

Abstract
Higher affinity for TnI explains how troponin C (TnC) carrying a causative hypertrophic cardiomyopathy mutation, TnCA8V, sensitizes muscle cells to Ca2+. Muscle fibers reconstituted with TnCA8V require ~2.3-fold less [Ca2+] to achieve 50% maximum-tension compared to fibers reconstituted with wild-type TnC (TnCWT). Binding measurements rule out a significant change in N-terminus Ca2+-affinity of isolated TnCA8V, and TnCA8V binds the switch-peptide of troponin-I (TnIsp) ~1.6-fold more strongly than TnCWT; thus we model the TnC-TnIsp interaction as competing with the TnI-actin interaction. Tension data are well-fit by a model constrained to conditions in which the affinity of TnCA8V for TnIsp is 1.5-1.7-fold higher than that of TnCWT at all [Ca2+]. Mean ATPase rates of reconstituted cardiac myofibrils is greater for TnCA8V than TnCWT at all [Ca2+], with statistically significant differences in the means at higher [Ca2+]. To probe TnC-TnI interaction in low Ca2+, displacement of bis-ANS from TnI was monitored as a function of TnC. Whereas Ca2+-TnCWT displaces significantly more bis-ANS than Mg2+-TnCWT, Ca2+-TnCA8V displaces probe equivalently to Mg2+-TnCA8V and Ca2+-TnCWT, consistent with stronger Ca2+-independent TnCA8V-TnIsp. A Matlab program for computing theoretical activation is reported. Our work suggests that contractility is constantly above normal in hearts made hypertrophic by TnCA8V.
Keywords: troponin I binding, modeling, cardiac troponin C, hypertrophic cardiomyopathy, myofibrillar ATPase, fluorescence
INTRODUCTION
Hypertrophic cardiomyopathy (HCM) is a disease of the myocardium in which the left ventricle wall becomes abnormally thick and stiff. Mutations in sarcomeric proteins have been reported as a primary source of dysfunction leading to the development of HCM in humans (1–5). Although sarcomeric protein mutations are considered “poison peptides” due to their negative impact on muscle regulation, little is known about the molecular mechanisms underlying changes in protein function. Within the sarcomere, TNNC1 (gene encoding cardiac troponin C, TnC) is a target for mutations associated with HCM (6–10). Of the missense mutations of TNNC1, A8V, causes the largest increase in Ca2+sensitivity of force development (7).
Cardiac troponin C function integrates with the exchange of Ca2+ within the cytosol. Aside from buffering Ca2+ in myocytes (11), TnC controls the kinetics of cardiac muscle activation and relaxation in response to association and dissociation of Ca2+ (12). As a component of myofibrils, TnC participates with the transport mechanisms of the sarcoplasmic reticulum and sarcolemma to regulate and respond to transient Ca2+ concentrations in the range of 10−7 to 10−5 M during rest and peak activation, respectively (13).
A divalent cation binding site (site II) in the N-terminal domain of TnC is specific for Ca2+ (14), which allows the N-terminal domain to exist in predominantly a ligand-free or ligand-bound form during resting or activating states, respectively (15). Ligand binding to site II is energetically coupled to an equilibrium of closed and open conformations of the N-terminal domain (16). Ca2+ binding destabilizes the secondary structure required for the closed conformation, thereby increasing the probability of the open conformation (17). The open conformation exposes a hydrophobic patch that interacts with H3(I) of cardiac troponin I (TnI) (Fig. 1). H3(I) together with H4(I) comprise a larger switch peptide (TnIsp) (16,18) that, along with the C-terminal domain of TnI, stabilizes troponin (Tn) and tropomyosin (Tm) in the position that blocks activation during rest (19–21). To activate contraction, TnIsp rotates away from actin to bind the open conformation of TnC (18). TnIsp binds the hydrophobic patch during the time that the N-terminal domain of TnC is in the open state, by a conformational selection mechanism (22–24). Baseline and maximum activation reflect the distributions of closed and open conformations, which have probabilities that depend on Ca2+ binding.
Figure 1. Ribbon diagram of the Ca2+-saturated structure of cardiac Tn showing the site of the mutation (magenta).

A. Peptide backbones are shown for TnC (green), TnI (red, black, gold and brown) and TnT (blue). Residues 150–159 corresponding to the switch region of TnI, H3(I), are shown (gold) at the site of interaction with the hydrophobic patch of TnC. B. The position of alanine 8 (purple) is shown in relation to the H3(I) helix and the lobes of TnC, which form Ca2+-dependent (N-Domain) and Ca2+-independent (C-Domain) contacts with TnI.
Although force measurements suggest that the A8V mutation acts by increasing the Ca2+ binding to N-terminal of domain of TnC as the primary cause of the disease (7), structural and functional measurements do not support this mechanism. No significant difference in N-terminal domain Ca2+ affinity is observed between isolated TnCA8V and wild-type (WT) TnC (TnCWT) (25). Instead, the A8V mutation shifts the conformation of the N-terminal domain in favor of the open conformations of ligand- bound and ligand- free TnC (26). The A8V mutation is located in the N-helix of the crystal structure of TnCWT and is situated between site II for Ca2+ and the H3(I) interaction site (Fig. 1) (18). A synthetic peptide corresponding to TnISP binds to TnCA8V ~1.6-fold more strongly than to TnCWT (27). Based on these observations, we hypothesize that altered interactions between TnC and TnISP is the primary source of the changes in function observed for myofilaments carrying TnCA8V (e.g., increase myofilament Ca2+ sensitivity). We present a model to reconcile this hypothesis with the paradoxical increased sensitivity to Ca2+ observed in force measurements (7).
MATERIALS AND METHODS
Three-dimensional Visualization
The location of the TnCA8V mutation within the 52- kDa Tn complex was visualized in the 1J1E Protein Data Bank file using PyMol software. PyMol is an open source molecular visualization program that allows manipulation of PDB files containing molecular coordinates obtained from structures based on x-ray crystallography or nuclear magnetic resonance.
Proteins
Human TnCWT, TnCA8V, and TnI were recombinantly expressed and purified according to established protocols (25,28).
Myofibrillar preparation
Porcine cardiac myofibrils were prepared as described previously (29) and stored in 50% glycerol at −20°C. Glycerol was removed by centrifugation (7 min at 800 × g and 4°C), and myofibrils were resuspended in wash buffer (10 mM MOPS, 10 mM KCl, pH 7.0) with 2 mM DTT.
Protein extraction and reconstitution
Native TnC was depleted from myofibrils using a CDTA extraction buffer (5 mM CDTA, 5 mM DTT, pH 8.4) for ~2.5 h at room temperature. After each 30 min of incubation, the myofibrils were centrifuged (5 min at 750 × g and 4°C), the supernatant was discarded, and a new CDTA extraction solution was added. To remove CDTA, myofibrils were centrifuged (5 min at 1,500 × g and 4°C) and resuspended in wash buffer three times. Native TnC-extracted myofibrils were reconstituted with TnCWT or TnCA8V (1.0 mg/ml) for 1 h at 4°C. Non-specifically bound TnC was removed from the reconstituted samples by several washes (centrifugation and resuspension) using wash buffer. Myofibril concentrations were determined using a PierceTM Coomassie Plus (Bradford) assay kit.
Myofibrillar ATPase activity
The ATPase assay was performed at various pCa values using solutions containing 2 mM EGTA, 3 mM NTA, 20 mM MOPS, 1 mM free Mg2+, ~2.5 mM MgATP2−, pH 7.0 with ionic strength fixed to 80 mM (adjusted with KCl) and selected Ca2+ concentrations. The free Ca2+ in the ATPase solutions were calculated using pCa calculator software (30). The pCa values of the solutions were 8.0, 7.0, 6.6, 5.8, and 5.0 (1.0 × 10−8 M, 1.0 × 10−7 M, 2.51 × 10−7 M, 1.58 × 10−6 M and 1.0 × 10−5 M free Ca+2, respectively). The myofibrillar concentration was 0.4 mg/ml, and the experiments were carried out at 25°C. The reaction was initiated by adding ATP and quenched after 7 min with the addition of ~4.6% trichloroacetic acid. The precipitated proteins were collected by centrifugation (7 min at 3,095 × g and 4°C). Inorganic phosphate released by ATP hydrolysis was measured in the supernatant using the colorimetric method of Fiske and Subbarow (31).
Fluorescence assay
TnCWT, TnCA8V, and TnI were dialyzed into fluorescence buffer containing 120 mM MOPS, 100 mM KCl, and 2 mM EGTA at pH 7.0. For experiments in the presence of Mg2+ and Ca2+, MgCl2 and CaCl2 were added before titration to yield final concentrations of 2 mM free Mg2+ and 0.1 mM free Ca2+. For experiments in the presence of Mg2+ only, MgCl2 was added to a final concentration of 2 mM free Mg2+. In addition, before titration, freshly prepared 1 mM DTT was added to the buffer. TnI and 4,4′-dianilino-1,1′- binaphthyl-5,5′-disulfonic acid, dipotassium salt (bis-ANS) were fixed at 0.5 mM. TnCWT or TnCA8V was titrated at every 0.05 μM until reaching 0.5 μM, followed by titration of every 0.1 μM until the final concentration of 1 μM was reached. TnC and TnI concentrations were determined by spectroscopy at 280 nm (NanoDrop ND-1000 spectrophotometer) with extension coefficients of 4,595 and 9,770 cm−1 M−1, respectively. The steady-state fluorescence measurements were performed in a Jasco FP-8300 spectrofluorometer at a constant photomultiplier (PMT) voltage of 340 V. The fluorescent probe bis-ANS was excited at 390 nm, emission was detected in a wavelength range of 450–520 nm, and the highest peak intensity was recorded. The experiments were carried out at 25°C and under constant stirring (600 rpm). A baseline experiment was performed in the absence of TnI to measure the fluorescence changes upon binding of TnC to bis-ANS. At the PMT voltage of 340V, minimum changes in fluorescence emission (A.U.) were observed in the baseline titration. The data obtained were then subtracted from the baseline. Data were analyzed and fit using linear regression.
Statistical analyses
The data were analyzed for significance using Student's t-test (unpaired). The results were considered statistically different when p < 0.05. The data are expressed as means ± S.E.
RESULTS
Modeling
We model regulation as modulation of Tm in the activated state (M) by multiple equilibria (32). In this model, M forms in the activated position of the thin filament as a ternary complex of myosin, Tm, and actin (33,34) from Tm in the central position of the thin filament (C) by a spontaneous process (equilibrium pathway). The equilibrium is perturbed by the crossbridge cycle in the physiologic muscle by an additional kinetic pathway. The kinetic pathway operates continuously with fixed non-zero probability to regenerate M without sampling C. Failure to regenerate returns Tm to the central position. The kinetic pathway is responsible for establishing an ensemble of Tm molecules that function as a unit in the manner predicted by theory (32) and consistent with the observation that an initial binding of two myosin heads establishes a regulatory unit that can accommodate binding by a much larger number of secondarily bound myosin heads (35). The combination of equilibrium and kinetic pathways are expressed in the model as (Fig. 2), where K0′ includes the number of Tm molecules functioning as an ensemble (n > 1). In addition, the odds of regenerating M without decay to C (α) is included in the mathematical expression of M,
| (1) |
that is derived for the steady-state (36). C is the only Ca2+-dependent variable of Eq. 1 with K0′, α, and n each having fixed values determined empirically from WT data (see Table 1).
Figure 2. Model accounting for results of functional studies.

The TnIsp is modeled as binding either TnC or actin and TnC is modeled as being in either the closed or open conformation. The model consists of two subsystems that partially overlap. The states of Tm (blue) include C, M, and Bi (i = 1, 2), corresponding to interactions with actin (green) in positions central, myosin dependent, and blocking, respectively. Steady-state constant, , governs the ensemble dynamic interactions of myosin with Tm that sustain M. The states of Tn (TnT, yellow; TnI, magenta, TnC, black-white) include Ti and Bi, corresponding to energetically coupled and uncoupled in the Ca2+-free state (i = 1) and Ca2+-bound state (i = 2), respectively. Most probable occupancy of the N-terminal domain regulatory site of TnC is shown as Ca2+-bound (black) or Ca2+-free (white). Equilibrium constants govern spontaneous interactions that occur between Tn and actin (K1 and K3), and Ca2+ and Tn (K2 and K4); K1 and K3 also include the equilibrium that favors Tm in the C position. For the Tn states, representative distributions of closed and open conformations of TnC are diagramed to show the ratios 9/1, 1/3, 3/1, and 1/9 for B1, B2, T1, T2, respectively, which are intended for relative comparison only.
Table 1.
Summary of Model Specific Constants
| Lower K2 Boundary1 |
Upper K2 Boundary |
|||||||
|---|---|---|---|---|---|---|---|---|
| WT | A8V | A8V | A8V | WT | A8V | A8V | A8V | |
| ΔK1 (note2) | 0.77 | 0.67 | 0.59 | 0.77 | 0.67 | 0.59 | ||
| K 0 ′ | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 3 α | 3.25 | 3.25 | 3.25 | 3.25 | 3.25 | 3.25 | 3.25 | 3.25 |
| n | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| K 1 | 275.00 | 211.54 | 183.33 | 161.76 | 1400.00 | 1076.92 | 933.33 | 823.53 |
| K2 (note3) | 1.67 | 1.67 | 1.67 | 1.67 | 10.00 | 10.00 | 10.00 | 10.00 |
| K3 (note4) | 27.55 | 21.19 | 18.37 | 16.21 | 14.00 | 10.77 | 9.33 | 8.24 |
| K4 | 0.17 | 0.17 | 0.17 | 0.17 | 0.10 | 0.10 | 0.10 | 0.10 |
| Range of Activation | ~10% to- 90% | ~10% to- 90% | ~10% to- 90% | ~10% to- 90% | ~5% to- 95% | ~5% to- 95% | ~5% to- 95% | ~5% to- 95% |
| Figures | 3A, 3C | 3A, 3C | 3A, 3C | 3A, 3C | 3B, 3D | 3B, 3D | 3B, 3D | 3B, 3D |
| Curve | Black | Red | Blue | Purple | Black | Red | Blue | Purple |
K2, K4, n, and α have values used previously (32). Values are dimensionless unless otherwise indicated.
Multiplier of WT K1
Multiple of 106 M−1
Not an adjustable parameter. Tabulated value calculated from K1K4/K2
Regulation is achieved by controlling the availability of C for M formation (Eq. 1). C is consumed in the reactions that transition Tm to the blocking position of the thin filament and form the interaction between actin and TnISP (Bi; Fig. 2), which is responsible for muscle relaxation. Expressed in mole fractions, C = 1 − M − B1− B2.
The reaction (i = 1, 2 and j = 1, 3; Fig. 2) represents two independent events required for transition to the blocking state (Bi), namely, thermal-elastic transfer of Tm from the central position to the blocking position, and interaction between actin and TnIsp. Dissociation of TnIsp and movement of Tm to either the central or activated positions chemically uncouples Tn from spontaneous interaction with actin. Letting Ti represent uncoupled Tn, mole fractions of coupled and uncoupled Tn are determined by the relation, 1 = B1 + B2 + T1 + T2.
By detailed balance (Fig. 2), the following relationship must hold,
| (2) |
Analysis of tension data
Fibers reconstituted with TnCA8V require less Ca2+ to achieve a given fraction of maximum tension than fibers reconstituted with TnCWT (7). Because site II of TnCWT and TnCA8V have indistinguishable affinities for Ca2+ (25), enhanced sensitivity to Ca2+ is more likely explained by strengthened interaction between TnC and TnIsp. We model no change in Ca2+ affinity by holding K2 and K4 constant and an increase in TnIsp affinity as a reduction in the apparent affinity of TnIsp for actin, i.e. reduction in K1 and K3 (Fig. 2). Holding K2/K4 constant implies that K1/K3 is constant (Eq. 2).
To vary only K1 and keeping K1/K3 constant, values for the other parameters must be fixed in Eq. 1. For simplicity, K0′ is set to unity and n, and α have values used previously ((32); see also Table 1). Because K2 governs the binding of Ca2+ to uncoupled Tn (Fig. 2), K2 is set approximately equal to the measured affinity constant for isolated Tn, and, hence, K4 is given by K2K3/K1 (Eq. 2). We compare K2 and K1/K3 values derived previously for skeletal muscle (32) with substantially greater values of each as a means of testing sensitivity (Table 1). For both lower and upper boundary conditions, parameter K1 is adjusted to achieve theoretical activations that fit Ca2+-dependent tension data normalized between 0% and 100% maximum (Fig. 3A, B). A 1.5- to 1.7- fold decrease in the bench-mark K1 shifts the theoretical Ca2+ activation curve to align with the tension data collected for fibers reconstituted with TnCA8V (Fig. 3A, B). By Eq. 2 and the constraint imposed on K2 and K4, the model predicts that K3 also must decrease 1.5- to 1.7- fold. Although a unique solution is not established, this analysis reconciles the shift observed in Ca2+ sensitive force (7) and the absence of measured change in Ca2+ binding affinity in isolated TnC (25). A Matlab program to reproduce and extend the modeling shown here is included in Supplementary Material (Fig. S1). As a control, we fit a 2- fold increase in Ca2+ activation data published for the L48Q mutation of TnC by holding K1 constant and increasing K2, consistent with the increase in affinity of Tn reported for this mutation (Supplementary Material, Fig. S2).
Figure 3. Tension data fit with model.

Data from chemically skinned and TnC-depleted cardiac fibers are reproduced from an earlier study (7) for TnCWT (circles) or TnCA8V (squares). Normalized theoretical curves are shown for the fit of WT data (black) and for varying one parameter, K1, by a multiplier (ΔK1; Table 1), where 1/ΔK1 is 1.3, 1.5, and 1.7 (purple, blue, red). A and B. Lower and upper K2 boundary conditions (Table 1) are plotted respectively on normalized scales. C and D. Theoretical curves from A and B shown on absolute scales.
On an absolute scale, the predicted activation is greater for all Ca2+ concentration between baseline and maximum fractional activations of 0.1–0.9 and 0.05–0.95 for lower and upper boundary conditions, respectively (Fig 3C, D; Table 1). Based on the shift to the open conformation described for TnCA8V (26), we interpret the decrease in both K1 and K3 as owing to increased competition for TnISP by the more probable open conformation of TnCA8V. Hence, the model predicts that a modest increase in contractility at all Ca2+ concentrations causes the leftward shift in the relative force curve (Fig. 3A, B).
Activation of myofibrillar ATPase
To test the model's prediction on an absolute scale, we measured Ca2+-dependent ATPase rate of cardiac myofibrils reconstituted with TnCA8V and TnCWT. Given midrange Ca2+ concentration, i.e., pCa 5.8, ATPase rates of myofibrils reconstituted with TnCWT and TnCA8V are 48.7% and 62.5% of maximum, respectively (Table 2). This difference is consistent with the enhancement observed in the tension record (Fig. 3A). Given various Ca2+ concentrations, all conditions stimulated higher mean ATPase rates for myofibrils reconstituted with TnCA8V than myofibrils reconstituted with TnCWT (Fig. 4). Differences in the means are statistically significant for Ca2+ concentrations above the midrange (Table 2). Elevation in ATPase rate at the extremes of Ca2+ concentration is consistent with a general increase in contractility brought about by the interaction between the more open TnCA8V and TnI conformations.
Table 2.
Summary of Cardiac Myofibrillar ATPase Activity
| TnC-WT | TnC-A8V | |
|---|---|---|
|
|
||
| Cardiac Myofibrils | nmol Pi·mg−1·min−1 | |
| pCa 8.0 | 32.65 ± 5.48 | 38.51 ± 3.55 |
| pCa 7.0 | 44.01 ± 4.51 | 48.06 ± 5.07 |
| pCa 6.6 | 46.79 ± 3.42 | 56.11 ± 4.90 |
| pCa 5.8 | 58.29 ± 6.07 | 82.52 ± 7.85* |
| pCa 5.0 | 85.25 ± 3.22 | 108.96 ± 10.65* |
|
|
||
| Ratios | ||
| (pCa 5.8–8.0) / (pCa 5.0–8.0) × 100 | 48.7% | 62.5% |
p < 0.05 vs WT
Figure 4. Ca2+-dependent ATPase measurement of reconstituted TnC-depleted myofibrils.
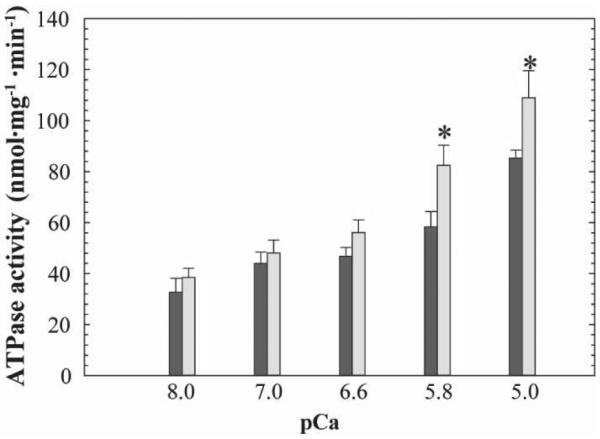
Myofibrils were reconstituted with TnCWT (black) and TnCA8V (gray). Mean ± SEM; n = 9. *p < 0.05 vs WT at same pCa. The specific ATPase activity measured in native cardiac myofibrils were 5.56 ± 2.06 and 55.15 ± 3.27 nmol Pi/mg/min at pCa 8.0 and pCa 5.0 (n = 7), respectively. The specific ATPase activity measured in TnC-extracted myofibrils were 12.03 ± 1.02 and 25.54 ± 2.65 nmol Pi/mg/min at pCa 8.0 and pCa 5.0 (n = 12), respectively.
Titration of fluorescence associated with TnI
To detect subtle differences in the interaction between TnC and TnI, we monitored the fluorescence of bis-ANS. Compared with bis-ANS bound to TnI, unbound bis-ANS has much reduced fluorescence intensity. We interpret reduction in fluorescence intensity as owing to the displacement of bis-ANS that results from complex formation between TnC and TnI. Rather than a typical hyperbolic binding curve, we observe a linear decrease in fluorescence as a function of added TnC (Fig. 5). Specific binding of TnC is evident from the saturation of the fluorescence decrease for additions that exceed 1 mol TnC/mol TnI (Fig. 5 insets). The appearance of irreversible binding may be explained by a combination6 of high affinity mass action binding (37) and persistent weak interactions that slow the diffusive separation of unbound proteins. To distinguish between the functions of the domains of TnC, we conducted the assay in two Ca2+ concentrations. Letting Mg-TnCWT and Ca-TnCWT represent the structures of TnC in low or saturating Ca2+ concentrations (see introduction), both structures are expected to form high-affinity interactions between the C-terminal domain of TnC and TnI. However, only Ca-TnCWT is expected to promote a secondary interaction between the N-terminal domain of TnC and TnI. As expected, the more extensively bound Ca-TnCWT displaces proportionately more bis-ANS than Mg-TnCWT (Fig. 5A). By contrast, Ca-TnCA8V and Mg-TnCA8V displace nearly the same amount of probe (Fig. 5B) and have nearly the same slope as Ca-TnCWT. Thus, compared with Mg-TnCWT, the N-terminal domain of Mg-TnCA8V appears to have a more stable association with TnI, consistent with having a more open N-terminal domain conformation independent of Ca2+-binding at site II.
Figure 5. TnC titration of bis-ANS fluorescence associated with TnI.

Plotted are titration of fluorescence change (arbitrary units) curves for TnCWT (A) and TnCA8V (B) with Ca2+ added (solid) or not added (open). Mean ± SEM; n = 4. Titrations are biphasic (insets) with the linear change saturating at 1 mol/mol TnC added. In A. linear regression of slopes corresponding to Ca2+-bound (solid line) and Ca2+-free (dashed line) are −1075 and −703, respectively. In B. linear regression of slopes corresponding to Ca2+-bound (solid line) and Ca2+ free (dashed line) are −922 and −937, respectively.
DISCUSSIONS
We find that a modest increase in affinity between the N-terminal domain of TnC and the switch region of TnI causes the observed shift in midrange tension output to lower Ca2+ concentration observed in cardiac muscle containing TnCA8V. Evidence in support of this hypothesis comes from the combination of experimental and modeling approaches. Measurement of binding rules out a significant change in Ca2+ binding affinity, and structural modeling rules out mutagenesis directed to the site of interaction between TnC and TnI. Based on a kinetic model of muscle regulation, we determined that the observed enhancement in Ca2+-sensitivity is explained by 1.5- to 1.7-fold greater affinity between TnC and TnISP, which matches the experimental measurement of 1.6 fold (27). Average myofibril ATPase rates are higher for myofibrils reconstituted with TnCA8V at all Ca2+ concentrations tested, and are significantly higher for Ca2+ concentrations above the titration midpoint. Compared with TnCWT, TnCA8V displaced more fluorescent probe from TnI in the absence of saturating Ca2+, consistent with a stronger interaction.
Our model is distinct in the manner by which Ca2+ participates in attaining the activated state. Standard models derived from Hill et al. (38) are fundamentally allosteric and hence propose structural changes in Tm owing to a series of structural changes initiated by Ca2+ binding to site II of TnC. Accordingly, the states of Tm corresponding to the B, C, and M positions result from Ca2+-free and myosin unbound; Ca2+-bound and myosin weakly bound to actin; and, Ca2+-bound and myosin strongly bound to actin, respectively (39–41). In our model, Ca2+-binding acts only to shift the equilibrium between closed and open states of TnC and TnIsp selects the open conformation of TnC independent of cation occupancy of TnC site II. Competition for TnIsp between actin and the open conformation of TnC occurs only in the B position. By moving to the C and M positions, Tm prevents the actin-TnIsp interaction, but not the interaction between the open conformation of TnC and TnIsp. In these uncoupled positions, TnIsp binding stabilizes the open conformation and hence increases site II affinity for Ca2+. We diagram a representative distribution of closed and open conformations of TnC for each Tn state to show how the competition by actin for TnIsp in state B shifts the equilibrium most strongly in favor of the closed conformation of TnC and the lack of competition in state T shifts the equilibrium most strongly in favor of the open conformation of TnC (Fig. 2).
Whereas the activated states of allosteric models require contribution of binding by both Ca2+ and strong-binding crossbridges, the activated state of our model requires no direct participation of Ca2+ binding. Instead, the activated state requires only Tm in the C position and force-bearing cycling crossbridges, consistent with consensus observations (41). Cooperative activation (39) without direct participation of ligand, i.e., non-allosteric cooperativity, is a hallmark of a second chance mechanism (36). The statistical basis of a second chance mechanism can be linked to fundamental thermodynamic properties of systems that couple equilibrium and kinetic forces, such as forces acting on Tm by cycling crossbridges (42). Absent experimental proof of a Ca2+-driven conformation of Tm being required for the activated state of the physiologic muscle, the simplest explanation is that Ca2+ binding is required only to perturb the equilibrium of closed and open states of TnC.
To date, three distinct probands carrying the Ala8 to Val mutation in TnC have been reported ((7,43) and personal communication). Although one of the probands reported a parent who was also positively genotyped for the mutation, the available genetic data remain difficult to interpret. In one clinical report, the mutation arose de novo; in another report, the proband carried two mutations in different sarcomeric proteins (7,43). The functional characterization is crucial to ascertain whether the A8V mutation in TnC is a true pathogenic variant and how it exerts its negative effect on muscle regulation.
Functional assays conducted with reconstituted proteins have shown that when the A8V mutant TnC is incorporated in the thin filament, it increases Ca2+ sensitivity of the myofilament. Kinetic measurements in reconstituted thin filament have suggested that a slower Ca2+ off rate might be the cause of the increased Ca2+ binding affinity to the N-terminal domain of TnCA8V (28,44). Activation and deactivation of muscle is controlled by Ca2+ binding and dissociating from the TnC N-terminal domain, which subsequently interacts with the C-terminal domain of TnI. Therefore, not only Ca2+ kinetics but also TnC and TnI affinity are crucial in the regulation of cardiac muscle contraction. Little is known about how TnCA8V delays the Ca2+ off rate and whether this involves an abnormal interaction between TnC and TnI. One simplistic explanation supported by our model is that the N-terminal domain of TnCA8V binds with greater affinity to the TnI C-terminal domain. The fluorescence binding studies reported here are in agreement with a previous report (27) and support the hypothesis that TnCA8V and TnI bind to each other at a higher affinity during resting conditions, when the muscle should be fully relaxed.
The consequences of increased binding affinity between TnCA8V and TnI may have broader physiological implications. The functional consequences of the A8V substitution in TnC were evaluated in a knock-in (KI) mouse (45). Intact cardiomyocytes isolated from both heterozygous and homozygous KI mice had slower sarcomere length relaxation time and Ca2+ decay at different frequencies of stimulation. Furthermore, homozygous mice showed signs of diastolic dysfunction and a hypercontractile phenotype, e.g., increased ejection fraction (45). The in vivo data can be correlated with the myofibrillar ATPase findings since higher myofibrillar ATPase activation can indicate a greater degree of muscle activation. Increased actomyosin ATPase activation was also observed in an early report (44). Here, we observed increased mean myofibrillar ATPase activity at all low Ca2+ concentrations tested, although statistical significance could not be demonstrated for all Ca2+ concentrations, reflecting sensitivity limits of the assay. We interpret an effect of the TnC-TnI interaction on myofibrillar ATPase as contributing to the contractility of cardiac myocytes. Hence, even a small but persistent increase in myofibrillar ATPase at low Ca2+ levels may be sufficient to cause hypertrophy of the muscle over time.
In conclusion, these results suggest that the effect of the A8V mutation in delaying the Ca2+ off rate from TnC would be secondary to altering the interaction between TnC and TnI. This is an important observation because new therapies could be tailored to specifically target binding between TnC and TnI, rather than intracellular Ca2+ concentration or Ca2+ binding to TnC. The results obtained in this work do not allow us to determine in exact nature of the altered interactions between TnC and TnI; however, if the defect lies in the kinetics of binding to TnI rather than dissociation from TnI, this could be an important site for the development of a therapeutic target.
Supplementary Material
Highlights.
-
1)
TnCA8V increases Ca2+-dependent tension without altering N-domain Ca2+-binding affinity in isolated protein
-
2)
TnCA8V enhances myofibrillar ATPase rates and TnI-TnC interaction
-
3)
Theoretical predictions of enhanced TnI-TnC interaction recapitulate experimental published data
-
4)
Stronger TnI-TnCA8V interaction may destabilizes blocking caused by TnI-actin interaction
-
5)
Permanent increase in contractility is the presumptive cause of hypertrophic cardiomyopathy
ACKNOWLEDGEMENTS
J.R.P. acknowledges support from the National Heart, Lung, and Blood Institute (NIH) Grant HL103840, Florida Heart Research Institute, and American Heart Association (AHA) 15GRNT25280004. We thank Dr. Anita Zot for expert editing assistance during revision.
ABBREVIATIONS
- TnC
Cardiac troponin C
- TnI
Cardiac troponin I
- Tm
Cardiac tropomyosin
- Tn
Cardiac troponin
- A8V
Alanine to valine replacement at position 8 of cardiac troponin C
- TnIsp
Switch peptide of cardiac TnI
- HCM
Hypertrophic cardiomyopathy
- TNNC1
Gene encoding cardiac troponin C
- CDTA
1,2-Diaminocyclohexanetetraacetic acid disodium
- DTT
Dithiothreitol
- EGTA
Ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid
- NTA
Nitrilotriacetic acid
- MOPS
3-(N-Morpholino)propanesulfonic acid, 4-Morpholinepropanesulfonic acid
- ATP
Adenosine 5′-triphosphate
- Bis-ANS
4,4′-Dianilino-1,1′-Binaphthyl-5,5′-Disulfonic Acid, Dipotassium Salt
- pCa
10−[Ca2+]
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Konno T, Chang S, Seidman JG, Seidman CE. Genetics of hypertrophic cardiomyopathy. Current opinion in cardiology. 2010;25:205–209. doi: 10.1097/HCO.0b013e3283375698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marian AJ. Hypertrophic cardiomyopathy: from genetics to treatment. European journal of clinical investigation. 2010;40:360–369. doi: 10.1111/j.1365-2362.2010.02268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Willott RH, Gomes AV, Chang AN, Parvatiyar MS, Pinto JR, Potter JD. Mutations in Troponin that cause HCM, DCM AND RCM: what can we learn about thin filament function? Journal of molecular and cellular cardiology. 2010;48:882–892. doi: 10.1016/j.yjmcc.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 4.Force T, Bonow RO, Houser SR, Solaro RJ, Hershberger RE, Adhikari B, Anderson ME, Boineau R, Byrne BJ, Cappola TP, Kalluri R, LeWinter MM, Maron MS, Molkentin JD, Ommen SR, Regnier M, Tang WH, Tian R, Konstam MA, Maron BJ, Seidman CE. Research priorities in hypertrophic cardiomyopathy: report of a Working Group of the National Heart, Lung, and Blood Institute. Circulation. 2010;122:1130–1133. doi: 10.1161/CIRCULATIONAHA.110.950089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013;381:242–255. doi: 10.1016/S0140-6736(12)60397-3. [DOI] [PubMed] [Google Scholar]
- 6.Hoffmann B, Schmidt-Traub H, Perrot A, Osterziel KJ, Gessner R. First mutation in cardiac troponin C, L29Q, in a patient with hypertrophic cardiomyopathy. Hum Mutat. 2001;17:524. doi: 10.1002/humu.1143. [DOI] [PubMed] [Google Scholar]
- 7.Landstrom AP, Parvatiyar MS, Pinto JR, Marquardt ML, Bos JM, Tester DJ, Ommen SR, Potter JD, Ackerman MJ. Molecular and functional characterization of novel hypertrophic cardiomyopathy susceptibility mutations in TNNC1-encoded troponin C. Journal of molecular and cellular cardiology. 2008;45:281–288. doi: 10.1016/j.yjmcc.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung WK, Kitner C, Maron BJ. Novel frameshift mutation in Troponin C (TNNC1) associated with hypertrophic cardiomyopathy and sudden death. Cardiology in the young. 2011;21:345–348. doi: 10.1017/S1047951110001927. [DOI] [PubMed] [Google Scholar]
- 9.Parvatiyar MS, Landstrom AP, Figueiredo-Freitas C, Potter JD, Ackerman MJ, Pinto JR. A Mutation in TNNC1-encoded Cardiac Troponin C, TNNC1-A31S, Predisposes to Hypertrophic Cardiomyopathy and Ventricular Fibrillation. The Journal of biological chemistry. 2012;287:31845–31855. doi: 10.1074/jbc.M112.377713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalyva A, Parthenakis FI, Marketou ME, Kontaraki JE, Vardas PE. Biochemical characterisation of Troponin C mutations causing hypertrophic and dilated cardiomyopathies. J Muscle Res Cell Motil. 2014;35:161–178. doi: 10.1007/s10974-014-9382-0. [DOI] [PubMed] [Google Scholar]
- 11.Bers DM. Calcium sources and sinks. 2nd ed Kluwer Academic Publishers; Boston/London: 2001. [Google Scholar]
- 12.Biesiadecki BJ, Davis JP, Ziolo MT, Janssen PM. Tri-modal regulation of cardiac muscle relaxation; intracellular calcium decline, thin filament deactivation, and cross-bridge cycling kinetics. Biophysical reviews. 2014;6:273–289. doi: 10.1007/s12551-014-0143-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bers DM. Calcium fluxes involved in control of cardiac myocyte contraction. Circulation research. 2000;87:275–281. doi: 10.1161/01.res.87.4.275. [DOI] [PubMed] [Google Scholar]
- 14.Holroyde MJ, Robertson SP, Johnson JD, Solaro RJ, Potter JD. The calcium and magnesium binding sites on cardiac troponin and their role in the regulation of myofibrillar adenosine triphosphatase. The Journal of biological chemistry. 1980;255:11688–11693. [PubMed] [Google Scholar]
- 15.Davis JP, Tikunova SB. Ca(2+) exchange with troponin C and cardiac muscle dynamics. Cardiovascular research. 2008;77:619–626. doi: 10.1093/cvr/cvm098. [DOI] [PubMed] [Google Scholar]
- 16.Gagne SM, Tsuda S, Li MX, Smillie LB, Sykes BD. Structures of the troponin C regulatory domains in the apo and calcium-saturated states. Nature structural biology. 1995;2:784–789. doi: 10.1038/nsb0995-784. [DOI] [PubMed] [Google Scholar]
- 17.Vinogradova MV, Stone DB, Malanina GG, Karatzaferi C, Cooke R, Mendelson RA, Fletterick RJ. Ca2+-regulated structural changes in troponin. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:5038–5043. doi: 10.1073/pnas.0408882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 19.Li MX, Spyracopoulos L, Beier N, Putkey JA, Sykes BD. Interaction of cardiac troponin C with Ca(2+) sensitizer EMD 57033 and cardiac troponin I inhibitory peptide. Biochemistry. 2000;39:8782–8790. doi: 10.1021/bi000473i. [DOI] [PubMed] [Google Scholar]
- 20.Luo Y, Wu JL, Li B, Langsetmo K, Gergely J, Tao T. Photocrosslinking of benzophenone-labeled single cysteine troponin I mutants to other thin filament proteins. Journal of molecular biology. 2000;296:899–910. doi: 10.1006/jmbi.1999.3495. [DOI] [PubMed] [Google Scholar]
- 21.Tripet B, Van Eyk JE, Hodges RS. Mapping of a second actin-tropomyosin and a second troponin C binding site within the C terminus of troponin I, and their importance in the Ca2+-dependent regulation of muscle contraction. Journal of molecular biology. 1997;271:728–750. doi: 10.1006/jmbi.1997.1200. [DOI] [PubMed] [Google Scholar]
- 22.Paakkonen K, Annila A, Sorsa T, Pollesello P, Tilgmann C, Kilpelainen I, Karisola P, Ulmanen I, Drakenberg T. Solution structure and main chain dynamics of the regulatory domain (Residues 1–91) of human cardiac troponin C. The Journal of biological chemistry. 1998;273:15633–15638. doi: 10.1074/jbc.273.25.15633. [DOI] [PubMed] [Google Scholar]
- 23.Paakkonen K, Sorsa T, Drakenberg T, Pollesello P, Tilgmann C, Permi P, Heikkinen S, Kilpelainen I, Annila A. Conformations of the regulatory domain of cardiac troponin C examined by residual dipolar couplings. European journal of biochemistry / FEBS. 2000;267:6665–6672. doi: 10.1046/j.1432-1327.2000.01763.x. [DOI] [PubMed] [Google Scholar]
- 24.Gaponenko V, Abusamhadneh E, Abbott MB, Finley N, Gasmi-Seabrook G, Solaro RJ, Rance M, Rosevear PR. Effects of troponin I phosphorylation on conformational exchange in the regulatory domain of cardiac troponin C. The Journal of biological chemistry. 1999;274:16681–16684. doi: 10.1074/jbc.274.24.16681. [DOI] [PubMed] [Google Scholar]
- 25.Pinto JR, Parvatiyar MS, Jones MA, Liang J, Ackerman MJ, Potter JD. A functional and structural study of troponin C mutations related to hypertrophic cardiomyopathy. The Journal of biological chemistry. 2009;284:19090–19100. doi: 10.1074/jbc.M109.007021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cordina NM, Liew CK, Gell DA, Fajer PG, Mackay JP, Brown LJ. Effects of calcium binding and the hypertrophic cardiomyopathy A8V mutation on the dynamic equilibrium between closed and open conformations of the regulatory N-domain of isolated cardiac troponin C. Biochemistry. 2013;52:1950–1962. doi: 10.1021/bi4000172. [DOI] [PubMed] [Google Scholar]
- 27.Swindle N, Tikunova SB. Hypertrophic cardiomyopathy-linked mutation D145E drastically alters calcium binding by the C-domain of cardiac troponin C. Biochemistry. 2010;49:4813–4820. doi: 10.1021/bi100400h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pinto JR, Reynaldo DP, Parvatiyar MS, Dweck D, Liang J, Jones MA, Sorenson MM, Potter JD. Strong cross-bridges potentiate the Ca(2+) affinity changes produced by hypertrophic cardiomyopathy cardiac troponin C mutants in myofilaments: a fast kinetic approach. The Journal of biological chemistry. 2011;286:1005–1013. doi: 10.1074/jbc.M110.168583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Solaro RJ, Pang DC, Briggs FN. The purification of cardiac myofibrils with Triton X-100. Biochimica et biophysica acta. 1971;245:259–262. doi: 10.1016/0005-2728(71)90033-8. [DOI] [PubMed] [Google Scholar]
- 30.Dweck D, Reyes-Alfonso A, Jr., Potter JD. Expanding the range of free calcium regulation in biological solutions. Anal Biochem. 2005;347:303–315. doi: 10.1016/j.ab.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 31.Fiske CH, Subbarow Y. The colorimetric determination of phosphorous. Journal of Biological Chemistry. 1925;66:375–400. [Google Scholar]
- 32.Zot HG, Hasbun JE, Van Minh N. Striated muscle regulation of isometric tension by multiple equilibria. PloS one. 2009;4:e8052. doi: 10.1371/journal.pone.0008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eaton BL. Tropomyosin binding to F-actin induced by myosin heads. Science. 1976;192:1337–1339. doi: 10.1126/science.131972. [DOI] [PubMed] [Google Scholar]
- 34.Tobacman LS, Butters CA. A new model of cooperative myosin-thin filament binding. The Journal of biological chemistry. 2000;275:27587–27593. doi: 10.1074/jbc.M003648200. [DOI] [PubMed] [Google Scholar]
- 35.Desai R, Geeves MA, Kad NM. Using fluorescent myosin to directly visualize cooperative activation of thin filaments. The Journal of biological chemistry. 2015;290:1915–1925. doi: 10.1074/jbc.M114.609743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zot HG, Hasbun JE, Minh NV. Second-chance signal transduction explains cooperative flagellar switching. PloS one. 2012;7:e41098. doi: 10.1371/journal.pone.0041098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calvert MJ, Ward DG, Trayer HR, Trayer IP. The importance of the carboxyl-terminal domain of cardiac troponin C in Ca2+-sensitive muscle regulation. The Journal of biological chemistry. 2000;275:32508–32515. doi: 10.1074/jbc.M005764200. [DOI] [PubMed] [Google Scholar]
- 38.Hill TL, Eisenberg E, Greene L. Theoretical model for the cooperative equilibrium binding of myosin subfragment 1 to the actin-troponin-tropomyosin complex. Proceedings of the National Academy of Sciences of the United States of America. 1980;77:3186–3190. doi: 10.1073/pnas.77.6.3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKillop DF, Geeves MA. Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophysical journal. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lehman W, Hatch V, Korman V, Rosol M, Thomas L, Maytum R, Geeves MA, Van Eyk JE, Tobacman LS, Craig R. Tropomyosin and actin isoforms modulate the localization of tropomyosin strands on actin filaments. Journal of molecular biology. 2000;302:593–606. doi: 10.1006/jmbi.2000.4080. [DOI] [PubMed] [Google Scholar]
- 41.Manning EP, Tardiff JC, Schwartz SD. A model of calcium activation of the cardiac thin filament. Biochemistry. 2011;50:7405–7413. doi: 10.1021/bi200506k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zot HG, Hasbun JE, Van Mihn N. Common Basis for Cellular Motility. ArXiv Doc. 2015 arxiv.org/abs/1511.00123.
- 43.Jaafar N, Girolami F, Zairi I, Kraiem S, Hammami M, Olivotto I. Genetic profile of hypertrophic cardiomyopathy in Tunisia: Is it different? Global Cardiology Science and Practice. 2015;2015:16. doi: 10.5339/gcsp.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Albury AN, Swindle N, Swartz DR, Tikunova SB. Effect of hypertrophic cardiomyopathy-linked troponin C mutations on the response of reconstituted thin filaments to calcium upon troponin I phosphorylation. Biochemistry. 2012;51:3614–3621. doi: 10.1021/bi300187k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martins AS, Parvatiyar MS, Feng HZ, Bos JM, Gonzalez-Martinez D, Vukmirovic M, Turna RS, Sanchez-Gonzalez MA, Badger CD, Zorio DA, Singh RK, Wang Y, Jin JP, Ackerman MJ, Pinto JR. In Vivo Analysis of Troponin C Knock-In (A8V) Mice: Evidence that TNNC1 Is a Hypertrophic Cardiomyopathy Susceptibility Gene. Circulation. Cardiovascular genetics. 2015;8:653–664. doi: 10.1161/CIRCGENETICS.114.000957. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.