

Abstract
Flavonoids are assumed to exert beneficial effects in different types of cancers at high concentrations. Yet, their molecular mechanisms of action remain unknown. The present study aimed to examine the effect of quercetin on proliferation and apoptosis in HER2-expressing breast cancer cells. The anti-proliferative effects of quercetin were examined by proliferation, MTT and clonogenic survival assays. The effect of quercetin on expression of apoptotic molecules was determined by western blotting. Luciferase reporter assay was performed to measure signal transducer and activator of transcription 3 (STAT3) transcriptional activity. ELISA assay was performed to measure intracellular MMP-9 levels. Immunocytochemistry was performed to evaluate the nuclear STAT3 level. The results revealed that quercetin inhibited the proliferation of BT-474 cells in a dose- and time-dependent manner. Quercetin also inhibited clonogenic survival (anchorage-dependent and -independent) of BT-474 cells in a dose-dependent manner. These growth inhibitions were accompanied with an increase in sub-G0/G1 apoptotic populations. Quercetin induced caspase-dependent extrinsic apoptosis upregulating the levels of cleaved caspase-8 and cleaved caspase-3, and inducing the cleavage of poly(ADP-ribose) polymerase (PARP). In contrast, quercetin did not induce apoptosis via intrinsic mitochondrial apoptosis pathway since this compound did not decrease the mitochondrial membrane potential and did not affect the levels of B-cell lymphoma 2 (Bcl-2) and Bcl-2-associated X protein (BAX). Quercetin reduced the expression of phospho-JAK1 and phospho-STAT3 and decreased STAT3-dependent luciferase reporter gene activity in the BT-474 cells. Quercetin inhibited MMP-9 secretion and decreased the nuclear translocation of STAT3. Our study indicates that quercetin induces apoptosis at concentrations >20 µM through inhibition of STAT3 signaling and could serve as a useful compound to prevent or treat HER2-overexpressing breast cancer.
Keywords: breast cancer, quercetin, apoptosis, HER2, p53, STAT3
Introduction
Consumption of particular fruits and vegetables is considered to prevent or even treat various diseases including cancer. For example, the consumption of broccoli (250 g/day) and Brussels sprouts (250 g/day) may decrease colorectal cancer risk (1). Significant efforts have been made to develop plant-derived dietary agents which have beneficial effect on cancer. Quercetin (3,3′,4′,5,6-pentahydroxyflavone) is a flavonoid that is found in many plants and foods, such as onions, green tea, apples, berries, broccoli, red wine and others (2,3). Quercetin exerts anti-oxidant (4), anti-inflammatory (5), anti-mutagenic (6), and anti-angiogenic activities (2). Moreover, in vitro and in vivo studies have shown that quercetin exhibits various anticancer activities. It was reported that quercetin-3-O-gluoside induced human DNA topoisomerase II inhibition, cell cycle arrest and apoptosis in hepatocelluar carcinoma cells (7). It was also reported that quercetin derivatives demonstrated anti-oxidant activity [monochloropivaloyl quercetin (IC50=27 µM)] and cytotoxicity in HeLa [chloronaphtoquinone quercetin (IC50=13.2 µM)] and NIH-3T3 [tri(diacetylcaffeoyl) quercetin (IC50=10.6 µM)] cells (8). Quercetin (40 mg/ml) is reported to inhibit the growth of MCF-7 breast cancer cells and to promote apoptosis by inducing G0/G1 phase arrest (9). Quercetin (100 µM) inhibited the growth of colorectal cancer cells, by up-regulation of the expression of tumor-suppressor genes and modulation of cell cycle-related and apoptosis genes (10,11). Moreover, quercetin (2%) inhibited carcinogen-induced rat mammary tumor growth (12).
Apoptosis is a vital component of various processes including normal cell turnover, proper development and functioning of the immune system, hormone-dependent atrophy, embryonic development and chemical-induced cell death (13). The process of apoptosis is associated with various caspases, which are aspartate-specific cysteine proteases and members of the interleukin-1β-converting enzyme family (14,15). Caspases, once activated, play a key role in the intracellular signal cascade for undergoing apoptosis. In most tumor cells, apoptosis occurs via two different signaling pathways: the extrinsic and intrinsic apoptosis pathways. The extrinsic pathway is related to the activation of death receptors, such as Fas and tumor necrosis factor receptors (TNFRs) and the cleavage of caspase-8 and caspase-3 (16–18). The intrinsic pathway is related to changes in the mitochondrial membrane potential (ΔΨ m), the mitochondrial permeability transition, and the cleavage of caspase-9 and caspase-3 (19). In both the extrinsic and intrinsic pathways, caspase-3 is responsible for the cleavage of poly(ADP-ribose) polymerase (PARP) during cell death (20).
Overexpression of HER2 is encountered in approximately 25% of invasive breast cancers (21). HER2-positive breast cancers tend to grow more quickly than HER2-negative breast cancers. HER2-positive cancers are associated with frequent recurrence and reduced overall survival, compared to HER2-negative tumor subtypes. The most widely used chemotherapeutic agent is Herceptin (trastuzumab), which acts by attaching itself to HER2 receptors on breast cancer cells and blocking them from receiving growth signals (22,23). Herceptin also causes arrest at the G1 phase of the cell cycle and inhibits the phosphorylation of p27Kip1, resulting in the suppression of cdk2 activity and reduced proliferation (24). Herceptin suppresses angiogenesis by both the induction of anti-angiogenic factors and the repression of pro-angiogenic factors. However, many women do not respond to Herceptin or develop resistance to this drug (25). This has resulted in significant efforts to identify other compounds that can effectively treat HER2-overexpressing breast cancer.
Previously, we reported that phytoestrogen suppresses cell growth and induces apoptosis by inhibiting signal transducer and activator of transcription 3 (STAT3) and/or NF-κB signaling in HER2-overexpressing breast cancer cells (26,27). In the present study, we investigated whether quercetin displays growth-suppressive activity in HER2-overexpressing BT-474 breast cancer cells. For this purpose, we tested the effects of quercetin on the proliferation and apoptosis of BT-474 cells. We also investigated the mechanism by which quercetin regulates the growth of BT-474 cells by analyzing the cell cycle and measuring the levels of apoptotic molecules and intracellular signaling molecules. We also aimed to ascertain whether quercetin inhibits the STAT3 signaling pathway, leading to the growth suppression of HER2-overexpressing breast cancer cells. Our study may advance human health by clarifying the efficacy of quercetin for the prevention and treatment of HER2-positive breast cancer.
Materials and methods
Compounds
Quercetin (3,3′,4′,5,6-pentahydroxyflavone) and carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). These compounds were dissolved in dimethyl sulfoxide (DMSO), and the final concentration of DMSO in the controls and each sample did not exceed 0.1%. We found that 0.1% DMSO did not affect the cell growth rate compared to 0% DMSO (no treatment) in the breast cancer cells (data not shown). JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide) was obtained from Molecular Probes (Invitrogen, Carlsbad, CA, USA). The caspase-8 inhibitor Z-IETD-fmk and the caspase-9 inhibitor Z-LEHD-fmk were obtained from R&D Systems, Inc. (Minneapolis, MN, USA). The STAT3 inhibitor S3I-201 was obtained from Calbiochem (San Diego, CA, USA). An EZ-western chemiluminescent detection kit was purchased from Daeil Lab Service Co. (Seoul, Korea).
Cell cultures
BT474 human breast cancer cells (ATCC, American Type Culture Collection; Manassas, VA, USA) were cultured in RPMI-1640 medium containing 50 U/ml penicillin, 50 mg/ml streptomycin and 10% fetal bovine serum (FBS; Welgene, Daegu, Korea) at 37°C in an atmosphere of 5% CO2.
Antibodies
Monoclonal or polyclonal antibodies (mouse or rabbit) directed against FAS, cleaved caspase-8, caspase-3, cleaved caspase-3 and PARP [poly(ADP-ribose) polymerase] were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). Monoclonal or polyclonal antibodies (mouse or rabbit) directed against Bcl-2, BAX, p53, phospho-p53 (Ser15), p21 and VEGF were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Monoclonal or polyclonal antibodies (mouse or rabbit) against Bcl-XL and HIF-1α were purchased from BD Biosciences (Franklin Lakes, NJ, USA). Monoclonal or polyclonal antibodies (mouse or rabbit) directed against STAT3, phospho-STAT3 (Tyr705), and phospho-JAK1 (Tyr1022/Tyr1023) were obtained from upstate-Millipore (Billerica, MA, USA). The anti-tubulin antibody was from Sigma Chemical Co. Horseradish peroxidase (HRP)-conjugated secondary antibodies (mouse and rabbit) were purchased from Calbiochem and anti-goat secondary antibody was from Jackson ImmunoResearch (West Grove, PA, USA).
Cell proliferation assay
Cells were seeded in 12-well culture plates at a density of 5×104 cells/well. After the cells were exposed to different concentrations of quercetin (20–60 µM) and incubated for 3 days, they were harvested by trypsinization, resuspended in 1–2 ml of medium, and counted using a hemocytometer.
MTT assay
Cells were seeded in 96-multi-well culture plates at a density of 3×103 cells/well and incubated for 24 h at 37°C. Then, they were treated with different concentrations of quercetin (20–60 µM) for 24, 48, or 72 h. After incubation, MTT reagents (0.5 mg/ml) were added to the each well and the plates were incubated in the dark at 37°C for 2 h. At the end of the incubation, the medium was removed, the resulting formazan was dissolved in DMSO, and the optical density was measured at 570 nm using an ELISA plate reader (fluorescence readers; Molecular Devices, Sunnyvale, CA, USA).
Clonogenic survival assays (anchorage-dependent and -independent)
For anchorage-dependent colony formation assay, cells were seeded into 6-well culture plates at a density of 5×102 cells/well. After overnight incubation, they were treated with different concentrations of quercetin (20–60 µM) or vehicle and maintained for 10 days at 37°C. Cells were fed every 3 days by removing old medium and adding fresh medium containing quercetin. Finally, the plates were stained with hematoxylin and the colony number was determined. For anchorage-independent colony formation assay, soft agar was used. Cells (1×103) were suspended in 1 ml of 0.6% soft agar that was layered on top of 1 ml of 1% solidified agar in each well of 12-well plates. The plates were then incubated for 15 to 21 days in complete RPMI medium containing quercetin (20–60 µm). The medium was changed every 3 days during this period. At the end of the experiment, tumor cell colonies measuring at least 30 µm were counted using a dissection microscope.
Cell cycle analyses by flow cytometry
Cells were harvested with 0.25% trypsin and washed once with phosphate-buffered saline (PBS). After centrifugation, the cells were fixed in cold 95% ethanol with 0.5% Tween-20, and stored at −20°C for at least 30 min. The cells were incubated in 50 µg/ml of propidium iodide (PI) (including 1% of sodium citrate and 50 µg/ml of RNase A) at room temperature in the dark for 30 min. The analysis of apoptotic cells was performed on a FACScan flow cytometer (Becton Dickinson, Mountain View, CA, USA) and the data were analyzed using CellQuest software.
Analysis of mitochondrial transmembrane potential (ΔΨm)
Cells were seeded at a density of 1×106 cells/dish in 100-mm dishes and incubated for 24 h at 37°C. After stabilization, the cells were treated with quercetin (20–60 µm) and vehicle for 72 h. After harvest by treatment with trypsin-EDTA, the cells were washed with cold PBS, centrifuged at 1,500 rpm for 5 min and stained with 4 µg/ml JC-1 for 15 min at 37°C in the dark. The data were analyzed by FACSCalibur flow cytometry (BD Biosciences) measuring the green fluorescence and red fluorescence at 514/529 nm (FL-1) and 585/590 nm (FL-2), respectively.
Western blot analysis
Cells were lysed in modified RIPA buffer [150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 50 mM Tris (pH 8.0), 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM NaF, 1 mM Na3VO4 and protease inhibitor mixture]. The lysates were cleared by centrifugation at 10,000 × g for 15 min and the supernatants were collected. The protein concentration was quantified using a Bio-Rad Bradford protein assay (Bio-Rad, Hercules, CA, USA). Equal amounts of protein lysates were used for western blot analysis with the indicated antibodies. Immunoreactive protein bands were detected with an EZ-Western detection kit (Daeil Lab Service Co., Ltd., Seoul, Korea).
Immunocytochemistry
Cells (2×104 cells/well) were seeded in 8-well chamber slides, incubated for 24 h at 37°C and treated with quercetin (60 µM) in the presence or absence of CoCl2 for another 24 h. The cells were fixed with 4% paraformaldehyde for 30 min and treated with 3% hydrogen peroxide (H2O2) in methanol for 20 min to quench endogenous peroxidase activity. The cells were washed with PBS, blocked with 5% BSA in PBS for 1 h and incubated with the anti-STAT3 primary antibody (1:100 dilution) overnight at 4°C. After washing with PBS, the cells were incubated with anti-rabbit biotin-conjugated secondary antibody for 1 h at room temperature. Then, the cells were treated with Vectastain ABC reagent (Vector Laboratories, Inc. Burlingame, CA, USA) for 30 min at 4°C and stained with diaminobenzidine tetrachloride (DAB) and hematoxylin. The cells were mounted with mounting medium and subsequently analyzed using microscopy.
Measurement of MMP-9 secreted from BT-474 cells by ELISA
To assess the level of MMP-9 in the BT-474 cell supernatants, the cells were treated with quercetin (20-60 µM). After 24 h, the media were collected, centrifuged to remove the cellular debris, and stored at −70°C until assay for MMP-9. The amount of MMP-9 secreted into the culture medium was measured by ELISA according to the manufacturer's instructions (R&D Systems). Briefly, 96-well plates were coated with capture antibody in ELISA coating buffer and incubated overnight at 4°C. The plates were then washed with PBS with 0.05% Tween-20 (PBS-T) and subsequently blocked with 10% FBS in PBS for 1 h at 20°C. Serial dilutions of standard antigen or sample in dilution buffer (10% FBS in PBS) were added to the plates, and the plates were incubated for 2 h at 20°C. After washing, biotin-conjugated anti-mouse IgE and streptavidin-conjugated horseradish peroxidase (SAv-HRP) were added to the plates, and the assay plates were incubated for 1 h at 20°C. Finally, the tetramethylbenzidine (TMB) substrate was added to the plates, and after 15 min of incubation in the dark, 2 NH2SO4 was added to stop the reaction. The optical density was measured at 450 nm on an automated ELISA reader.
STAT3 luciferase reporter assay
BT-474 cells were plated and allowed to attach by overnight incubation at 37°C. Cells were transiently transfected with p4xM67-TK-luc plasmid (Addgene plasmid 8688; Addgene, Cambridge, MA, USA) containing four copies of the STAT-binding site (TTCCCGTAA). The next day, the cells were treated with different concentrations of quercetin (20–60 µM) for 24 h and then submitted to the luciferase assays. Luciferase assays were performed using a dual-luciferase assay kit according to the manufacturer's instructions (Promega, Madison, WI, USA). Finally, luciferase activities were determined using a luminometer (BMG Labtech, Ortenberg, Germany).
Statistical analysis
All experiments were performed in triplicate. The data for the cell proliferation, MTT, ELISA, and STAT3 luciferase reporter assays are expressed as the mean ± standard deviation (SD). The standard deviations for all of the measured biological parameters are displayed in the appropriate figures. A Student's t-test was used for single variable comparisons, and a p-value of <0.05 was considered to be indicative of a statistically significant result.
Results
Quercetin suppresses the growth of BT-474 cells
The effects of quercetin on cell growth were measured by cell proliferation and MTT assays in the BT-474 cells. As shown in Fig. 1A, quercetin and genistein significantly inhibited BT-474 cell proliferation in a dose-dependent manner (20–100 µM) after 72 h of treatment (proliferation assay). Between two phytoestrogens, quercetin had the stronger growth suppressive activity compared to genistein in the BT-474 cells. Therefore, we chose quercetin for our experimental study. In addition, the time-dependent growth suppressive activity of quercetin was measured by the MTT assay (Fig. 1B). As shown in Fig. 1A and B, the proliferation assay appeared to be more sensitive than the MTT assay with respect to measuring the intensity of the cell growth inhibition. Moreover, the growth inhibition induced by quercetin was verified by microscopic observation. As shown in Fig. 1C, quercetin effectively inhibited the growth rate of BT-474 monolayer cells after 72 h of treatment. Of note, quercetin also induced morphological changes in these cells (Fig. 1C). Since lower concentrations of phytoestrogen could stimulate the growth of breast cancer cells through nuclear and membrane estrogen receptors, we performed an MTT assay to measure the growth rate of BT-474 cells under lower concentrations of quercetin (0–20 µM). We found that lower concentrations of quercetin did not affect the cell growth rate (Fig. 1D).
Figure 1.
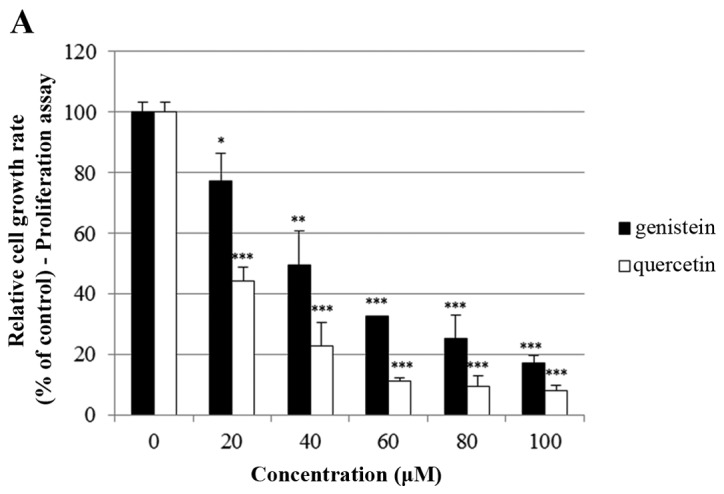
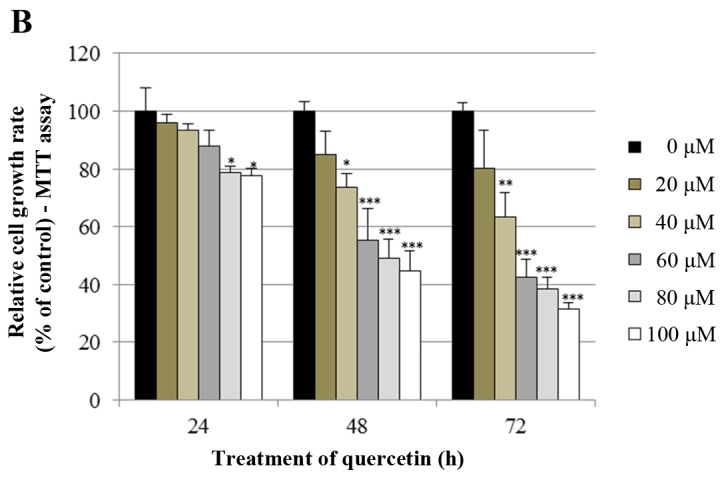

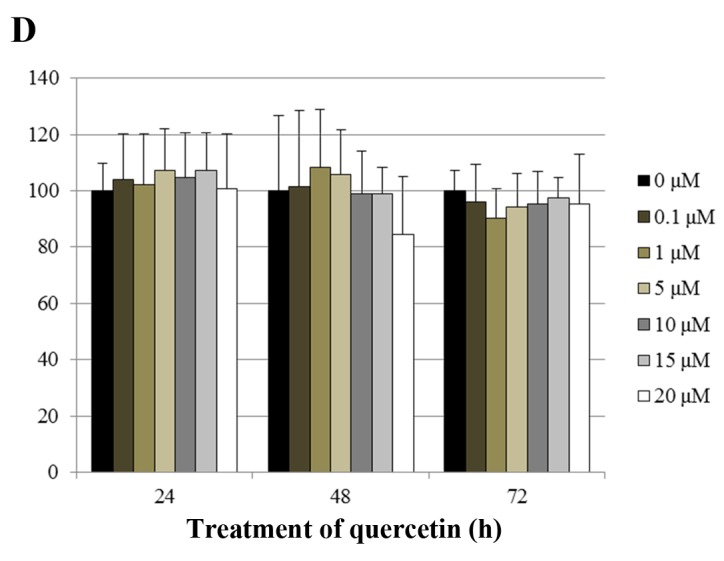
Effect of quercetin on BT-474 cell growth. (A) BT-474 cells were treated with different doses of quercetin and genistein (0–100 µM). After 72 h, cell viability was assessed using a cell proliferation assay. (B) BT-474 cells were treated with different doses of quercetin (0–100 µM). The relative cell growth rate was measured by MTT assay after 24, 48 and 72 h. The growth rate of the vehicle-treated cells was set to 100%, and the relative decrease in cell viability resulting from the phytoestrogen treatment is expressed as a percentage of the control. (C) BT-474 cells were treated with different doses of quercetin (0–60 µM) for 72 h and photographed by phase-contrast microscopy (original magnification, x40). (D) BT-474 cells were treated with different doses of quercetin (0–20 µM). The relative cell growth rate was measured by MTT assay after 24, 48 and 72 h. Control cells were treated with DMSO alone. Data are shown as the means of three independent experiments (error bars denote SD). *P<0.05, **P<0.01, ***P<0.001.
Quercetin inhibits clonogenic survival of BT-474 cells
Next, we investigated the effect of quercetin on clonogenic. survival of BT-474 cells using clonogenic survival assays (anchorage-dependent and -independent). As shown in Fig. 2A, quercetin significantly inhibited anchorage-dependent colony formation dose-dependently in the BT-474 cells. Consistently with this result, quercetin strongly decreased anchorage-independent colony formation in the BT-474 cells (Fig. 2B). These results suggest that quercetin inhibits clonogenic survival of the BT-474 cells.
Figure 2.


Quercetin inhibits anchorage-dependent and -independent clonogenic survival of BT-474 cells. (A) BT-474 cells were seeded into 6-well culture plates at a density of 5×102 cells/well. After overnight incubation, the cells were treated with different concentrations of quercetin (0–60 µM) and maintained for 10 days at 37°C. Finally, the plates were stained with hematoxylin and the colony number was counted. (B) Cells (1×103) were suspended in 1 ml of 0.6% soft agar that was layered on top of 1 ml of 1% solidified agar in each well of 12-well plates. The plates were then incubated for 15 to 21 days in complete RPMI medium containing quercetin. Colonies were stained with crystal violet. ***P<0.001.
The growth-suppressive activity of quercetin is accompanied by an increase in the sub-G0/G1 apoptotic population in BT-474 cells
To investigate whether quercetin inhibits cell proliferation by promoting changes in cell cycle progression, the effect of quercetin on the cell cycle profile was assessed in the BT-474 cells. For this purpose, the cells were treated with quercetin (20–60 µM) for 72 h and then analyzed for cell cycle stage by flow cytometry. The results demonstrated that quercetin induced an increase in the sub-G0/G1 apoptotic population in the BT-474 cells (Fig. 3).
Figure 3.

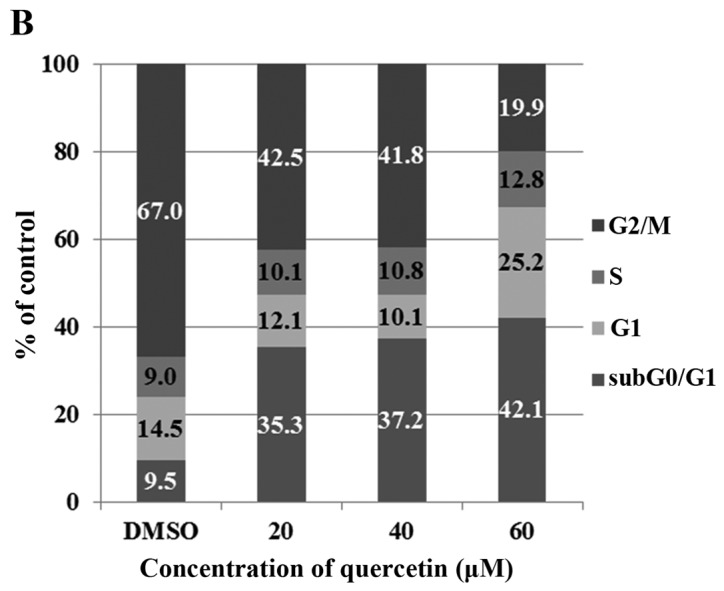
Effect of quercetin on the cell cycle and sub-G0/G1 apoptotic population of BT-474 cells. (A) BT-474 cells were treated with quercetin (0–60 µM) and fixed 72 h later for flow cytometry. Propidium iodide-labeled nuclei were analyzed for DNA content. (B) The sub-G0/G1 apoptotic population and the G1, S and G2/M phase populations were quantified using DNA histograms. The data shown are representative of three independent experiments that gave similar results.
Quercetin induces extrinsic apoptosis in BT-474 cells
Next, we investigated whether apoptosis induced by quercetin occurs via the extrinsic apoptosis pathway in the BT-474 cells. For this purpose, we measured the loss of mitochondrial transmembrane potential (ΔΨm) within the cells using JC-1. JC-1 is able to selectively enter mitochondria and reversibly transforms the color from red to green when the membrane potential decreases. In non-apoptotic cells with high mitochondrial ΔΨm, JC-1 spontaneously forms complexes known as J-aggregates with intense red fluorescence. In contrast, in apoptotic cells (particularly mitochondrial-mediated apoptotic cells) with low ΔΨm, JC-1 remains in the monomeric form, which shows only green fluorescence. In our study, quercetin did not induce a low mitochondrial transmembrane potential (ΔΨm), showing relatively weak green fluorescence (DMSO, 3.0%; quer 20 µM, 12.6%; quer 40 µM, 12.2%; quer 60 µM, 14.8%) compared to FCCP (positive control, 87.6%) (Fig. 4A). We also measured the levels of Bcl-2 family members (BAX and Bcl-2) which are important in the intrinsic mitochondrial apoptosis pathway. We found that quercetin failed to decrease the level of Bcl-2 or increase the level of BAX as shown in Fig. 4B and C. These results demonstrate that quercetin does not induce apoptosis via the intrinsic mitochondrial pathway but induces apoptosis via the extrinsic pathway in BT-474 breast cancer cells.
Figure 4.

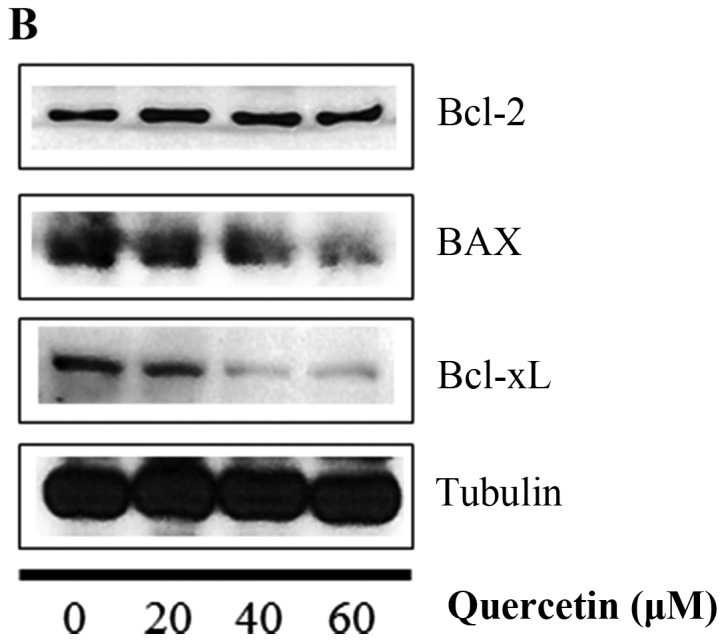
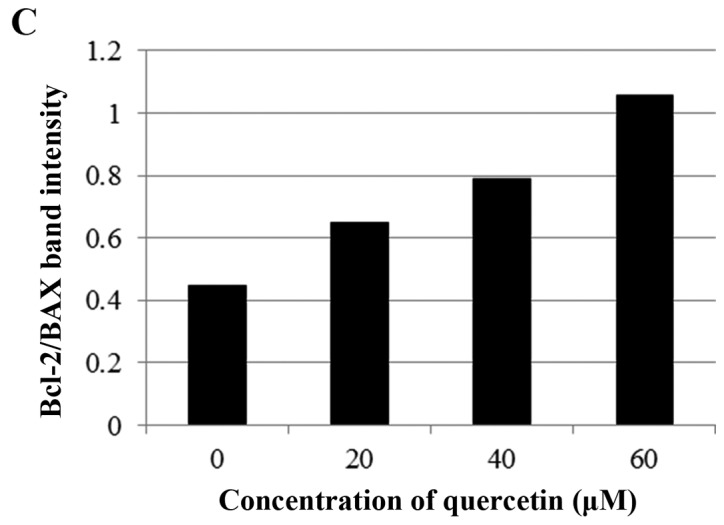
Quercetin induces apoptosis via the extrinsic pathway in BT-474 cells. (A) BT-474 cells were incubated with quercetin (0–60 µM) for 72 h and were dyed with JC-1 (4 µg/ml). The data were analyzed by FACSCalibur flow cytometry measuring the green fluorescence and red fluorescence at 514/529 nm (FL-1) and 585/590 nm (FL-2), respectively. The data shown are representative of three independent experiments that gave similar results. (B and C) Analysis of intrinsic apoptosis-related molecules. BT-474 cells were treated with quercetin (0–60 µM) for 24 h. Total proteins were analyzed by western blotting with anti-Bcl-2, anti-BAX, anti-Bcl-xL and anti-tubulin antibodies.
Quercetin induces apoptosis via the caspase-dependent apoptosis pathway in BT-474 cells
In this step, we investigated whether quercetin activates the caspase-dependent apoptosis pathway by measuring the expression of caspase-8, caspase-3, and PARP. We observed that quercetin upregulated the levels of cleaved caspase-8 and caspase-3, and induced the cleavage of PARP in the BT-474 cells (Fig. 5A). We also found that the cleavage of caspase-8, caspase-3 and PARP was inhibited by the caspase-8 inhibitor Z-IETD-fmk and the caspase-9 inhibitor Z-LEHD-fmk (Fig. 5B), but quercetin prevented this inhibition and was able to induce the cleavage of caspase-8, caspase-3 and PARP in the presence of Z-IETD-fmk and Z-LEHD-fmk (Fig. 5B). Moreover, the caspase-8 and caspase-9 inhibitors did not suppress cell growth, while quercetin was able to induce apoptosis even in their presence (Fig. 5C). These results confirm that quercetin strongly promoted apoptosis via a caspase-dependent mechanism in the BT-474 cells.
Figure 5.
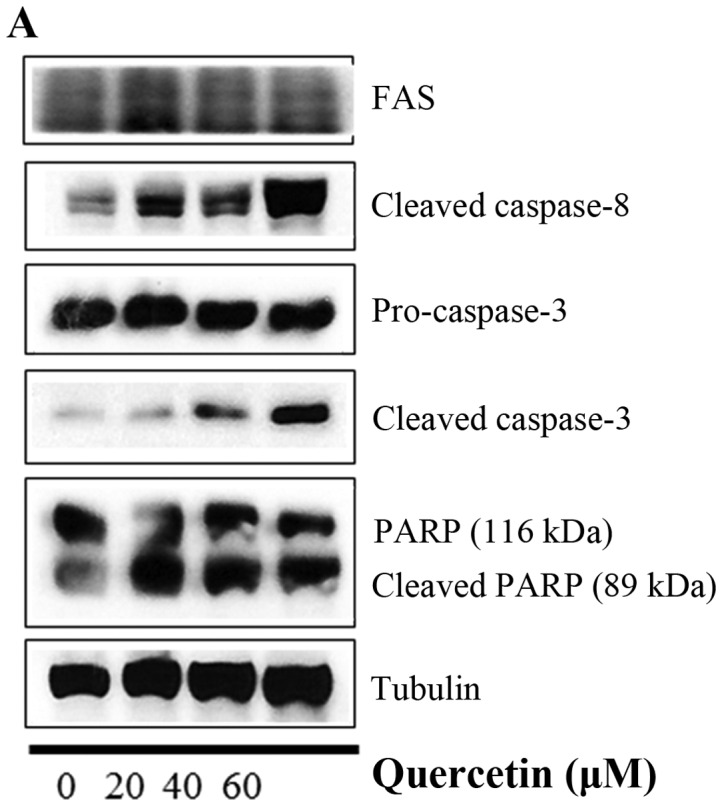
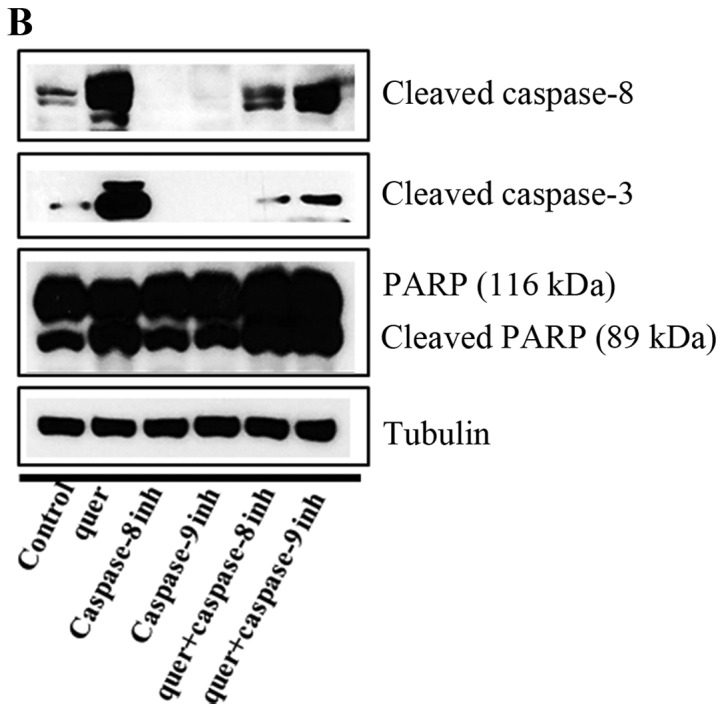

Quercetin induces caspase-dependent apoptosis in BT-474 cells. (A) Quercetin induces apoptosis via a caspase-dependent apoptosis pathway in the BT-474 cells. BT-474 cells were treated with quercetin (0–60 µM) for 24 h. Whole cell lysates were analyzed by western blotting with anti-FAS, anti-cleaved caspase-8, anti-caspase-3, anti-cleaved caspase-3, anti-PARP and anti-tubulin antibodies. The data shown are representative of three independent experiments that gave similar results. (B) Effect of caspase-8 and caspase-9 inhibitors on quercetin-induced apoptosis in BT-474 cells. BT-474 cells were exposed to 60 µM quercetin with or without the caspase-8 inhibitor (40 µM) or the caspase-9 inhibitor (40 µM) for 24 h, the cell lysates were separated by SDS-PAGE, and western blotting with specific antibodies was performed (anti-cleaved caspase-8, anti-cleaved caspase-3, anti-cleaved PARP, and anti-tubulin). The data shown are representative of three independent experiments that gave similar results. (C) Effect of caspase-8 and caspase-9 inhibitors on BT-474 cell proliferation. BT-474 cells were exposed to 60 µM quercetin with or without the caspase-8 inhibitor (40 µM) or the caspase-9 inhibitor (40 µM) for 72 h and photographed by phase contrast microscopy (original magnification, x40).
Effect of quercetin on STAT3 activation in BT-474 cells
Quercetin upregulated phospho-p53 (p-p53) and p21 (p53 target gene) (Fig. 6A). Quercetin did not affect the p53 level. As shown in Fig. 6B, we aimed to ascertain whether quercetin affects STAT3 signaling measuring levels of p-STAT3 and VEGF (STAT-3 target gene). We found that quercetin reduced the expression of p-STAT3 as well as p-JAK1 (an upstream kinase of STAT3) (Fig. 6B). Quercetin also reduced the level of VEGF (Fig. B). Since STAT3 is a potential modulator of HIF-1α (28), we observed the relationship between STAT3 and HIF-1α. We found that quercetin suppressed the expression of p-STAT3 and HIF-1α that was upregulated by CoCl2 (hypoxia mimic) (Fig. 6C). Immunocytochemical staining indicated that quercetin decreased the nuclear localization of STAT3 in the presence and absence of CoCl2 (Fig. 6D). Fig. 6E showed that quercetin strongly decreased STAT3 transcriptional activity as revealed by transient transfection and luciferase assays. As shown in Fig. 6F, quercetin suppressed the production of STAT3 target gene, MMP-9, as revealed by ELISA assay. These results suggest that quercetin decreases HER2-positive breast cancer cell growth rate at higher concentrations (>20 µM) by inhibiting the STAT3 signaling pathway.
Figure 6.
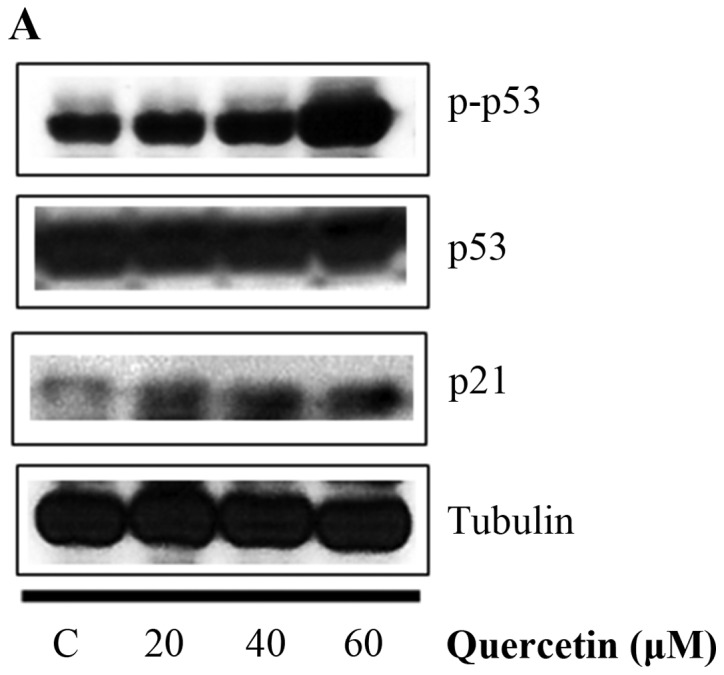
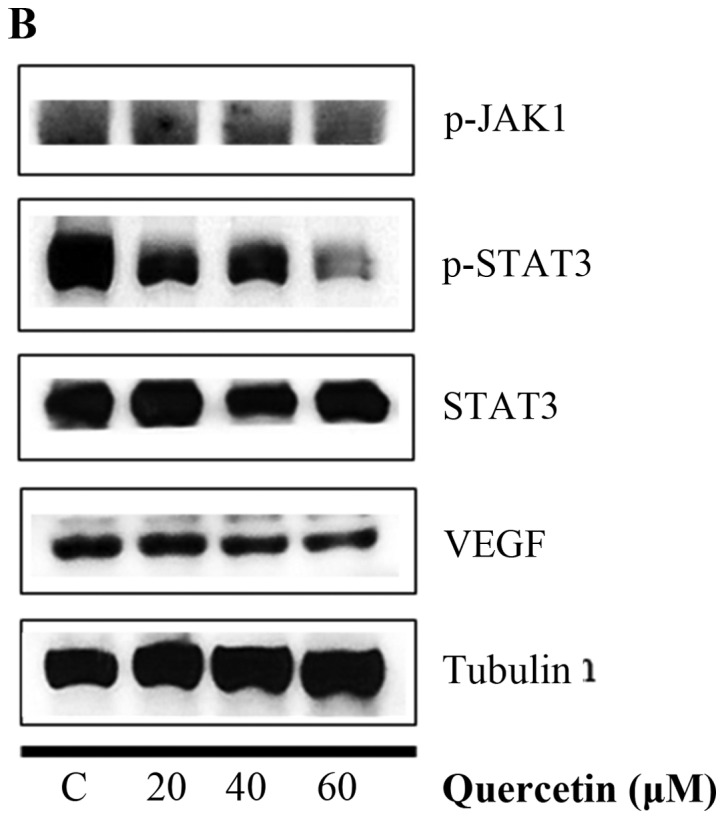
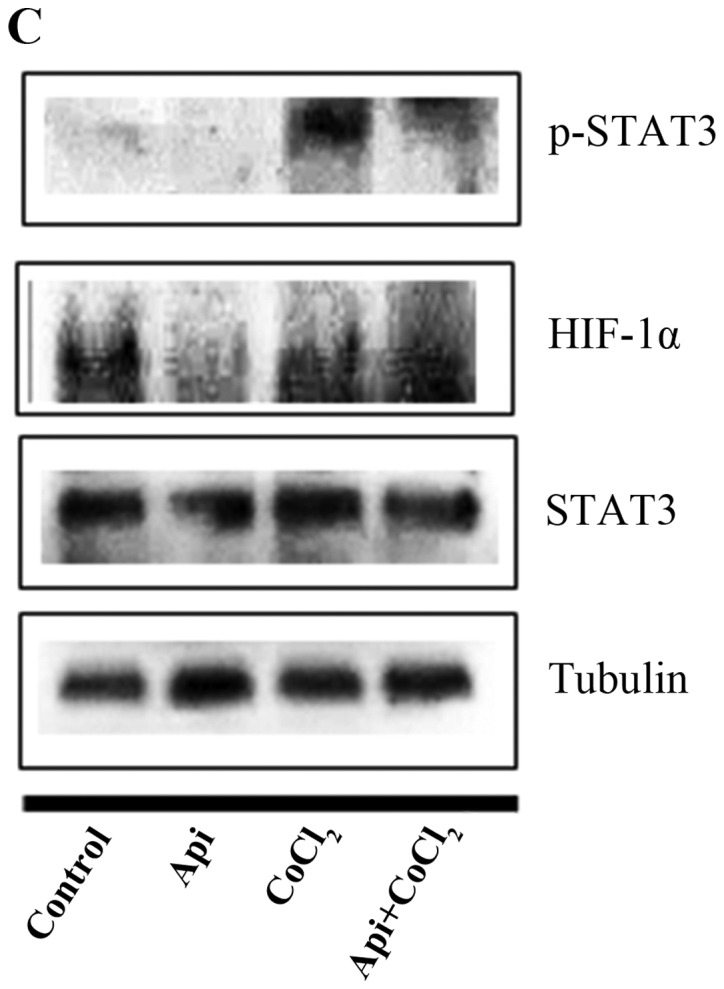

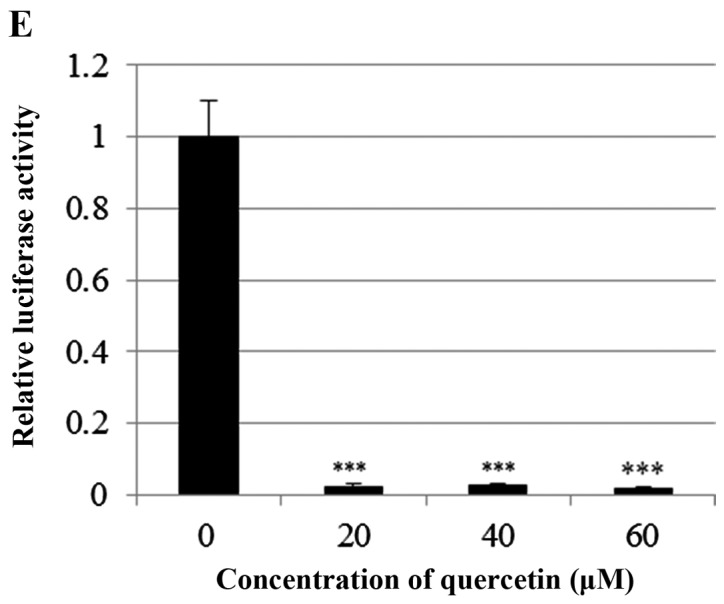
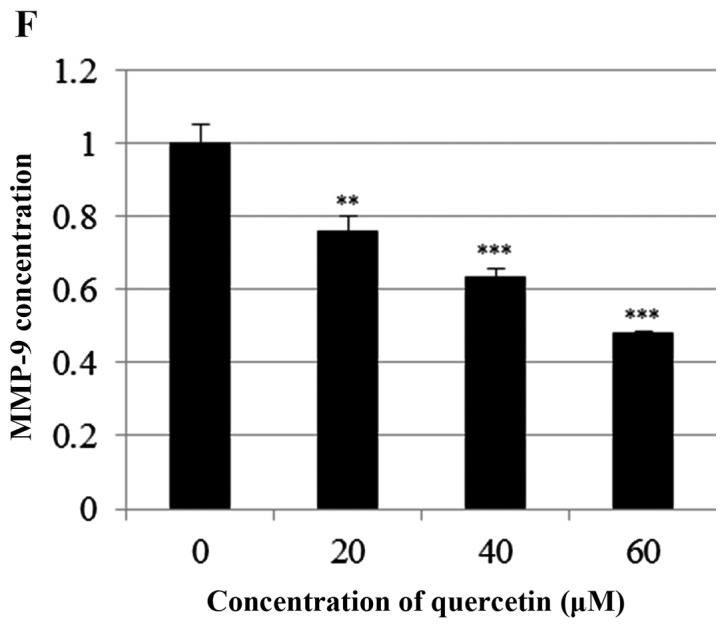
Effect of quercetin on STAT3 activation in BT-474 cells. (A) BT-474 cells were treated with quercetin (0–60 µM) for 24 h. Whole cell lysates were analyzed by western blotting with anti-p-p53, anti-p53, anti-p21 and anti-tubulin antibodies. (B) BT-474 cells were treated with quercetin (0–60 µM) for 24 h. Whole cell lysates were analyzed by western blotting with anti-p-JAK1, anti-p-STAT3, anti-STAT3, anti-VEGF, and anti-tubulin antibodies. (C) BT-474 cells were treated with quercetin (60 µM) for 24 h in the presence or absence of CoCl2 (4 h). Whole cell lysates were analyzed by western blotting with anti-phospho-STAT3, anti-HIF-1α, anti-STAT3, and anti-tubulin antibodies. (D) BT-474 cells were treated with quercetin (60 µM) for 24 h in the presence or absence of CoCl2 and then submitted to immunocytochemistry for detection of nuclear STAT3. The data shown are representative of three independent experiments that gave similar results. (E) BT-474 cells were transiently transfected with p4xM67-TK-luc plasmid containing four copies of the STAT-binding site, treated with quercetin (0–60 µM) and submitted to dual-luciferase assay. (F) BT-474 cells were treated with quercetin (0–60 µM) for 24 h and the intracellular MMP-9 concentration was measured by ELISA. Data are shown as the means of three independent experiments (error bars denote SD). *P<0.05, **P<0.01, ***P<0.001.
Effect of S3I-201 on STAT3 activation in BT-474 cells
Finally, we investigated whether the STAT3 inhibitor S3I-201 inhibits cell proliferation and STAT3 activation in BT-474 cells. As shown in Fig. 7A and B, S3I-201 decreased cell growth in a dose-and time-dependent manner. Furthermore, S3I-201 reduced the expression of p-STAT3, STAT-3 and VEGF (Fig. 7C). These results demonstrate that STAT3 inhibition induced cell growth inhibition and repressed the expression of oncogenic molecules.
Figure 7.
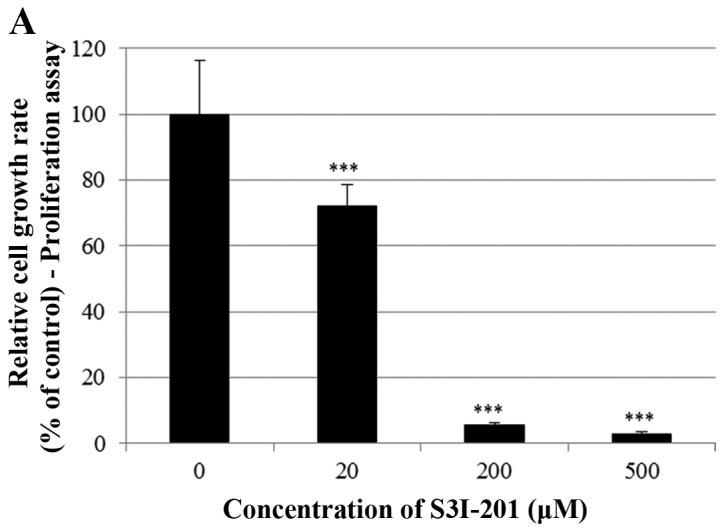
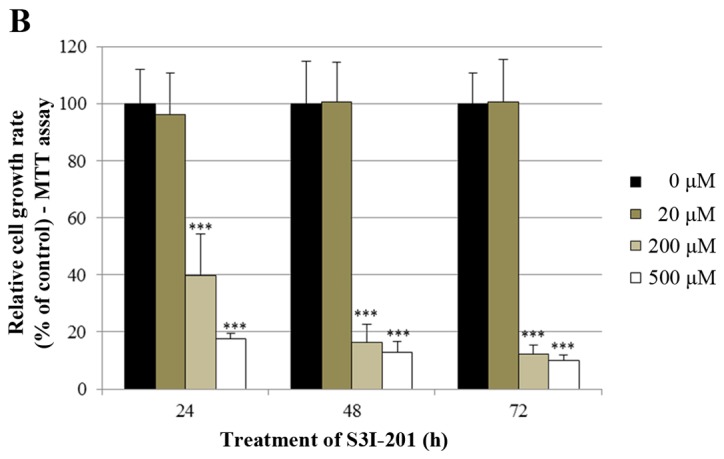
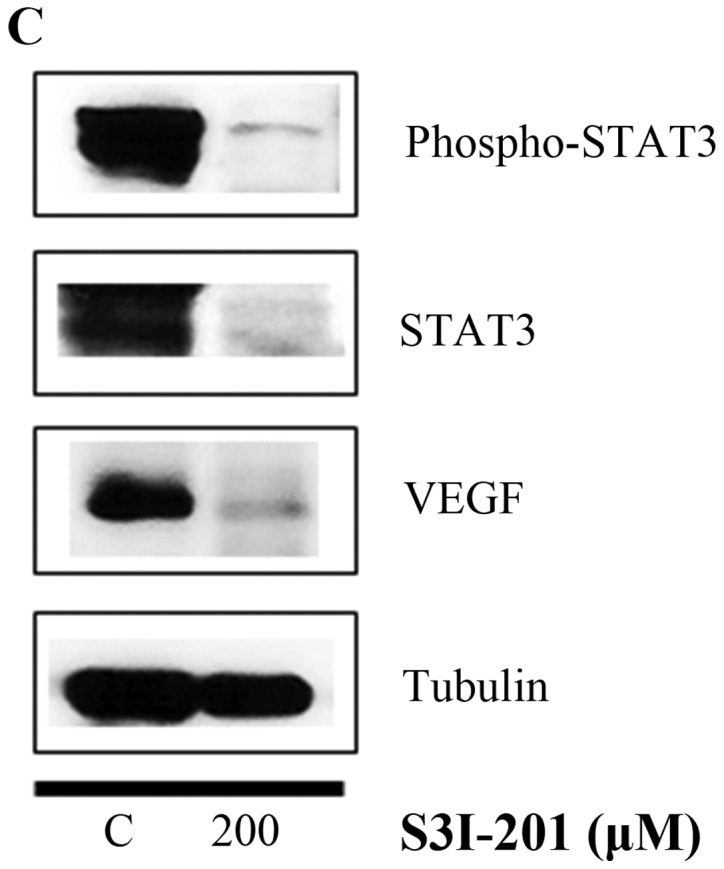
Effect of the STAT3 inhibitor S3I-201 on the growth of BT-474 cells. (A) BT-474 cells were treated with different doses of the STAT3 inhibitor S3I-201 (0–500 µM). After 72 h, the cell viability was assessed using a cell proliferation assay. (B) BT-474 cells were treated with different doses of the STAT3 inhibitor S3I-201 (0-500 µM). The relative cell growth rate was measured by MTT assay after 24, 48 and 72 h. The growth rate of the vehicle-treated cells was set to 100%, and the relative decrease in cell viability resulting from the S3I-201 treatment was expressed as a percentage of the control. Data are shown as the means of three independent experiments (error bars denote SD). ***P<0.001. (C) BT-474 cells were treated with the STAT3 inhibitor S3I-201 for 24 h. Whole cell lysates were analyzed by western blotting with anti-p-STAT3, anti-STAT3, anti-VEGF and anti-tubulin antibodies. The data shown are representative of three independent experiments that gave similar results.
Discussion
In the present study, we investigated the mechanism by which quercetin inhibits cell growth and induces apoptosis in HER2-overexpressing BT-474 breast cancer cells. Quercetin significantly inhibited BT-474 cell growth in a dose- and time-dependent manner. Clonogenic survival assays demonstrated that quercetin inhibited anchorage-dependent and -independent colony formation in a dose-dependent manner. These growth inhibitions were related with an increase in t he sub-G0/G1 apoptotic population in BT-474 cells. Quercetin increased the number of apoptotic cells in a dose-dependent manner, as assessed by FACS analysis. Interestingly, quercetin did not induce apoptosis via the intrinsic mitochondrial apoptosis pathway in the BT-474 cells as revealed by JC-1 dyeing of the cells and western blot analysis; quercetin did not reduce mitochondrial transmembrane potential (ΔΨm) maintaining red flouorescence, and failed to decrease the level of Bcl-2 or increase the level of BAX. Whereas, quercetin induced apoptosis via the caspase-dependent extrinsic apoptosis pathway since quercetin increased the cleavage of caspase-8, caspase-3 and PARP. Moreover, quercetin reversed inhibition of the cleavage of caspase-8, caspase-3 and PARP induced by caspase-8 inhibitor Z-IETD-fmk and the caspase-9 inhibitor Z-LEHD-fmk. These results suggest that quercetin contains a strong apoptotic capacity. The caspases, a family of cysteine-dependent aspartate-directed proteases, are common death proteases (29). Caspases are synthesized as relatively inactive zymogens that become activated by scaffold-mediated transactivation or by cleavage via upstream proteases in an intracellular cascade (29). Once activated, they cleave a variety of intracellular polypeptides, including major structural elements of the cytoplasm and nucleus, components of the DNA repair machinery, and a number of protein kinases (29).
Quercetin increased the expression of active p53 (p-p53) and p21 (p53 target gene), suggesting that this compound suppresses HER2-overexpressing breast cancer cell growth via a p53-dependent manner. In agreement with our data, quercetin has been shown to increase the levels of p-p53 and p21 in human lung carcinoma cells (30). The p53 tumor suppressor inhibits cellular proliferation by inducing cell cycle arrest and apoptosis in response to cellular stresses including DNA damage, growth factor deprivation, hypoxia and oncogene activation (31,32). p53-dependent apoptosis is produced by the caspase proteinases and related to pro-apoptotic proteins such as BAX, NOXA and PUMA (33).
Interestingly, quercetin decreased the expression of p-JAK1 (upstream kinase of STAT3), p-STAT3 and VEGF (STAT3 target gene) suggesting its negative regulation of STAT3 pathway in BT-474 cells. Elevated p-STAT3 expression by CoCl2 was also reduced by quercetin. Quercetin inhibited nuclear localization of STAT3 in the presence or absence of CoCl2 as revealed by immunocytochemistry. Quercetin inhibited the production of MMP-9 as revealed by ELISA assay. The STAT3 inhibitor S3I-201 decreased the cell growth and expression of p-STAT3, STAT-3 and VEGF in the BT-474 cells. These results clearly indicate that quercetin induces growth-suppressive activity by inhibiting the STAT3 signaling pathway. STAT3 is a transcription factor that regulates the gene expression in response to various cellular stimuli and plays an important role in cell growth and apoptosis. STAT3 usually acts as a tumor-promoter, although its role as a tumor-suppressor has been recently reported (33,34). STAT3 accelerates cell proliferation and angiogenesis, inhibits apoptosis, and drives invasion and metastasis (33–35). STAT3 in melanoma tumors is associated with poor prognosis (35–37). Constitutive STAT3 phosphorylation is mediated by several upstream kinases (Jak and Src) and is thought to be a key component of the oncogenic process (38,39). Phytoestrogen (resveratrol) is known to inhibit STAT3 signaling and induces the apoptosis of malignant cells containing activated STAT3 (40). The VEGF promoter contains various transcription factor binding sites, including sites for STAT3 (41) and HIF-1 (42). The physical interaction of STAT3 with HIF-1 controls VEGF transcriptional activation by their binding to the VEGF promoter (28).
Breast cancers with HER2 gene amplification or HER2 protein overexpression are HER2-positive. Approximately, 20-25% of invasive breast carcinomas reveal HER2 overexpression (43). A normal breast cell has 20,000 HER2 receptors, while a breast cancer cell may have up to 1.5 million. HER2 enhances the aggressiveness of breast cancer and is associated with recurrence when compared to HER2-negative breast cancer. HER2 is a member of the HER/ErbB2/Neu protein family, which also includes HER1/EGFR, HER3 and HER4. HER2 crosstalks with the estrogen receptor (ER) signal transduction pathway (44), and its expression level can be regulated by ER. In the present study, we found that quercetin significantly inhibited the growth and induced apoptosis in HER2-overexpressing breast cancer cells. This indicates that quercetin could be a useful natural therapy that inhibits HER2-overexpressing breast cancer. Quercetin could be a promising target for the treatment and prevention of HER2-overexpressing breast cancer.
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (NRF-2015R1C1A2A01051539). This research was also supported by a grant funded by the Traditional Korean Medicine R&D Project of the Ministry of Health and Welfare (HI12C1889 and HI11C2110).
References
- 1.Walters DG, Young PJ, Agus C, Knize MG, Boobis AR, Gooderham NJ, Lake BG. Cruciferous vegetable consumption alters the metabolism of the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in humans. Carcinogenesis. 2004;25:1659–1669. doi: 10.1093/carcin/bgh164. [DOI] [PubMed] [Google Scholar]
- 2.Igura K, Ohta T, Kuroda Y, Kaji K. Resveratrol and quercetin inhibit angiogenesis in vitro. Cancer Lett. 2001;171:11–16. doi: 10.1016/S0304-3835(01)00443-8. [DOI] [PubMed] [Google Scholar]
- 3.Hertog MG, Hollman PC. Potential health effects of the dietary flavonol quercetin. Eur J Clin Nutr. 1996;50:63–71. [PubMed] [Google Scholar]
- 4.Coskun O, Kanter M, Korkmaz A, Oter S. Quercetin, a flavonoid antioxidant, prevents and protects streptozotocin-induced oxidative stress and beta-cell damage in rat pancreas. Pharmacol Res. 2005;51:117–123. doi: 10.1016/j.phrs.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 5.Boots AW, Wilms LC, Swennen EL, Kleinjans JC, Bast A, Haenen GR. In vitro and ex vivo anti-inflammatory activity of quercetin in healthy volunteers. Nutrition. 2008;24:703–710. doi: 10.1016/j.nut.2008.03.023. [DOI] [PubMed] [Google Scholar]
- 6.Geetha T, Malhotra V, Chopra K, Kaur IP. Antimutagenic and antioxidant/prooxidant activity of quercetin. Indian J Exp Biol. 2005;43:61–67. [PubMed] [Google Scholar]
- 7.Sudan S, Rupasinghe HP. Quercetin-3-O-glucoside induces human DNA topoisomerase II inhibition, cell cycle arrest and apoptosis in hepatocellular carcinoma cells. Anticancer Res. 2014;34:1691–1699. [PubMed] [Google Scholar]
- 8.Danihelová M, Veverka M, Sturdík E, Jantová S. Antioxidant action and cytotoxicity on HeLa and NIH-3T3 cells of new quercetin derivatives. Interdiscip Toxicol. 2013;6:209–216. doi: 10.2478/intox-2013-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deng XH, Song HY, Zhou YF, Yuan GY, Zheng FJ. Effects of quercetin on the proliferation of breast cancer cells and expression of survivin in vitro. Exp Ther Med. 2013;6:1155–1158. doi: 10.3892/etm.2013.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Erk MJ, Roepman P, van der Lende TR, Stierum RH, Aarts JM, van Bladeren PJ, van Ommen B. Integrated assessment by multiple gene expression analysis of quercetin bioactivity on anticancer-related mechanisms in colon cancer cells in vitro. Eur J Nutr. 2005;44:143–156. doi: 10.1007/s00394-004-0503-1. [DOI] [PubMed] [Google Scholar]
- 11.Murtaza I, Marra G, Schlapbach R, Patrignani A, Künzli M, Wagner U, Sabates J, Dutt A. A preliminary investigation demonstrating the effect of quercetin on the expression of genes related to cell-cycle arrest, apoptosis and xenobiotic metabolism in human CO115 colon-adenocarcinoma cells using DNA microarray. Biotechnol Appl Biochem. 2006;45:29–36. doi: 10.1042/BA20060044. [DOI] [PubMed] [Google Scholar]
- 12.Verma AK, Johnson JA, Gould MN, Tanner MA. Inhibition of 7,12-dimethylbenz(a)anthracene- and N-nitrosomethylurea-induced rat mammary cancer by dietary flavonol quercetin. Cancer Res. 1988;48:5754–5758. [PubMed] [Google Scholar]
- 13.Elmore S. Apoptosis: A review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan TJ, Han LH, Cong RS, Liang J. Caspase family proteases and apoptosis. Acta Biochim Biophys Sin (Shanghai) 2005;37:719–727. doi: 10.1111/j.1745-7270.2005.00108.x. [DOI] [PubMed] [Google Scholar]
- 15.Bosch M, Poulter NS, Vatovec S, Franklin-Tong VE. Initiation of programmed cell death in self-incompatibility: Role for cytoskeleton modifications and several caspase-like activities. Mol Plant. 2008;1:879–887. doi: 10.1093/mp/ssn053. [DOI] [PubMed] [Google Scholar]
- 16.Zhang A, Wu Y, Lai HWL, Yew DT. Apoptosis - a brief review. Neuroembryology. 2004;3:47–59. doi: 10.1159/000085404. [DOI] [Google Scholar]
- 17.Waring P, Müllbacher A. Cell death induced by the Fas/Fas ligand pathway and its role in pathology. Immunol Cell Biol. 1999;77:312–317. doi: 10.1046/j.1440-1711.1999.00837.x. [DOI] [PubMed] [Google Scholar]
- 18.Gupta S. Molecular signaling in death receptor and mitochondrial pathways of apoptosis (Review) Int J Oncol. 2003;22:15–20. [PubMed] [Google Scholar]
- 19.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 20.Boulares AH, Yakovlev AG, Ivanova V, Stoica BA, Wang G, Iyer S, Smulson M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J Biol Chem. 1999;274:22932–22940. doi: 10.1074/jbc.274.33.22932. [DOI] [PubMed] [Google Scholar]
- 21.Wilson CA, Cajulis EE, Green JL, Olsen TM, Chung YA, Damore MA, Dering J, Calzone FJ, Slamon DJ. HER-2 overexpression differentially alters transforming growth factor-beta responses in luminal versus mesenchymal human breast cancer cells. Breast Cancer Res. 2005;7:R1058–R1079. doi: 10.1186/bcr1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joshi JP, Brown NE, Griner SE, Nahta R. Growth differentiation factor 15 (GDF15)-mediated HER2 phosphorylation reduces trastuzumab sensitivity of HER2-overexpressing breast cancer cells. Biochem Pharmacol. 2011;82:1090–1099. doi: 10.1016/j.bcp.2011.07.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Favoni RE, Daga A, Malatesta P, Florio T. Preclinical studies identify novel targeted pharmacological strategies for treatment of human malignant pleural mesothelioma. Br J Pharmacol. 2012;166:532–553. doi: 10.1111/j.1476-5381.2012.01873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tokunaga E, Oki E, Nishida K, Koga T, Egashira A, Morita M, Kakeji Y, Maehara Y. Trastuzumab and breast cancer: Developments and current status. Int J Clin Oncol. 2006;11:199–208. doi: 10.1007/s10147-006-0575-4. [DOI] [PubMed] [Google Scholar]
- 25.Dean-Colomb W, Esteva FJ. Her2-positive breast cancer: Herceptin and beyond. Eur J Cancer. 2008;44:2806–2812. doi: 10.1016/j.ejca.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 26.Seo HS, Choi HS, Choi HS, Choi YK, Um JY, Choi I, Shin YC, Ko SG. Phytoestrogens induce apoptosis via extrinsic pathway, inhibiting nuclear factor-kappaB signaling in HER2-overexpressing breast cancer cells. Anticancer Res. 2011;31:3301–3313. [PubMed] [Google Scholar]
- 27.Seo HS, Choi HS, Kim SR, Choi YK, Woo SM, Shin I, Woo JK, Park SY, Shin YC, Ko SG. Apigenin induces apoptosis via extrinsic pathway, inducing p53 and inhibiting STAT3 and NFκB signaling in HER2-overexpressing breast cancer cells. Mol Cell Biochem. 2012;366:319–334. doi: 10.1007/s11010-012-1310-2. [DOI] [PubMed] [Google Scholar]
- 28.Jung JE, Lee HG, Cho IH, Chung DH, Yoon SH, Yang YM, Lee JW, Choi S, Park JW, Ye SK, et al. STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB J. 2005;19:1296–1298. doi: 10.1096/fj.04-3099fje. [DOI] [PubMed] [Google Scholar]
- 29.Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: Structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- 30.Kuo PC, Liu HF, Chao JI. Survivin and p53 modulate quercetin-induced cell growth inhibition and apoptosis in human lung carcinoma cells. J Biol Chem. 2004;279:55875–55885. doi: 10.1074/jbc.M407985200. [DOI] [PubMed] [Google Scholar]
- 31.Schuler M, Green DR. Mechanisms of p53-dependent apoptosis. Biochem Soc Trans. 2001;29:684–688. doi: 10.1042/bst0290684. [DOI] [PubMed] [Google Scholar]
- 32.Shen Y, White E. p53-dependent apoptosis pathways. Adv Cancer Res. 2001;82:55–84. doi: 10.1016/S0065-230X(01)82002-9. [DOI] [PubMed] [Google Scholar]
- 33.de la Iglesia N, Konopka G, Puram SV, Chan JA, Bachoo RM, You MJ, Levy DE, Depinho RA, Bonni A. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 2008;22:449–462. doi: 10.1101/gad.1606508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lewis HD, Winter A, Murphy TF, Tripathi S, Pandey VN, Barton BE. STAT3 inhibition in prostate and pancreatic cancer lines by STAT3 binding sequence oligonucleotides: Differential activity between 5′ and 3′ ends. Mol Cancer Ther. 2008;7:1543–1550. doi: 10.1158/1535-7163.MCT-08-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kortylewski M, Jove R, Yu H. Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev. 2005;24:315–327. doi: 10.1007/s10555-005-1580-1. [DOI] [PubMed] [Google Scholar]
- 36.Niu G, Bowman T, Huang M, Shivers S, Reintgen D, Daud A, Chang A, Kraker A, Jove R, Yu H. Roles of activated Src and Stat3 signaling in melanoma tumor cell growth. Oncogene. 2002;21:7001–7010. doi: 10.1038/sj.onc.1205859. [DOI] [PubMed] [Google Scholar]
- 37.Xie TX, Huang FJ, Aldape KD, Kang SH, Liu M, Gershenwald JE, Xie K, Sawaya R, Huang S. Activation of stat3 in human melanoma promotes brain metastasis. Cancer Res. 2006;66:3188–3196. doi: 10.1158/0008-5472.CAN-05-2674. [DOI] [PubMed] [Google Scholar]
- 38.Sellers LA, Feniuk W, Humphrey PP, Lauder H. Activated G protein-coupled receptor induces tyrosine phosphorylation of STAT3 and agonist-selective serine phosphorylation via sustained stimulation of mitogen-activated protein kinase. Resultant effects on cell proliferation. J Biol Chem. 1999;274:16423–16430. doi: 10.1074/jbc.274.23.16423. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, Turkson J, Carter-Su C, Smithgall T, Levitzki A, Kraker A, Krolewski JJ, Medveczky P, Jove R. Activation of Stat3 in v-Src-transformed fibroblasts requires cooperation of Jak1 kinase activity. J Biol Chem. 2000;275:24935–24944. doi: 10.1074/jbc.M002383200. [DOI] [PubMed] [Google Scholar]
- 40.Kotha A, Sekharam M, Cilenti L, Siddiquee K, Khaled A, Zervos AS, Carter B, Turkson J, Jove R. Resveratrol inhibits Src and Stat3 signaling and induces the apoptosis of malignant cells containing activated Stat3 protein. Mol Cancer Ther. 2006;5:621–629. doi: 10.1158/1535-7163.MCT-05-0268. [DOI] [PubMed] [Google Scholar]
- 41.Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, Zhang S, Wang T, Sinibaldi D, Coppola D, et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21:2000–2008. doi: 10.1038/sj.onc.1205260. [DOI] [PubMed] [Google Scholar]
- 42.Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/MCB.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tolaney SM, Krop IE. Mechanisms of trastuzumab resistance in breast cancer. Anticancer Agents Med Chem. 2009;9:348–355. doi: 10.2174/1871520610909030348. [DOI] [PubMed] [Google Scholar]
- 44.Buzdar AU. Role of biologic therapy and chemotherapy in hormone receptor- and HER2-positive breast cancer. Ann Oncol. 2009;20:993–999. doi: 10.1093/annonc/mdn739. [DOI] [PubMed] [Google Scholar]