

Abstract
Connexin43 (Cx43) containing gap junctions play an important role in bone homeostasis, yet little is known about the second messengers communicated by Cx43 among bone cells. Here, we used MC3T3-E1 pre-osteoblasts and UMR106 rat osteosarcoma cells to test the hypothesis that cAMP is a second messenger communicated by bone cells through Cx43 containing gap junctions in a manner that is sufficient to impact osteoblast function. Overexpression of Cx43 markedly enhanced the activity of a cAMP-response element driven transcriptional luciferase reporter (CRE-luc) and increased phospho-CREB and phospho-ERK1/2 levels following expression of a constitutively active Gsα or by treatment with prostaglandin E2 (PGE2), 3-Isobutyl–1-methyl xanthine (IBMX) or forskolin. The Cx43-dependent potentiation of signaling in PGE2 treated cells was not accompanied by a further increase in cAMP levels, suggesting that the cAMP was shared between cells rather than Cx43 enhancing cAMP production. To support this, we developed a novel assay in which one set of cells expressing constitutively active Gsα (donor cells) were co-cultured with a second set of cells expressing a CRE-luc reporter (acceptor cells). Using this assay, activation of a CRE-luc reporter in the acceptor cells was both Cx43- and cell contact-dependent, indicating communication of cAMP among cells. Finally, we showed that Cx43 increased the cAMP-dependent mRNA expression of receptor activator of nuclear factor kappa B ligand (RANKL) and enhanced the repression of the sclerostin mRNA, implying a potential mechanism for the modulation of tissue remodeling. In total, these data demonstrate that Cx43 can communicate cAMP between cells and, more importantly, that the communicated cAMP is sufficient to impact signal transduction cascades and the expression of key bone effector molecules between interconnected cells.
Keywords: Gap junction, prostaglandin E2, osteoblast, G protein, cyclic AMP (cAMP), connexin
1. Introduction
Cell-to-cell transfer of small molecules through gap junctions regulates diverse biologic processes (1,2). The size and charge selectivity of the small molecules that can be communicated by gap junctions is dictated by the connexin composition of the channel. A connexin is the monomeric protein subunit of a gap junction, and six connexin monomers form a hemichannel. When hemichannels on adjacent cells dock, they form a gap junction channel that interconnects the two cells.
In bone, the gap junction-dependent intercellular communication of signals is important to tissue function (3-6). Indeed, bone is formed through the coordinated action of hundreds of connexin expressing osteoblasts, acting in concert to lay down a collagen-rich extracellular matrix that subsequently becomes mineralized. An elaborate network of gap junction-expressing osteoblasts and osteocytes (7-9) plays an important role in the activation of both the osteoblast and the bone resorbing osteoclasts (10). Numerous studies have implicated Cx43-containing gap junctions in a complex array of biologic responses that influence bone quality in vivo, including regulation of mechanosensitivity, hormonal and growth factor responsiveness, and control of the balance of bone resorption and bone formation (3-5,11). Despite the array of Cx43-sensitive affects on osteoblast and osteocyte function, little is known about the specific, biologically relevant molecules that are being communicated throughout this cellular network to impact bone homeostasis.
Previous studies have shown in relatively non-physiological contexts (e.g., cAMP added via patch pipet or photo-release of caged cAMP) that the biophysical properties of Cx43 permit the passage of cAMP between cells (12,13). However, whether or not Cx43-dependent communication of cAMP occurs at a level sufficient to elicit a biological response in coupled bone cells has not been demonstrated. Similarly, PGE2 has been shown to regulate the abundance of Cx43 in bone cells (14-17), but whether or not cAMP produced by PGE2 is then communicated through this gap junction-coupled network of cells and is capable of eliciting a biological response has not been explicitly tested. Indeed, if cAMP is a biologically relevant second messenger that permits coordination of osteoblast activity, then it is essential that not only must cAMP pass between gap junction coupled cells but also must impact signaling and cell function. Here, we identify cAMP as a biologically relevant second messenger communicated by bone cells via Cx43 containing gap junctions. Further, we show that Cx43-dependent communication of cAMP impacts the expression of sclerostin and RANKL, two critically important factors for the regulation of bone homeostasis.
2. Materials and methods
2.1. Chemicals, Antibodies and Reagents
Prostaglandin E2 (PGE2), 3-Isobutyl–1-methyl xanthine (IBMX) and H89 dihydrochloride were purchased from Sigma. Forskolin was purchased from Calbiochem. Tissue culture media and fetal bovine serum were purchased from Hyclone. Antibodies were purchased as follows: rabbit anti-connexin43 (#C6219) antibody was purchased from Sigma; rabbit anti-phospho-CREB (Ser133, #9198), total-CREB (#9197), rabbit anti-phospho-ERK1/2 (Thr202/Tyr404; #9101), rabbit anti-total-ERK1/2 (#9102) and horseradish peroxidase-conjugated secondary antibodies were purchased from Cell Signaling Technology. The mouse anti-GAPDH antibody (#MAB 374) was purchased from Millipore. All other chemicals were purchased from Sigma, unless indicated otherwise.
2.2. Cell Culture
MC3T3-E1 clone 4 mouse pre-osteoblasts and UMR106 rat osteosarcoma cells were purchased from the ATCC. UMR106 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin, and 50 μg/ml gentamycin. MC3T3 cells were cultured as previously described (18). Gja1flox/flox mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and maintained in the animal care facility at the University of Maryland School of Medicine. All animal studies were performed with approval by the Animal Care and Use Committee at the University of Maryland School of Medicine. Primary murine osteoblasts were isolated from the long bones of 4 week old Gja1flox/flox mice by collagenase digestion, as described previously (19,20). To facilitate Cx43 gene deletion, cells were transduced with an eGFP adenovirus (Ad-GFP) or a Cre recombinase expressing adenovirus (Ad-Cre) at an moi of 5 in the presence of tissue culture grade poly-l-lysine (0.5 μg/ml). Cells were maintained in a tissue culture incubator at 37°C, 5% CO2. Media was replaced every 2-3 days and cells were passaged upon reaching confluence. Cell viability was routinely monitored by a CCK-8 (cell counting kit-8) assay (Dojindo), as described previously (21).
2.3. Plasmids
The constitutively active pcDNA3.1-Gsα long Q227L plasmid, which has reduced GTPase activity resulting in a constitutively active function, was obtained from the Missouri S&T cDNA Resource Center. pcDNA3 was purchased from Invitrogen. The cAMP-response element luciferase reporter plasmid (CRE-Luc) was obtained from Clontech. The pSFFV-Cx43 construct, which contains the full-length rat Cx43 open reading frame cloned into the EcoR1 site of the pSFFV-neo plasmid, was provided by Dr. Thomas Steinberg (Washington University, St Louis, MO). The pSFFV-neo empty vector (22) was provided by Dr. Gabriel Nunez (University of Michigan, Ann Arbor, MI). The pSFFV-Cx43 G138R was generated by Mutagenex from the pSFFV-Cx43 backbone vector, by a G>C mutation at nucleotide 608 (NCBI Reference Sequence: NM_012567.2). We find that the use of the pSFFV-neo vector, which has modest level of expression relative to CMV/CAG based expression vectors, generates more consistent responses than more robust overexpression vectors. Further, unlike pSFFV-Cx43, we have routinely observed that using stronger promoters (CMV/CAG) to drive Cx43 have effects that mimic loss of function rather than gain of function with respect to signaling and gene expression (data not shown). We have observed similar effects of modest Cx43 expression in human synovial cells (23). The promoterless pRL-null vector was from Promega. The PKI plasmid was provided by Dr. Raymond Penn (Thomas Jefferson University, Philadelphia, PA) (24). The cAMP responsive nano-lantern luminescence reporter plasmid Nano-lantern(cAMP-1.6)/pcDNA3 was a gift from Takeharu Nagai (Addgene plasmid # 53594) (25). Plasmid DNA was prepared using either a PureYield endotoxin-free plasmid maxi prep kit (Promega) or a HiSpeed maxi prep kit (Qiagen).
2.4. Transient Transfections
One day prior to transfection, cells were seeded at 25,000-30,000 cells/cm2 in the appropriate multiwell tissue culture plate. Cells were co-transfected with up to three plasmids with JetPrime reagent (Polypus), according to manufacturer's directions. Luciferase reporter plasmids were used at 0.13 μg/cm2. The Cx43 expression vector (pSFFV-Cx43) and its negative control (pSFFV-neo) were used at 0.27 μg/cm2. The constitutively active GSα plasmid (pcDNA3.1- GSα long Q227L) and empty pcDNA3 vectors were used at 0.13 μg/cm2. JetPrime reagent was added at 4:1 ratio (μl JetPrime:μg of DNA).
Cell treatments were begun 48 hours post-transfection. Where indicated, cells were pre-treated for 30 minutes with H89 (10 μM) prior to exposure to PGE2 (2 μM, 4 hours), forskolin (10 μM, 4 hours) or IBMX (1 mM, 4 hours). Subsequently, the cells were rinsed in Hank's balanced salt solution (HBSS) and then lysed in 1× passive lysis buffer (Promega). Luciferase activity in the lysate was then monitored on a Berthold Centro LB960 luminometer, as described previously (26). Transfection efficiency was routinely monitored by co-transfection with a promotorless renilla luciferase construct (pRL-null) using a dual luciferase assay (27). For calcium free culture conditions, cells were placed in calcium free-Ringer's solution (10mM HEPES, 140 mM NaCl, 4 mM KCl, 1 mM MgSO4, 10 mM glucose, 5 mM NaHCO3, pH 7.4) and loaded with BAPTA-AM (10 μM) just prior to treatment with PGE2 (2 μM, 4 hours). Experiments were conducted on six replicate wells and repeated a minimum of three times. Data from a representative experiment are shown.
2.5. Modified Parachute Assay
This assay is a modified version of the classic gap junction assay known as the parachute assay. The original parachute assay for gap junctions used a population of “donor” cells that were loaded with calcium green AM ester, a gap junction permeable, low molecular weight fluorescent dye (28,29). The donor cells were then seeded onto a monolayer of unlabeled “acceptor” cells, and gap junction communication determined by the diffusion of the calcium green from the donor cell to the acceptor cell. In our variation, we co-transfect the donor cells (UMR106) with the pcDNA3.1- GSα long Q227L plasmid and pSFFV-neo (empty vector) or pSFFV-Cx43. Acceptor cells were co-transfected with a CRE-luc reporter plasmid and pSFFV-neo or pSFFV-Cx43. Forty-eight hours post-transfection, the cells were rinsed in HBSS, trypsinized and co-cultured at a 1:2 donor:acceptor cell ratio in a 48-well plate, and incubated for 24 hours. The cells were then rinsed twice with HBSS and lysed in reporter lysis buffer and luciferase activity measured as above. To ensure that gap junction communication was involved in the communication of signals from the donor cells to the acceptor cells, the experiment was repeated with acceptor cells seeded in the well of a multiwell plate and the donor cells seeded into a transwell chamber (Corning Life Sciences, 5 μm pore size), which shared the tissue culture media, but prevented direct cell-to-cell contact. These experiments were performed in sample size of six per group and repeated a minimum of three times.
In parallel, we performed an immunofluorescence-based version of this modified parachute assay. MC3T3 cells were separated into donor and acceptor cell populations and transfected with or without Cx43 and GSα, as indicated. Forty-eight hours post-transfection, the donor cell population was stained with DiI and then seeded onto a confluent monolayer of the acceptor cells. Immunofluorescence for phospho-CREB was performed to examine activation of signaling in the acceptor cells, as described (30). To quantify positive signals, the phospho-CREB staining image (12-bit) was converted into a binary image using a threshold value of 780. The number of objects above this threshold (size range from 0.004 to 0.4 mm) was quantified using Nikon NIS Elements software (v4.3). Data are from three fields of view per slide and three slides per group.
2.6. Western Blotting
Whole cell extracts were prepared using modified RIPA buffer containing 50 mM Tris, pH 8.0, 150 mM NaCl, 10 mM sodium pyrophosphate, 10 mM sodium fluoride, 10 mM β-glycerophosphate, 1 mM EGTA, 1 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS and 1X HALT protease and phosphatase inhibitor cocktail (Thermo Scientific) as described (31,32). Equal amounts of cell lysates were electrophoresed on SDS-PAGE gels and transferred to PVDF membranes. Membranes were blocked in 5% non-fat dry milk in phosphate buffered saline containing 0.1% Tween-20 and probed with primary and secondary antibodies as described previously (23). After incubation in ECL detection reagent (Clarity ECL Western Blotting Reagent, Bio-Rad), blots were imaged and analyzed using an EpiChem gel documentation system (UVP Bio imaging Systems). Quantitation of western blots was performed using ImageJ software (Fiji v2.0).
2.7. cAMP ELISA Assay
Cyclic AMP ELISA kits were purchased from Cayman Chemicals. Briefly, cells were transfected with pSFFV-neo or pSFFV-Cx43. Forty-eight hours post-transfection, the cells were treated with vehicle (0.1% ethanol) or PGE2 (2 μM, 4 hours). Subsequently, the cells were incubated in 0.1 M HCl for 20 minutes at room temperature, scraped and pelleted by centrifugation. The cAMP levels in the resultant cell supernatants were determined according to manufacturer's directions using a μQuant spectrophotometer (Biotek). Data were determined by comparison to known concentrations of cAMP standards.
2.8. RNA Isolation and Quantitative Reverse Transcription and Real-Time PCR
RNA extraction was done by Directzol RNA mini prep (Zymo). RNA was reverse transcribed with iScript (BioRad) reverse transcription master mix, according to the manufacturer directions. Quantitative real time PCR was carried out by SYBR green master mix from Quanta using an Applied Biosystems 7300 sequence detection system. A melting curve was performed to ensure amplification of a single PCR product. For each sample, the relative gene expression was determined by simultaneously normalizing the gene of interest with three housekeeping genes (Rpl13, Hprt and Gapdh) by the 2-ΔΔCt method, using GeNorm v3.5 software as described (23,26). The primer sets used for quantitative real time PCR are: Sclerostin/Sost - GGA ATG ATG CCA CAG AGG TCA T and CCC GGT TCA TGG TCT GGT T; RANKL/Tnfsf11 - ACC AGC ATC AAA ATC CCA AGT T and TCA GAA TTG CCC GAC CAG TT; Cx43/Gja1 - CAG GCC GGA AGC ACC AT and GCT GTC GTC AGG GAA ATC AAA; Rpl13 - CGA AAC AAG TCC ACG GAG TCA and GAG CTT GGA GCG GTA CTC CTT; Hprt - AGC AGT ACA GCC CCA AAA TGG and AAC AAA GTC TGG CCT GTA TCC AA; and Gapdh - CGT GTT CCT ACC CCC AAT GT and TGT CAT CAT ACT TGG CAG GTT TCT.
2.9. Statistical Analysis
Experiments were repeated a minimum of three times and each experiment was done in six replicate wells, unless indicated otherwise. Graphs show averages with error bars indicating standard deviations. Samples were compared by a two-way ANOVA using Prism 6 software. A p-value <0.05 was used as a threshold for statistical significance and for interaction of the variables.
3. Results
3.1. Alteration of Cx43 expression levels modulates basal phospho-CREB levels in osteoblasts
We observed that modest (2- to 4-fold) overexpression of Cx43 increased the basal phospho-CREB levels in MC3T3 pre-osteoblasts and UMR106 osteoblast-like osteosarcoma cells (Fig. 1A; quantitation in Supl Fig 1A). Conversely, deletion of Cx43 in primary osteoblasts isolated from Gja1floxflox mice reduced basal phospho-CREB levels. These observations prompted us to ask if Cx43 could communicate cAMP-dependent signals between cells to facilitate a coordinated osteoblast response to these factors converging on phospho-CREB.
Figure 1. Connexin43 overexpression synergistically enhances cAMP-dependent signaling.
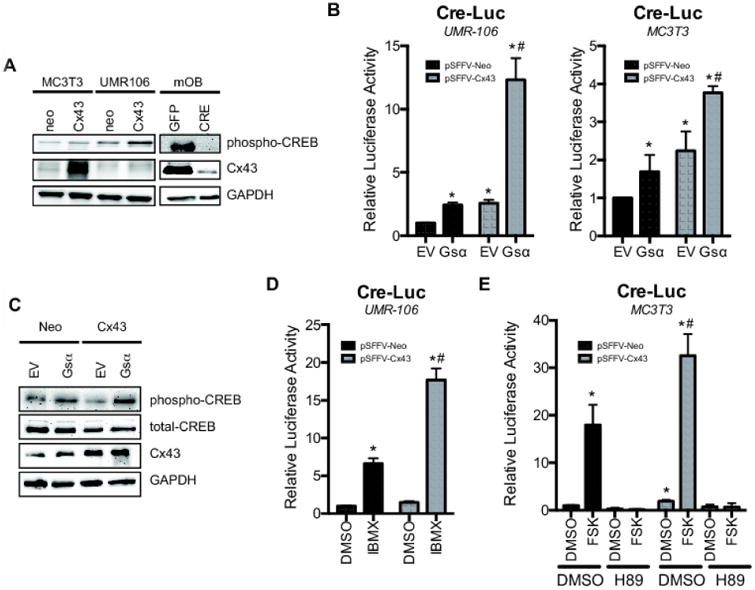
A, Basal phospho-CREB, Cx43 and GAPDH were examined in whole cell extracts of MC3T3, UMR106 and murine primary osteoblasts (mOB) from Gja1flox/flox mice treated with GFP-encoding (GFP) or CRE recombinase-encoding (CRE) adenovirus particles by western blotting. B, CRE-Luc activity was determined in UMR106 (left) or MC3T3 (right) cells co-transfected with pSFFV-Neo (neo) or pSFFV-Cx43 (Cx43) and pcDNA (EV) or pcDNA3.1-Gsα long Q227L (Gsα). C, Western blotting of whole cell extracts from transfected UMR106 cells showing levels of phospho- and total CREB, Cx43 and GAPDH 48 hours post-transfection. D, CRE-Luc activity was determined in UMR106 cells co-transfected with pSFFV-Neo or pSFFV-Cx43 and treated with IBMX (1 mM, 4 h) E, CRE-Luc activity was determined in MC3T3 cells co-transfected with pSFFV-Neo or pSFFV-Cx43 and treated with forskolin (FSK, 10 μM, 4h) in the presence or absence of H89 (10 μM). DMSO was used as a negative control. Bar graphs display mean ± standard deviation. *, p-value < 0.05 versus empty vector transfected control. #, indication of an interaction of the two variables (p-value < 0.05) by two-way ANOVA.
3.2. Connexin43 enhances the osteoblast response to cAMP generating factors
To test if Cx43 expression could influence cAMP-dependent signaling, we overexpressed Cx43 in UMR106 cells, which have very little endogenous Cx43 (Fig1A; quantitation in Supl Fig 1A) (33), along with a constitutively active Gsα (pcDNA3.1- Gsα long Q227L plasmid) and assessed their influence on a cAMP/protein kinase A (PKA)-dependent CRE-Luc reporter. When Cx43 and GSα long Q227L were co-expressed, there was a marked potentiation of CRE-luc activity (Fig. 1B, left). Analogous results were obtained in MC3T3 pre-osteoblasts cells (Fig. 1B, right), which have a moderate amount of endogenous Cx43 expression (Fig. 1A). Western blots with anti-phospho-CREB antibodies confirmed that Cx43 overexpression enhanced the phosphorylation of CREB, consistent with increased cAMP/PKA-dependent signaling (Fig. 1C; quantitation in Supl Fig 1B). A similar effect of Cx43 on cAMP/PKA-dependent signaling was observed after treating osteoblastic cells with the phosphodiesterase inhibitor, IBMX, for 4 h (Fig. 1D), or with forskolin (adenylate cyclase agonist) for 4 h (Fig. 1E), as determined by a CRE-luc pathway specific luciferase reporter assay. The activation of CRE-Luc by forskolin was blocked by the PKA-inhibitor H89 (Fig. 1E).
In order to examine a biologically relevant stimulator of cAMP production, we treated MC3T3 and UMR106 cells with PGE2, which acts through EP2 and EP4 receptors to stimulate Gsα and cAMP accumulation. PGE2 is a bone anabolic factor that plays an important role in bone mechanotransduction, including modulation of the osteoanabolic Wnt/β-catenin signaling pathway (34-38). In both cell types, PGE2 activated CRE-Luc reporter activity, and overexpression of Cx43 synergistically enhanced this response (Fig. 2A). The PKA inhibitors, H89 (Fig. 2B) and PKI (Fig. 2C), blocked this effect. Western blots probed with anti-phospho-CREB antibodies confirmed that Cx43 overexpression further enhanced the phosphorylation of CREB following treatment with 2 μM PGE2 in MC3T3 cells (Fig. 2D; quantitation in Supl Fig 2A). In addition, phospho-ERK1/2 levels were similarly elevated in MC3T3 cells treated with both PGE2 and overexpressing Cx43 (Fig. 2D; quantitation in Supl Fig 2A). Both CREB and ERK1/2 phosphorylation were blocked by H89, indicating that they are activated downstream of PKA activity. Because PGE2 can also act through Gq/calcium dependent pathways, we treated UMR106 cells with forskolin to confirm involvement of the cAMP pathway. As was seen with PGE2, forskolin increased both CREB and ERK1/2 (Fig. 2E; quantitation in Supl Fig 2B) phosphorylation in a PKA-dependent manner, and this effect was enhanced by Cx43 overexpression. As observed for PGE2 treatment, both CREB and ERK1/2 phosphorylation were blocked by H89 in forskolin treated cells. Similarly, overexpression of the PKA inhibitor PKI reduced the combined effects of Cx43 and PGE2 on phospho-CREB and phospho-ERK1/2 levels (Fig. 2F; quantitation in Supl Fig 2C). These data indicate that Cx43 can synergistically enhance the osteoblast response to cAMP generating factors to increase cAMP/PKA-dependent signaling and ERK1/2 activation.
Figure 2. Connexin43 potentiates PGE2 signaling in a cAMP PKA-dependent manner.
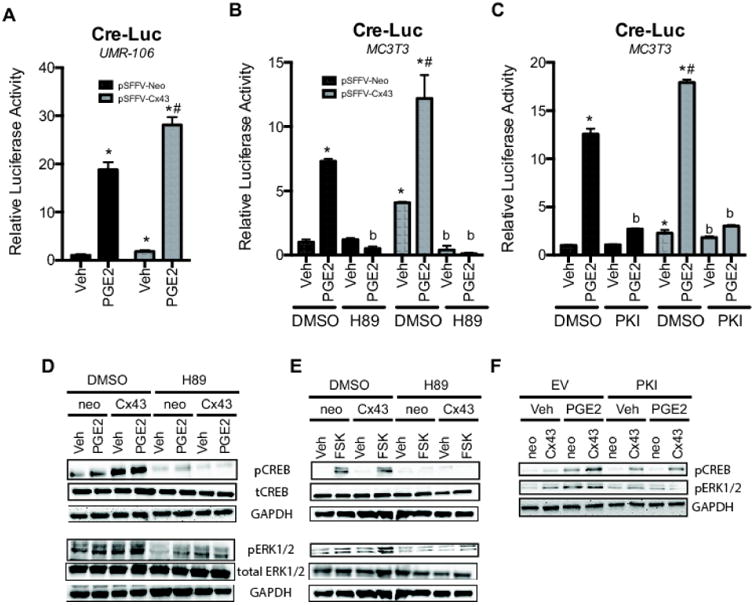
A-B, CRE-Luc activity was determined in UMR106 (A) and MC3T3 (B-C) cells co-transfected with pSFFV-Neo or pSFFV-Cx43, and then treated with PGE2 (2 μM, 4 h) or vehicle (Veh, 0.1% DMSO) 48 hours post-transfection. (B), For H89 (10 μM) treated samples, the cells were pre-treated for 30 minutes prior to PGE2 application. H89 remained in the media during the course of PGE2 exposure. (C), samples were co-transfected with PKI plasmid or empty vector control. Bar graphs display mean ± standard deviation. *, p-value < 0.05 versus empty vector transfected control. #, Indication of an interaction of the two variables (p-value < 0.05) by two-way ANOVA. b, p-value < 0.05 versus corresponding control (non-inhibitor treated) sample. D-F, Western blotting of whole cell extracts from transfected (D,F) MC3T3 and (E) UMR106 cells probed for phospho- and total CREB, phospho and total ERK1/2 and GAPDH. D, 48 hours post-transfection the cells were treated with PGE2 (2 μM, 4 h) in the presence or absence of H89 (10 μM). E, 48 hours post-transfection the cells were treated with forskolin (FSK, 10 μM, 4 h) in the presence or absence of H89 (10 μM). F, MC3T3 cells were co-transfected with the PKA inhibitor, PKI and the indicated plasmids. 48 hours post-transfection the cells were treated with PGE2 (2 μM, 4 h). Note the altered load order in F relative to D and E.
3.3. Connexin43 permits cAMP sharing among cells
Based on the synergistic activation of the CRE-Luc reporter and signal pathway activation by Cx43 and cAMP stimulating factors, we hypothesized that if Cx43 were involved in sharing cAMP between cells, then the total abundance of cAMP in the cell culture would remain static, despite Cx43 overexpression. In contrast, a previous study reported that loss of Cx43 function through antisense RNA reduced cAMP levels in parathyroid hormone treated ROS17/2.8 osteosarcoma cells (39). This result would suggest that the action of Cx43 lies upstream of cAMP production in response to parathyroid hormone, as the amount of cAMP present in the cell population was inhibited by Cx43 knockdown. Indeed, in this report forskolin-induced cAMP levels were unaffected by Cx43 antisense RNA, consistent with a defect in signaling upstream of adenylate cyclase activation and cAMP production. In addition, several studies have shown roles for Cx43 in various aspects of signaling that are gap junction/communication-independent (40,41). Thus, it remained possible that the mechanisms of Cx43 potentiation of cAMP-dependent signaling involved Cx43 directly or indirectly increasing the cAMP response in cells, rather than sharing cAMP between cells. Therefore, we examined the abundance of cAMP in UMR106 cells using a cAMP ELISA and bioluminescence cAMP reporter (nano-lantern (cAMP1.6)/pcDNA3). We chose UMR106 cells for this study as their low level of Cx43 expression minimized the effects of the endogenously expressed Cx43 containing gap junctions, and these cells have a higher transfection efficiency than MC3T3 cells. Though PGE2 treatment increased cAMP levels in the cultured cells, the combination of Cx43 overexpression and PGE2 treatment did not further increase cAMP levels (Fig. 3A-B). Rather, the total amount of cAMP is slightly reduced in Cx43 transfected cells treated with PGE2. The failure of Cx43 to increase total cAMP levels, despite synergistically activating phospho-CREB, phospho-ERK1/2 and a CRE-luc reporter, is consistent with a model of cAMP being shared between Cx43-expressing cells.
Figure 3. PGE2 does not synergistically stimulate cAMP levels in UMR106 cells, despite synergistically activating signaling that converges on a CRE-Luc reporter.
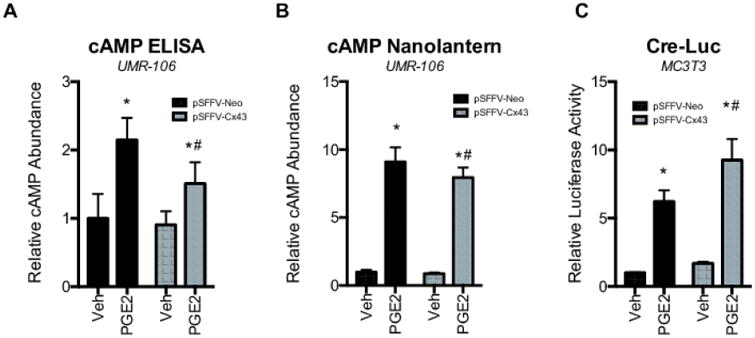
A-B, Relative cAMP abundance is shown as determined by a (A) cAMP ELISA or (B) as Renilla luciferase activity in cells transfected with the bioluminescent Nano-lantern (cAMP1.6)/pcDNA3 cAMP reporter in extracts from cells transfected with pSFFV-Neo or pSFFV-Cx43 and subsequently treated with PGE2 (2 μM, 4 h). Bar graphs display mean ± standard deviation. *, p-value < 0.05 versus the vehicle (Veh) treated control. #, indication of an interaction of the two variables (p-value < 0.05) by two-way ANOVA. C, MC3T3 cells were co-transfected with pSFFV-Neo or pSFFV-Cx43 and a CRE-Luc reporter. 48 hours post-transfection the cells were placed in calcium-free Ringer's solution and loaded with BAPTA-AM (10 μM) prior to treatment with with PGE2 (2 μM, 4 h) or vehicle (Veh, 0.1% ethanol). The bar graph displays mean relative CRE-luciferase activity ± standard deviation. *, p-value < 0.05 versus the vehicle (Veh) treated control. #, indication of an interaction of the two variables (p-value < 0.05) by two-way ANOVA.
Calcium dependent activation of cAMP-dependent signaling occurs in many cell types, where cAMP primes elevation of intracellular calcium influx, which then activates calcium sensitive adenylate cyclases to re-initiate cAMP dependent signaling (42,43). To address whether the effects of Cx43 on PGE2-dependent cAMP-signaling are an indirect effect of shared calcium rather than cAMP, we performed the Cre-Luc reporter under calcium free conditions. MC3T3 cells were cultured in calcium free Ringer's solution and loaded with the intracellular calcium chelator BAPTA-AM (dose) prior to treatment with PGE2 (Fig. 3C). Despite the removal of extracellular calcium and the sequestration of intracellular calcium, the synergistic amplification of CRE-luc activity by PGE2 and Cx43 was still observed, implying that the cells may directly share cAMP rather than a re-initiation of cAMP production in the gap junction coupled cells.
To confirm that Cx43 channels are required for this effect, we used a plasmid construct that encodes for a mutant Cx43 (G138R) that forms hypomorphic or dominant negative gap junctional communication and hyperactive hemichannels (44-46). Unlike the wild type Cx43, when this construct was overexpressed it was unable to enhance PGE2-stimulated CRE-luciferase activity (Fig. 4). Instead, the ability of the Cx43 G138R mutant to affect PGE2 responses was indistinguishable from the pSFFV-neo empty vector transfection controls. This suggests that functional Cx43 gap junctions, not just the structure or Cx43 hemichannel activity, are required for the Cx43-dependent communication of cAMP signaling in bone cells.
Figure 4. Wild type Cx43, but not a mutant Cx43 with reduced gap junctional communication and hyperactive hemichannel activity, potentiates PGE2 signaling.
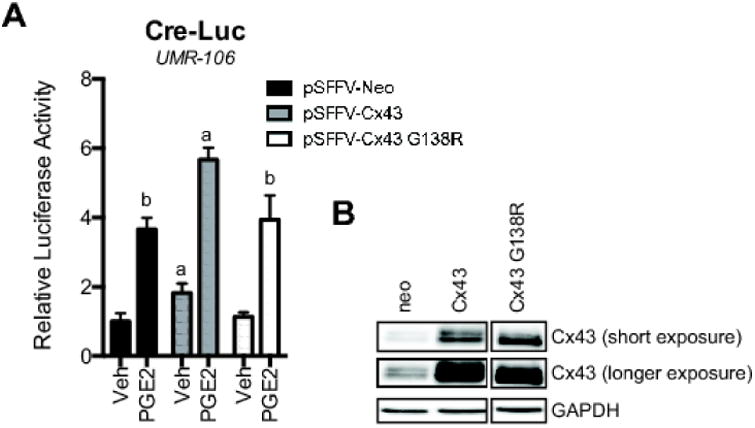
A, CRE-Luc activity was determined in UMR106 cells co-transfected with pSFFV-Neo, pSFFV-Cx43 or pSFFV-Cx43 G138R and then treated with PGE2 (2 μM, 4 h) or vehicle (Veh) 48 hours post-transfection. Bar graphs display mean ± standard deviation. a, p-value < 0.05 versus empty vector transfected control. b, p-value < 0.05 versus PGE2 treated wild type Cx43 transfected cells. B, Western blotting of whole cell extracts from transfected cells showing levels of Cx43 and GAPDH 48 hours post-transfection. The images are from the same blot and same exposure but unrelated wells were removed.
To further confirm the observation of the requirement for functional gap junctions and sharing of cAMP-dependent signals, we developed a modified version of the classic parachute assay for examining cell-to-cell communication (28,29). For this novel assay, we expressed the constitutively active Gsα construct in one set of UMR106 cells, which we refer to as the donor cell. Accordingly, these donor cells generate cAMP. In a second population of UMR106 cells, which we term the acceptor cells, we expressed a CRE-Luc reporter. Thus, these acceptor cells have the potential to respond to increases in cAMP levels. Next, we co-cultured the donor and acceptor cells. When the donor cells and acceptor cells both overexpressed Cx43 and the donor cell expressed the constitutively active Gsα construct, we observed a nearly two-fold increase in CRE-Luc activity (Fig. 5A). This suggested that cAMP generated in the donor cells was communicated to the acceptor cell and activated the CRE-luc reporter. To confirm, we co-cultured the UMR106 donor and acceptor cells but did not introduce exogenous Cx43 into the acceptor cell population (Fig. 5B). In this context, the donor cells were unable to stimulate CRE-luc activity in the acceptor cells. Further, when Cx43 overexpressing donor and acceptor UMR106 cells were separated by a transwell chamber, the donor cells were unable to stimulate CRE-luc activity in the acceptor cell population (Fig. 5C). In total, these data strongly indicate that cAMP generated in one cell population can influence CRE-luc activity in another cell population in a Cx43 and cell-to-cell contact dependent manner, strongly supporting the notion of intercellular communication of cAMP between cells to elicit a signaling response. Similar data were obtained using an immunofluorescence-based approach to this assay, in which donor cells (MC3T3) were transfected with empty vector or the constitutively active Gsα construct and labeled with DiI and then seeded onto a monolayer of cells transfected with pSFFV-neo or pSFFV-Cx43 and subsequently stained with phospho-CREB antibody (Fig. 5D). The number of phospho-CREB positive cells was quantitated in cells whose fluorescently intensity exceeded background threshold levels. There was increase in phospho-CREB positive cells in Gsα expressing cultures, which was synergistically increased by Cx43 overexpression. Importantly, an increase in phospho-CREB positive cells adjacent to the DiI labeled donor cells was observed consistent with a model of shared signals between cells resulting in an amplified the number of cells responding to a cue. Indeed, this finding is consistent with our previous findings of a Cx43-dependent increase in the percentage of phospho-ERK and phospho-PKCδ positive osteoblasts when stimulated with FGF2 (18,30).
Figure 5. Cx43 overexpression and cell-cell contact is necessary for constitutively active Gsα expressing cells to activate a CRE-luc reporter in another cell population.
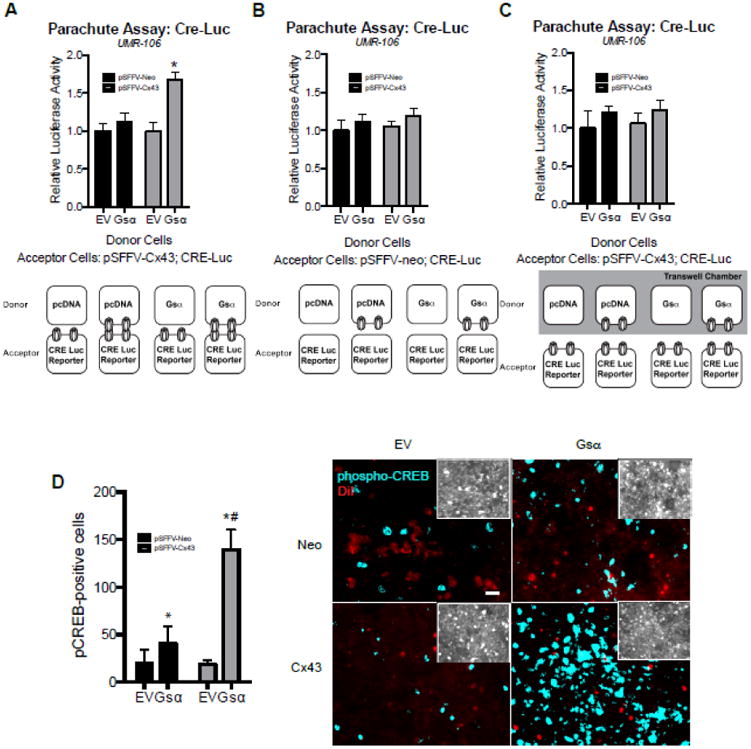
A-C, CRE-Luc activity was determined in co-cultured UMR106 donor and acceptor cells after 24 h of co-culture. A, Donor cells were transfected with pcDNA (EV) or pcDNA-Gsα (Gsα). Acceptor cells were co-transfected with a CRE-luc reporter and pSFFV-Cx43. 48 h post-transfection, the cells were co-cultured together for 24 hours as indicated in the cartoon below. B, Donor cells were transfected with pcDNA (EV) or pcDNA-Gsα (Gsα). Acceptor cells were co-transfected with a CRE-luc reporter and pSFFV-neo. 48 h post-transfection, the cells were co-cultured together for 24 hours as indicated in the cartoon below. C, Donor cells were transfected with pcDNA (EV) or pcDNA-Gsα (Gsα). Acceptor cells were co-transfected with a CRE-luc reporter and pSFFV-Cx43. 48 h post-transfection, the acceptor cells were seeded in the bottom of a tissue culture plate, while the donor cells were seeded into the upper chamber of a transwell chamber. The cells were co-cultured together for 24 hours sharing the same media, but without direct cell-to-cell contact, as indicated in the cartoon below. Bar graphs display mean ± standard deviation. *, p-value < 0.05 versus empty vector transfected control. D, Left, quantitation of phospho-CREB positive cells (fluorescence intensity above a threshold of 780) per field of view following co-culture of DiI labeled donor cells expressing pcDNA (EV) or pcDNA-Gsα (Gsα) and pSFFV-Neo or pSFFV-Cx43 seeded on to a confluent monolayer of unlabeled MC3T3 cells. Bar graphs display mean number of cells per field of view n=9 fields of view from three slides per condition. *, p-value < 0.05 versus the vehicle (Veh) treated control. #, indication of an interaction of the two variables (p-value < 0.05) by two-way ANOVA. Right, representative images of the binarized phospho-CREB immunofluorescence (blue) and DiI labeled donor cells (red). Scale bar equals 20 μm. Insets, show a brightness-enhanced phospho-CREB image showing similar cellular densities between fields of view among each treatment groups.
3.4. Connexin43-dependent communication of cAMP differentially regulates the expression of key regulators of bone homeostasis
To determine the consequence of Cx43-communicated cAMP, we examined the expression of two key regulators of bone homeostasis, RANKL (gene name, Tnfsf11) and sclerostin (gene name, Sost), by quantitative real time RT-PCR (Fig. 6). We also confirmed Cx43 (gene name, Gja1) overexpression by quantitative real time RT-PCR. These data show that Cx43 amplified the effects of PGE2 and Gsα expression on these genes. Induction of cAMP signaling with either PGE2 or Gsα, synergistically combined with the effect of Cx43 overexpression to amplify RANKL/Tnfsf11 expression, a key factor in the activation of osteoclasts and bone resorption. Cx43 and either PGE2 or Gsα synergistically reduced the expression Sclerostin/Sost, a repressor of the Wnt/β-catenin cascade in bone cells and inhibitor of osteoblastogenesis. Alteration of the balance of these two vital factors in bone remodeling underscores the potential importance of cAMP as a second messenger communicated by bone cells.
Figure 6. Cx43 overexpression synergistically enhances the Gsα and PGE2-dependent effects on RANKL/Tnfsf11 and Sclerostin/Sost mRNA expression.
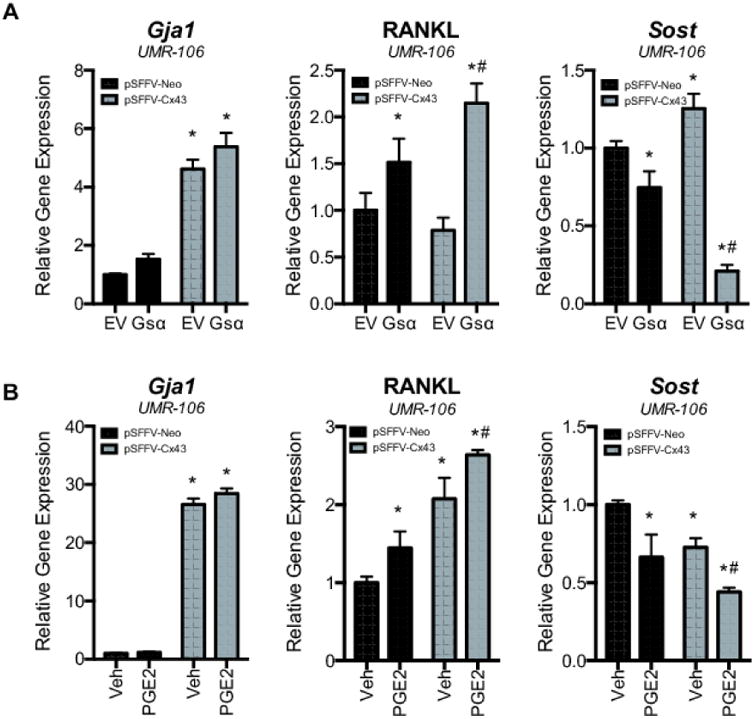
A-C, Relative gene expression is shown for Cx43/Gja1, RANKL/Tnfsf11 and Sclerostin/Sost as determined by quantitative real time RT-PCR of RNA from UMR106 cells transfected with pSFFV-neo or pSFFV-Cx43 and, co-transfected with (A) pcDNA or pcDNA-Gsα or (B), treated with vehicle (Veh) or PGE2 (2 μM, 4 h). Bar graphs display mean ± standard deviation. *, p-value < 0.05 versus empty vector transfected control. #, indication of an interaction of the two variables (p-value < 0.05) by two-way ANOVA.
4. Discussion
In this study, we show that Cx43 synergistically enhanced the ability of osteoblasts to respond to cAMP-dependent signals by propagating signals through interconnected cells (Fig. 7). Specifically, we activated cAMP-dependent signaling using a physiologically relevant factor, PGE2, as well as with more direct cAMP-activation by overexpression of a constitutively active Gsα, treatment with the adenylate cyclase agonist, forskolin or treatment with the phosphodiesterase inhibitor, IBMX. We also used a novel, modified version of the parachute assay to demonstrate that cAMP-production in one cell can synergistically activate cAMP- signal pathway specific luciferase reporters in an adjacent cell. Indeed, in this modified parachute assay, the fact that separating the donor and acceptor cells from each other via transwell chambers, suggest that it is direct cell-to-cell communication, not Cx43 hemichannel activity or other communication-independent functions, that is critical to the transmission of cAMP-dependent signals between the cells. Our immunofluorescence data further underscores that the overexpression of Cx43 permits cells that are not expressing the Gsα to activate CREB, increasing the total percentage of cells responding to the signal. These findings are similar to our previous findings showing a synergistic increase in FGF2-dependent phospho-ERK and phospho-PKCδ activation to stimulate Runx2 transcriptional activity in bone cells, a response that also included an increased percentage of responding cells in the presence of Cx43 expression, the need for physical interaction and functional channel activity (18,30). Recently published work used cellular and mathematical models to show that cell-to-cell communication through gap junctions can enhance the sensitivity of cell populations to a weak stimuli, such as a shallow epidermal growth factor concentration gradient, by collectively coordinating and averaging noisy signals sensed by single cells (47).
Figure 7. Model of intercellular communication of cAMP among bone cells to modulate signaling and gene expression.

Activation of PGE2 receptors triggers the generation of cAMP in the responding osteoblasts. Subsequently, the Cx43-dependent communication of cAMP among interconnected permits these cells to activate downstream PKA and ERK1/2-dependent signaling to modulate the expression of the sclerostin and RANKL encoding genes.
In addition, this is the first study to show the potentiation of PGE2 signaling by Cx43 in osteoblastic cells and is the first to show that Cx43 and cAMP interact to synergistically regulate the gene expression of two key regulators of bone turnover, namely RANKL/Tnfsf11 and sclerostin/Sost. Others have previously shown that cAMP can regulate RANKL/Tnfsf11 and sclerostin/Sost expression (36,38,48), but we now provide evidence that Cx43 propagates cAMP to regulate these factors. This is a key concept as these data demonstrate that not only can Cx43 gap junctions communicate cAMP between cells, but more importantly, that the communicated cAMP is sufficient to elicit a biological response in gap junction coupled cells. Explicitly connecting these dots in bone is particularly interesting as Cx43, PGE2, sclerostin and RANKL are all intimately involved in how bone responds to mechanical cues, as well as for maintenance of skeletal homeostasis (5,49-52).
These findings of cAMP sharing between cells are consistent with similar observations made in other cell types, using less biologically relevant models. For example, in HeLa and N2A cells, cAMP can be passed through Cx43 containing gap junction channels by introducing cAMP into a patch pipet and showing connexin-dependent transfer from the patched cells into an adjacent cell using a reporter gene readout (12). In this model, Cx43 was up to 10 times more permeable to cAMP than Cx40 or Cx26. Similarly, Cx43-dependent transfer of a photo-activatable cAMP was shown using an EPAC-based FRET sensor in Rat1 fibroblasts and Cx43 siRNA-mediated knockdown (13). Interestingly, our data is in contrast to a report in retinal pigment epithelial cells, which shows Cx43 regulates cAMP-dependent signaling in a structural context involving the Cx43 C-terminus, and not by direct cell-to-cell communication (53). While we and others have shown an important role in the Cx43 C-terminus in signaling cascade activation in bone cells (54-57), the data from both the modified parachute assay and the Cx43 G138R mutant suggest that gap junctional communication is required for the cAMP effects we observed here. This Cx43 mutant forms hypomorphic or dominant negative gap junctions, yet has hyperactive hemichannel function (44-46). Thus, the inability of this mutant Cx43 to enhance PGE2-stimulated CRE-luciferase activity supports the notion that functional Cx43 gap junctions, not just the structure or Cx43 hemichannels, are required for the Cx43-dependent communication of cAMP signaling in bone cells. In addition, our finding are distinct from those reported for the intersection of Cx43 and parathyroid hormones in ROS17/2.8 osteosarcoma cells (39). In that study, the generation of cAMP was blunted by Cx43 knockdown by antisense RNA suggesting an action upstream of cAMP production, while in our study we show that Cx43 overexpression does not enhance cAMP levels, but rather permits its communication among cells, and Cx43 gene deletion reduces phospho-CREB levels.
We cannot fully rule out the possibility that the increase in cAMP-dependent signaling does not involve the direct communication of cAMP from cell-to-cell, but rather involves the re-amplification of cAMP-dependent signaling in the second cell. Re-amplification of cAMP-dependent signaling occurs in many cell types, where cAMP primes elevation of intracellular calcium influx, which then activates adenylate cyclase to re-initiate cAMP dependent signaling (42,43). However, the cAMP ELISA and nanolantern data do not show a synergistic increase in cAMP levels by Cx43, rather a statistically significant decrease in cAMP levels is observed in PGE2 treated cells expressing Cx43. This would seem to suggest that cAMP levels are perhaps diffusing between adjacent cells and the total amount of cAMP on a per cell basis is reduced. This possibility of direct cAMP sharing is underscored by the data in which removal of calcium did not prevent the luciferase driven synergy between Cx43 and PGE2. However, future experiments are planned for examining these possibilities in more depth with more sensitivity probes and greater spatial and temporal resolution. These future experiments will also help us determine the “range” of cAMP transmission among these cells.
Our data may help to explain the skeletal phenotype of Cx43 knockout mice, at least in part. In vivo studies have shown osteoblast-osteocyte expressed Cx43 plays an important role in bone homeostasis and mechanotransduction (3-6). PGE2 is a bone anabolic factor involved in mechanotransduction (34-38). Disruption of the gap junction coupled cellular network, which permits the coordinated action of bone cells in response to mechanotransduction signals like PGE2, may explain the complex partitioning of the cortical bone formation and resorption at the periosteal and endosteal bone surfaces, respectively, of the Cx43 osteoblast-lineage specific conditional knockout models (3-6). These conditional knockout models have reduced sclerostin expression, perhaps contributing to increased periosteal bone apposition, and an increased RANKL to osteoprotegerin ratio, leading to increased endosteal osteoclastic bone resorption (58-60). The data presented here support the involvement of Cx43 gap junction communicated cAMP in the regulation of these both sclerostin and RANKL. Additionally, osteoblast-lineage Cx43 deficiency has been shown to decrease the anabolic response of bone to intermittent parathyroid hormone (PTH) treatment in vivo (61). Like PGE2, PTH acts through a G-protein coupled receptor to stimulate cAMP. Accordingly, our data suggest that perhaps it is the inability of Cx43 deficient cells to effectively communicate cAMP second messengers that underpins key aspects of this phenotype. In addition to the Cx43-dependent communication of cAMP, the C-terminus of Cx43 can bind to β-arrestin, sequestering it away from its role in attenuating Gsα activation by the PTH receptor (56). However, a Cx43 C-terminus truncation mutant could not fully recapitulate the effect of Cx43 deletion on the anabolic action of PTH on bone (57), suggesting that β-arrestin sequestration is not the only mechanism by which Cx43 can facilitate cAMP-dependent signaling.
Importantly, the present data demonstrate only a single second messenger communicated by gap junctions. This is not to suggest that cAMP is the only second messenger communicated by gap junctions to affect bone function. We think many second messengers are exchanged through gap junctions in bone. The specific cell context will influence the nature of these messages, and, in some cases, these exchanged signals may be conflicting. Such an explanation is obligated by the diverse set of cellular responses that can occur in Cx43-deleted bone cells during loading, unloading, and hormone stimulation (5,49). Previously, we have demonstrated that FGF2 stimulation of osteoblasts can lead to the Cx43-dependent exchange of inositol phosphates to regulate bone cell function through Runx2 (26). While our experiments with the Cx43 G138R mutant do not support a role for Cx43 hemichannels in the amplification of cAMP-dependent signals in this system, this does not preclude a role for hemichannels to function through other pathways to influence bone. In fact, Cx43 hemichannels have been shown to be involved in the release of PGE2 from mechanically stimulated osteocytes (55,62,63). Further, PGE2 has been shown to increase Cx43 expression and regulate gap junctional communication among osteoblasts and osteocytes (15,17,64). The subsequent gap junction dependent communication of the signals downstream of PGE2 release, including cAMP, may be an important regulator network for controlling the autocrine and paracrine action of PGE2 in bone formation.
5. Conclusion
In total, our data demonstrate the Cx43-dependent cell-to-cell exchange of cAMP between osteoblasts downstream of PGE2, and show that this communicated second messenger is sufficient to regulate the expression of key modulators of bone turnover. These data implicate cAMP as a biologically relevant second messenger communicated by bone cells through Cx43 gap junctions. By understanding the nature of the signals communicated among bone cell networks, we can begin to unravel the regulatory circuits leading to bone anabolic and catabolic responses.
Supplementary Material
Highlights.
Cx43 potentiates cAMP-dependent signaling in osteoblasts in a physiologic context
Cx43 can communicate cAMP between osteoblasts in a Cx43- and cell contact-dependent manner
Communication of cAMP by Cx43 is sufficient to affect signaling and osteoblast gene expression
Acknowledgments
This work was supported by a grant from the National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01-AR063631) to JPS and (F31-AR064673) to AMB. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of Interest: The authors declare that they have no conflicts of interest with the contents of this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vinken M. Introduction: connexins, pannexins and their channels as gatekeepers of organ physiology. Cell Mol Life Sci. 2015;72:2775–2778. doi: 10.1007/s00018-015-1958-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nielsen MS, Axelsen LN, Sorgen PL, Verma V, Delmar M, Holstein-Rathlou NH. Gap junctions. Compr Physiol. 2012;2:1981–2035. doi: 10.1002/cphy.c110051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buo AM, Stains JP. Gap junctional regulation of signal transduction in bone cells. FEBS Lett. 2014;588:1315–1321. doi: 10.1016/j.febslet.2014.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stains JP, Watkins MP, Grimston SK, Hebert C, Civitelli R. Molecular mechanisms of osteoblast/osteocyte regulation by connexin43. Calcif Tissue Int. 2014;94:55–67. doi: 10.1007/s00223-013-9742-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lloyd SA, Loiselle AE, Zhang Y, Donahue HJ. Shifting paradigms on the role of connexin43 in the skeletal response to mechanical load. J Bone Miner Res. 2014;29:275–286. doi: 10.1002/jbmr.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plotkin LI, Bellido T. Beyond gap junctions: Connexin43 and bone cell signaling. Bone. 2013;52:157–166. doi: 10.1016/j.bone.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones SJ, Gray C, Sakamaki H, Arora M, Boyde A, Gourdie R, Green C. The incidence and size of gap junctions between the bone cells in rat calvaria. Anatomy and embryology. 1993;187:343–352. doi: 10.1007/BF00185892. [DOI] [PubMed] [Google Scholar]
- 8.Shapiro F. Variable conformation of GAP junctions linking bone cells: a transmission electron microscopic study of linear, stacked linear, curvilinear, oval, and annular junctions. Calcif Tissue Int. 1997;61:285–293. doi: 10.1007/s002239900337. [DOI] [PubMed] [Google Scholar]
- 9.Doty SB. Morphological evidence of gap junctions between bone cells. Calcif Tissue Int. 1981;33:509–512. doi: 10.1007/BF02409482. [DOI] [PubMed] [Google Scholar]
- 10.Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26:229–238. doi: 10.1002/jbmr.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plotkin LI. Connexin 43 hemichannels and intracellular signaling in bone cells. Front Physiol. 2014;5:131. doi: 10.3389/fphys.2014.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanaporis G, Mese G, Valiuniene L, White TW, Brink PR, Valiunas V. Gap junction channels exhibit connexin-specific permeability to cyclic nucleotides. The Journal of general physiology. 2008;131:293–305. doi: 10.1085/jgp.200709934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ponsioen B, van Zeijl L, Moolenaar WH, Jalink K. Direct measurement of cyclic AMP diffusion and signaling through connexin43 gap junctional channels. Exp Cell Res. 2007;313:415–423. doi: 10.1016/j.yexcr.2006.10.029. [DOI] [PubMed] [Google Scholar]
- 14.Donahue HJ, McLeod KJ, Rubin CT, Andersen J, Grine EA, Hertzberg EL, Brink PR. Cell-to-cell communication in osteoblastic networks: cell line-dependent hormonal regulation of gap junction function. J Bone Miner Res. 1995;10:881–889. doi: 10.1002/jbmr.5650100609. [DOI] [PubMed] [Google Scholar]
- 15.Civitelli R, Ziambaras K, Warlow PM, Lecanda F, Nelson T, Harley J, Atal N, Beyer EC, Steinberg TH. Regulation of connexin43 expression and function by prostaglandin E2 (PGE2) and parathyroid hormone (PTH) in osteoblastic cells. J Cell Biochem. 1998;68:8–21. doi: 10.1002/(sici)1097-4644(19980101)68:1<8::aid-jcb2>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 16.Xia X, Batra N, Shi Q, Bonewald LF, Sprague E, Jiang JX. Prostaglandin promotion of osteocyte gap junction function through transcriptional regulation of connexin 43 by glycogen synthase kinase 3/beta-catenin signaling. Mol Cell Biol. 2010;30:206–219. doi: 10.1128/MCB.01844-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cherian PP, Cheng B, Gu S, Sprague E, Bonewald LF, Jiang JX. Effects of mechanical strain on the function of Gap junctions in osteocytes are mediated through the prostaglandin EP2 receptor. J Biol Chem. 2003;278:43146–43156. doi: 10.1074/jbc.M302993200. [DOI] [PubMed] [Google Scholar]
- 18.Niger C, Buo AM, Hebert C, Duggan BT, Williams MS, Stains JP. ERK acts in parallel to PKCdelta to mediate the connexin43-dependent potentiation of Runx2 activity by FGF2 in MC3T3 osteoblasts. Am J Physiol Cell Physiol. 2012;302:C1035–1044. doi: 10.1152/ajpcell.00262.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Castro CH, Shin CS, Stains JP, Cheng SL, Sheikh S, Mbalaviele G, Szejnfeld VL, Civitelli R. Targeted expression of a dominant-negative N-cadherin in vivo delays peak bone mass and increases adipogenesis. J Cell Sci. 2004;117:2853–2864. doi: 10.1242/jcs.01133. [DOI] [PubMed] [Google Scholar]
- 20.Bakker AD, Klein-Nulend J. Osteoblast isolation from murine calvaria and long bones. Methods Mol Biol. 2012;816:19–29. doi: 10.1007/978-1-61779-415-5_2. [DOI] [PubMed] [Google Scholar]
- 21.Niger C, Howell FD, Stains JP. Interleukin-1beta increases gap junctional communication among synovial fibroblasts via the extracellular-signal-regulated kinase pathway. Biol Cell. 2010;102:37–49. doi: 10.1042/BC20090056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuhlbrigge RC, Fine SM, Unanue ER, Chaplin DD. Expression of membrane interleukin 1 by fibroblasts transfected with murine pro-interleukin 1 alpha cDNA. Proc Natl Acad Sci U S A. 1988;85:5649–5653. doi: 10.1073/pnas.85.15.5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta A, Niger C, Buo AM, Eidelman ER, Chen RJ, Stains JP. Connexin43 enhances the expression of osteoarthritis-associated genes in synovial fibroblasts in culture. BMC musculoskeletal disorders. 2014;15:425. doi: 10.1186/1471-2474-15-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morgan SJ, Deshpande DA, Tiegs BC, Misior AM, Yan H, Hershfeld AV, Rich TC, Panettieri RA, An SS, Penn RB. beta-Agonist-mediated relaxation of airway smooth muscle is protein kinase A-dependent. J Biol Chem. 2014;289:23065–23074. doi: 10.1074/jbc.M114.557652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saito K, Chang YF, Horikawa K, Hatsugai N, Higuchi Y, Hashida M, Yoshida Y, Matsuda T, Arai Y, Nagai T. Luminescent proteins for high-speed single-cell and whole-body imaging. Nature communications. 2012;3:1262. doi: 10.1038/ncomms2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niger C, Luciotti MA, Buo AM, Hebert C, Ma V, Stains JP. The regulation of runt-related transcription factor 2 by fibroblast growth factor-2 and connexin43 requires the inositol polyphosphate/protein kinase Cdelta cascade. J Bone Miner Res. 2013;28:1468–1477. doi: 10.1002/jbmr.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dyer BW, Ferrer FA, Klinedinst DK, Rodriguez R. A noncommercial dual luciferase enzyme assay system for reporter gene analysis. Analytical biochemistry. 2000;282:158–161. doi: 10.1006/abio.2000.4605. [DOI] [PubMed] [Google Scholar]
- 28.Ziambaras K, Lecanda F, Steinberg TH, Civitelli R. Cyclic stretch enhances gap junctional communication between osteoblastic cells. J Bone Miner Res. 1998;13:218–228. doi: 10.1359/jbmr.1998.13.2.218. [DOI] [PubMed] [Google Scholar]
- 29.Yellowley CE, Li Z, Zhou Z, Jacobs CR, Donahue HJ. Functional gap junctions between osteocytic and osteoblastic cells. J Bone Miner Res. 2000;15:209–217. doi: 10.1359/jbmr.2000.15.2.209. [DOI] [PubMed] [Google Scholar]
- 30.Lima F, Niger C, Hebert C, Stains JP. Connexin43 potentiates osteoblast responsiveness to fibroblast growth factor 2 via a protein kinase C-delta/Runx2-dependent mechanism. Mol Biol Cell. 2009;20:2697–2708. doi: 10.1091/mbc.E08-10-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niger C, Hebert C, Stains JP. Interaction of connexin43 and protein kinase C-delta during FGF2 signaling. BMC Biochem. 2010;11:14. doi: 10.1186/1471-2091-11-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu S, Niger C, Koh EY, Stains JP. Connexin43 Mediated Delivery of ADAMTS5 Targeting siRNAs from Mesenchymal Stem Cells to Synovial Fibroblasts. PLoS One. 2015;10:e0129999. doi: 10.1371/journal.pone.0129999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steinberg TH, Civitelli R, Geist ST, Robertson AJ, Hick E, Veenstra RD, Wang HZ, Warlow PM, Westphale EM, Laing JG, et al. Connexin43 and connexin45 form gap junctions with different molecular permeabilities in osteoblastic cells. EMBO J. 1994;13:744–750. doi: 10.1002/j.1460-2075.1994.tb06316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Forwood MR. Inducible cyclo-oxygenase (COX-2) mediates the induction of bone formation by mechanical loading in vivo. J Bone Miner Res. 1996;11:1688–1693. doi: 10.1002/jbmr.5650111112. [DOI] [PubMed] [Google Scholar]
- 35.Chow JW, Fox SW, Lean JM, Chambers TJ. Role of nitric oxide and prostaglandins in mechanically induced bone formation. J Bone Miner Res. 1998;13:1039–1044. doi: 10.1359/jbmr.1998.13.6.1039. [DOI] [PubMed] [Google Scholar]
- 36.Genetos DC, Yellowley CE, Loots GG. Prostaglandin E2 signals through PTGER2 to regulate sclerostin expression. PLoS One. 2011;6:e17772. doi: 10.1371/journal.pone.0017772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310:1504–1510. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- 38.Lara-Castillo N, Kim-Weroha NA, Kamel MA, Javaheri B, Ellies DL, Krumlauf RE, Thiagarajan G, Johnson ML. In vivo mechanical loading rapidly activates beta-catenin signaling in osteocytes through a prostaglandin mediated mechanism. Bone. 2015;76:58–66. doi: 10.1016/j.bone.2015.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vander Molen MA, Rubin CT, McLeod KJ, McCauley LK, Donahue HJ. Gap junctional intercellular communication contributes to hormonal responsiveness in osteoblastic networks. J Biol Chem. 1996;271:12165–12171. doi: 10.1074/jbc.271.21.12165. [DOI] [PubMed] [Google Scholar]
- 40.Kameritsch P, Pogoda K, Pohl U. Channel-independent influence of connexin 43 on cell migration. Biochim Biophys Acta. 2012;1818:1993–2001. doi: 10.1016/j.bbamem.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 41.Vinken M, Decrock E, Leybaert L, Bultynck G, Himpens B, Vanhaecke T, Rogiers V. Non-channel functions of connexins in cell growth and cell death. Biochim Biophys Acta. 2012;1818:2002–2008. doi: 10.1016/j.bbamem.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 42.Hofer AM. Interactions between calcium and cAMP signaling. Curr Med Chem. 2012;19:5768–5773. doi: 10.2174/092986712804143286. [DOI] [PubMed] [Google Scholar]
- 43.Cooper DM. Store-operated Ca(2)(+)-entry and adenylyl cyclase. Cell Calcium. 2015;58:368–375. doi: 10.1016/j.ceca.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 44.Gong XQ, Shao Q, Langlois S, Bai D, Laird DW. Differential potency of dominant negative connexin43 mutants in oculodentodigital dysplasia. J Biol Chem. 2007;282:19190–19202. doi: 10.1074/jbc.M609653200. [DOI] [PubMed] [Google Scholar]
- 45.Roscoe W, Veitch GI, Gong XQ, Pellegrino E, Bai D, McLachlan E, Shao Q, Kidder GM, Laird DW. Oculodentodigital dysplasia-causing connexin43 mutants are nonfunctional and exhibit dominant effects on wild-type connexin43. J Biol Chem. 2005;280:11458–11466. doi: 10.1074/jbc.M409564200. [DOI] [PubMed] [Google Scholar]
- 46.Dobrowolski R, Sommershof A, Willecke K. Some oculodentodigital dysplasia-associated Cx43 mutations cause increased hemichannel activity in addition to deficient gap junction channels. J Membr Biol. 2007;219:9–17. doi: 10.1007/s00232-007-9055-7. [DOI] [PubMed] [Google Scholar]
- 47.Ellison D, Mugler A, Brennan MD, Lee SH, Huebner RJ, Shamir ER, Woo LA, Kim J, Amar P, Nemenman I, Ewald AJ, Levchenko A. Cell-cell communication enhances the capacity of cell ensembles to sense shallow gradients during morphogenesis. Proc Natl Acad Sci U S A. 2016;113:E679–688. doi: 10.1073/pnas.1516503113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park HJ, Baek K, Baek JH, Kim HR. The cooperation of CREB and NFAT is required for PTHrP-induced RANKL expression in mouse osteoblastic cells. J Cell Physiol. 2015;230:667–679. doi: 10.1002/jcp.24790. [DOI] [PubMed] [Google Scholar]
- 49.Grimston SK, Watkins MP, Stains JP, Civitelli R. Connexin43 modulates post-natal cortical bone modeling and mechano-responsiveness. BoneKEy reports. 2013;2:446. doi: 10.1038/bonekey.2013.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thompson WR, Rubin CT, Rubin J. Mechanical regulation of signaling pathways in bone. Gene. 2012;503:179–193. doi: 10.1016/j.gene.2012.04.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burgers TA, Williams BO. Regulation of Wnt/beta-catenin signaling within and from osteocytes. Bone. 2013;54:244–249. doi: 10.1016/j.bone.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O'Brien CA, Nakashima T, Takayanagi H. Osteocyte control of osteoclastogenesis. Bone. 2013;54:258–263. doi: 10.1016/j.bone.2012.08.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kojima A, Nakahama K, Ohno-Matsui K, Shimada N, Mori K, Iseki S, Sato T, Mochizuki M, Morita I. Connexin 43 contributes to differentiation of retinal pigment epithelial cells via cyclic AMP signaling. Biochem Biophys Res Commun. 2008;366:532–538. doi: 10.1016/j.bbrc.2007.11.159. [DOI] [PubMed] [Google Scholar]
- 54.Hebert C, Stains JP. An intact connexin43 is required to enhance signaling and gene expression in osteoblast-like cells. J Cell Biochem. 2013;114:2542–2550. doi: 10.1002/jcb.24603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Batra N, Burra S, Siller-Jackson AJ, Gu S, Xia X, Weber GF, DeSimone D, Bonewald LF, Lafer EM, Sprague E, Schwartz MA, Jiang JX. Mechanical stress-activated integrin alpha5beta1 induces opening of connexin 43 hemichannels. Proc Natl Acad Sci U S A. 2012;109:3359–3364. doi: 10.1073/pnas.1115967109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bivi N, Lezcano V, Romanello M, Bellido T, Plotkin LI. Connexin43 interacts with betaarrestin: a pre-requisite for osteoblast survival induced by parathyroid hormone. J Cell Biochem. 2011;112:2920–2930. doi: 10.1002/jcb.23208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pacheco-Costa R, Davis HM, Sorenson C, Hon MC, Hassan I, Reginato RD, Allen MR, Bellido T, Plotkin LI. Defective cancellous bone structure and abnormal response to PTH in cortical bone of mice lacking Cx43 cytoplasmic C-terminus domain. Bone. 2015;81:632–643. doi: 10.1016/j.bone.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Watkins M, Grimston SK, Norris JY, Guillotin B, Shaw A, Beniash E, Civitelli R. Osteoblast connexin43 modulates skeletal architecture by regulating both arms of bone remodeling. Mol Biol Cell. 2011;22:1240–1251. doi: 10.1091/mbc.E10-07-0571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Y, Paul EM, Sathyendra V, Davison A, Sharkey N, Bronson S, Srinivasan S, Gross TS, Donahue HJ. Enhanced osteoclastic resorption and responsiveness to mechanical load in gap junction deficient bone. PLoS One. 2011;6:e23516. doi: 10.1371/journal.pone.0023516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bivi N, Condon KW, Allen MR, Farlow N, Passeri G, Brun LR, Rhee Y, Bellido T, Plotkin LI. Cell autonomous requirement of connexin 43 for osteocyte survival: consequences for endocortical resorption and periosteal bone formation. J Bone Miner Res. 2012;27:374–389. doi: 10.1002/jbmr.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chung DJ, Castro CH, Watkins M, Stains JP, Chung MY, Szejnfeld VL, Willecke K, Theis M, Civitelli R. Low peak bone mass and attenuated anabolic response to parathyroid hormone in mice with an osteoblast-specific deletion of connexin43. J Cell Sci. 2006;119:4187–4198. doi: 10.1242/jcs.03162. [DOI] [PubMed] [Google Scholar]
- 62.Siller-Jackson AJ, Burra S, Gu S, Xia X, Bonewald LF, Sprague E, Jiang JX. Adaptation of connexin 43-hemichannel prostaglandin release to mechanical loading. J Biol Chem. 2008;283:26374–26382. doi: 10.1074/jbc.M803136200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cherian PP, Siller-Jackson AJ, Gu S, Wang X, Bonewald LF, Sprague E, Jiang JX. Mechanical strain opens connexin 43 hemichannels in osteocytes: a novel mechanism for the release of prostaglandin. Mol Biol Cell. 2005;16:3100–3106. doi: 10.1091/mbc.E04-10-0912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cheng B, Kato Y, Zhao S, Luo J, Sprague E, Bonewald LF, Jiang JX. PGE(2) is essential for gap junction-mediated intercellular communication between osteocyte-like MLO-Y4 cells in response to mechanical strain. Endocrinology. 2001;142:3464–3473. doi: 10.1210/endo.142.8.8338. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.