

Abstract
Cardiac troponin (cTn) acts as a pivotal regulator of muscle contraction and relaxation and is composed of three distinct subunits (cTnC: a highly conserved Ca2+ binding subunit, cTnI: an actomyosin ATPase inhibitory subunit, and cTnT: a tropomyosin binding subunit). In this mini-review, we briefly summarize the structure-function relationship of cTn and its subunits, its modulation by PKA-mediated phosphorylation of cTnI, and what is known about how these properties are altered by hypertrophic cardiomyopathy (HCM) associated mutations of cTnI. This includes recent work using computational modeling approaches to understand the atomic-based structural level basis of disease-associated mutations. We propose a viewpoint that it is alteration of cTnC-cTnI interaction (rather than the Ca2+ binding properties of cTn) per se that disrupt the ability of PKA-mediated phosphorylation at cTnI Ser-23/24 to alter contraction and relaxation in at least some HCM-associated mutations. The combination of state of the art biophysical approaches can provide new insight on the structure-function mechanisms of contractile dysfunction resulting cTnI mutations and exciting new avenues for the diagnosis, prevention, and even treatment of heart diseases.
Keywords: Cardiac troponin, PKA phosphorylation, hypertrophic cardiomyopathy, mutation, cTnC-cTnI interaction, structure-function relationship
Graphical abstract
Familial hypertrophic cardiomyopathy (HCM) is a common autosomal dominant disease, resulting in increased ventricular wall size that can cause outflow tract obstruction and unexpected sudden cardiac death [1, 2]. It was recently estimated that the prevalence of HCM-associated mutations is 1 per 500 individuals [3, 4]. To date, over 900 mutations associated with HCM have been identified in 24 genes [5, 6]. A large number of these HCM-related mutations reside in thick filament associated proteins (cardiac β-myosin heavy chain, cardiac myosin binding protein C (cMyBP-C), and titin) with fewer in the thin filament proteins actin, tropomyosin (Tm), and cardiac troponin (cTn)) [7-20]. For cTn, the effect of HCM-related mutations has been investigated using various biochemical and physiological approaches. For instance, recombinant cTn subunits containing mutations have been incorporated into reconstituted cTn complexes or thin filaments to study the effects on protein-protein interaction and ATPase activity in vitro [21-23]; adenovirus containing mutation genes have been constructed and transduced into isolated cardiomyocytes to examine effects on intact cardiomyocyte shortening and intracellular Ca2+ handling [24]; and transgenic animal models have been developed to study the disease progression [25-33]. In general, functional studies of many of the HCM-related mutations have been shown to increase the Ca2+ sensitivity of contractile (tension) and ATPase activity, with some exceptions for mutations which produced complex and contradictory results [23, 34].
There is growing evidence that the Ca2+ dependence of contractile properties may not be the only modulatory mechanism affected, for at least some mutations. During β-adrenergic stimulation, protein kinase A (PKA) phosphorylates multiple myofilament proteins, including the residues Ser-23 and Ser-24 of cardiac troponin I (cTnI) [35]. This phosphorylation reduces the Ca2+ binding affinity of cTn and the strength of cTnI interaction with cardiac troponin C (cTnC) [36]. In this review, we specifically focus on HCM-associated mutations of cTnI, and recent studies indicating disruption of the role of PKA-mediated modulation of contraction and relaxation. We speculate that introduction of particular mutations in cTnI may either directly affect the cTnC-cTnI (C-I) interaction (mutations located at the inhibitory/switch peptide regions) or cTnI-actin interaction (mutations located at the C-terminal domain), and thus modulate myosin binding (cross-bridge formation), tension generation and relaxation. We further propose that for at least some mutations, it disrupts the ability of PKA-mediated phosphorylation at Ser-23/Ser-24 of cTnI to alter these contractile properties. Below we first review the structure-function relationship of the cTn and its subunits, and the effects of cTnI phosphorylation by PKA. We then focus on recent studies of how mutations in cTnI that are associated with HCM may alter the regulation of cTnC-cTnI interaction and contractile function. We finish with a short perspective on how a combination of state of the art biophysical approaches can provide new insight on the structure-function mechanisms of contractile dysfunction resulting cTnI mutations and exciting new avenues for studying familial/genetic based disease development in the heart.
Cardiac Troponin – The Gate-Keeper of Contraction
Heart muscle cells (cardiomyocytes) contain a high density of the organelles of contraction, myofibrils, which consist of repeating structural units-named as sarcomeres. Sarcomeres, the smallest contractile units of muscle, are composed of thick filaments that contain the motor protein myosin, and thin filaments composed of double helical strands of actin monomers, a coiled-coil dimer strand of tropomyosin (Tm) and the troponin ternary complex [37]. The cardiac troponin complex (cTn) is a critical regulator of contraction and is composed of three distinct subunits named according to their functions: a highly conserved Ca2+ binding subunit (cTnC); an actomyosin ATPase inhibitory subunit (cTnI) and a Tm binding subunit (cTnT) [37].
Troponin Structure
Troponin C (TnC)
Troponin C (TnC, the Ca2+ binding subunit), is a dumbbell-shaped protein with N- and C-terminal globular domains connected by a long central helix linker, and belongs to the super-family of calmodulin [38-41]. The two globular domains each consist of two helix-loop-helix motifs that bind divalent metal ions [39, 42]. The C-terminal domain of TnC (CTnC) has two high-affinity Ca2+/Mg2+ binding sites (site III and site IV), which are primarily occupied by Mg2+ when muscle cells are at rest. The affinity for these sites for Ca2+ is 107 M-1 [43, 44], which is higher than that for Mg2+ (103 M-1) [43], but the 5 × 103-5 × 104-fold greater intracellular Mg2+ levels (0.5-5 mM) [45], compared with Ca2+ (< 100 nM) in resting cardiac muscle insures that mostly Mg2+ is bound to sites III and IV. During the cardiac twitch, when intracellular Ca2+ levels can rise to 1-10 µM [46], there is likely some transient exchange of Mg2+ for Ca2+. The CTnC domain primarily plays a structural role in anchoring the proteins within the whole Tn complex and to the thin filament [47].
The N-terminal domain of cTnC, found in cardiac and slow-twitch skeletal muscle, only has one active Ca2+-binding site (site II), compared with two active sites in fast-twitch skeletal TnC (sTnC) [48]. These sites are lower-affinity (105 M-1) metal ion binding sites that are highly specific for Ca2+ [43, 44]. N-terminal Ca2+ binding is responsible for initiating thin filament activation and the subsequent generation of force and contraction [48-50]. Several loop residue substitutions in site I of cTnC are responsible for the loss of the ability to bind Ca2+. Studies using demembranated cardiac muscle preparations have demonstrated that replacement of native cTnC with a variant that eliminates site II Ca2+ binding (via mutagenesis) also eliminates the ability of Ca2+ to initiate contraction [51]. Interestingly, replacing native cTnC with an engineered cTnC that has a dysfunctional site II, but repaired Ca2+ binding at site I does not restore the ability of Ca2+ to activate force generation, suggesting site II (other than site I) is critical for the regulatory function of cTnC [50]. However, when cTnC is engineered such that both site I and site II bind Ca2+, it increases Ca2+ sensitivity of contraction over that of native or wild-type cTnC, suggesting site II is not completely effective in activation of cardiac muscle [50].
The structure of the regulatory domain of TnC has been determined in both Ca2+-saturated and Ca2+-free states, as well as in complex with the switch-peptide of TnI [52-56]. Information obtained from these different structures is critical to understanding how Ca2+ binding changes the conformation within the N-terminus domain that initiates the signaling process leading to thin filament activation. In the apo-state, the regulatory domain stays in a closed conformation with most of the hydrophobic residues buried [52]. When Ca2+ binds to the regulatory domain of TnC it induces an opened structure of the hydrophobic patch, facilitating increased interaction with TnI [54]. Interestingly, structural changes in the N-terminal domain of cTnC (NcTnC) in response to Ca2+ binding are much smaller than for NsTnC, such that most of the hydrophobic patch residues remain un-exposed [52]. It has been demonstrated that both Ca2+ binding at site II and interaction with the cTnI switch peptide are required to maintain NcTnC in a stable open state [53, 57]. Ca2+ binding to NcTnC induces a conformational change in the A- and B- helices (“open” conformation) that increases the affinity of this region for the switch peptide of cTnI. The cTnI switch-peptide is adjacent to the cTnI inhibitory region, which binds to actin in the absence of Ca2+ and is release when Ca2+ binds to TnC, allowing increased Tm mobility and myosin interaction with actin to form force generating cross-bridges [37].
Troponin I (TnI)
Troponin I (TnI, the inhibitory subunit) is a highly flexible protein that is able to adapt favorable conformations to interact with both TnC and TnT, as well as with actin [58]. There are three isoforms of TnI expressed in vertebrate striated muscles: the fast and slow skeletal isoforms (sTnI) and the cardiac-specific isoform [59-61]. For cardiac muscle, slow sTnI (ssTnI) is expressed during early embryonic development, but there is a progressive increase in cTnI which completely replaces ssTnI shortly after birth [62]. cTnI is the sole isoform present in the adult heart and it does not switch under pathological conditions [63]. Several studies of rodents and rabbits have demonstrated that the presence of ssTnI results in a higher Ca2+ binding affinity of Tn compared with cTnI [64-67]. Reiser et al. reported that ssTnI is expressed in neonatal myocardium and transitions into cTnI in adult myocardium, and it is associated with the decreased Ca2+ sensitivity in adult myocardium [64]. Westfall et al. reported that ssTnI plays an important role in the reduced pH sensitivity observed in fetal cardiac and slow skeletal muscle with respect to the adult cardiac myocytes, and further demonstrated that the isoform transition of TnI during development may result in decreased Ca2+ sensitivity and increased pH responsiveness of tension development of cardiomyocytes [65]. Using cardiomyocytes from adult transgenic mice, F'entzke et al. found that the overexpression of slow sTnI in the cardiac muscle impaired relaxation (diastolic function) and suggested this was due to enhanced Ca2+ binding affinity [67].
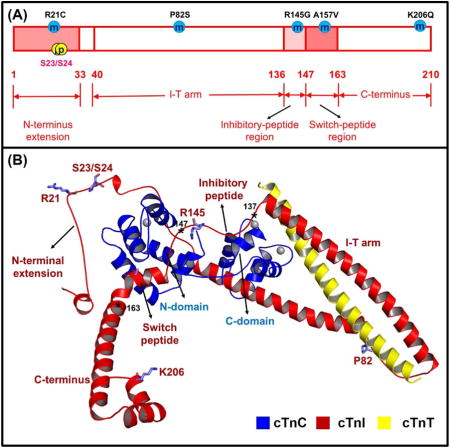
In terms of the structure-function relationship, human cTnI (NCBI protein ID: NP_000354.4) can be divided into five functional regions [36, 59]: (1) the cardiac-specific N-terminal extension (residues 1-33), containing two PKA-dependent phosphorylation sites not present in either fast and slow sTnI; (2) the I-T arm region (residues 40-136), which is composed of two α–helices that interact with the α–helices of C-terminal domain of TnT and the C-lobe of TnC; (3) the inhibitory-peptide region (residues 137-146), that interact with actin-Tm and perhaps C-lobe of TnC; (4) the switch-peptide region (residues 147-163), that interact with the hydrophobic patch of NcTnC; and (5) the C-terminus mobile domain (residues 164-210), which has a second actin-Tm binding site [68-71]. Although the solution nuclear magnetic resonance (NMR) and/or X-ray crystal structures of each cTnI fragment is available, the entire structure of cTnI in the whole cTn complex has still has not been solved.
The cardiac-specific N-terminal extension
There is no crystal structure available for the cardiac-specific N-terminal extension of cTnI (cTnI1-32), however Howarth et al. [72] used solution NMR to determine the structures of both non-phosphorylated and bis-phosphorylated species. In the absence of phosphorylation, the N-terminus extension is less structured, and interacts with the NcTnC. Upon the bis-phosphorylation on Ser-23/Ser-24, the C-terminal α–helix (residues 21-30) is stabilized by electrostatic interactions between the negatively charged phosphorylated serine and the neighboring basic residues. This weakens the interaction between cTnI1-32 and NcTnC, allowing it to be more mobile and increasing potential interaction with other regions of cTn.
The I-T arm region
The I-T arm region (cTnI40-136), which has been considered to play a more structural rather than regulatory function on anchoring the troponin complex onto the thin filament, may form a helical coiled-coil with a portion of C-terminal domain of TnT [73]. The first half part of the I-T arm region (cTnI40-80) has been demonstrated to bind with the hydrophobic cleft of the C-lobe of cTnC [74].
The inhibitory-peptide region
The inhibitory-peptide region (cTnI137-146), is considered a key region of cTnI as it interacts strongly with actin in the absence of Ca2+ and stabilizes Tm in a position that blocks strong myosin binding to actin that is required for contraction [75, 76]. The structure of cTnI137-146 is missing in the core crystal structure of cTn complex solved by Takeda et al. [77], and studies on the structures of this region are not in good agreement [78-80]. Using site-directed spin labeling electron paramagnetic resonance (SDSL-EPR), Brown et al. determined the structure of the inhibitory region of cTnI in the intact cardiac troponin ternary structure, which demonstrated that cTnI129-137 forms a regular 3.6 residues/turn a–helix, and the following region cTnI138-145 displayed no secondary structure [78]. Using the cross-linking and fluorescence-detected resonance energy transfer (FRET) approaches, Tung et al. proposed the inhibitory region is in a (3-hairpin structure [79]. In contrast, Lindhout et al. reported the NMR structure of cTnI128-147 with cTnC89-161, in which cTnI134-139 forms ɑ–helix structure, and cTnI140-147 displayed an extended conformation that potentially interacts with the C-lobe of cTnC [80].
The switch-peptide region
The switch-peptide region (cTnI147-163) is located just next to the inhibitory-peptide region, and acts as another crucial region in cTnI, since it is required to stabilize NcTnC in the “open” conformation. In contrast to the regulatory N-domain of sTnC, binding of Ca2+ to cTnC does not induce an “open” state of the regulatory domain in order to interact with cTnI [52, 55]. Using multinuclear multidimensional NMR spectroscopy, Li et al. determined the solution structure of the NcTnCCa2+cTnI147-163 complex [53]. In this NMR structure, the N-terminus of cTnI147-163 binds to the hydrophobic patch (interface of helices A and B) of NcTnC, thus stabilizing the “open” conformation of NcTnC, similar to that of the Ca2+-saturated NsTnC [54, 81]. Despite the relative position of the switch-peptide region in this binary cTn complex [53] being different than that in the ternary cTn crystal structure (PDB: 1J1E) provided by Takeda et al. [77], the orientation and conformation of switch-peptide with respect to the NcTnC are in a very good agreement.
The C-terminus region
The C-terminus region (cTnI164-210), also known as the mobile region, serves as the second actin-Tm binding site [68, 69]. This region is the most conserved part in TnI among different species and isoforms [56, 70, 71]. According to the core domain of X-ray structure of the whole troponin complex, the C-terminus has an α–helical structure and is free of interaction with either cTnC or cTnT.
Troponin T (TnT)
Troponin T (TnT, the Tm binding subunit), a striated-muscle specific protein with ∼250-300 amino acids with a molecular weight range from 31-kDa to 36-kDa, serves as the structural “glue” that holds the Tn-Tm-actin complex together [82]. Similar to TnI, TnT also has three homologous genes evolved in mammalian striated muscle: the fast and slow skeletal isoforms and the cardiac specific isoform [71, 83]. The expression of the three isoforms in adult striated muscle is strictly regulated by a specific manner according to muscle-fiber types [83]. In addition to expression in cardiomyocytes [84, 85], cardiac TnT (cTnT) also presents in the embryonic skeletal muscle, Duchenne muscular dystrophy and myopathic skeletal muscle [86].
TnT is regarded as a highly asymmetric elongated protein, and can be divided into two functionally distinct regions: the N-terminus T1 region that interacts with Tm [87], and the globular C-terminus T2 region that is integrated into the Tn complex, interacting with both TnC and TnI and anchoring the Tn complex onto the thin filament [82, 83, 88, 89]. The sequences of the middle and the C-terminus portions of TnT are highly conserved among species for all three isoforms, however, the size/length of the N-terminus region is variant among species and isoforms [83]. There is no solved crystal/NMR structure for whole TnT, and only limited structural data are available for partial regions of TnT. The high resolution structure of the T2 region is present in Tn complex structures, showing that the main TnI-TnT interface is the coiled-coil α-helices bundle containing residues Phe-90 to Arg-136 of cTnI and residues Leu-224 to Val-274 of cTnT in the human cTn complex [77], or residues Gly-55 to Leu-102 of fast sTnI and residues Glu-199 to Gln-245 of fast sTnT in the chicken fast skeletal muscle troponin complex [90]. For the T1 region, Ertz-Berger el al. computationally built up an α-helix structure for residues 70-170 (part of the T1 region) of murine cTnT [91]. Manning el al. then docked this cTnT structure onto a known Tm structure and performed large-scaled computational modeling studies [92].
Cardiac Troponin Function
In the absence of Ca2+ (diastole), cTnC exists in its “closed” conformation, and cTnI binds actin tightly, inhibiting actin-myosin interaction (Fig. 1A) [37, 55]. During systole, depolarization of cardiomyocyte membranes results in Ca2+-induced Ca2+ release from the sarcoplasmic reticulum (SR). In turn, the rise in intracellular Ca2+ results in Ca2+ binding to cTnC and this initiates a chain of events involving dynamic and structural changes in cTn, Tm, actin and myosin (Fig. 1B-D) [37]. Ca2+ binding to site II of cTnC reduces the stability of the “closed” conformation of the N-terminus half of the protein and increases interaction with the cTnI switch-peptide [37, 52]. In turn, this results in an “open” conformation of cTnC, exposing hydrophobic residues that increase interaction with the switch-peptide of cTnI, while decreasing the interaction between the (adjacent) inhibitory-peptide domain of cTnI with actin (Fig. 1B) [37]. Consequently, this allows increased Tm mobility and exposure of myosin binding sites on actin, allowing the formation of actin-myosin “cross-bridges” (Fig. 1C) [37]. In cardiac muscle, strong cross-bridge binding increases Ca2+ binding to cTn, allowing additional myosin binding, resulting in strong allosteric (cooperative) cross-bridge formation, cycling and force generation (Fig. 1D) [93-96]. When Ca2+ dissociates from cTn, it is primarily re-sequestrated into the SR, thus when myosin dissociates from thin filaments there may be a negative feedback decrease in cTn Ca2+ affinity that accelerates relaxation. Thus, cTn acts as a gate-keeper of contraction and a logical point for fine control of cardiac contractile performance, so investigating its structure and function relationship can be very informative for understanding contractile dysfunction with at least some mutations associated with cardiac disease.
Figure 1.
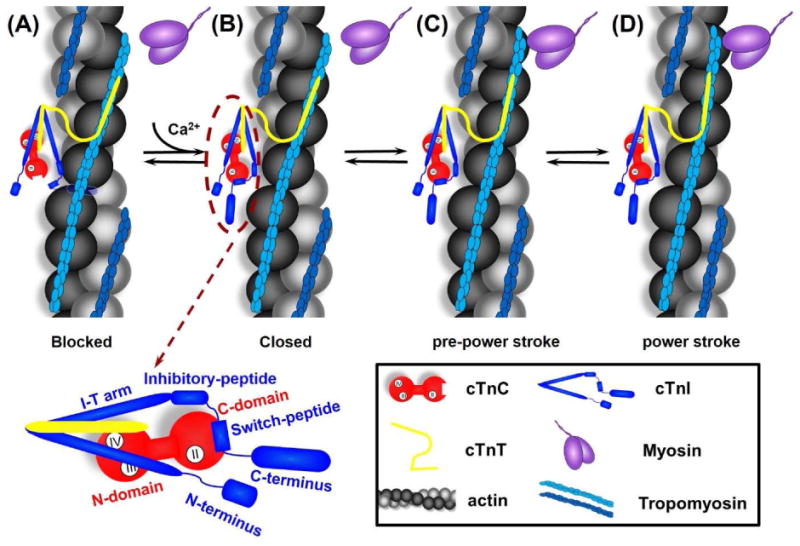
Cartoon model of the signaling pathway for thin filament activation. From left to right are: (A) In diastole (in the absence of Ca2+), cTnC exists in its “closed” conformation, and the inhibitory-peptide of cTnI binds actin tightly, inhibiting actin-myosin interaction. (B) In systole, Ca2+ binding to site II of cTnC induces an “open” conformation and increases interaction between NcTnC and cTnI switch-peptide, which pulls the adjacent inhibitory-peptide of cTnI from binding with actin. (C) Consequently, this leads to increased Tm mobility, exposes the myosin binding site on actin and myosin weakly interaction with actin (pre-power stroke), and (D) This weakly interaction further pushes Tm, and myosin strongly binds with actin, allowing the formation of cross-bridges and thus force generation (power stroke). The left-bottom is the close-up of cTn complex with the labeling of specific region of cTnC and cTnI.
Post-translational Modification of cTnI
One method of modulating contraction via cTn is phosphorylation of key amino acids. Post-translational modification of cTnI by phosphorylation plays a key role in regulating the function of cTnI by precipitating structural changes that alter its interaction with cTnC or actin and thus influence cardiac muscle function. It is important to point out that the sequence and length of cTnI varies among species, thus the discrepancy in residue number may actually refer to the same residue. For example, in rodent, there is one redundant residue (Ala-25) in the N-terminus of cTnI, thus the residue number of cTnI after this residue will change (+1) correspondingly. There are multiple sites on human cTnI (NCBI protein ID: NP_000354.4) that can be phosphorylated by various kinases (Fig. 2B), including Ser-5/Ser-6, Ser-23/Ser-24, Tyr-26, Thr-31, Ser-42/Ser-44, Thr-51, Ser-77/Thr-78, Thr-143, Ser-150, Ser-166, Thr-181 and Ser-199 (the numbering includes the initial Met-1), in which Thr-31 can only be phosphorylated in vitro, and phosphorylation of Ser-150 has never been observed in the human heart. Ser-42/Ser-44, Thr-143 and Ser-199 are all substrates of protein kinase C (PKC). Phosphorylation of Ser-42/Ser-44 has been reported to depress maximal tension, decrease the cross-bridge cycling kinetics, and reduce thin filament sliding velocity in the motility assay and ATPase rate [97-99]. Wang et al. reported that phosphorylation of Thr-144 in mouse heart (NCBI protein ID: NP_033432.1) increase the Ca2+ sensitivity of contraction [100], while Burkart et al. and Lu et al. reported Thr-144 phosphorylation or glutamic acid substitution (T144E) in mouse heart did not change the thin filament Ca2+ sensitivity [98, 101]. It has also been reported that phosphorylation of this site depressed the acto-myosin ATPase activity and contractility [102, 103], and depressed the cooperative activation of the thin filament [101]. Ser-150 can be phosphorylated by either P21-activatedkinase (Pak) or AMP-activated Protein Kinase (AMPK), which increased the Ca2+ sensitivity of force development [104], prolonged the relaxation [105], and increased the adrenergic-induced myocardial hypertrophy [106]. Thr-31, Thr-51 and Thr-181 of human cardiac cTnI can all be phosphorylated by mammalian sterile 20-like kinase 1 (Mst1) in vitro, and Thr-31 serves as the preference site [107]. Recently, Zhang et al. identified several novel phosphorylation sites on human cTnI (Ser-5/Ser-6, Tyr-26, Thr-51, Thr-181 and Ser-199), and found that in heart failure patient there was a decrease in the extent of phosphorylation sites Ser-5/Ser-6/Tyr-26 that located in the N-terminus extension, and an increase in the phosphorylation sites Ser-77/Thr-78 at the I-T arm region and Ser-166/Thr-181/Ser-199 resided at the C-terminal domain [108]. Henze et al. reported that aspartic acid substitution of Ser-6 (S6D) in mouse heart significantly depressed the maximal force, ATPase rate, kinetics of force redevelopment and did not change the Ca2+ sensitivity of force development, whereas aspartic acid substitution of Ser-5 (S5D) had no effect on all those parameters [109]. Salhi et al. demonstrated that Tyr-26 phosphorylation, glutamic acid substitution (Y26E) or aspartic acid substitution (Y26D) decreased the Ca2+ binding to thin filament, decreased the Ca2+ sensitivity of force development (by exchanging the recombinant human cTn into skinned rat myocyte), and accelerated the Ca2+ dissociation rate from cTnC [110]. Wijnker et al. found that pseudo-phosphorylation of Ser-199 (S199D) in human cardiomyocytes increased the Ca2+ sensitivity of force development, and did not affect the maximal and passive forces, and the rate of force redevelopment [111].
Figure 2.
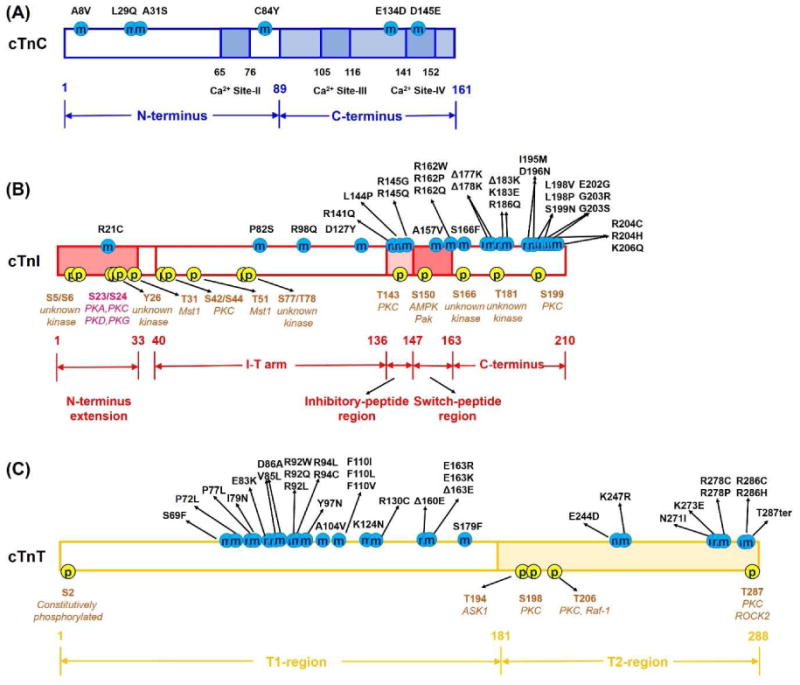
The structural/functional domain and HCM-associated mutations in human (A) cTnC, (B) cTnI and (C) cTnT subunits. Here, the residue number of cTnI corresponds to that human sequence including Met-1.
Among all the phosphorylation sites, Ser-23/Ser-24 (S23/S24) in the cardiac-specific N-terminus of cTnI have been the most studied and their modulatory function is best understood. β-adrenergic stimulation serves as a major physiological mechanism to meet the increase in circulatory demand, acting through positive inotropic and lusitropic effects [35, 112]. It is well established that during β-adrenergic stimulation, cTnI is phosphorylated by PKA at sites S23/S24, and that this affects Ca2+-mediated contraction of cardiac muscle [35]. However, S23/S24 can be phosphorylated by other kinases in vitro, including PKC [113], protein kinase D (PKD) [114-116] and GMP-dependent protein kinase (PKG) [117]. Phosphorylation at these sites has been shown to increase the Ca2+ dissociation rate from cTnC, weaken the interaction between cTnI and the N-terminus of cTnC, reduce the Ca2+ sensitivity (pCa50) of cardiac muscle tension production, increase cross-bridge cycling kinetics, and accelerate cardiac muscle cell relaxation [35, 36, 118-124]. Zhang et al. demonstrated that PKA phosphorylation of a cardiac skinned muscle results in decreased Ca2+ sensitivity of the contractile apparatus, and increased the rate of relaxation [35]. Kentish et al. also reported that PKA phosphorylation accelerated myofibrillar relaxation rate and suggested this was due, at least in part, to faster cross-bridge cycle kinetics [119]. In support of this, we reported that either PKA phosphorylation of isolated rat cardiac myofibrils or exchange of cTn containing cTnI with phosphor-mimetic S23D/S24D accelerates and shortens the initial slow-phase of relaxation that is thought to reflect the rate cross-bridge detachment, the rate limiting transition in loaded cross-bridge cycling [125]. Additionally, we have demonstrated that reduction of cTn Ca2+ binding affinity and/or cTnC-cTnI binding affinity reduces the duration of the initial slow-phase of relaxation [125]. Thus, together the data suggests that accelerating relaxation is a primary functional role of phosphorylation at Ser-23/Ser-24.
cTn Mutations Associated with Familial Hypertrophic Cardiomyopathy (HCM)
Since the first mutations associated with HCM were detected in 1993 [10, 20], there has been a dramatic increase in the number of cTn mutations reported. Thus far, a total of at least 69 mutations have been identified in cTn subunit proteins that have been reported to be associated with HCM, including 6 in cTnC (Fig. 2A) [19, 126, 127], 30 in cTnI (Fig. 2B) [5, 16, 128-137], and 34 in cTnT (Fig. 2C) [9, 15, 129, 137-143]. All of the mutations are single nucleotide polymorphisms (SNPs or variants), and the degree of cardiac pathology associated with them is highly variable as is verification of their causal nature. There is also considerable variation in the extent to which these variants have been studied. Functional studies have been performed using tissue from patients, transgenic animal models and recombinant proteins in solution or exchanged into cardiac tissue. Measurements have primarily been made of Ca2+ binding affinity and ATPase activity in solution and the Ca2+ sensitivity of steady-state contraction in demembranated cardiac muscle, though an increasing number of studies are reporting on the kinetics of contractile activation and relaxation. Many (but not all) of the HCM-associated cTn mutations increase the Ca2+ binding affinity of cTn (KCa), paralleled with enhanced Ca2+ sensitivity of ATPase activity and thin filament activation (force development). Considering the central role of cTn in muscle regulation and contraction, introduction of a mutation into cTn may potentially influence interactions between cTnC-cTnI-cTnT, and the activation signal pathway transmitted from cTn to the whole thin filament. In this section, we focus primarily on HCM-associated mutations identified in cTnI. It is interesting that most of the cTnI mutations are located at the inhibitory/switch-peptide region and the C-terminus segment of cTnI, which suggests mutations at the inhibitory/switch-peptide regions may directly influence the interaction of cTnI with NcTnC, while the mutations at the C-terminal domain may directly affect the cTnI-actin interaction (thus indirectly influencing cTnC-cTnI interaction). As mentioned above, when Ca2+ binds to the regulatory domain of cTnC, it strengthens the cTnC-cTnI interaction, and weakens the cTnI-actin interaction, thus allowing the formation of actin-myosin cross-bridge and force generation. As such, all the mutations in the inhibitory/switch-peptide and C-terminus regions of cTnI could directly influence the equilibrium between cTnC-cTnI and cTnI-actin interaction, and thus modulate myosin binding (cross-bridge formation), tension generation and relaxation.
It is important to mention that the sequence and length of cTn subunits vary among species, thus the discrepancy in residue number may actually refer to the same residue. For example, in rodent, there is one redundant residue (Ala-25) in the N-terminus of cTnI, thus the residue number of cTnI after this residue will change (+1) correspondingly. Using skinned muscle fibers, Takahashi-Yanaga et al. studied the function of six HCM-associated cTnI mutations (R145G, R145Q, R162W, ΔK183, G203S, and K206Q) and found that, with the exception of cTnIG203S (which showed a tendency, but without statistical significance), all the other mutations significantly increased the Ca2+ sensitivity of myofibrillar ATPase activity and force generation [144]. Elliott et al. reported that both cTnIR145G and cTnIR162W mutations reduced the inhibition of actin-Tm-activated myosin ATPase, and significantly increased the Ca2+ sensitivity of ATPase activity, suggesting both mutations may impair relaxation in addition to enhancing contraction [145]. Using the in-vitro motility assay with recombinant rat cTn subunits, Kohler et al. investigated the effect of three HCM-associated cTnI mutations (ΔK183, G203S, and K206Q) on the Ca2+ regulation, and found that all three mutants enhanced the Ca2+ sensitivity and maximal speed of filament sliding [146]. Among all HCM-related cTnI mutations, the cTnIR145G (cTnIR146G in rodent), located in the inhibitory-peptide of cTnI, has received the most prominent attention [29, 102, 144, 145, 147-156]. There are complex and sometimes contradictory results from the literatures for the effects of cTnIR145G on the maximal force production and the Ca2+ sensitivity of force generation. For example, James et al. reported that cTnIR146G Tg mice showed a significant increase in the Ca2+ sensitivity of force and a significant depress in maximal tension when compared with the NTG littermate controls [156]. Takahashi-Yanaga et al. also reported that the cTnIR145G resulted in an increase in the Ca2+ sensitivity of force generation and myofibrillar ATPase activity in skinned muscle fibers [144]. However, incorporation of recombinant human cTnIR145G into guinea-pig cardiac trabeculae skinned fibres had no effect on Ca2+ sensitivity [155]. Similarly, using recombinant human cTnIR145G exchanged into murine myofibrils, Kruger et al. reported no change in the Ca2+ sensitivity of tension development [151]. They also reported a slightly decreased Ca2+ sensitivity of force in myofibrils from transgenic cTnIR146G mice [151]. The reasons for different results reported for this mutation (cTnIR145G) on the Ca2+ sensitivity of tension are unclear. However, many factors can affect the Ca2+ sensitivity of tension, such as species-specific differences, exchange efficiency (mutant to wild-type ratio), solution pH and temperature. Additionally, using the reconstituted actin-tropomyosin activated myosin ATPase assay, Lang et al. [148], Takahashi-Yanaga et al. [144], and Elliot et al. [145] reported that cTnIR145G decreased maximal ATPase in the presence of Ca2+, and reduced inhibition of actomyosin ATPase activity in the absence of Ca2+. cTnIR21C is the only identified HCM-associated mutation located at the cardiac-specific N-terminus of cTnI [30, 157-159]. Using transgenic mice, Wang el al. reported that the cTnIR21C mutation prevented PKA-mediated phosphorylation in vivo [30]. By replacing the recombinant human cTnI into porcine muscle, Gomes et al. reported that cTnIR21C significantly increased the Ca2+ sensitivity of force development with respect to the cTnIWT [157]. He also reported that compared to cTnIWT, PKA phosphorylation of fibers containing cTnIR21C resulted in a significantly smaller decrease in the Ca2+ sensitivity of force generation, which is attribute to the decreased ability of PKA to phosphorylated cTnIR21C at S23/S24 [157]. Dweck et al. found that the isolated cardiac myocytes from R21C transgenic mice (with ages older than 12 months) significantly delayed Ca2+ transient decay and relaxation [158]. The phenotype of cTnIR21C mutation supports the regulatory role of cTnI N-terminus in diastolic function of the heart. Very recently, Ramirez-Correa et al. studied the cTnIP82S mutation, which was initially considered as a disease-causing mutation, however, later studies suggested the contrary [28]. Using transgenic mice, they found that cTnIP83S prolonged the isovolumetric relaxation time, impaired ejection and relaxation time, as well as blunted the β–adrenergic response and impaired myofilament cooperativity [28]. Similar to these cTnI mutations, several (but not all) HCM-associated mutations in cTnC (A8V, L29Q, A31S, C84Y and D145E) and cTnT (F110I, R92L/W/Q, R94L, A104V, R130C, Δ160E, E163R, S179F and E244D) have been demonstrated to increase Ca2+ sensitivity of ATPase activity and/or force development [31-33, 126, 160-184]. Pinto et al. reported that cTnCA8V and cTnCD145E significantly slowed Ca2+ dissociation kinetics (koff) in solution, suggesting both mutations may alter muscle relaxation properties [178]. Considering that most of the findings of these HCM mutations were focused on the Ca2+ sensitivity of ATPase activity and/or force development, it is of importance to also investigate how they influence the muscle relaxation.
Recently, we reported that both cTnIR146G and cTnIR21C mutations (based on rat sequence) alter not only normal contractile properties, but also blunted the modulatory capacity of S23/S24 PKA-mediated phosphorylation during β-adrenergic stimulation [185]. We [185] and others [30, 158] have demonstrated that cTnIR21C disrupts the PKA phosphorylation on S23/S24 of cTnI, and thus results in a “blunted” β-adrenergic stimulation effects, which may be the actual physiologic/pathogenic mechanism of cTnIR21C. So, to understand whether it is the cTnIR21C per se that is altering function or just the inability to get S23/S24 phosphorylated, we constructed the bis-phosphomimic substitutions S23D/S24D into cTnIR21C (cTnIR21C/S23D/S24D) to mimic effect of PKA phosphorylation. Using steady-state fluorescence measurements, we found that both mutations significantly increased Ca2+ binding affinity to cTn (KCa) and the affinity of cTnC for cTnI (KC-I). PKA phosphorylation of cTnI (or introduction of bis-phosphomimic substitutions on cTnI, cTnIS23D/S24D) resulted in a similar reduction of KCa for all complexes, but the reduction in KC-I seen with cTnIWT did not occur for either mutation in cTnI. When recombinant cTn containing either mutation was exchanged into isolated adult rat cardiac myofibrils they caused an increased Ca2+ sensitivity of tension (pCa50) as reported by others [29, 144, 145] and prolonged the duration of the early, slow-phase of relaxation. Interestingly, PKA phosphorylation of cTnI resulted in decreased pCa50 for cTnIWT exchanged myofibrils, but not for either mutation. PKA phosphorylation also accelerated the early, slow-phase relaxation for myofibrils with cTnIWT, especially at Ca2+-levels that the heart operates in-vivo. Importantly, this effect was blunted for cTnIR146G and cTnIR21C exchanged myofibrils. Interestingly, this blunting of PKA-mediated effects has also been reported for another HCM-associated mutation, cTnCL29Q. Dong et al. found that cTnCL29Q abolished the enhancement of closing rate of NcTnC (that is triggered by Ca2+ dissociation) upon PKA phosphorylation of cTnI [186]. In addition, Schmidtmann et al. [181] and Li et al. [179] reported that cTnCL29Q hindered the effect of PKA phosphorylation of cTnI on transduction the signal from cTnC to cTnI. Together these findings suggest that, for at least some HCM-related mutations, an additional contractile abnormality may be blunting (or uncoupling) of myofilament response to the PKA-mediated phosphorylation of cTnI during β-adrenergic stimulation. The solution measurements of cTnI affinity for cTnC suggest this may be due to an inability of S23/S24 phosphorylation to weaken this interaction, an idea that is supported by molecular dynamics modeling studies of cTn discussed in the next section.
Computational Studies on the cTn Structure and HCM-associated cTn Mutations
Understanding the structural changes in proteins that result in changing function is beneficial in gaining a firm understanding of the molecular mechanisms involved in modulating contraction and its dysfunction with disease. The correlation between specific alterations in the cTn structure by introduction of a single amino-acid mutation that results in complex functional phenotypes remains poorly understood. NMR and X-ray crystallography have shed light on the atomic level structures of the cTnC regulatory domain and the cTn complex [52-56, 77]. Li et al. solved the NMR structures of the regulatory domain of cTnC (NcTnC, residues 1-89) in both Ca2+-saturated and Ca2+-free states, and the regulatory domain of cTnC in complex with the switch-peptide of cTnI [53]. Based on these NMR structures, Wang et al. applied integrated experimental-computational approaches to study the interactions of cTnC variants with altered Ca2+ binding affinities with the switch peptide of cTnI [187, 188]. Lindert and Kekenes-Huskey et al. studied the dynamics and Ca2+ association to NcTnC via both conventional and accelerated molecular dynamics (MD) simulations [189, 190], as well as the exposure dynamics and kinetics of cTnC hydrophobic residues via micro-second MD simulations [191]. In 2003, Takeda et al. determined the first crystal structure of the human cTn complex (with residues cTnC 1-161, cTnI 40-191, cTnT 202-276) in the Ca2+-saturated form [77], making it possible for the first time to study detailed interactions between cTn subunit proteins. Using FRET techniques in combination with MD simulations, Jayasundar et al. studied the molecular details of how a Ca2+ signal, received at cTnC, is transmitted to cTnI [192]. Varughese et al. also applied this structure to study the binding of the drug Bepridil to cTnC [193], designed a set of potential cTnC-binding ligands [194], and studied the interactions and correlated motions among the three components of cTn [195].
All of the above studies used either partial models of cTnC in the presence/absence of Ca2+ and/or the cTnI switch-peptide, or the cTn model without the N-terminus of cTnI. Recently, using solution NMR, Howarth et al. [72] solved the structure of the N-terminus of cTnI, for both the non-phosphorylated and bis-phosphorylated (at position Ser-23/Ser-24) species, thus making it feasible to study PKA-mediated phosphorylation of cTnI employing computational models. We recently built up the structure of the cTn complex (including residues cTnC 1-161, cTnI 1-210, and cTnT 236-285) containing the cardiac-specific, N-terminus of cTnI (Fig. 3) [196]. This structure also included the inhibitory-peptide (residues 138-148) and the C-terminus (residues 192-210) of cTnI, and the C-terminal residues (residues 282-285) of cTnT that were not present in Takeda's crystal structure 1J1E or Howarth's NMR structure. Using this constructed model, we performed triplicate MD simulations of cTnI-WT and cTnI-S23D/S24D containing cTn models to elucidate the effect of PKA phosphorylation on cTn structure and Ca2+ binding. The most significant finding was that introduction of the bis-phosphomimic substitutions onto Ser-23/Ser-24 (S23D/S24D) led to formation of an intra-subunit interaction between the N-terminus and the inhibitory-peptide of cTnI that may destabilize the interaction of NcTnC hydrophobic residues with the switch-peptide residues of cTnI [196].
Figure 3.
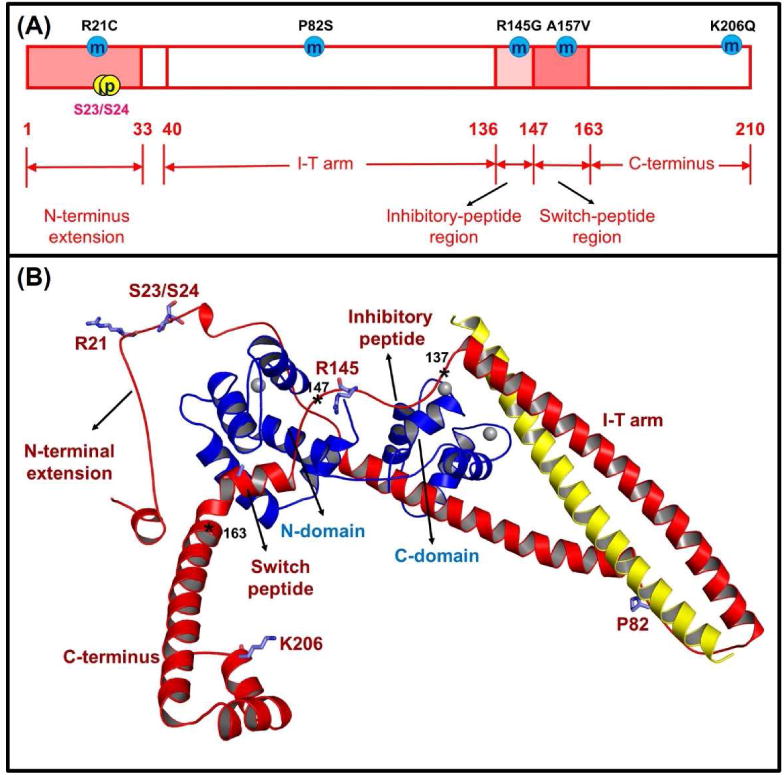
Human cTnI (A) sequence and (B) ternary structure with the some HCM-related mutation sites located at different regions of cTnI (R21C, P82S, R145G, A157V and K206Q) and PKA phosphorylation sites (S23/S24). In the ternary structure, the cTnC (1-161) is shown in blue, the cTnI (1-210) is in red, and the cTnT (236-285) is in yellow. The asterisks indicate three key positions in human cTnI, for which residues 137-146 is the inhibitory-peptide region of cTnI, residues 147-163 is the switch-peptide region of cTnI, and residues 164-210 belong to the C-terminus of cTnI.
This intra-subunit interaction of cTnI has previously been suggested by Solaro and colleagues based on solution biochemical and spectroscopic studies [36, 72, 197]. Howarth et al. applied the comparative docking to determine the low energy structure between the NMR structure of cTnI1-32 in the bis-phosphorylated state and the Takeda's crystal structure of cTn [72]. They proposed that in the low energy structure, cTnI1-32 resulted in weakening interaction with NcTnC and re-positioning the cTnI1-32 for favorable interactions with basic regions of cTnI, and most likely the inhibitory-peptide region of cTnI [72]. Specifically, the acidic amino acids (Asp-3, Glu-4, Asp-7 and Glu-11) of N-terminus interacted with residues Arg-142 and Arg-146 of the inhibitory-peptide region [72]. Sadayappan et al. generated a cardiac-specific transgenic mice in which residues 2-11 of cTnI (cTnI (Δ2-11)) were deleted, and tested the hypothesis that the acidic N' region of cTnI help regulate myocardial function [197]. Compared to the non-transgenic heart, the cTnI (Δ2-11) transgenic mice had significantly decreased contraction and relaxation under basal and β-adrenergic stress, accompanied by a reduction in the maximal Ca2+-dependent tension and Ca2+-activated Mg2+-ATPase activity. However, the Ca2+-sensitivity of force development and PKA phosphorylation of cTnI-S23/S24 were not affected, suggesting that the acidic N' region of cTnI does not play a direct role in the Ca2+-induced transition in NcTnC. The cTnI (Δ2-11) transgenic mice also had decrease in myocardial contractility, demonstrating the importance of acidic N' region in modulating contractility and mediating the response of the heart to the β-adrenergic stimulation. These results provide further support for a role of the acidic N-terminus in regulating cardiac contractility and mediating the response of the heart to β-adrenergic stimulation by interacting directly with the inhibitory-peptide region of cTnI. Based on these finding and the NMR structural data, Solaro et al. predicted that the phosphorylated N-terminal extension of cTnI can interact with the inhibitory-peptide region of cTnI [36].
Our MD simulations also suggest a bending at the N-terminal extension of cTnI and a more compact cTn structure upon introducing the bis-phosphomimic substitutions that is consistent with previous biochemical studies studying bis-phosphorylation of cTnI S23/S24 (cTnIpS23/pS24) on overall cTn structure [198-200]. Using fluorescence studies, Dong et al. demonstrated that bis-phosphorylation resulted in a reduction of the axial ratio of cTnI and the formation of a more compact structure [198]. Using small-angle neutron scattering, Heller et al. found that cTnIpS23/pS24 induced a dramatic bending of the rod-like cTnI at its N-terminus that binds with cTnC, resulting in a dramatic decrease in the axial ratio of cTnI and the cTn complex overall [199]. In addition, using surface plasmon resonance (SPR), Reiffert et al. determined that upon PKA phosphorylation, the shape of cTnI changed from an asymmetrical structure to a more symmetrical one, which is consistent with the bending that results in a shorter and broader structure [200]. All of the above experimental findings suggest the formation of an alternative binding pattern for the bis-phosphorylated cardiac-specific N-terminal extension.
The relationship between cTnC-cTnI interaction and the blunted PKA effects for some HCM-related cTnI mutations
As mentioned above, cTnC-cTnI (C-I) interaction plays a ‘gatekeeper’ role in transmitting the Ca2+ signal to other myofilament proteins to initiate cardiac muscle contraction. Structural changes of the N-domain in cTnC in response to Ca2+ binding are different from those found for sTnC [52-54, 57]. For the sTnC, Ca2+ binding to its regulatory domain induces a transition from “closed” to “open” structure of the hydrophobic patch, facilitating sTnI binding and muscle activation [54]. However, Ca2+ binding to cTnC does not result in a similar structural alteration (hydrophobic patch exposure is minimal), and interaction with the cTnI switch-peptide is also required to expose and stabilize the hydrophobic patch [52, 53, 57]. Studies of C-I interaction also offer insight into how mutations in troponin may result in cardiac contractile dysfunction, and provides insight into potential targets for therapeutic agents that could mediate the Ca2+ sensitivity of the myofilaments in diseased hearts.
Using steady-state fluorescence measurements, we determined the effects of PKA phosphorylation (or bis-phosphomimic substitutions) on cTn-Ca2+ binding affinity (KCa) and cTnC-cTnI affinity (KC-I) in the absence of confounding influences of other myofilament proteins [125]. We found that both KCa and KC-I were decreased upon PKA phosphorylation of cTnI or introduction of the bis-phosphomimic substitutions on cTnI (cTnIS23D/S24D) [125]. By exchanging the recombinant cTn complex into demembranated trabeculae and isolated myofibrils, we studied the effects on myofibril activation/relaxation kinetics [125, 201]. We demonstrated that PKA phosphorylation of cTnI-S23/S24 (or bis-phosphomimic substitutions) decreased the Ca2+ sensitivity of force production and, most importantly, accelerated the initial slow-phase relaxation for rat left ventricular myofibrils, especially at the sub-maximal Ca2+ levels that heart operates in vivo [125, 201]. These findings suggest the strength of cTnC-cTnI interaction modulates the kinetics of thin filament activation, the magnitude of tension development and the initial phase of myofibril relaxation. Our computational results suggested that this may be the result of formation of an intra-subunit interaction between the N-terminus and the inhibitory-peptide of cTnI [196]. We hypothesized that this intra-subunit interaction may weaken interactions between the switch-peptide of cTnI and NcTnC, allowing stronger interaction between the cTnI inhibitory-peptide and actin (move the equilibrium towards thin filament deactivation), and this may provide the structural basis for how PKA phosphorylation of cTnI modulates cross-bridge activity and relaxation.
Based on our findings, we further hypothesized that introduction of a mutation located in either the inhibitory-peptide or the N-terminus of cTnI may disrupt the formation of this intra-subunit interaction and blunt the effects of PKA-mediated phosphorylation during β-adrenergic stimulation. Recently, we tested this hypothesis by studying two HCM-associated mutations, cTnIR146G and cTnIR21C that are located in the inhibitory-peptide and the N-terminus of cTnI (respectively) using combined steady-state fluorescence measurements, myofibril kinetics/mechanics measurements and computational modeling [185, 202]. We found that both mutants increased KCa and KC-I in solution, increased the Ca2+ sensitivity of myofibril tension development (pCa50), and also prolonged the early, slow-phase of relaxation. Importantly, both mutants blunted the ability of PKA to reduce KC-I and pCa50 and speed relaxation of myofibrils [125]. It is worth mentioning that cTnIR21C disrupts the PKA phosphorylation on S23/S24 of cTnI [30, 158, 185], and thus results in a “blunted” β-adrenergic stimulation response, which may be the actual physiologic/pathogenic mechanism of cTnIR21C. Therefore, to further understand whether it is the cTnIR21C per se that is altering function or just the inability to get S23/S24 phosphorylated, we introduced the bis-phosphomimic substitutions S23D/S24D into cTnIR21C (cTnIR21C/S23D/S24D) to mimic effect of PKA phosphorylation. Our results indicated that even with bis-phosphomimic substitutions (‘forced phosphorylation’), the phosphorylation mediated effects on KC-I and myofibril relaxation were still blunted, suggesting that the cTnIR21C mutation per se results in the cardiac dysfunction of modulation by phosphorylation, similar to our results for the cTnIR146G mutation. Our computational results of cTn suggested that introduction of either mutation inhibited the formation of the intra-subunit interaction between the N-terminus and the inhibitory-peptide of cTnI that normally seen for cTn with the bis-phosphomimic substitutions of S23/S24 [196]. Additionally, both mutations abrogated the destabilization of contacts between the cTnI switch-peptide and hydrophobic residues in the N-terminal lobe of cTnC. This suggests a potential structure-based mechanism of how both mutations can impair PKA regulation of contraction and relaxation. Furthermore, it suggests that the degree of cTnC-cTnI interaction may act as a regulator that is modulated by changes in either Ca2+ or cTnI phosphorylation during β-adrenergic stimulation of the heart.
Summary and Future Directions
The cTn ternary complex is the key regulatory protein complex of myofilament contraction and tension generation. Studying how disease-related mutations affect conformational changes in cTn provides insight as to how Ca2+-mediated signal transduction may be altered and result in contractile dysfunction associated with cardiac disease (the correlations between genotype and phenotype). Mounting evidence obtained from the biochemical/biophysical, physiological and computational studies have shed light on understanding the structure-function relationships, regulation, post-translational modification and pathogenicity of mutations from cTn subunits.
Our recent findings suggest that for at least two HCM-associated cTnI mutations (cTnIR146G and cTnIR21C) there is a blunting of the ability of PKA-mediated phosphorylation to modulate both contraction and relaxation [185]. Considering the specific locations of both mutations (cTnIR146G in the inhibitory-peptide and cTnIR21C in the N-terminus), it was not surprising that both mutations inhibited formation of the intra-subunit interaction between the N-terminus and the inhibitory-peptide of cTnI that normally seen with phosphorylation. Whether this is the case for other mutations in cTnI or the other cTn subunits remains to be determined. We are currently studying three additional HCM-associated mutations (cTnIP82S in the I-T arm region, cTnIA157V in the switch-peptide region and cTnIK206Q in the C-terminus region) using the combined protein biochemistry, myofibril kinetics/mechanics measurements, and computational modelling approach.
Computational models with detailed structural and temporal information can provide invaluable mechanistic interpretations of experimental data, enabling extended understanding of how disease-associated mutations result in functional changes of the thin filament or whole sarcomere at an atomic-based structural level. Even more complete models, such as the models of Schwartz and colleagues [92] that computationally study the whole thin filament (with cTn, Tm, and actin) and potential future models with atomistic details and dynamics of multiple thin and thick filaments in a sarcomere can be even more enlightening. This more complete overall picture, based on the view from the structural basis, will assist in the interpretation of the physiological findings.
Computationally studies can also act as a powerful strategy to assist and guide the design of engineered troponin variants as a strategy to treat muscle disease. Based on the current computational models, we and others have designed engineered cTnC variants with altered Ca2+ binding affinities (to cTn) and demonstrated that those variants alter the contractile function in cardiomyocytes and correct or prevent disease-related aberrant Ca2+ binding and contractile function [163, 187, 188, 203-209]. These studies also provide important implications with respect to the design of Ca2+ sensitizing or desensitizing small molecules or drugs. Altering cTnC-cTnI interaction may be a promising target to reduce or reverse the hypertrophic signaling that results from the pathological myofilament protein mutations, or strengthen contraction to counter reduced systolic function in hearts with dilated cardiomyopathy. Taken together, considering the central role of troponin in muscle contraction and regulation, further elucidation the structure-function relationship can provide powerful strategies for the diagnosis, prevention, and even treatment of heart diseases.
Highlights.
cTn, one component of the thin filament, plays a central role of muscle regulation and contraction.
During β-adrenergic stimulation, cTnI is phosphorylated by PKA at sites S23/S24 located in the cardiac-specific N-terminus.
This phosphorylation weakens the interaction between cTnI and the N-terminus of cTnC, reduces the Ca2+ sensitivity (pCa50) of tension production, increases crossbridge cycling kinetics and accelerates cardiac muscle cell relaxation.
Mutations in cTnI that have been identified as associated with hypertrophic cardiomyopathy may disrupt some or all of these PKA mediated modulatory effects.
For at least some HCM mutations, the “blunting” of effects that PKA-mediated phosphorylation on contraction and relaxation is associated with a loss of the ability of cTnI Ser-23/24 phosphorylation to reduce the affinity of cTnI for cTnC.
Acknowledgments
MR is an Established Investigator of the American Heart Association (AHA). This work was supported by HL111197 (MR), and AHA award 15POST25080292 (YC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Maron BJ, Roberts WC, McAllister HA, Rosing DR, Epstein SE. Circulation. 1980;62:218–229. doi: 10.1161/01.cir.62.2.218. [DOI] [PubMed] [Google Scholar]
- 2.Teare D. Br Heart J. 1958;20:1–8. doi: 10.1136/hrt.20.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Circulation. 1995;92:785–789. doi: 10.1161/01.cir.92.4.785. [DOI] [PubMed] [Google Scholar]
- 4.Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Hl UKR, Maisch B, McKenna WJ, Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L, Keren A. Eur Heart J. 2008;29:270–276. doi: 10.1093/eurheartj/ehm342. [DOI] [PubMed] [Google Scholar]
- 5.Mogensen J, Murphy RT, Kubo T, Bahl A, Moon JC, Klausen IC, Elliott PM, McKenna WJ. J Am Coll Cardiol. 2004;44:2315–2325. doi: 10.1016/j.jacc.2004.05.088. [DOI] [PubMed] [Google Scholar]
- 6.Tester DJ, Ackerman MJ. Annu Rev Med, Annual Reviews, Palo Alto. 2009:69–84. doi: 10.1146/annurev.med.60.052907.103838. [DOI] [PubMed] [Google Scholar]
- 7.Bonne G, Carrier L, Bercovici J, Cruaud C, Richard P, Hainque B, Gautel M, Labeit S, James M, Beckmann J, Weissenbach J, Vosberg HP, Fiszman M, Komajda M, Schwartz K. Nat Genet. 1995;11:438–440. doi: 10.1038/ng1295-438. [DOI] [PubMed] [Google Scholar]
- 8.Watkins H, Conner D, Thierfelder L, Jarcho JA, Macrae C, McKenna WJ, Maron BJ, Seidman JG, Seidman CE. Nat Genet. 1995;11:434–437. doi: 10.1038/ng1295-434. [DOI] [PubMed] [Google Scholar]
- 9.Ho CY, Lever HM, DeSanctis R, Farver CF, Seidman JG, Seidman CE. Circulation. 2000;102:1950–1955. doi: 10.1161/01.cir.102.16.1950. [DOI] [PubMed] [Google Scholar]
- 10.Thierfelder L, Macrae C, Watkins H, Tomfohrde J, Williams M, McKenna W, Bohm K, Noeske G, Schlepper M, Bowcock A, Vosberg HP, Seidman JG, Seidman C. Proc Natl Acad Sci U S A. 1993;90:6270–6274. doi: 10.1073/pnas.90.13.6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geisterfer-Lowrance AAT, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. Cell. 1990;62:999–1006. doi: 10.1016/0092-8674(90)90274-i. [DOI] [PubMed] [Google Scholar]
- 12.Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC, Rayment I, Sellers JR, Fananapazir L, Epstein ND. Nat Genet. 1996;13:63–69. doi: 10.1038/ng0596-63. [DOI] [PubMed] [Google Scholar]
- 13.Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, Odonoghue A, Spirito P, Matsumori A, Moravec CS, Seidman JG, Seidman CE. New Engl J Med. 1995;332:1058–1064. doi: 10.1056/NEJM199504203321603. [DOI] [PubMed] [Google Scholar]
- 14.Moolman JC, Corfield VA, Posen B, Ngumbela K, Seidman C, Brink PA, Watkins H. J Am Coll Cardiol. 1997;29:549–555. doi: 10.1016/s0735-1097(96)00530-x. [DOI] [PubMed] [Google Scholar]
- 15.Nakajima Taniguchi C, Matsui H, Fujio Y, Nagata S, Kishimoto T, Yamauchi Takihara K. J Mol Cell Cardiol. 1997;29:839–843. doi: 10.1006/jmcc.1996.0322. [DOI] [PubMed] [Google Scholar]
- 16.Kimura A, Harada H, Park JE, Nishi H, Satoh M, Takahashi M, Hiroi S, Sasaoka T, Ohbuchi N, Nakamura T, Koyanagi T, Hwang TH, Choo TA, Chung KS, Hasegawa A, Nagai R, Okazaki O, Nakamura H, Matsuzaki M, Sakamoto T, Toshima H, Koga Y, Imaizumi T, Sasazuki T. Nat Genet. 1997;16:379–382. doi: 10.1038/ng0897-379. [DOI] [PubMed] [Google Scholar]
- 17.Mogensen J, Klausen IC, Pedersen AK, Egeblad H, Bross P, Kruse TA, Gregersen N, Hansen PS, Baandrup U, Borglum AD. J Clin Invest. 1999;103:R39–R43. doi: 10.1172/JCI6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Satoh M, Takahashi M, Sakamoto T, Hiroe M, Marumo F, Kimura A. Biochem Biophys Res Commun. 1999;262:411–417. doi: 10.1006/bbrc.1999.1221. [DOI] [PubMed] [Google Scholar]
- 19.Hoffmann B, Schmidt-Traub H, Perrot A, Osterziel KJ, Geßner R. Hum Mutat. 2001;17:524–524. doi: 10.1002/humu.1143. [DOI] [PubMed] [Google Scholar]
- 20.Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, Seldman JG, Seidman CE. Cell. 77:701–712. doi: 10.1016/0092-8674(94)90054-x. [DOI] [PubMed] [Google Scholar]
- 21.Albury ANJ, Swindle N, Swartz DR, Tikunova SB. Biochemistry. 2012;51:3614–3621. doi: 10.1021/bi300187k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu B, Tikunova SB, Kline KP, Siddiqui JK, Davis JP. Plos One. 2012;7:11. doi: 10.1371/journal.pone.0038259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson P, Griffiths PJ, Watkins H, Redwood CS. Circul Res. 2007;101:1266–1273. doi: 10.1161/CIRCRESAHA.107.156380. [DOI] [PubMed] [Google Scholar]
- 24.Rust EM, Albayya FP, Metzger JM. J Clin Invest. 1999;103:1459–1467. doi: 10.1172/JCI6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tardiff JC, Factor SM, Tompkins BD, Hewett TE, Palmer BM, Moore RL, Schwartz S, Robbins J, Leinwand LA. J Clin Invest. 1998;101:2800–2811. doi: 10.1172/JCI2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tardiff JC, Hewett TE, Palmer BM, Olsson C, Factor SM, Moore RL, Robbins J, Leinwand LA. J Clin Invest. 1999;104:469–481. doi: 10.1172/JCI6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Javadpour MM, Tardiff JC, Pinz I, Ingwall JS. J Clin Invest. 2003;112:768–775. doi: 10.1172/JCI15967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramirez-Correa GA, Frazier AH, Zhu GS, Zhang PB, Rappold T, Kooij V, Bedja D, Snyder GA, Lugo-Fagundo NS, Hariharan R, Li YJ, Shen XX, Gao WD, Cingolani OH, Takimoto E, Foster DB, Murphy AM. J Appl Physiol. 2015;118:212–223. doi: 10.1152/japplphysiol.00463.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wen YH, Pinto JR, Gomes AV, Xu YY, Wang YC, Wang Y, Potter JD, Kerrick WGL. Journal of Biological Chemistry. 2008;283:20484–20494. doi: 10.1074/jbc.M801661200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang YC, Pinto JR, Solis RS, Dweck D, Liang JS, Diaz-Perez Z, Ge Y, Walker JW, Potter JD. Journal of Biological Chemistry. 2012;287:2156–2167. doi: 10.1074/jbc.M111.294306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guinto PJ, Haim TE, Dowell-Martino CC, Sibinga N, Tardiff JC. Am J Physiol-Heart Circul Physiol. 2009;297:H614–H626. doi: 10.1152/ajpheart.01143.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jimenez J, Tardiff JC. Am J Physiol-Heart Circul Physiol. 2011;300:H627–H635. doi: 10.1152/ajpheart.00247.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moore RK, Grinspan LT, Jimenez J, Guinto PJ, Ertz-Berger B, Tardiff JC. J Mol Cell Cardiol. 2013;58:188–198. doi: 10.1016/j.yjmcc.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Willott RH, Gomes AV, Chang AN, Parvatiyar MS, Pinto JR, Potter JD. J Mol Cell Cardiol. 2010;48:882–892. doi: 10.1016/j.yjmcc.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 35.Zhang R, Zhao J, Mandveno A, Potter JD. Circul Res. 1995;76:1028–1035. doi: 10.1161/01.res.76.6.1028. [DOI] [PubMed] [Google Scholar]
- 36.Solaro RJ, Rosevear P, Kobayashi T. Biochem Biophys Res Commun. 2008;369:82–87. doi: 10.1016/j.bbrc.2007.12.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gordon AM, Homsher E, Regnier M. Physiol Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 38.Kawasaki H, Nakayama S, Kretsinger RH. BioMetals. 1998;11:277–295. doi: 10.1023/a:1009282307967. [DOI] [PubMed] [Google Scholar]
- 39.Herzberg O, James MNG. Nature. 1985;313:653–659. doi: 10.1038/313653a0. [DOI] [PubMed] [Google Scholar]
- 40.Li MX, Wang X, Sykes BD. J Muscle Res Cell Motil. 2004;25:559–579. doi: 10.1007/s10974-004-5879-2. [DOI] [PubMed] [Google Scholar]
- 41.Dvoretsky A, Abusamhadneh EM, Howarth JW, Rosevear PR. Journal of Biological Chemistry. 2002;277:38565–38570. doi: 10.1074/jbc.M205306200. [DOI] [PubMed] [Google Scholar]
- 42.Collins JH. J Muscle Res Cell Motil. 1991;12:3–25. doi: 10.1007/BF01781170. [DOI] [PubMed] [Google Scholar]
- 43.Holroyde MJ, Robertson SP, Johnson JD, Solaro RJ, Potter JD. Journal of Biological Chemistry. 1980;255:1688–1693. [PubMed] [Google Scholar]
- 44.Potter JD, Gergely J. Journal of Biological Chemistry. 1975;250:4628–4633. [PubMed] [Google Scholar]
- 45.Brinley FJ, Scarpa A, Tiffert T. J Physiol-London. 1977;266:545–565. doi: 10.1113/jphysiol.1977.sp011781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Evenas J, Malmendal A, Forsen S. Curr Opin Chem Biol. 1998;2:293–302. doi: 10.1016/s1367-5931(98)80072-0. [DOI] [PubMed] [Google Scholar]
- 47.Zot HG, Potter JD. Journal of Biological Chemistry. 1982;257:7678–7683. [PubMed] [Google Scholar]
- 48.Van Eerd JP, Takahashi K. Biochem Biophys Res Commun. 1975;64:122–127. doi: 10.1016/0006-291x(75)90227-2. [DOI] [PubMed] [Google Scholar]
- 49.Sheng ZL, Strauss WL, Francois JM, Potter JD. Journal of Biological Chemistry. 1990;265:21554–21560. [PubMed] [Google Scholar]
- 50.Sweeney HL, Brito RMM, Rosevear PR, Putkey JA. Proc Natl Acad Sci U S A. 1990;87:9538–9542. doi: 10.1073/pnas.87.24.9538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Putkey JA, Sweeney HL, Campbell ST. Journal of Biological Chemistry. 1989;264:12370–12378. [PubMed] [Google Scholar]
- 52.Spyracopoulos L, Li MX, Sia SK, Gagne SM, Chandra M, Solaro RJ, Sykes BD. Biochemistry. 1997;36:12138–12146. doi: 10.1021/bi971223d. [DOI] [PubMed] [Google Scholar]
- 53.Li MX, Spyracopoulos L, Sykes BD. Biochemistry. 1999;38:8289–8298. doi: 10.1021/bi9901679. [DOI] [PubMed] [Google Scholar]
- 54.Gagne SM, Tsuda S, Li MX, Smillie LB, Sykes BD. Nat Struct Biol. 1995;2:784–789. doi: 10.1038/nsb0995-784. [DOI] [PubMed] [Google Scholar]
- 55.Sia SK, Li MX, Spyracopoulos L, Gagne SM, Liu W, Putkey JA, Sykes BD. Journal of Biological Chemistry. 1997;272:18216–18221. doi: 10.1074/jbc.272.29.18216. [DOI] [PubMed] [Google Scholar]
- 56.Vassylyev DG, Takeda S, Wakatsuki S, Maeda K, Maeda Y. Proc Natl Acad Sci U S A. 1998;95:4847–4852. doi: 10.1073/pnas.95.9.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dong WJ, Xing J, Villain M, Hellinger M, Robinson JM, Chandra R, Solaro RJ, Umeda PK, Cheung HC. Journal of Biological Chemistry. 1999;274:31382–31390. doi: 10.1074/jbc.274.44.31382. [DOI] [PubMed] [Google Scholar]
- 58.Martins SM, Chapeaurouge A, Ferreira ST. Eur J Biochem. 2002;269:5484–5491. doi: 10.1046/j.1432-1033.2002.03248.x. [DOI] [PubMed] [Google Scholar]
- 59.Perry SV. Mol Cell Biochem. 1999;190:9–32. [PubMed] [Google Scholar]
- 60.Hastings KEM. Cell Struct Funct. 1997;22:205–211. doi: 10.1247/csf.22.205. [DOI] [PubMed] [Google Scholar]
- 61.Chong SM, Jin JP. J Mol Evol. 2009;68:448–460. doi: 10.1007/s00239-009-9202-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saggin L, Gorza L, Ausoni S, Schiaffino S. Journal of Biological Chemistry. 1989;264:16299–16302. [PubMed] [Google Scholar]
- 63.Sasse S, Brand NJ, Kyprianou P, Dhoot GK, Wade R, Arai M, Periasamy M, Yacoub MH, Barton PJR. Circul Res. 1993;72:932–938. doi: 10.1161/01.res.72.5.932. [DOI] [PubMed] [Google Scholar]
- 64.Reiser PJ, Westfall MV, Schiaffino S, Solaro RJ. Am J Physiol-Heart Circul Physiol. 1994;267:H1589–H1596. doi: 10.1152/ajpheart.1994.267.4.H1589. [DOI] [PubMed] [Google Scholar]
- 65.Westfall MV, Lee AM, Robinson DA. Journal of Biological Chemistry. 2005;280:41324–41331. doi: 10.1074/jbc.M506043200. [DOI] [PubMed] [Google Scholar]
- 66.Solaro RJ, Lee JA, Kentish JC, Allen DG. Circul Res. 1988;63:779–787. doi: 10.1161/01.res.63.4.779. [DOI] [PubMed] [Google Scholar]
- 67.F'Entzke RC, Buck SH, Patel JR, Lin H, Wolska BM, Stojanovic MO, Martin AF, Solaro RJ, Moss RL, Leiden JM. J Physiol-London. 1999;517:143–157. doi: 10.1111/j.1469-7793.1999.0143z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rarick HM, Tu XH, Solaro RJ, Martin AF. Journal of Biological Chemistry. 1997;272:26887–26892. doi: 10.1074/jbc.272.43.26887. [DOI] [PubMed] [Google Scholar]
- 69.Foster DB, Noguchi T, VanBuren P, Murphy AM, Van Eyk JE. Circul Res. 2003;93:917–924. doi: 10.1161/01.RES.0000099889.35340.6F. [DOI] [PubMed] [Google Scholar]
- 70.Jin JP, Yang FW, Yu ZB, Ruse CI, Bond M, Chen A. Biochemistry. 2001;40:2623–2631. doi: 10.1021/bi002423j. [DOI] [PubMed] [Google Scholar]
- 71.Jin JP, Zhang ZL, Bautista JA. Crit Rev Eukaryot Gene Expression. 2008;18:93–124. doi: 10.1615/critreveukargeneexpr.v18.i2.10. [DOI] [PubMed] [Google Scholar]
- 72.Howarth JW, Meller J, Solaro RJ, Trewhella J, Rosevear PR. J Mol Biol. 2007;373:706–722. doi: 10.1016/j.jmb.2007.08.035. [DOI] [PubMed] [Google Scholar]
- 73.Pearlstone JR, Smillie LB. Canadian Journal of Biochemistry and Cell Biology. 1985;63:212–218. doi: 10.1139/o85-030. [DOI] [PubMed] [Google Scholar]
- 74.Gasmi-Seabrook GMC, Howarth JW, Finley N, Abusamhadneh E, Gaponenko V, Brito RMM, Solaro RJ, Rosevear PR. Biochemistry. 1999;38:8313–8322. doi: 10.1021/bi9902642. [DOI] [PubMed] [Google Scholar]
- 75.Syska H, Wilkinson JM, Grand RJA, Perry SV. Biochem J. 1976;153:375–&. doi: 10.1042/bj1530375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Talbot JA, Hodges RS. Journal of Biological Chemistry. 1981;256:2798–2802. [PubMed] [Google Scholar]
- 77.Takeda S, Yamashita A, Maeda K, Maeda Y. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 78.Brown LJ, Sale KL, Hills R, Rouviere C, Song LK, Zhang XJ, Fajer PG. Proc Natl Acad Sci U S A. 2002;99:12765–12770. doi: 10.1073/pnas.202477399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tung CS, Wall ME, Gallagher SC, Trewhella J. Protein Sci. 2000;9:1312–1326. doi: 10.1110/ps.9.7.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lindhout DA, Sykes BD. Journal of Biological Chemistry. 2003;278:27024–27034. doi: 10.1074/jbc.M302497200. [DOI] [PubMed] [Google Scholar]
- 81.Slupsky CM, Sykes BD. Biochemistry. 1995;34:15953–15964. doi: 10.1021/bi00049a010. [DOI] [PubMed] [Google Scholar]
- 82.Perry SV. J Muscle Res Cell Motil. 1998;19:575–602. doi: 10.1023/a:1005397501968. [DOI] [PubMed] [Google Scholar]
- 83.Wei B, Jin JP. Archives of Biochemistry and Biophysics. 2011;505:144–154. doi: 10.1016/j.abb.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Anderson PAW, Malouf NN, Oakeley AE, Pagani ED, Allen PD. Circul Res. 1991;69:1226–1233. doi: 10.1161/01.res.69.5.1226. [DOI] [PubMed] [Google Scholar]
- 85.Jin JP, Wang J, Zhang JY. Gene. 1996;168:217–221. doi: 10.1016/0378-1119(95)00803-9. [DOI] [PubMed] [Google Scholar]
- 86.Ricchiuti V, Apple FS. Clin Chem. 1999;45:2129–2135. [PubMed] [Google Scholar]
- 87.Jin JP, Chong SM. Archives of Biochemistry and Biophysics. 2010;500:144–150. doi: 10.1016/j.abb.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Heeley DH, Golosinska K, Smillie LB. Journal of Biological Chemistry. 1987;262:9971–9978. [PubMed] [Google Scholar]
- 89.Schaertl S, Lehrer SS, Geeves MA. Biochemistry. 1995;34:15890–15894. doi: 10.1021/bi00049a003. [DOI] [PubMed] [Google Scholar]
- 90.Vinogradova MV, Stone DB, Malanina GG, Karatzaferi C, Cooke R, Mendelson RA, Fletterick RJ. Proc Natl Acad Sci U S A. 2005;102:5038–5043. doi: 10.1073/pnas.0408882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ertz-Berger BR, He HM, Dowell C, Factor SM, Haim TE, Nunez S, Schwartz SD, Ingwall JS, Tardiff JC. Proc Natl Acad Sci U S A. 2005;102:18219–18224. doi: 10.1073/pnas.0509181102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Manning EP, Tardiff JC, Schwartz SD. Biochemistry. 2011;50:7405–7413. doi: 10.1021/bi200506k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Martyn DA, Regnier M, Xu D, Gordon AM. Biophys J. 2001;80:360–370. doi: 10.1016/S0006-3495(01)76020-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Martyn DA, Freitag CJ, Chase PB, Gordon AM. Biophys J. 76:1480–1493. doi: 10.1016/S0006-3495(99)77308-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fuchs F, Wang YP. American Journal of Physiology - Cell Physiology. 1991;261:C787–C792. doi: 10.1152/ajpcell.1991.261.5.C787. [DOI] [PubMed] [Google Scholar]
- 96.Wang X, Li MX, Spyracopoulos L, Beier N, Chandra M, Solaro RJ, Sykes BD. Journal of Biological Chemistry. 2001;276:25456–25466. doi: 10.1074/jbc.M102418200. [DOI] [PubMed] [Google Scholar]
- 97.Pyle WG, Sumandea MP, Solaro RJ, De Tombe PP. Am J Physiol-Heart Circul Physiol. 2002;283:H1215–H1224. doi: 10.1152/ajpheart.00128.2002. [DOI] [PubMed] [Google Scholar]
- 98.Burkart EM, Sumandea MP, Kobayashi T, Nili M, Martin AF, Homsher E, Solaro RJ. Journal of Biological Chemistry. 2003;278:11265–11272. doi: 10.1074/jbc.M210712200. [DOI] [PubMed] [Google Scholar]
- 99.Mathur MC, Kobayashi T, Chalovich JM. Biophys J. 2008;94:542–549. doi: 10.1529/biophysj.107.113944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang H, Grant JE, Doede CM, Sadayappan S, Robbins J, Walker JW. J Mol Cell Cardiol. 2006;41:823–833. doi: 10.1016/j.yjmcc.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 101.Lu QW, Hinken AC, Patrick SE, Solaro RJ, Kobayashi T. Journal of Biological Chemistry. 2010;285:11810–11817. doi: 10.1074/jbc.M109.055657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lindhout DA, Li MX, Schieve D, Sykes BD. Biochemistry. 2002;41:7267–7274. doi: 10.1021/bi020100c. [DOI] [PubMed] [Google Scholar]
- 103.Li MX, Wang X, Lindhout DA, Buscemi N, Van Eyk JE, Sykes BD. Biochemistry. 2003;42:14460–14468. doi: 10.1021/bi035408y. [DOI] [PubMed] [Google Scholar]
- 104.Nixon BR, Thawornkaiwong A, Jin J, Brundage EA, Little SC, Davis JP, Solaro RJ, Biesiadecki BJ. Journal of Biological Chemistry. 2012;287:19136–19147. doi: 10.1074/jbc.M111.323048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Oliveira SM, Zhang YH, Solis RS, Isackson H, Bellahcene M, Yavari A, Pinter K, Davies JK, Ge Y, Ashrafian H, Walker JW, Carling D, Watkins H, Casadei B, Redwood C. Circul Res. 2012;110:1192–1201. doi: 10.1161/CIRCRESAHA.111.259952. [DOI] [PubMed] [Google Scholar]
- 106.Taglieri DM, Monasky MM, Knezevic I, Sheehan KA, Lei M, Wang X, Chernoff J, Wolska BM, Ke YB, Solaro RJ. J Mol Cell Cardiol. 2011;51:988–996. doi: 10.1016/j.yjmcc.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.You B, Yan GJ, Zhang ZL, Yan L, Li J, Ge QY, Jin JP, Sun JX. Biochem J. 2009;418:93–101. doi: 10.1042/BJ20081340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang PB, Kirk JA, Ji WH, dos Remedios CG, Kass DA, Van Eyk JE, Murphy AM. Circulation. 2012;126:1828–U1123. doi: 10.1161/CIRCULATIONAHA.112.096388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Henze M, Patrick SE, Hinken A, Scruggs SB, Goldspink P, de Tombe PP, Kobayashi M, Ping PP, Kobayashi T, Solaro RJ. Biochimica Et Biophysica Acta-Molecular Cell Research. 2013;1833:823–832. doi: 10.1016/j.bbamcr.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Salhi HE, Walton SD, Hassel NC, Brundage EA, de Tombe PP, Janssen PML, Davis JP, Biesiadecki BJ. J Mol Cell Cardiol. 2014;76:257–264. doi: 10.1016/j.yjmcc.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wijnker PJM, Li YJ, Zhang PB, Foster DB, dos Remedios C, Van Eyk JE, Stienen GJM, Murphy AM, van der Velden J. J Mol Cell Cardiol. 2015;82:93–103. doi: 10.1016/j.yjmcc.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Solaro RJ, Moir AJG, Perry SV. Nature. 1976;262:615–617. doi: 10.1038/262615a0. [DOI] [PubMed] [Google Scholar]
- 113.Kobayashi T, Yang XF, Walker LA, Van Breemen RB, Solaro RJ. J Mol Cell Cardiol. 2005;38:213–218. doi: 10.1016/j.yjmcc.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 114.Haworth RS, Cuello F, Herron TJ, Franzen G, Kentish JC, Gautel M, Avkiran M. Circul Res. 2004;95:1091–1099. doi: 10.1161/01.RES.0000149299.34793.3c. [DOI] [PubMed] [Google Scholar]
- 115.Cuello F, Bardswell SC, Haworth RS, Yin XK, Lutz S, Wieland T, Mayr M, Kentish JC, Avkiran M. Circul Res. 2007;100:864–873. doi: 10.1161/01.RES.0000260809.15393.fa. [DOI] [PubMed] [Google Scholar]
- 116.Bardswell SC, Cuello F, Rowland AJ, Sadayappan S, Robbins J, Gautel M, Walker JW, Kentish JC, Avkiran M. Journal of Biological Chemistry. 2010;285:5674–5682. doi: 10.1074/jbc.M109.066456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Layland J, Li JM, Shah AM. J Physiol-London. 2002;540:457–467. doi: 10.1113/jphysiol.2001.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kirkpatrick KP, Robertson AS, Klaiman JM, Gillis TE. J Exp Biol. 2011;214:1981–1988. doi: 10.1242/jeb.052860. [DOI] [PubMed] [Google Scholar]
- 119.Kentish JC, McCloskey DT, Layland J, Palmer S, Leiden JM, Martin AF, Solaro RJ. Circul Res. 2001;88:1059–1065. doi: 10.1161/hh1001.091640. [DOI] [PubMed] [Google Scholar]
- 120.Pi YQ, Kemnitz KR, Zhang DH, Kranias EG, Walker JW. Circul Res. 2002;90:649–656. doi: 10.1161/01.res.0000014080.82861.5f. [DOI] [PubMed] [Google Scholar]
- 121.Chandra M, Dong WJ, Pan BS, Cheung HC, Solaro RJ. Biochemistry. 1997;36:13305–13311. doi: 10.1021/bi9710129. [DOI] [PubMed] [Google Scholar]
- 122.Turnbull L, Hoh JFY, Ludowyke RI, Rossmanith GH. J Physiol-London. 2002;542:911–920. doi: 10.1113/jphysiol.2002.022707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nixon BR, Walton SD, Zhang B, Brundage EA, Little SC, Ziolo MT, Davis JP, Biesiadecki BJ. J Mol Cell Cardiol. 2014;72:177–185. doi: 10.1016/j.yjmcc.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hanft LM, Biesiadecki BJ, McDonald KS. J Physiol-London. 2013;591:4535–4547. doi: 10.1113/jphysiol.2013.258400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rao V, Cheng YH, Lindert S, Wang D, Oxenford L, McCulloch AD, McCammon JA, Regnier M. Biophys J. 2014;107:1196–1204. doi: 10.1016/j.bpj.2014.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Landstrom AP, Parvatiyar MS, Pinto JR, Marquardt ML, Bos JM, Tester DJ, Ornmen SR, Potter JD, Ackerman MJ. J Mol Cell Cardiol. 2008;45:281–288. doi: 10.1016/j.yjmcc.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rani DS, Nallari P, Priyamvada S, Narasimhan C, Singh L, Thangaraj K. BMC Med Genet. 2012;13:8. doi: 10.1186/1471-2350-13-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ, Potter JD, Knollmann BC. J Clin Invest. 2008;118:3893–3903. doi: 10.1172/JCI36642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ashrafian H, Redwood C, Blair E, Watkins H. Trends Genet. 2003;19:263–268. doi: 10.1016/S0168-9525(03)00081-7. [DOI] [PubMed] [Google Scholar]
- 130.Alcalai R, Seidman JG, Seidman CE. J Cardiovasc Electrophysiol. 2008;19:104–110. doi: 10.1111/j.1540-8167.2007.00965.x. [DOI] [PubMed] [Google Scholar]
- 131.Fatkin D, Graham RM. Physiol Rev. 2002;82:945–980. doi: 10.1152/physrev.00012.2002. [DOI] [PubMed] [Google Scholar]
- 132.Niimura H, Patton KK, McKenna WJ, Soults J, Maron BJ, Seidman JG, Seidman CE. Circulation. 2002;105:446–451. doi: 10.1161/hc0402.102990. [DOI] [PubMed] [Google Scholar]
- 133.Konno T, Shimizu M, Ino H, Fujino N, Hayashi K, Uchiyama K, Kaneda T, Inoue M, Masuda E, Mabuchi H. J Intern Med. 2005;258:216–224. doi: 10.1111/j.1365-2796.2005.01539.x. [DOI] [PubMed] [Google Scholar]
- 134.Van Driest SL, Ellsworth EG, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Circulation. 2003;108:445–451. doi: 10.1161/01.CIR.0000080896.52003.DF. [DOI] [PubMed] [Google Scholar]
- 135.Arad M, Penas-Lado M, Monserrat L, Maron BJ, Sherrid M, Ho CY, Barr S, Karim A, Olson TM, Kamisago M, Seidman JG, Seidman CE. Circulation. 2005;112:2805–2811. doi: 10.1161/CIRCULATIONAHA.105.547448. [DOI] [PubMed] [Google Scholar]
- 136.Frazier A, Judge DP, Schulman SP, Johnson N, Holmes KW, Murphy AM. Pediatr Cardiol. 2008;29:846–850. doi: 10.1007/s00246-007-9177-9. [DOI] [PubMed] [Google Scholar]
- 137.Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, Benaiche A, Isnard R, Dubourg O, Burban M, Gueffet JP, Millaire A, Desnos M, Schwartz K, Hainque B, Komajda M, Project EHF. Circulation. 2003;107:2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 138.Varnava AM, Elliott PM, Baboonian C, Davison F, Davies MJ, McKenna WJ. Circulation. 2001;104:1380–1384. doi: 10.1161/hc3701.095952. [DOI] [PubMed] [Google Scholar]
- 139.Anan R, Shono H, Kisanuki A, Arima S, Nakao S, Tanaka H. Circulation. 1998;98:391–397. doi: 10.1161/01.cir.98.5.391. [DOI] [PubMed] [Google Scholar]
- 140.Torricelli F, Girolami F, Olivotto I, Passerini I, Frusconi S, Vargiu D, Richard P, Cecchi F. Am J Cardiol. 2003;92:1358–1362. doi: 10.1016/j.amjcard.2003.08.031. [DOI] [PubMed] [Google Scholar]
- 141.Ackerman MJ, VanDriest SL, Ommen SR, Will ML, Nishimura RA, Tajik AJ, Gersh BJ. J Am Coll Cardiol. 2002;39:2042–2048. doi: 10.1016/s0735-1097(02)01900-9. [DOI] [PubMed] [Google Scholar]
- 142.Song L, Zou YB, Wang JH, Wang ZM, Zhen YS, Lou KJ, Zhang Q, Wang XJ, Wang H, Li J, Hui R. Clin Chim Acta. 2005;351:209–216. doi: 10.1016/j.cccn.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 143.Van Driest SL, Ackerman MJ, Ommen SR, Shakur R, Will ML, Nishimura RA, Tajik AJ, Gersh BJ. Circulation. 2002;106:3085–3090. doi: 10.1161/01.cir.0000042675.59901.14. [DOI] [PubMed] [Google Scholar]
- 144.Takahashi-Yanaga F, Morimoto S, Harada K, Minakami R, Shiraishi F, Ohta M, Lu UW, Sasaguri T, Ohtsuki I. J Mol Cell Cardiol. 2001;33:2095–2107. doi: 10.1006/jmcc.2001.1473. [DOI] [PubMed] [Google Scholar]
- 145.Elliott K, Watkins H, Redwood CS. Journal of Biological Chemistry. 2000;275:22069–22074. doi: 10.1074/jbc.M002502200. [DOI] [PubMed] [Google Scholar]
- 146.Köhler J, Chen Y, Brenner B, Gordon AM, Kraft T, Martyn DA, Regnier M, Rivera AJ, Wang CK, Chase PB. Physiological Genomics. 2003;14 doi: 10.1152/physiolgenomics.00101.2002. [DOI] [PubMed] [Google Scholar]
- 147.Deng Y, Schmidtmann A, Redlich A, Westerdorf B, Jaquet K, Thieleczek R. Biochemistry. 2001;40:14593–14602. doi: 10.1021/bi0115232. [DOI] [PubMed] [Google Scholar]
- 148.Lang R, Gomes AV, Zhao JJ, Housmans PR, Miller T, Potter JD. Journal of Biological Chemistry. 2002;277:11670–11678. doi: 10.1074/jbc.M108912200. [DOI] [PubMed] [Google Scholar]
- 149.Westfall MV, Borton AR, Albayya FP, Metzger JM. Circul Res. 2002;91:525–531. doi: 10.1161/01.res.0000034710.46739.c0. [DOI] [PubMed] [Google Scholar]
- 150.Reis S, Littwitz C, Preilowski S, Mugge A, Stienen GJM, Pott L, Jaquet K. Pflugers Arch. 2008;457:17–24. doi: 10.1007/s00424-008-0487-4. [DOI] [PubMed] [Google Scholar]
- 151.Kruger M, Zittrich S, Redwood C, Blaudeck N, James J, Robbins J, Pfitzer G, Stehle R. J Physiol-London. 2005;564:347–357. doi: 10.1113/jphysiol.2004.079095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kobayashi T, Solaro RJ. Journal of Biological Chemistry. 2006;281:13471–13477. doi: 10.1074/jbc.M509561200. [DOI] [PubMed] [Google Scholar]
- 153.Zhou ZQ, Rieck D, Li KL, Ouyang YX, Dong WJ. Archives of Biochemistry and Biophysics. 2013;535:56–67. doi: 10.1016/j.abb.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Brunet NM, Chase PB, Mihajlovic G, Schoffstall B. Archives of Biochemistry and Biophysics. 2014;552:11–20. doi: 10.1016/j.abb.2013.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Burton D, Abdulrazzak H, Knott A, Elliott K, Redwood C, Watkins H, Marston S, Ashley C. Biochem J. 2002;362:443–451. doi: 10.1042/0264-6021:3620443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.James J, Zhang Y, Osinska H, Sanbe A, Klevitsky R, Hewett TE, Robbins J. Circul Res. 2000;87:805–811. doi: 10.1161/01.res.87.9.805. [DOI] [PubMed] [Google Scholar]
- 157.Gomes AV, Harada K, Potter JD. J Mol Cell Cardiol. 2005;39:754–765. doi: 10.1016/j.yjmcc.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 158.Dweck D, Sanchez-Gonzalez MA, Chang AN, Dulce RA, Badger CD, Koutnik AP, Ruiz EL, Griffin B, Liang JS, Kabbaj M, Fincham FD, Hare JM, Overton M, Pinto JR. Journal of Biological Chemistry. 2014;289:23097–23111. doi: 10.1074/jbc.M114.561472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Liang JS, Kazmierczak K, Rojas AI, Wang YC, Szczesna-Cordary D. Biomed Res Int. 2015:9. doi: 10.1155/2015/742536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pinto JR, Parvatiyar MS, Jones MA, Liang JS, Ackerman MJ, Potter JD. Journal of Biological Chemistry. 2009;284:19090–19100. doi: 10.1074/jbc.M109.007021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Liang B, Chung F, Qu Y, Pavlov D, Gillis TE, Tikunova SB, Davis JP, Tibbits GF. Physiological Genomics. 2008;33:257–266. doi: 10.1152/physiolgenomics.00154.2007. [DOI] [PubMed] [Google Scholar]
- 162.Parvatiyar MS, Landstrom AP, Figueiredo-Freitas C, Potter JD, Ackerman MJ, Pinto JR. Journal of Biological Chemistry. 2012;287:31845–31855. doi: 10.1074/jbc.M112.377713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Parvatiyar MS, Pinto JR, Liang J, Potter JD. Journal of Biological Chemistry. 2010;285:27785–27797. doi: 10.1074/jbc.M110.112326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Nakaura H, Yanaga F, Ohtsuki I, Morimoto S. J Biochem. 1999;126:457–460. doi: 10.1093/oxfordjournals.jbchem.a022473. [DOI] [PubMed] [Google Scholar]
- 165.Harada K, Potter JD. Journal of Biological Chemistry. 2004;279:14488–14495. doi: 10.1074/jbc.M309355200. [DOI] [PubMed] [Google Scholar]
- 166.Harada K, Takahashi-Yanaga F, Minakami R, Morimoto S, Ohtsuki I. J Biochem. 2000;127:263–268. doi: 10.1093/oxfordjournals.jbchem.a022603. [DOI] [PubMed] [Google Scholar]
- 167.Manning EP, Guinto PJ, Tardiff JC. Journal of Biological Chemistry. 2012;287:14515–14523. doi: 10.1074/jbc.M111.257436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Cuinto PJ, Manning EP, Schwartz SD, Tardiff JC. J Theor Comput Chem. 2007;6:413–419. doi: 10.1142/S0219633607003271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Haim TE, Dowell C, Diamanti T, Scheuer J, Tardiff JC. J Mol Cell Cardiol. 2007;42:1098–1110. doi: 10.1016/j.yjmcc.2007.03.906. [DOI] [PubMed] [Google Scholar]
- 170.Chandra M, Tschirgi ML, Tardiff JC. Am J Physiol-Heart Circul Physiol. 2005;289:H2112–H2119. doi: 10.1152/ajpheart.00571.2005. [DOI] [PubMed] [Google Scholar]
- 171.Schober T, Huke S, Venkataraman R, Gryshchenko O, Kryshtal D, Hwang HS, Baudenbacher FJ, Knollmann BC. Circul Res. 2012;111:170–U120. doi: 10.1161/CIRCRESAHA.112.270041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hernandez OM, Szczesna-Cordary D, Knollmann BC, Miller T, Bell M, Zhao JJ, Sirenko SG, Diaz Z, Guzman G, Xu YY, Wang Y, Kerrick WGL, Potter JD. Journal of Biological Chemistry. 2005;280:37183–37194. doi: 10.1074/jbc.M508114200. [DOI] [PubMed] [Google Scholar]
- 173.Hinkle A, Tobacman LS. Journal of Biological Chemistry. 2003;278:506–513. doi: 10.1074/jbc.M209194200. [DOI] [PubMed] [Google Scholar]
- 174.Szczesna D, Zhang R, Zhao JJ, Jones M, Guzman G, Potter JD. Journal of Biological Chemistry. 2000;275:624–630. doi: 10.1074/jbc.275.1.624. [DOI] [PubMed] [Google Scholar]
- 175.Yanaga F, Morimoto S, Ohtsuki I. Journal of Biological Chemistry. 1999;274:8806–8812. doi: 10.1074/jbc.274.13.8806. [DOI] [PubMed] [Google Scholar]
- 176.Moore RK, Abdullah S, Tardiff JC. Archives of Biochemistry and Biophysics. 2014;552:21–28. doi: 10.1016/j.abb.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Matsumoto F, Maeda K, Chatake T, Maeda Y, Fujiwara S. Biochem Biophys Res Commun. 2009;382:205–209. doi: 10.1016/j.bbrc.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 178.Pinto JR, Reynaldo DP, Parvatiyar MS, Dweck D, Liang J, Jones MA, Sorenson MM, Potter JD. Journal of Biological Chemistry. 2011;286:1005–1013. doi: 10.1074/jbc.M110.168583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Li AY, Stevens CM, Liang B, Rayani K, Little S, Davis J, Tibbits GF. Plos One. 2013;8:13. doi: 10.1371/journal.pone.0079363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Dweck D, Hus N, Potter JD. Journal of Biological Chemistry. 2008;283:33119–33128. doi: 10.1074/jbc.M804070200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Schmidtmann A, Lindow C, Villard S, Heuser A, Mugge A, Gessner R, Granier C, Jaquet K. FEBS J. 2005;272:6087–6097. doi: 10.1111/j.1742-4658.2005.05001.x. [DOI] [PubMed] [Google Scholar]
- 182.Pinto JR, Siegfried JD, Parvatiyar MS, Li DX, Norton N, Jones MA, Liang JS, Potter JD, Hershberger RE. Journal of Biological Chemistry. 2011;286:34404–34412. doi: 10.1074/jbc.M111.267211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Swindle N, Tikunova SB. Biochemistry. 2010;49:4813–4820. doi: 10.1021/bi100400h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Little SC, Biesiadecki BJ, Kilic A, Higgins RSD, Janssen PML, Davis JP. Journal of Biological Chemistry. 2012;287:27930–27940. doi: 10.1074/jbc.M111.337295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Cheng Y, Rao V, Tu Ay, Lindert S, Wang D, Oxenford L, Mc Culloch AD, Mc Cammon JA, Regnier M. Journal of Biological Chemistry. 2015;290:27749–27766. doi: 10.1074/jbc.M115.683045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Dong WJ, Xing J, Ouyang YX, An JL, Cheung HC. Journal of Biological Chemistry. 2008;283:3424–3432. doi: 10.1074/jbc.M703822200. [DOI] [PubMed] [Google Scholar]
- 187.Wang D, Robertson IM, Li MX, Mc Cully ME, Crane ML, Luo Z, Tu AY, Daggett V, Sykes BD, Regnier M. Biochemistry. 2012;51:4473–4487. doi: 10.1021/bi3003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wang D, McCully ME, Luo Z, McMichael J, Tu AY, Daggett V, Regnier M. Archives of Biochemistry and Biophysics. 2013;535:75. doi: 10.1016/j.abb.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Lindert S, Kekenes-Huskey PM, McCammon JA. Biophys J. 2012;103:1784–1789. doi: 10.1016/j.bpj.2012.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kekenes-Huskey PM, Lindert S, McCammon JA. PLoS Comp Biol. 2012;8:11. doi: 10.1371/journal.pcbi.1002777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lindert S, Kekenes-Huskey P, McCammon JA. Biophys J. 2013;104:331A–332A. [Google Scholar]
- 192.Jayasundar JJ, Xing J, Robinson JM, Cheung HC, Dong WJ. PLoS ONE. 2014;9:e87135. doi: 10.1371/journal.pone.0087135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Varughese JF, Baxley T, Chalovich JM, Li YM. J Phys Chem B. 2011;115:2392–2400. doi: 10.1021/jp1094504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Varughese JF, Li YM. Journal of Biomolecular Structure & Dynamics. 2011;29:135. doi: 10.1080/07391102.2011.10507378. [DOI] [PubMed] [Google Scholar]
- 195.Varughese JF, Chalovich JM, Li YM. Journal of Biomolecular Structure & Dynamics. 2010;28:159–173. doi: 10.1080/07391102.2010.10507350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Cheng YH, Lindert S, Kekenes-Huskey P, Rao VS, Solaro RJ, Rosevear PR, Amaro R, McCulloch AD, McCammon JA, Regnier M. Biophys J. 2014;107:1675–1685. doi: 10.1016/j.bpj.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sadayappan S, Finley N, Howarth JW, Osinska H, Klevitsky R, Lorenz JN, Rosevear PR, Robbins J. The FASEB Journal. 2008;22:1246–1257. doi: 10.1096/fj.07-9458com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Dong WJ, Chandra M, Xing J, She MD, Solaro RJ, Cheung HC. Biochemistry. 1997;36:6754–6761. doi: 10.1021/bi9622276. [DOI] [PubMed] [Google Scholar]
- 199.Heller WT, Finley NL, Dong WJ, Timmins P, Cheung HC, Rosevear PR, Trewhella J. Biochemistry. 2003;42:7790–7800. doi: 10.1021/bi0341509. [DOI] [PubMed] [Google Scholar]
- 200.Reiffert SU, Jaquet K, Heilmeyer LMG, Herberg FW. Biochemistry. 1998;37:13516–13525. doi: 10.1021/bi980280j. [DOI] [PubMed] [Google Scholar]
- 201.Rao VS, Korte FS, Razumova MV, Feest ER, Hsu H, Irving TC, Regnier M, Martyn DA. The Journal of Physiology. 2013;591:475–490. doi: 10.1113/jphysiol.2012.241604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Lindert S, Cheng YH, Kekenes-Huskey P, Regnier M, McCammon JA. Biophys J. 2015;108:395–407. doi: 10.1016/j.bpj.2014.11.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Feest ER, Korte FS, Tu AY, Dai J, Razumova MV, Murry CE, Regnier M. J Mol Cell Cardiol. 2014;72:219–227. doi: 10.1016/j.yjmcc.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Korte FS, Feest ER, Razumova MV, Tu AY, Regnier M. Am J Physiol-Heart Circul Physiol. 2012;303:H863–H870. doi: 10.1152/ajpheart.00395.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Kreutziger KL, Piroddi N, McMichael JT, Tesi C, Poggesi C, Regnier M. J Mol Cell Cardiol. 2011;50:165–174. doi: 10.1016/j.yjmcc.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Longyear TJ, Turner MA, Davis JP, Lopez J, Biesiadecki B, Debold EP. J Appl Physiol. 2014;116:1165–1174. doi: 10.1152/japplphysiol.01161.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Liu B, Lee RS, Biesiadecki BJ, Tikunova SB, Davis JP. Journal of Biological Chemistry. 2012;287:20027–20036. doi: 10.1074/jbc.M111.334953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Norman C, Rall JA, Tikunova SB, Davis JP. Am J Physiol-Heart Circul Physiol. 2007;293:H2580–H2587. doi: 10.1152/ajpheart.00039.2007. [DOI] [PubMed] [Google Scholar]
- 209.Alves ML, Dias FAL, Gaffin RD, Simon JN, Montminy EM, Biesiadecki BJ, Hinken AC, Warren CM, Utter MS, Davis RT, Sadayappan S, Robbins J, Wieczorek DF, Solaro RJ, Wolska BM. Circ-Cardiovasc Genet. 2014;7:132–143. doi: 10.1161/CIRCGENETICS.113.000324. [DOI] [PMC free article] [PubMed] [Google Scholar]