genes copy‐number changes in FFPE tissues have been successfully conducted 21, 22. As suggested 21, 22, only high‐ quality DNA template was accepted: (1) with an OD260/280 ratio range of 1.7–1.9, as measured by the NanoDrop 1000 instrument (NanoDrop Technologies, Wilmington, DE); (2) without DNA degradation (tested by Gel electrophoresis, Fig. S1). All DNA working solutions were diluted to 5 ng/μL for final storage and usage.
Primer design
By searching the DGV database (http://projects.tcag.ca/variation/?source=hg19), we found the landmark of BCL2L1 CNV (chr20:30,252,261 .. 30,310,656), and then DNA sequence was downloaded from NCBI (http://www.ncbi.nlm.nih.gov/). Primer premier v5.0 software (Applied Biosystems, Foster City, CA, USA) was used to design primers for BCL2L1. The primers used for MCL1 19 were previously described.
Single‐copy genes RPPH1 and β‐globin were used as reference genes. The primers for RPPH1 were also designed by Primer premier, and the primers for β‐globin were extracted from published papers 23, 24, 25.
All primer sequences used in this study were available in Table S2. We subsequently conducted BLAST search (http://www.blast.ncbi.nlm.nih.gov) and UCSC In‐Silico PCR (http://genome.ucsc.edu) to evaluate and confirm the specificity of these primers.
Real‐Time Quantitative Polymerase Chain Reaction
BCL2L1 and MCL1 CNVs were tested by real‐time quantitative polymerase chain reaction (RT‐qPCR) relative to two sing‐copy reference genes (RPPH1 and β‐globin). To screen the CNV, combined use of two reference genes was suggested to produce a robust, reliable, and accurate quantification 26, 27, 28.
All PCRs were done on a 7900HT Sequence Detection System (Applied Biosystems) with Toyobo Thunderbird SYBR@qPCR Mix. The PCR reaction mixture (10 μL) contained 5 μL 2×SYBR mix, 3 μmol/L each of the forward and reverse primers, and 3 μL (5 ng/μL) template DNA. All PCR reaction mixes were prepared in bulk with 10% extra volume, allowing excess for pipetting waste and were transferred to the 384‐well plate with multichannel pipette. The PCR conditions were 95°C for 5 min, followed by 40 cycles of 95°C for 15 sec, 58°C for 30 sec, and 72°C for 45 sec. Melting (dissociation) curve analysis was performed on every run to verify specificity and identify the PCR products. Test for BCL2L1,MCL1,RPPH1, and β‐globin were done in triplicate in the same 384‐well plate. The threshold cycle number (Ct) values were automatically determined by the ABI system.
Samples were successfully genotyped when Ct values for the triplicate fell within 0.3 units of each other; otherwise, the sample was retested. Triplicate wells of simplex (BCL2L1, MCL1, RPPH1 or β‐globin) reactions containing a series of twofold dilutions of pooled germline DNA samples (50–1.0625 ng/reaction) from 50 randomly selected healthy individuals were used to determine the PCR efficiencies of each assay. Each plate also contained three negative controls (water) and calibrator DNA (this calibrator was verified to be without variations in these tested four genes by a whole genome copy‐number array in China). The r2 correlation for each standard curve was ≥0.98.
Statistical analyses
OS was the primary outcome measure which was calculated from the date of operation to the date of death from any cause or the date of last follow‐up. Relative quantification of BCL2L1, MCL1 was performed by the 2 ‐∆∆Ct method 29 with little modifications as described by Lazar et al. 26, and the equation is shown in Appended Method. Consequently, BCL2L1 or MCL1 CNVs were dichotomized into two groups: “non‐amplification” and “amplification”. 2‐∆∆Ct value >1 was interpreted as “amplification”. Multivariate Cox proportional hazards regression analysis was used to evaluate the effect of CNV status and clinicopathological variables on OS, illustrated as hazard ratio (HR) with their corresponding 95% confidence interval (95% confidence interval [CI]). The Kaplan–Meier method was used to compare OS among different CNV status. Statistical analyses were conducted with SAS software (version 9.1.3; SAS Institute, Cary, NC). Statistical significance of the improvement in area under curve (AUC) after adding an explanatory factor was calculated by the Delong's test 30. All statistical tests were two‐sided with a significance level of a = 0.05.
Bioinformatics analysis
CNV data and corresponding normalized gene expression data from tumor tissues for lung adenocarcinoma (LUAD) and Lung squamous cell carcinoma (LUSC) were obtained from The Cancer Genome Atlas (TCGA) data portal (http://cancergenome.nih.gov/, November 2014). This study meets the publication guidelines proved by TCGA (http://cancergenome.nih.gov/publications). The level 3 CNV segmentation data were retrieved and processed using TCGA‐assembler 31. If the CNV value was higher than 0.2, we defined it as “amplification”; otherwise, “non‐amplification”, as suggested by others scholars 32. Differences in gene expression levels between CNV status were assessed by a Wilcoxon signed‐rank test.
Results
Basic characteristics and genotyping data
The demographic and clinical variables of 516 patients with NSCLC are shown in Table 1. The patients were aged between 22 and 80 years at diagnosis with a mean of 58.01 years and standard deviation of 9.56 years. There were more men than women (389 vs. 127) and more ever smokers than non‐smokers (358 vs. 158); 250 of the patients had early‐stage lung cancer (stages Ia, Ib, IIa, and IIb). A total of 242 patients had lung squamous carcinoma, and 249 had lung adenocarcinoma. The median follow‐up time (MFT) for overall patients was 28 months, during when 234 (45.3%) patients died.
Table 1.
Associations of patient demographic and tumor‐related characters with OS
| Parameter | Patients | Death | MST | Univariate analysisa | Multivariate analysisb | ||
|---|---|---|---|---|---|---|---|
| No. | No. | (months) | HR (95% CI) | P | HR (95% CI) | P | |
| Age (years) | |||||||
| ≤58 | 259 | 111 | 65.5 | 1.00 | 1.00 | ||
| >58 | 257 | 123 | 57.6 | 1.16 (0.9–1.50) | 0.254 | 1.28 (0.97–1.69) | 0.079 |
| Sex | |||||||
| Male | 389 | 173 | 63.3 | 1.00 | 1.00 | ||
| Female | 127 | 61 | 46.7 | 1.07 (0.80–1.43) | 0.657 | 1.08 (0.67–1.76) | 0.752 |
| Smoking status | |||||||
| Never | 158 | 76 | 46.5 | 1.00 | 1.00 | ||
| Ever | 358 | 158 | 63.9 | 0.91 (0.69–1.20) | 0.494 | 1.05 (0.65–1.69) | 0.842 |
| Alcohol intake | |||||||
| Never | 311 | 139 | 61.6 | 1.00 | 1.00 | ||
| Ever | 205 | 95 | 57.7 | 0.98 (0.75–1.27) | 0.871 | 0.99 (0.72–1.36) | 0.946 |
| Family history of cancer | |||||||
| No | 440 | 202 | 59.1 | 1.00 | 1.00 | ||
| Yes | 76 | 32 | NA | 0.89 (0.61–1.29) | 0.538 | 0.86 (0.58–1.27) | 0.451 |
| Histological types | |||||||
| Squamous Carcinoma | 242 | 106 | 67.7 | 1.00 | 1.00 | ||
| Adenocarcinoma | 249 | 124 | 46.5 | 1.22 (0.94–1.58) | 0.137 | 1.37 (0.99–1.89) | 0.055 |
| Others | 25 | 4 | NA | 0.29 (0.11–0.79) | 0.015 | 0.26 (0.06–1.09) | 0.065 |
| TNM stage | |||||||
| Ia | 38 | 5 | NA | 1.00 | 1.00 | ||
| Ib | 87 | 23 | NA | 2.34 (0.89–6.16) | 0.085 | 2.63 (0.99–6.99) | 0.053 |
| IIa | 62 | 24 | NA | 3.78 (1.44–9.92) | 0.007 | 4.2 (1.59–11.13) | 0.004 |
| IIb | 63 | 26 | NA | 4.70 (1.80–12.24) | 0.002 | 5.24 (1.98–13.87) | 0.001 |
| IIIa | 168 | 102 | 23.3 | 8.35 (3.40–20.53) | <.0001 | 8.68 (3.47–21.69) | <.0001 |
| IIIb | 28 | 21 | 16 | 12.64 (4.75–33.64) | <.0001 | 11.13 (4.07–30.48) | <.0001 |
| IV | 39 | 26 | 17 | 10.74 (4.12–28.05) | <.0001 | 10.21 (3.86–27.03) | <.0001 |
| Missing | 31 | ||||||
| Laterality | |||||||
| Left | 236 | 117 | 41.9 | 1.00 | 1.00 | ||
| Right | 275 | 113 | 66.3 | 0.79 (0.61–1.03) | 0.077 | 0.93 (0.71–1.22) | 0.589 |
| Others | 5 | ||||||
| Surgical approach | |||||||
| Lobectomy or sublobectomy | 442 | 181 | 85.3 | 1.00 | 1.00 | ||
| Pneumonectomy | 74 | 53 | 18.6 | 2.56 (1.88–3.49) | <.0001 | 1.82 (1.28–2.58) | 0.001 |
| Chemotherapyc | |||||||
| No | 209 | 87 | NA | 1.00 | 1.00 | ||
| Yes | 307 | 147 | 48.9 | 1.07 (0.82–1.40) | 0.613 | 0.87 (0.64–1.20) | 0.398 |
| Radiotherapyc | |||||||
| No | 372 | 156 | 84.8 | 1.00 | 1.00 | ||
| Yes | 144 | 78 | 38.7 | 1.24 (0.95–1.63) | 0.120 | 1.04 (0.75–1.44) | 0.812 |
| DNA source | |||||||
| Fresh | 126 | 33 | NA | 1.00 | 1.00 | ||
| FFPE | 390 | 201 | 57.6 | 1.34 (0.92–1.95) | 0.132 | 1.42 (0.97–2.09) | 0.073 |
FFPE, formalin‐fixed paraffin‐embedded; MST, median survival time; OS, overall survival; HR, hazards ratio; 95% confidence interval, 95% CI; not available, NA.
univariate analysis.
Adjusted by other variables in Table 1.
Chemotherapy or Radiotherapy after operation.
In multivariable analysis, TNM stage and surgical approach were found to be independent prognostic factors for NSCLC patients’ OS.
CNV of BCL2L1, MCL1, and OS
In this study, 64 (12.4%), 173 (33.5%) of 516 NSCLC patients were identified to carry amplifications of BCL2L1 and MCL1, respectively. Patients with amplifications of BCL2L1 exhibited significantly increased hazards of death, compared with those without amplifications (adjusted HR = 1.62, 95% CI = 1.10–2.40, P = 0.015, Table 2 ). For MCL1 CNVs, amplifications showed a strong association with shorter OS (amplification vs. non‐amplification, adjusted HR = 1.39, 95% CI = 1.05–1.84, P = 0.020). For illustrative purpose, Kaplan–Meier curves of the associations with OS and CNV status are shown in Figure 1.
Table 2.
The Associations between CNVs of BCL2L1, MCL1, and NSCLC OS
| CNVs | Number of | MST | Univariate analysisa | Multivariate analysisb | |||
|---|---|---|---|---|---|---|---|
| Patients | Death | (months) | HR (95% CI) | P | HR (95% CI) | P | |
| BCL2L1 | |||||||
| Overall | |||||||
| Non‐amplification | 452 | 200 | 65.3 | 1.00 | 1.00 | ||
| Amplification | 64 | 34 | 45.6 | 1.41 (0.98–2.03) | 0.064 | 1.62 (1.10–2.40) | 0.015 |
| Squamous carcinoma | |||||||
| Non‐amplification | 214 | 89 | NA | ||||
| Amplification | 28 | 17 | 20.1 | 1.88 (1.12–3.17) | 0.018 | 1.35 (0.76–2.39) | 0.307 |
| Adenocarcinoma | |||||||
| Non‐amplification | 221 | 108 | 45.3 | ||||
| Amplification | 28 | 16 | 37.6 | 1.36 (0.81–2.30) | 0.251 | 1.62 (0.91–2.88) | 0.102 |
| MCL1 | |||||||
| Overall | |||||||
| Non‐amplification | 343 | 140 | 66.5 | 1.00 | 1.00 | ||
| Amplification | 173 | 94 | 46.7 | 1.29 (0.99–1.67) | 0.060 | 1.39 (1.05–1.84) | 0.020 |
| Squamous carcinoma | |||||||
| Non‐amplification | 170 | 69 | NA | ||||
| Amplification | 72 | 37 | 36.7 | 1.27 (0.85–1.89) | 0.249 | 1.17 (0.77–1.80) | 0.458 |
| Adenocarcinoma | |||||||
| Non‐amplification | 158 | 68 | 57.2 | ||||
| Amplification | 91 | 56 | 38.0 | 1.31 (0.92–1.87) | 0.139 | 1.40 (0.93–2.09) | 0.105 |
CNVs, Copy‐number variation; MST, median survival time; OS, overall survival; HR, hazards ratio; 95% CI, 95% confidence interval.
univariate analysis.
Adjusted by age, gender, smoking status, alcohol intake, family history of cancer, histological types, TNM stages, laterality, surgical approach, chemotherapy, radiotherapy, and DNA source.
Figure 1.
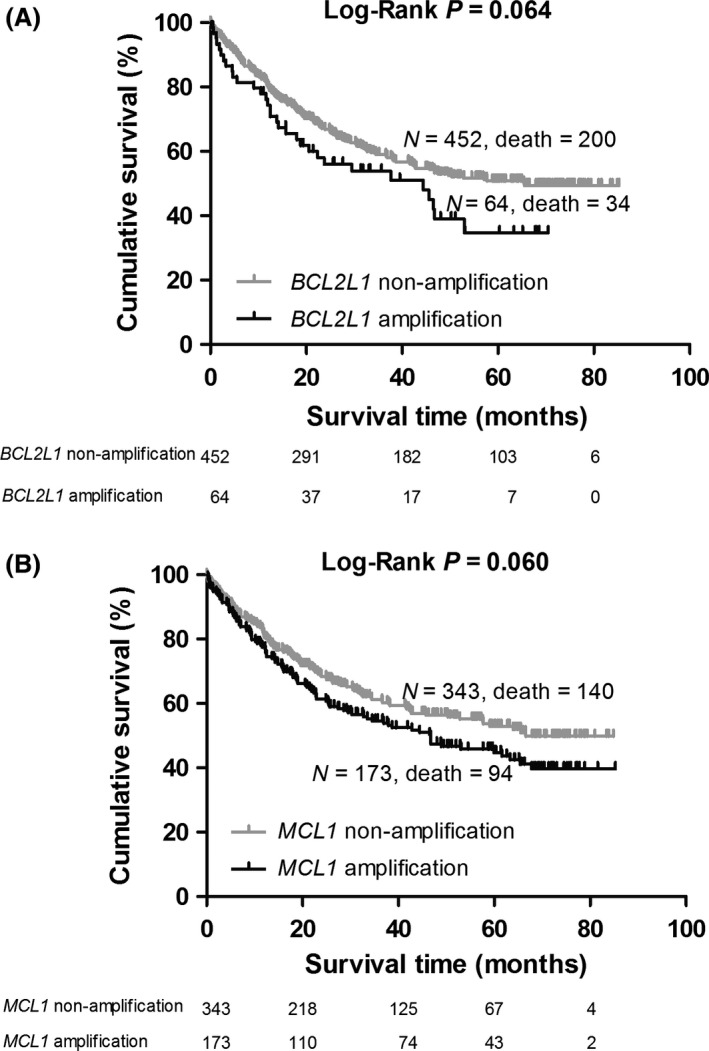
(A) Kaplan–Meier overall survival (OS) curve for patients without and with BCL2L1 amplification; (B) Kaplan–Meier OS curve for patients without and with MCL1 amplification.
Different histological subtypes (LAUD and LUSC) have equal distribution of amplifications of BCL2L1 (chi‐squared test, P = 0.910) and MCL1 (chi‐squared test, P = 0.110). Subgroup analyses stratified by different histological subtypes showed that the prognostic effects of BCL2L1 and MCL1 failed to reach significance in the both groups of LAUD and LUSC (Table 2).
Bioinformatics analysis
To explore the effect of CNVs in MCL1 and BCL2L1 on the corresponding genes expressions, we derived CNV data (in level 3) for tumor tissues from 491 LAUD and 487 LUSC patients in September, 2014., and normalized gene expression data (in level 3) from 1084 LAUD and 1033 LUSC tissues in TCGA database (performed on 17 November 2014). Overall, there were 986 NSCLC patients had data on both CNV and gene expression. As shown in Figure 2, the amplification of MCL1 was shown to be associated with higher expression levels of MCL1 mRNA (P = 0.005), compared with non‐amplification. Nevertheless, no significant correlation was found between BCL2L1 CNV and mRNA expression levels (P = 0.382).
Figure 2.
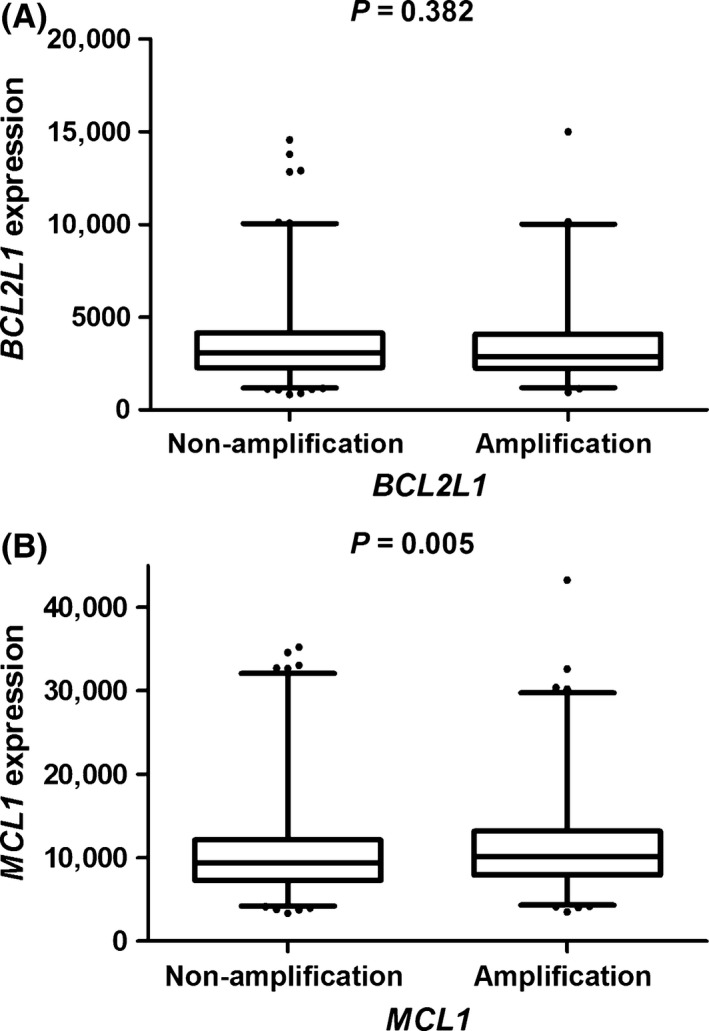
Analyses of BCL2L1 (A) and MCL1 (B) mRNA expression levels by corresponding CNV status in 986 NSCLC patients’ tumor tissues from the TCGA Project. CNV, copy‐number variation; TCGA, The Cancer Genome Atlas.
Receiver operating characteristic curve
The capacity of CNVs for the classification of OS in NSCLC patients was evaluated by the multivariate logistic regression model and receiver operating characteristic curve. As shown in Figure 3, with MCL1 CNVs, the AUC for predicted model was significantly improved to 74.1%, which is higher than the 72.2% AUC for the model only included age, sex and the independent prognostic variables (TNM stage and surgical approach) (P = 0.042, DeLong's test). But there is no similar finding for the BCL2L1 CNV status (72.2% vs. 72.5%, P = 0.451, DeLong's test).
Figure 3.
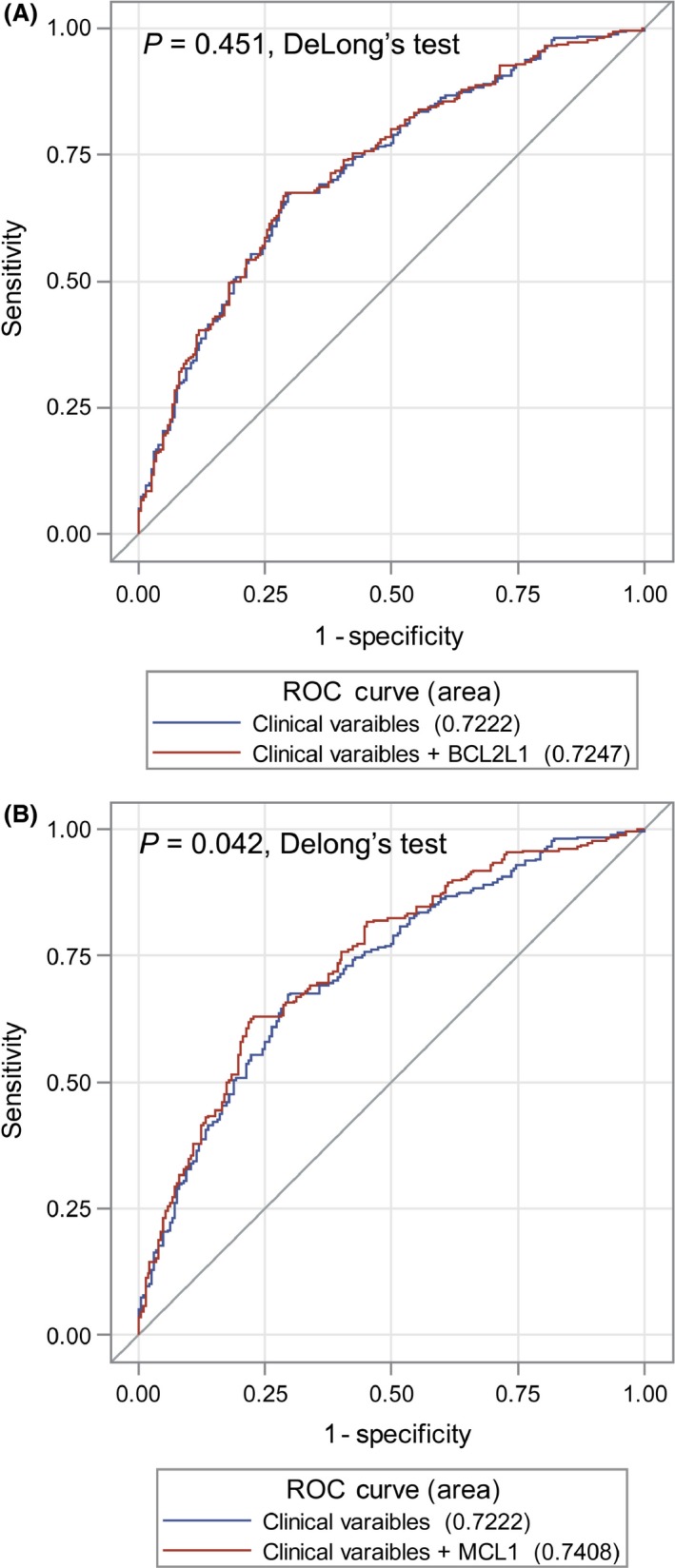
Receiver operating characteristic curves for prediction of OS rate based on clinical variables (age, sex, TNM stage, and surgical approach) and clinical variables plus BCL2L1 copy‐number variation (CNV) (A) or MCL1 CNV (B). OS, overall survival.
Discussion
In this study, we found that amplifications of MCL1 and BCL2L1 were likely to modulate OS of NSCLC patients in a southern Chinese population. However, only MCL1 CNVs were found to be capable of influencing corresponding mRNA expression levels. The observed improvement of discrimination of NSCLC OS by MCL1 CNV status supports the prognostic impact of associations and potential clinical applications.
CNV is proposed to influence gene expression, possibly by altering gene dosage, disrupting coding sequences, or perturbing long‐range gene regulation 33. Meanwhile, a study found that 53% of the expression probes associated with a comparative genomic hybridization (CGH) clone, were located outside the CNVs that encompass the specific clone 34. This suggested that gene expressions might be modulated by CNVs which were long‐range apart. Therefore, it is biologically possible that BCL2L1 CNVs were not associated with corresponding mRNA expression levels.
MCL1 CNV was found to influence mRNA expression levels of MCL1. MCL1 can bind and sequester the pro‐apoptotic proteins, Bax and Bak, and consequently suppress the release of cytochrome c from mitochondria into the cytoplasm 35. In cytoplasm, cytochrome c could induce the activation of caspases and then launch macromolecular degradation, which are the classic steps during mitochondrial pathway (intrinsic pathway) and apoptosis 36. MCL1 plays a key role in cell immortalization, malignant transformation, and chemoresistance 16. Silencing the expression of MCL1 with small interfering RNA (siRNA) potently killed a subgroup of NSCLC cell lines 17. Over expression of MCL1 protein was also found in a subset of human NSCLC cells. And high level of MCL1 may protect lung cancer cells from death induced by a variety of pro‐apoptotic stimuli 37. MCL1 was suggested to be a critical molecule for chemoresistance in A549 cells associated with TGF‐β‐induced EMT 38. The expression of MCL1 mRNA or protein has been associated with tumor progression and adverse patient outcome in multiple cancer types 39, 40, 41, 42, 43. BCL2 inhibitors could induce apoptosis; this has been explored as a therapeutic approach in NSCLC 44. A matter of interest is that, studies have suggested that MCL‐1 expression could be related to resistance to BCL2 inhibitors (such as ABT‐737) in lung cancer cell lines 45, 46. Therefore, MCL1 CNV status might be considered for clinical decision of treatment using apoptotic inhibitor correlated with the Bcl‐2 proteins.
We are aware that this study has some limitations. First, relatively small sample size and short median follow‐up time were the major concern. However, by conducting power and sample size calculation 47, our study assessed MCL1 CNVs in 516 NSCLC patients, achieving a power of 0.964 to detect the potentially clinical significant differences in OS. Second, patients in this study may have received a wide variety of therapies, often sequentially or simultaneously. Only information about whether patients received postoperational chemotherapy /radiation therapy was available. In this study, chemotherapy and radiation therapy did not independently affect NSCLC OS and there were no significant differences among results of subgroups that accepted variant chemotherapy and radiation therapy (data not shown). Third, the prognosis predicting model was only built in a Sothern Chinese population; the application and interpretation of our findings should be cautious and still needs further investigation. Fourth, lacking of information on resection margin status (R0 or R1) was another limitation. Finally, we acknowledge that this study will be more clinically critical if the association between the gene status and NSCLC‐specific death are explored, on which we continue to work.
Our findings suggested that the amplifications of MCL1 might be associated with unfavorable NSCLC OS. However, the functional consequences of MCL1 amplifications and external validations need to be extensively investigated in the future.
Conflicts of Interest
The author(s) indicated no potential conflicts of interest.
Supporting information
Figure S1. DNA degradation tested by Gel electrophoresis
Table S1. Characteristics of overall patients were recruited and patients were used in the current study
Table S2. Primer Sequence
Acknowledgments
This work was supported by the National Science Foundation of China (NSF 81172754 and NSF 81172752) and Natural Science Foundation of Hubei Province (2011CDB203). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Cancer Medicine 2016; 5(9):2171–2179
References
- 1. Torre, L. A. , Bray F., Siegel R. L., Ferlay J., Lortet‐Tieulent J., and Jemal A.. 2015. Global cancer statistics, 2012. CA Cancer J. Clin. 65:87–108 [DOI] [PubMed] [Google Scholar]
- 2. Zhou, M. , Wang H., Zhu J., Chen W., Wang L., Liu S., et al. 2015. Cause‐specific mortality for 240 causes in China during 1990–2013: a systematic subnational analysis for the Global Burden of Disease Study 2013. Lancet 387:251–272. [DOI] [PubMed] [Google Scholar]
- 3. Global Burden of Disease Cancer Collaboration : Fitzmaurice, C. , Dicker D., Pain A., Hamavid H., Moradi‐Lakeh M., MacIntyre M. F., et al. 2015. The Global Burden of Cancer 2013. JAMA oncol. 1:505–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chansky, K. , Sculier J. P., Crowley J. J., Giroux D., Van Meerbeeck J., and Goldstraw P.. 2009. The International Association for the Study of Lung Cancer Staging Project: prognostic factors and pathologic TNM stage in surgically managed non‐small cell lung cancer. J. Thorac. Oncol. 4:792–801 [DOI] [PubMed] [Google Scholar]
- 5. Cardarella, S. , and Johnson B. E.. 2013. The impact of genomic changes on treatment of lung cancer. Am. J. Respir. Crit. Care Med. 188:770–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zinkel, S. , Gross A., and Yang E.. 2006. BCL2 family in DNA damage and cell cycle control. Cell Death Differ. 13:1351–1359 [DOI] [PubMed] [Google Scholar]
- 7. Levine, B. , Sinha S. C., and Kroemer G.. 2008. Bcl‐2 family members: dual regulators of apoptosis and autophagy. Autophagy 4:600–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang, T. M. , Barbone D., Fennell D. A., and Broaddus V. C.. 2009. Bcl‐2 family proteins contribute to apoptotic resistance in lung cancer multicellular spheroids. Am. J. Respir. Cell Mol. Biol. 41:14–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Daniel, J. C. , and Smythe W. R.. 2004. The role of Bcl‐2 family members in non‐small cell lung cancer. Semin. Thorac. Cardiovasc. Surg. 16:19–27 [DOI] [PubMed] [Google Scholar]
- 10. Wertz, I. E. , Kusam S., Lam C., Okamoto T., Sandoval W., Anderson D. J., et al. 2011. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 471:110–114 [DOI] [PubMed] [Google Scholar]
- 11. Smith, L. T. , Mayerson J., Nowak N. J., Suster D., Mohammed N., Long S., et al. 2006. 20q11.1 amplification in giant‐cell tumor of bone: array CGH, FISH, and association with outcome. Genes Chromosomes Cancer 45:957–966. [DOI] [PubMed] [Google Scholar]
- 12. Postma, C. , Terwischa S., Hermsen M. A., van der Sijp J. R., and Meijer G. A.. 2007. Gain of chromosome 20q is an indicator of poor prognosis in colorectal cancer. Cell Oncol. 29:73–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tsujimoto, Y. , and Shimizu S.. 2000. Bcl‐2 family: life‐or‐death switch. FEBS Lett. 466:6–10. [DOI] [PubMed] [Google Scholar]
- 14. Wu, H. , Xue D., Chen G., Han Z., Huang L., Zhu C., et al. 2014. The BCL2L1 and PGAM5 axis defines hypoxia‐induced receptor‐mediated mitophagy. Autophagy 10:1712–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sillars‐Hardebol, A. H. , Carvalho B., Belien J. A., de Wit M., Delis‐van Diemen P. M., Tijssen M., et al. 2012. BCL2L1 has a functional role in colorectal cancer and its protein expression is associated with chromosome 20q gain. J. Pathol. 226:442–450. [DOI] [PubMed] [Google Scholar]
- 16. Zhang, D. , Li F., Weidner D., Mnjoyan Z. H., and K. Fujise . 2002. Physical and functional interaction between myeloid cell leukemia 1 protein (MCL1) and Fortilin. The potential role of MCL1 as a fortilin chaperone. J. Biol. Chem. 277:37430–37438. [DOI] [PubMed] [Google Scholar]
- 17. Zhang, H. , Guttikonda S., Roberts L., Uziel T., Semizarov D., Elmore S. W., et al. 2011. Mcl‐1 is critical for survival in a subgroup of non‐small‐cell lung cancer cell lines. Oncogene 30:1963–1968. [DOI] [PubMed] [Google Scholar]
- 18. Psyrri, A. , Papageorgiou S., Liakata E., Scorilas A., Rontogianni D., Kontos C. K., et al. 2009. Phosphatidylinositol 3’‐kinase catalytic subunit alpha gene amplification contributes to the pathogenesis of mantle cell lymphoma. Clin. Cancer Res. 15:5724–5732. [DOI] [PubMed] [Google Scholar]
- 19. Beroukhim, R. , Mermel C. H., Porter D., Wei G., Raychaudhuri S., Donovan J., et al. 2010. The landscape of somatic copy‐number alteration across human cancers. Nature 463:899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Edge S. B., Byrd D., Compton C. C., Fritz A. G., Greene F. L., and Trotti A., eds. 2009. American Joint Committee on Cancer Staging Manual, 7th ed. Springer, New York. [Google Scholar]
- 21. Yu, J. , Miller R., Zhang W., Sharma M., Holtschlag V., Watson M. A., et al. 2008. Copy‐number analysis of topoisomerase and thymidylate synthase genes in frozen and FFPE DNAs of colorectal cancers. Pharmacogenomics 9:1459–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lake, S. L. , Kalirai H., Dopierala J., Damato B. E., and Coupland S. E.. 2012. Comparison of formalin‐fixed and snap‐frozen samples analyzed by multiplex ligation‐dependent probe amplification for prognostic testing in uveal melanoma. Invest. Ophthalmol. Vis. Sci. 53:2647–2652. [DOI] [PubMed] [Google Scholar]
- 23. Berwick, M. , Matullo G., Song Y. S., Guarrera S., Dominguez G., Orlow I., et al. 2004. Association between aryl hydrocarbon receptor genotype and survival in soft tissue sarcoma. J. Clin. Oncol. 22:3997–4001. [DOI] [PubMed] [Google Scholar]
- 24. Yeh, C. C. , Hsieh L. L., Tang R., Chang‐Chieh C. R., and Sung F. C.. 2005. Vegetable/fruit, smoking, glutathione S‐transferase polymorphisms and risk for colorectal cancer in Taiwan. World J. Gastroenterol. 11:1473–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lai, C. Y. , Hsieh L. L., Sung F. C., Tang R., Bai C. H., Wu F. Y., et al. 2013. Tumor site‐ and stage‐specific associations between allelic variants of glutathione S‐transferase and DNA‐repair genes and overall survival in colorectal cancer patients receiving 5‐fluorouracil‐based chemotherapy. PLoS ONE 8:e69039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lazar, V. , Ecsedi S., Szollosi A. G., Toth R., Vizkeleti L., Rakosy Z., et al. 2009. Characterization of candidate gene copy number alterations in the 11q13 region along with BRAF and NRAS mutations in human melanoma. Mod. Pathol. 22:1367–1378 [DOI] [PubMed] [Google Scholar]
- 27. De Preter, K. , Speleman F., Combaret V., Lunec J., Laureys G., Eussen B. H., et al. 2002. Quantification of MYCN, DDX1, and NAG gene copy number in neuroblastoma using a real‐time quantitative PCR assay. Mod. Pathol. 15:159–166 [DOI] [PubMed] [Google Scholar]
- 28. Hoebeeck, J. , van der Luijt R., Poppe B., De Smet E., Yigit N., Claes K., et al. 2005. Rapid detection of VHL exon deletions using real‐time quantitative PCR. Lab. Invest. 85:24–33. [DOI] [PubMed] [Google Scholar]
- 29. Hoebeeck, J. , Speleman F., and Vandesompele J.. 2007. Real‐time quantitative PCR as an alternative to Southern blot or fluorescence in situ hybridization for detection of gene copy number changes. Methods Mol. Biol. 353:205–226. [DOI] [PubMed] [Google Scholar]
- 30. DeLong, E. R. , DeLong D. M., and Clarke‐Pearson D. L.. 1988. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics 44:837–845. [PubMed] [Google Scholar]
- 31. Zhu, Y. , Qiu P., and Ji Y.. 2014. TCGA‐Assembler: open‐source software for retrieving and processing TCGA data. Nat. Methods 11:599–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Laddha, S. V. , Ganesan S., Chan C. S., and White E.. 2014. Mutational landscape of the essential autophagy gene BECN1 in human cancers. Mol. Cancer Res. 12:485–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Orozco, L. D. , Cokus S. J., Ghazalpour A., Ingram‐Drake L., Wang S., van Nas A., et al. 2009. Copy number variation influences gene expression and metabolic traits in mice. Hum. Mol. Genet. 18:4118–4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stranger, B. E. , Forrest M. S., Dunning M., Ingle C. E., Beazley C., Thorne N., et al. 2007. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315:848–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Adams, J. M. , and Cory S.. 1998. The Bcl‐2 protein family: arbiters of cell survival. Science 281:1322–1326. [DOI] [PubMed] [Google Scholar]
- 36. Thomas, L. W. , Lam C., and Edwards S. W.. 2010. Mcl‐1; the molecular regulation of protein function. FEBS Lett. 584:2981–2989. [DOI] [PubMed] [Google Scholar]
- 37. Song, L. , Coppola D., Livingston S., Cress W. D., and Haura E. B.. 2005. Mcl‐1 regulates survival and sensitivity to diverse apoptotic stimuli in human non‐small cell lung cancer cells. Cancer Biol. Ther. 4:267–276. [DOI] [PubMed] [Google Scholar]
- 38. Toge, M. , Yokoyama S., Kato S., Sakurai H., Senda K., Doki Y., et al. 2015. Critical contribution of MCL‐1 in EMT‐associated chemo‐resistance in A549 non‐small cell lung cancer. Int. J. Oncol. 46:1844–1848. [DOI] [PubMed] [Google Scholar]
- 39. Michels, J. , Obrist F., Vitale I., Lissa D., Garcia P., Behnam‐Motlagh P., et al. 2014. MCL‐1 dependency of cisplatin‐resistant cancer cells. Biochem. Pharmacol. 92:55–61. [DOI] [PubMed] [Google Scholar]
- 40. Lee, W. S. , Park Y. L., Kim N., Oh H. H., Son D. J., Kim M. Y., et al. 2015. Myeloid cell leukemia‐1 is associated with tumor progression by inhibiting apoptosis and enhancing angiogenesis in colorectal cancer. Am. J. Cancer Res. 5:101–113. [PMC free article] [PubMed] [Google Scholar]
- 41. Personeni, N. , Rimassa L., Pressiani T., Destro A., Ligorio C., Tronconi M. C., et al. 2013. Molecular determinants of outcome in sorafenib‐treated patients with hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 139:1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Likui, W. , Qun L., Wanqing Z., Haifeng S., Fangqiu L., and Xiaojun L.. 2009. Prognostic role of myeloid cell leukemia‐1 protein (Mcl‐1) expression in human gastric cancer. J. Surg. Oncol. 100:396–400. [DOI] [PubMed] [Google Scholar]
- 43. Shigemasa, K. , Katoh O., Shiroyama Y., Mihara S., Mukai K., Nagai N., et al. 2002. Increased MCL–1 Expression Is Associated with Poor Prognosis in Ovarian Carcinomas. Cancer Sci. 93:542–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Han, B. , Park D., Li R., Xie M., Owonikoko T. K., Zhang G., et al. 2015. Small‐Molecule Bcl2 BH4 Antagonist for Lung Cancer Therapy. Cancer Cell 27:852–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lin, X. , Morgan‐Lappe S., Huang X., Li L., Zakula D. M., Vernetti L. A., et al. 2007. ‘Seed’ analysis of off‐target siRNAs reveals an essential role of Mcl‐1 in resistance to the small‐molecule Bcl‐2/Bcl‐XL inhibitor ABT‐737. Oncogene 26:3972–3979. [DOI] [PubMed] [Google Scholar]
- 46. Tahir, S. K. , Yang X., Anderson M. G., Morgan‐Lappe S. E., Sarthy A. V., Chen J., et al. 2007. Influence of Bcl‐2 family members on the cellular response of small‐cell lung cancer cell lines to ABT‐737. Cancer Res. 67:1176–1183. [DOI] [PubMed] [Google Scholar]
- 47. Dupont, W. D. , and Plummer W. D.. 1990. Power and sample size calculations: a review and computer program. Control. Clin. Trials 11:116–128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. DNA degradation tested by Gel electrophoresis
Table S1. Characteristics of overall patients were recruited and patients were used in the current study
Table S2. Primer Sequence