

Summary
The phosphoenolpyruvate:carbohydrate phosphotransferase systems (PTS) are found in bacteria, where they play central roles in sugar uptake and regulation of cellular uptake processes. Little is known about how the membrane-embedded components (EIICs) selectively mediate the passage of carbohydrates across the membrane. Here we report the functional characterization and 2.55 Å resolution structure of a maltose transporter, bcMalT, belonging to the Glucose superfamily of EIIC transporters. bcMalT crystallized in an outward-facing occluded conformation, in contrast to the structure of another Glucose superfamily EIIC, bcChbC, which crystallized in an inward-facing occluded conformation. The structures differ in the position of a structurally conserved substrate-binding domain that is suggested to play a central role in sugar transport. Additionally, molecular dynamics simulations suggest a potential pathway for substrate entry from the periplasm into the bcMalT substrate-binding site. These results provide a mechanistic framework for understanding substrate recognition and translocation for the Glucose superfamily EIIC transporters.
Graphical Abstract
Introduction
The phosphoenolpyruvate:carbohydrate phosphotransferase system, PTS, is responsible for carrying out the energy-dependent transport of carbohydrates as well as a variety of regulatory functions in bacteria (Deutscher et al., 2014; Siebold et al., 2001). Active sugar uptake is driven by covalent modification of the translocated sugar with a phosphate group that derives from phopsphoenol pyruvate. This phosphate is transferred to the sugar through a series of soluble phosphocarrier proteins that include, in order of phosphorylation, enzyme I (EI), histidine-containing phosphocarrier protein (HPr), enzyme IIA (EIIA), and enzyme IIB (EIIB) (Kundig et al., 1964). The protein that transports the sugar across the membrane and also catalyzes the phosphotransfer from EIIB is the integral membrane protein enzyme IIC (EIIC). EIIC, EIIB, and EIIA can be encoded by the same gene or separately.
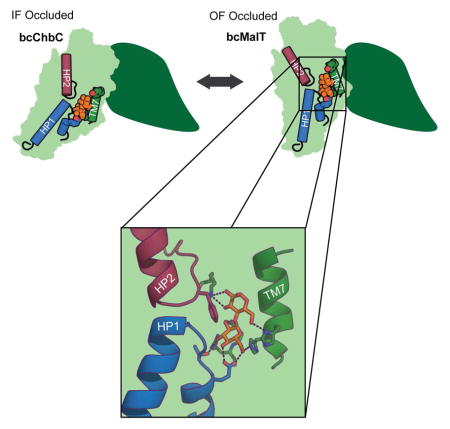
The EIIC components of the PTS are grouped in four different superfamilies, the largest of which is the Glucose superfamily (Nguyen et al., 2006; Saier et al., 2005). Members within the superfamily share little sequence identity and their substrate selectivity varies considerably. Recently, a crystal structure of the Glucose superfamily EIIC from Bacillus cereus, bcChbC, was crystallized in an inward-facing occluded conformation (Cao et al., 2011). The bound substrate, diacetylchitobiose ((GlcNAc)2), was located near the cytoplasmic surface of the protein but was shielded from the solvent by a conserved loop from the neighboring protomer in the dimer. However, many of the amino acids found to interact with the bound substrate (GlcNAc)2 are not conserved in other Glucose superfamily proteins. In addition, in the absence of an outward-facing structure, the conformational changes leading to carbohydrate transport remain unknown. In order to further understanding of both the mechanism of transport within EIIC proteins, as well as identify residues that confer selectivity to Glucose superfamily PTS EIIC transporters, we have undertaken structural studies on other members of the Glucose superfamily of EIIC transporters. To this end, we have solved the crystal structure of an EIIC in an outward-facing conformation and characterized its function. This structure possesses the same overall fold as the previous bcChbC structure, but a rigid body motion of the C-terminal domain moves the substrate binding site by roughly 20 Å.
Results
Functional Characterization of bcMalT
The EIIC domain from Bacillus cereus (Uniprot ID Q63GK8) shares greater than 60% amino acid sequence identity with that of functionally characterized maltose PTS transporters named MalT from Enterococcus faecalis (Le Breton et al., 2005), Streptococcus pyogenes (Shelburne et al., 2008) and Streptococcus mutans (Webb et al., 2007) (Figure S1). The operon that includes this gene also contains open reading frames predicted to encode for an alpha-glucosidase and maltose 6′-phosphate phosphatase. Consequently, the most likely function of this protein is maltose transport, and we refer to it as bcMalT from this point onward.
To verify that bcMalT is a maltose transporter, we measured radiolabeled maltose binding to full-length bcMalT (containing both the EIIB and EIIC domains) solubilized in lauryldimethylamine oxide (LDAO) using the scintillation proximity assay (SPA) (Figure 1A). Binding of maltose was abolished in the presence of the detergent decylmaltopyranoside (DM), which has a maltose-like headgroup (Figure 1B). No significant binding was observed for a number of other sugars tested, with the exception of sucrose (Figure 1C). It is notable that the glucose monosaccharide does not bind, indicating that interactions with both sugar moieties are necessary; furthermore, the 1,1-linked glucose disaccharide, trehalose, also does not bind, demonstrating sensitivity to the disaccharide linkage as well. In competitive binding experiments, unlabeled maltose competed for binding with [3H]maltose with a half-inhibition concentration (IC50) of 4.5 ± 1.1 μM (Figure 1A). We then reconstituted full-length bcMalT into liposomes to measure bcMalT-mediated [3H]maltose uptake by facilitated diffusion. The time-dependent accumulation of the radiolabeled sugar in bcMalT-containing proteoliposomes reached a steady-state level after ~10 minutes, which was about 5-fold higher than the accumulation determined in control liposomes (Figure 1D). To confirm that the measured radioactive signal does not merely reflect binding of radiolabeled maltose to the bcMalT molecules situated in the membrane of the proteoliposomes, experiments were performed in which bcMalT-containing proteoliposomes were incubated in the presence or absence of 5 mM LDAO. By measuring the protein retention on the filters used for the uptake studies we found that this concentration of LDAO causes the permeabilization of the proteoliposomes without abolishing their integrity. After determining the non-specific interaction of [3H]maltose with control liposomes, our data show that in detergent-free proteoliposomes approximately 7 molecules of maltose are translocated by one molecule of bcMalT, whereas 1 molecule of maltose is bound per bcMalT molecule as measured in LDAO-permeabilized proteoliposomes (Figure 1E). Transport was also measured for trypsinized bcMalT containing only the EIIC domain, which exhibited activity similar to the full-length protein (Figure S2). These results indicate that the EIIC domain of bcMalT can indeed mediate the facilitated diffusion of maltose. Facilitated sugar transport in the absence of the EIIB phosphotransferase domain has been shown for ChbC (Cao et al., 2011) and other PTS EIIC domains (Postma et al., 1993).
Figure 1. bcMalT is a maltose transporter.

a. Competitive binding to full-length bcMalT in LDAO between [3H]maltose and unlabeled maltose. b. Binding of [3H]maltose to full-length bcMalT solubilized in either LDAO or DM. c. Competitive binding to full-length bcMalT in LDAO between [3H]maltose and 500 μM of the indicated unlabeled sugars, measured by SPA. d. Uptake of [3H]maltose into proteoliposomes containing full-length bcMalT (black squares) or control liposomes without protein (white circles). Error bars are standard deviations of three technical replicates. e. Verification of [3H]maltose uptake by comparing accumulation in proteoliposomes incorporating full-length bcMalT or control liposomes without protein in the presence or absence of 5 mM LDAO.
Structure of bcMalT
We crystallized the trypsinized EIIC domain of selenomethionine-derivatized bcMalT using the lipidic cubic phase method. The best crystal diffracted to 2.55 Å and indexed to the I222 space group. Phases were solved by the single-wavelength anomalous dispersion method (Figure S3A–C, Table 1). The protein crystallized as a homodimer with one protomer in the asymmetric unit (Figure S3D). The final model contained residues 8 through 450 as well as a molecule of maltose.
Table 1.
Data collection and refinement statistics
| bcMalT | |
|---|---|
| Data collection | |
| Space group | I222 |
| Unit Cell (Å) | a=71.63, b=108.18, c=139.329 |
| Wavelength (Å) | 0.9787 |
| Resolution (Å) | 50.00 - 2.55 |
| Rsym | 0.11 (0.804) |
| I/σI | 11.6 (1.48) |
| Completeness (%) | 100.0 (99.9) |
| Redundancy | 4.6 (4.0) |
| CC1/2 | (0.632) |
| Refinement | |
| Resolution (Å) | 34.83 – 2.55 |
| No. reflections | 33939 |
| Rwork/Rfree | 23.7/28.2 |
| No. atoms | |
| Protein | 3402 |
| Ligand/ion | 23 |
| Water | 14 |
| B-factors (Å2) | |
| Protein | 52.4 |
| Ligand/ion | 45.3 |
| Water | 46.6 |
| R.m.s deviations | |
| Bond lengths (Å) | 0.002 |
| Bond angles (°) | 0.447 |
| Ramachandran Plot | |
| Favored (%) | 96.6 |
| Allowed (%) | 3.2 |
| Outliers (%) | 0.2 |
Although the EIIC portion of bcMalT is only 19% identical and 50% similar to bcChbC, the structural fold of the two proteins is similar and almost all of the individual secondary structure elements are conserved (Figure 2A, B). Both proteins contain two distinct structural domains formed by the N- and C-terminal halves of each protomer (Figure 2C). We define the N-terminal domain as the dimerization domain, and the C-terminal domain as the substrate-binding domain, for reasons to be discussed below. Like bcChbC, the N-terminal dimerization domain of bcMalT is initiated with a short cytoplasmic amphipathic helix (AH1) followed by five transmembrane helices (TM1 – TM5). The periplasmic loop between TM3 and TM4 is 13 residues longer in bcMalT and forms a second two-stranded antiparallel β-sheet in addition to the β-sheet present in bcChbC. TM1, TM2, TM3, and TM5 from the dimerization domain comprise most of the dimerization interface between the two protomers of the bcMalT homodimer (Figure 3A). The dimerization domain is connected to the substrate-binding domain through a periplasmic amphipathic helix (AH2). The dimerization domains of the two neighboring protomers form a large and mostly hydrophobic interface, with a buried surface area of ~2700 Å2.
Figure 2. Structure of bcMalT.

a–b. Topology diagrams of bcMalT (a) and bcChbC (b) oriented with the periplasmic side on top. The N-terminal dimerization domain and C-terminal transport domain are colored dark and light green, respectively; HP1 and HP2 are colored marine and raspberry. Structural elements in the transport domain not shared with bcChbC are indicated with red labels, and include a periplasmic loop following TM7 containing a two-stranded antiparallel β-sheet (β5 and β6) and two additional α-helices (periplasmic helices PH1 and PH2). c. Protomer of bcMalT in two orientations, colored according to the scheme in panel a. Hairpin loops HP1 and HP2 are shown in marine and raspberry.
Figure 3. Comparison of bcMalT and bcChbC.

a. Dimeric arrangement of bcMalT (left) and bcChbC (right). The N-terminal dimerization domains are dark blue and dark green. The C-terminal transport domains are colored light blue and light green. Maltose or (GlcNAc)2 is shown as red spheres. The approximate location of the membrane is marked with black lines. b. Superposition of the dimerization domains (TM1-5, left) and transport domains (TM6-TM10, right) of bcChbC (raspberry) and bcMalT (marine).
The bcMalT and bcChbC structures align poorly when superposed over the entire monomer. However, the two individual domains align very well when superposed independently (Figure 3B). The difference between the two structures can be described as a rigid body rotation of 44° and a translation of 9 Å of the bcMalT substrate-binding domain towards the periplasm (Figure 3A). In bcChbC, the substrate-binding domain contains 5 transmembrane helices (TM6 – TM10) and two re-entrant hairpin loops, one close to the cytoplasmic side following TM8 and another close to the periplasmic side following TM9. The bcMalT structure contains equivalents of each of these helices as well as the hairpin loops; however, due to the substantial shift of the substrate-binding domain towards the periplasm, the helices corresponding to TM6 and TM7 do not seem to extend to the cytoplasmic surface of the protein. Although these helices do not appear to span the membrane in the bcMalT structure, we continue to refer to them as TM6 and TM7 for consistency with the bcChbC structure. (Figure 2C). Another difference between the substrate-binding domains of bcMalT and bcChbC is the addition of two alpha-helices and two beta strands (β5, β6, PH1, and PH2, Figure 2) on the periplasmic side of bcMalT between TM7 and TM8.
Active Site Architecture
Each protomer contains a large cavity in which substrate is bound. While the substrate-binding cavity of bcChbC lies near the cytoplasmic surface of the protein, the change in position of the substrate-binding domain in bcMalT repositions the cavity close to the periplasmic surface (Figure 4A). A small opening to the cavity is located on the periplasmic side of the bcMalT crystal structure, but it appears to be too narrow to allow access of substrate from the extracellular side.
Figure 4. The substrate-binding cavity and active site in bcMalT.

a. The bcMalT (top) and bcChbC (bottom) dimer with the substrate binding cavities shown as pink surfaces, illustrating the change in position of the cavity from the periplasmic to cytoplasmic side. b. Fo-Fc electron density for the substrate molecule in bcMalT, contoured at 3 σ (left) and 2Fo-Fc electron density contoured at 1.5 σ (right). c–d. The substrate-binding site in bcMalT (c) and bcChbC (d) is shown with the bound maltose or (GlcNAc)2 as yellow and red sticks. Conserved interactions occur between the disaccharides and HP1 (marine), HP2 (raspberry), and TM6, 7, and 8.
The bcMalT protomer contains a clear disaccharide density in the substrate-binding cavity that closely matches maltose (Figure 4B). There was some additional positive Fo-Fc density on C2 of the reducing end of maltose corresponding to an additional atom. We suspect this additional density comes from a disordered acyl chain and the maltose bound to the active site comes from the head group of either the dodecylmaltopyranoside (DDM) or DM used during the course of the purification. The remainder of the acyl chain would extend outside the substrate cavity and is presumably unstructured. Consistent with this hypothesis, when the binding assays are performed with DM rather than LDAO, the protein does not bind maltose (Figure 1B).
Although the substrate-binding cavity lies near the interface with the dimerization domain, all the residues directly interacting with the substrate come from the substrate-binding domain. A number of polar and charged sidechains form hydrogen bonds with maltose (Figure 4C). Two highly conserved residues, Glu355 on HP1 and His240 on TM7, interact with the C6 hydroxyl on the non-reducing sugar that receives the phosphate from EIIB. Thr354 on HP1 and Arg232 and Glu231 on TM6 form additional hydrogen bonds to the non-reducing sugar, while Lys307 on TM8 and His241 on TM7 stabilize the reducing sugar. Additionally, the aromatic ring of Phe393 on HP2 contributes stacking interactions. The Glucose superfamily contains a number of subfamilies, of which bcMalT belongs to the glucose (Glc) subfamily. Within the Glc subfamily of EIIC transporters, Glu355, His240, His241, Arg232, and Thr354 appear to be strongly conserved (Figure S1). Mutation of residues equivalent to Glu355, His240, His241, and Arg232 in other Glc subfamily members has been shown to result in loss of binding or uptake activity (Lanz and Erni, 1998; Ruijter et al., 1992; Saraceni-Richards and Jacobson, 1997). Glu231 is slightly less conserved. In Glc subfamily members selective for N-acetyl glucosamine, Glu231 is replaced with an asparagine, which would presumably better accommodate the acetyl group at the sugar’s C2 position.
While sequence identity between bcChbC and bcMalT is less than 20%, there are similarities in the construction of the substrate-binding site. In particular, the same structural elements (HP1, HP2, and TM6-8) form the binding site in bcChbC, and the histidine and glutamate interacting with the C6-OH (His250 and Glu334 in bcChbC) are conserved (Figure 4C, D). These two residues appear to be conserved across the Glucose superfamily. The conserved glutamate in both proteins is preceded by a small polar residue (Thr354 in bcMalT, Asn333 in bcChbC) that forms a hydrogen bond with C3-OH on the non-reducing sugar. Trp382 in bcChbC is equivalent to Phe393 in bcMalT, providing stacking interactions with the disaccharide. Differences between the two binding sites may explain their respective sugar specificities. In bcMalT, Glu231 and Arg232 interact with C2-OH and C3-OH on the non-reducing sugar, whereas in bcChbC these residues are replaced with the shorter sidechains of Val241 and His242 in order to accommodate the bulky N-acetyl group on C2 of (GlcNAc)2.
Substrate access to the bcMalT binding site
In the bcMalT crystal structure, the substrate-binding cavity is close to the periplasmic side of the membrane, but does not appear to have an opening large enough to allow substrate to diffuse freely. The structure therefore corresponds to the outward-facing occluded state of the transport cycle. In order to gain insight into how the substrate-binding site is accessed from the periplasmic side, we used molecular dynamics (MD) simulations to search for potential pathways for substrate access in bcMalT. To observe fluctuations in the opening to the periplasm, we performed MD simulations of 3 independent systems of bcMalT embedded in a membrane environment, starting from the crystal structure. Interestingly, we observed conformational changes in two of the systems that widened the cavity opening enough to release bound substrate into the periplasm (Figure 5A). The cavity entrance was opened by a rotation of TM7 towards AH2 (Figure 5B). Tyr249 on TM7, which is moderately conserved across the Glc subfamily, is pulled away from the substrate enlarging the mouth of the cavity (Figure 5C, D). This suggests that the outward-facing occluded conformation observed in the crystal structure can be converted by a relatively small conformational movement to an outward-facing open state that allows substrate access.
Figure 5. Dynamics of substrate release.

a. Substrate release during the course of the MD simulations, as indicated by the distance between Glu355 and the sugar. Traces are shown for a total of six protomers from two 200-ns simulations and one 250-ns simulation. b. The bcMalT crystal structure viewed from the periplasmic side. The bound maltose is shown in orange. Inset on left shows a close-up view of TM7 in the bcMalT crystal (green) and in a frame from an extended MD simulation showing release of the maltose (blue). The maltose molecule from the bcMalT crystal structure is shown as orange sticks. c–d. Surface representation of the bcMalT crystal structure (c) and the simulation model (d) shown from the same orientation. The maltose molecule from the crystal structure is shown as orange sticks.
Discussion
We have reported the first crystal structure of a PTS EIIC from the Glucose superfamily in an outward-facing occluded conformation. Comparison with a previous inward-facing occluded structure allows us to propose a mechanism of transport in which a large rigid-body motion of the substrate-binding domain moves the substrate from one side of the bilayer to the other. We have also shown that the transporter selectively binds and transports maltose across the membrane. Although the rate observed for maltose transport is low, this is expected as the assay measures facilitated diffusion in the absence of the PTS components needed to phosphorylate the EIIB domain. In the cell, phosphorylation of the substrate when bound to EIIC would presumably accelerate substrate release and the overall rate of transport.
Previous reports using charge distribution analysis, hydropathy analysis, accessibility assays, and/or fusion assays have presented conflicting results in the predicted topology of different, and in some cases, the same Glucose superfamily EIIC proteins (Buhr and Erni, 1993; Lee and Saier, 1983; McCoy et al., 2015; Sugiyama et al., 1991; Vervoort et al., 2005; Yagur-Kroll and Amster-Choder, 2005; Yagur-Kroll et al., 2009). Although the sequence identity between bcMalT and bcChbC is low, their topologies are essentially identical, supporting the conclusions of Nguyen et al (Nguyen et al., 2006) that the EIICs of Glucose superfamily members share similar folds. The difference in the position of the C-terminal substrate-binding domain is large enough to change the position of some structural elements relative to the membrane, as seen for example with the partial displacement of TM6 and TM7 into the periplasm in bcMalT, which could explain the inconsistencies in characterizing the topology of these proteins.
The secondary structural elements and the overall structural fold of bcMalT and bcChbC are similar (Figure 3B), as is the architecture of the substrate-binding cavity (Figure 4C, D), but the location of the substrate-binding domain differs by a rigid body displacement (Fig. 4A). This difference converts the transporter from an inward-facing to an outwards-facing state, suggesting that the two proteins share a similar transport cycle but have crystallized in two different states. According to this hypothesis, we can propose an alternating access model for the bcMalT transport cycle beginning with an outward-facing open state (Fig. 6A) in which the substrate-binding cavity is accessible to substrate from the periplasmic side. A rotation of TM7 closes off the cavity to create the substrate-bound, outward-facing occluded state (Fig. 6B), represented by the bcMalT crystal structure. The substrate-binding domain, comprising helices TM6-10, HP1 and HP2, moves as a rigid body towards the cytoplasm to form the inward-occluded state (Fig. 6C), corresponding to the bcChbC crystal structure. Because all residues that interact directly with the bound disaccharide are from the substrate-binding domain, the substrate-binding domain can move between these inward and outward-facing states without requiring the disruption and re-formation of interactions with the substrate. This observation is consistent with an “elevator car” type transport mechanism, also observed in the transporters GltPh (Reyes et al., 2009), ASBT (Zhou et al., 2014), and NhaA (Lee et al., 2013), in which the ligand-binding site is carried intact from one side of the bilayer to the other by a rigid body motion of one domain relative to the bilayer. The TM4-5 loop, which blocks cytoplasmic access to the substrate-binding cavity in the bcChbC structure (Cao et al., 2011), then opens to allow phosphorylated EIIB access to the substrate (Fig. 6D), and the phosphorylated substrate is released into the cytoplasm.
Figure 6. Proposed mechanism of substrate transport.

Diagram showing a potential model for the Glucose superfamily EIIC transport cycle.
A crystal structure has recently been reported for another PTS EIIC, a vitamin C transporter from the Ascorbate superfamily, which catalyzes the conversion of L-ascorbic acid to L-ascorbate 6-phosphate (Luo et al., 2015). This protein, UlaA, has a unique fold in which each protomer contains 11 transmembrane helices and four reentrant hairpins. These are divided between distinct dimerization and substrate binding domains related by an inverted repeat. This fold cannot be superposed onto the Glucose superfamily members bcMalT and bcChbC (Luo et al., 2015). UlaA was crystallized in both an outward-open and outward-occluded state that involved a rigid body rotation of the substrate binding domain by 4.33° and left the substrate-binding cavity unchanged. Though not as large a difference as that observed between the bcChbC and bcMalT structures (44° and a translation of 9 Å), this indicates that the mechanism of transport may ultimately be conserved between different PTS superfamilies despite their difference in fold.
Methods
Expression and Purification
We initially screened 92 different EIIC homologs in multiple plasmids to find optimal candidates for protein crystallization based on expression levels and size exclusion chromatography profiles. The gene encoding the protein described in this report was cloned from the Bacillus cereus E33L chromosome into a pMCSG28 vector containing a C-terminal His-tag. Transformed cells were grown in selenomethionine-containing minimal media until reaching an optical density of 0.6 and then induced with 0.5 mM IPTG. Cells were then grown at 20°C overnight, spun down at 4500g for 15 minutes, and resuspended in a solution of 20 mM HEPES pH 7.5, 150 mM NaCl, and 10% glycerol.
Cell suspensions were supplemented with 2 mM β-mercaptoethanol (βME), 25 μg mL−1 DNase I, 5 mM MgCl2, and 1 mM PMSF and then sonicated until cells appeared fully lysed. Protein extraction was carried out by addition of 30 mM DDM (Anatrace) and gentle shaking for 2 hours at room temperature. After centrifugation for 45 minutes at 55,000g, the supernatant was run over cobalt-based affinity resin (Talon, Clontech) followed by washing with 20 mM imidazole, and elution with 300 mM imidazole. The full-length EIICB protein was unstable at high concentrations in detergent, so partially-proteolyzed protein with only the EIIC domain intact was used for crystallization trials. Protein was partially proteolyzed with trypsin (1:20 wt ratio of bcMalT to trypsin) at room temperature for 30 minutes and then run over a Superdex 200 Increase 10/300 GL size exclusion column equilibrated with 20 mM HEPES pH 7.5, 150 mM NaCl, 10 mM trehalose, 4 mM DM and 5 mM βME. MALDI mass spectrometry was carried out following the ultrathin layer methodology (Cadene and Chait, 2000) on a Bruker Microflex mass spectrometer, and indicated that the purified, trypsinized protein had a molecular weight of 50.84 KDa (data not shown), most likely corresponding to residues 2 through 472 of the 545-residue gene product. For functional characterization by SPA, full length His-tagged bcMalT was purified in the same way as described above except that 4 mM LDAO was used instead of DM for size exclusion chromatography and trehalose was removed from the buffer.
Crystallization
Protein for crystallization was concentrated to 50 – 55 mg ml−1 and 2:3 protein solution:molten monoolein (w:w) were mixed with a coupled syringe device. Drops of the protein lipid mixture were placed in 24-well sitting drop trays and covered with 4 μl of well solution composed of 39% polyethylene glycol 400, 450 mM NaCl, 3% ethanol, and 100 mM N-(2-Acetamido)iminodiacetic acid pH 6.7. Crystals were collected in approximately three weeks and flash frozen in liquid nitrogen.
X-ray data collection and processing
X-ray data were collected at beamline 17-ID (IMCA-CAT) at the Advanced Photon Source at Argonne National Laboratory. We collected a data set with a resolution of 2.55 Å at a wavelength of 0.9787 Å. The reflections were then processed with HKL2000 (Otwinowski and Zanolari, 1997). The crystal belonged to space group I222 with unit cell dimensions of 71.6 Å, 108.2 Å, and 139.3 Å.
Structure determination and refinement
SHELXD (Sheldrick, 2008) was used to find 15 of 16 selenium sites within the asymmetric unit. These sites were input into AUTOSHARP (Vonrhein et al., 2007) from which the initial phases were determined. The structure was completed through successive rounds of model building in COOT (Emsley et al., 2010) and refinement in PHENIX (Adams et al., 2010). The final asymmetric unit contained one protomer of bcMalT, 14 water molecules, and one molecule of maltose. While the 2.55 Å dataset was obtained from a crystal grown in the presence of trehalose, used as an additive, the substrate density clearly does not accommodate trehalose. Slightly lower resolution data were collected on crystals not containing additional sugar additives and these maps also contained Fo-Fc density corresponding to maltose. Our binding data also indicate that trehalose cannot bind to the transporter (Fig. 1C). In the final stages of refinement, TLS groups were determined using TLSMD (Painter and Merritt, 2006) and protein geometry was validated with MolProbity (Chen et al., 2010).
Scintillation Proximity Assay
Assays were done in 96-well format and a MicroBeta 1450 Microplate Counter (Perkin Elmer) was used for scintillation counting. Full-length bcMalT-His was purified in LDAO or DM-containing buffer as described above. The binding was measured after mixing protein, substrate and Copper HIS-Tag YSI beads (Perkin Elmer, RPNQ0096) and incubating for 1 hr at 4° C in 20 mM HEPES pH 7.5, 150 mM NaCl, 1 mM βME and 4 mM LDAO or DM. Each 100 μL reaction contained 300 ng bcMalT-His, 1 μM [3H]maltose (ARC, ART 0474), the indicated concentration of unlabeled substrate, and 2.5 mg/ml Copper HIS-Tag YSI beads. Background binding was subtracted after measuring scintillation in the absence of protein. Data were fitted and plotted in Origin 6.1 software.
[3H]Maltose uptake in proteoliposomes
Purified recombinant MalT (full-length or trypsinized) was reconstituted at a 1:100 (w/w) ratio in preformed, Triton X-100 (0.12 % [w/w])-destabilized liposomes that were prepared with E. coli polar lipid extract (Avanti, Inc.) in 100 mM potassium phosphate, pH 7.5 and 2 mM β-mercaptoethanol. Prior to the uptake measurements, frozen proteoliposomes were subjected three times to a freeze-thaw cycle followed by 2 × 10 s sonication in a Bioruptor®. The uptake reaction was initiated by the 20-fold dilution of the proteoliposome suspension (4.5 mg lipid/mL) into assay buffer composed of 100 mM potassium phosphate, pH 7.5 plus the 20 mM of [3H]maltose (1 Ci/mmol) at 23 °C. Reactions were stopped by the addition of ice-cold 100 mM potassium phosphate, pH 6.0 and 100 mM LiCl and filtered through Millipore 0.22 μm nitrocellulose filters. The radioactivity retained on the filters was determined with scintillation counting. Known amounts of radioactivity were used to convert counts per minute (cpm) to mol.
In order to distinguish between actual [3H]maltose accumulation on bcMalT-containing liposomes and binding to the membrane-embedded protein in the proteoliposomes, the (proteo)liposomes were incubated in the assay buffer in the presence or absence of 5 mM LDAO for 30 min at 23 °C. This concentration of detergent was chosen based on experiments in which the protein retention of LDAO-treated proteoliposomes on 0.22 μm nitrocellulose filters equaled that of proteoliposomes that were not subjected to the LDAO treatment and internally accumulated radiotracer was released. 20 μM 3H-maltose (1 Ci/mmol) was added and the samples were incubated for another 30 min (corresponding to the steady-state level of maltose accumulation (Fig. 1D). Accumulation of 3H-maltose was measured as described above.
Simulations
The bcMalT crystal structure was used to generate a protein-membrane complex system using CHARMM-GUI Membrane Builder (Brooks et al., 2009; Jo et al., 2007; Jo et al., 2008; Jo et al., 2009; Wu et al., 2014), http://www.charmm-gui.org/input/membrane, with a 1:3 ratio of POPG:POPE bilayer, around 50,000 water molecules (determined from 17.5 Å of water thickness above and below the membrane bilayer), and 0.15 M of K+ and Cl− ions. The simulation system was triplicated and independently simulated to get better sampling. All systems were equilibrated following Membrane Builder equilibration protocols, and 200 (replica 1 and 2) to 250 ns (replica 3) of production runs were performed with a 2-fs time-step under the constant pressure and temperature conditions (NPT) at 1 bar and 303.15 K. A semi-isotropic Langevin piston and Langevin temperature coupling method were adopted to maintain the pressure and temperature respectively. All simulations were performed using NAMD 2.9 software package (Phillips et al., 2005) with the all-atom CHARMM36 force field (Klauda et al., 2010).
Supplementary Material
Highlights.
bcMalT is a selective maltose transporter belonging to the EIIC Glucose superfamily
bcMalT is the first outward-facing structure of a Glucose superfamily EIIC
bcMalT and previously-characterized EIIC bcChbC share a similar fold
The bcMalT substrate-binding domain is displaced relative to that of bcChbC by 20 Å
Acknowledgments
This work was supported by the US National Institutes of Health (R01GM098878, R01HL086392, R01DK088057, U54GM095315, and U54GM087519), the American Heart Association (12EIA8850017), and the Cancer Prevention and Research Institute of Texas (R12MZ). Final data were collected at beamline 17-ID at the Advanced Photon Source, which is supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute. Crystals were also screened at Northeastern Collaborative Access Team (NE-CAT) beamlines, beamlines 8.2.2 and 5.0.2 at Berkeley Center for Structural Biology at the Lawrence Berkeley Laboratory. We thank K. Rajashankar for advice on X-ray crystallography, and K. Rajashankar, L. Keefe, E. Zoellner, K. Battaile, and A. Mulichak for beamline support. We thank R. Bruni and B. Kloss, both from the New York Consortium on Membrane Protein Structure, for EIIC homologs in expression vectors.
Footnotes
Accession Codes Atomic coordinates and structure factors have been deposited with the Protein Data Bank under accession ID 5IWS.
Author Contributions MZ, JGM, and ZR conceived the project and designed the experiments. JGM, ZR, SM and EJL expressed, purified, and crystallized bcMalT and solved its structure. VS, ZR, JGM and MQ conducted binding and transport studies. JL and WI performed molecular dynamics simulations. SP performed mass spectrometry. All authors analyzed data and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica Section D, Biological crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. CHARMM: the biomolecular simulation program. Journal of computational chemistry. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhr A, Erni B. Membrane topology of the glucose transporter of Escherichia coli. The Journal of biological chemistry. 1993;268:11599–11603. [PubMed] [Google Scholar]
- Cadene M, Chait BT. A robust, detergent-friendly method for mass spectrometric analysis of integral membrane proteins. Analytical chemistry. 2000;72:5655–5658. doi: 10.1021/ac000811l. [DOI] [PubMed] [Google Scholar]
- Cao Y, Jin X, Levin EJ, Huang H, Zong Y, Quick M, Weng J, Pan Y, Love J, Punta M, et al. Crystal structure of a phosphorylation-coupled saccharide transporter. Nature. 2011;473:50–54. doi: 10.1038/nature09939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta crystallographica Section D, Biological crystallography. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutscher J, Ake FM, Derkaoui M, Zebre AC, Cao TN, Bouraoui H, Kentache T, Mokhtari A, Milohanic E, Joyet P. The bacterial phosphoenolpyruvate:carbohydrate phosphotransferase system: regulation by protein phosphorylation and phosphorylation-dependent protein-protein interactions. Microbiology and molecular biology reviews : MMBR. 2014;78:231–256. doi: 10.1128/MMBR.00001-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta crystallographica Section D, Biological crystallography. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Im W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PloS one. 2007;2:e880. doi: 10.1371/journal.pone.0000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. Journal of computational chemistry. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Jo S, Lim JB, Klauda JB, Im W. CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophysical journal. 2009;97:50–58. doi: 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Jr, Pastor RW. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. The journal of physical chemistry B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundig W, Ghosh S, Roseman S. Phosphate Bound to Histidine in a Protein as an Intermediate in a Novel Phospho-Transferase System. Proceedings of the National Academy of Sciences of the United States of America. 1964;52:1067–1074. doi: 10.1073/pnas.52.4.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz R, Erni B. The glucose transporter of the Escherichia coli phosphotransferase system. Mutant analysis of the invariant arginines, histidines, and domain linker. The Journal of biological chemistry. 1998;273:12239–12243. doi: 10.1074/jbc.273.20.12239. [DOI] [PubMed] [Google Scholar]
- Le Breton Y, Pichereau V, Sauvageot N, Auffray Y, Rince A. Maltose utilization in Enterococcus faecalis. Journal of applied microbiology. 2005;98:806–813. doi: 10.1111/j.1365-2672.2004.02468.x. [DOI] [PubMed] [Google Scholar]
- Lee C, Kang HJ, von Ballmoos C, Newstead S, Uzdavinys P, Dotson DL, Iwata S, Beckstein O, Cameron AD, Drew D. A two-domain elevator mechanism for sodium/proton antiport. Nature. 2013;501:573–577. doi: 10.1038/nature12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CA, Saier MH., Jr Mannitol-specific enzyme II of the bacterial phosphotransferase system. III. The nucleotide sequence of the permease gene. The Journal of biological chemistry. 1983;258:10761–10767. [PubMed] [Google Scholar]
- Luo P, Yu X, Wang W, Fan S, Li X, Wang J. Crystal structure of a phosphorylation-coupled vitamin C transporter. Nature structural & molecular biology. 2015;22:238–241. doi: 10.1038/nsmb.2975. [DOI] [PubMed] [Google Scholar]
- McCoy JG, Levin EJ, Zhou M. Structural insight into the PTS sugar transporter EIIC. Biochimica et biophysica acta. 2015;1850:577–585. doi: 10.1016/j.bbagen.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TX, Yen MR, Barabote RD, Saier MH., Jr Topological predictions for integral membrane permeases of the phosphoenolpyruvate:sugar phosphotransferase system. Journal of molecular microbiology and biotechnology. 2006;11:345–360. doi: 10.1159/000095636. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Zanolari B. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW Jr, Sweet RM, editors. Methods in Enzymology. New York: Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta crystallographica Section D, Biological crystallography. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. Journal of computational chemistry. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postma PW, Lengeler JW, Jacobson GR. Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiological reviews. 1993;57:543–594. doi: 10.1128/mr.57.3.543-594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes N, Ginter C, Boudker O. Transport mechanism of a bacterial homologue of glutamate transporters. Nature. 2009;462:880–885. doi: 10.1038/nature08616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruijter GJ, van Meurs G, Verwey MA, Postma PW, van Dam K. Analysis of mutations that uncouple transport from phosphorylation in enzyme IIGlc of the Escherichia coli phosphoenolpyruvate-dependent phosphotransferase system. Journal of bacteriology. 1992;174:2843–2850. doi: 10.1128/jb.174.9.2843-2850.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saier MH, Hvorup RN, Barabote RD. Evolution of the bacterial phosphotransferase system: from carriers and enzymes to group translocators. Biochemical Society transactions. 2005;33:220–224. doi: 10.1042/BST0330220. [DOI] [PubMed] [Google Scholar]
- Saraceni-Richards CA, Jacobson GR. A conserved glutamate residue, Glu-257, is important for substrate binding and transport by the Escherichia coli mannitol permease. Journal of bacteriology. 1997;179:1135–1142. doi: 10.1128/jb.179.4.1135-1142.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelburne SA, 3rd, Keith DB, Davenport MT, Horstmann N, Brennan RG, Musser JM. Molecular characterization of group A Streptococcus maltodextrin catabolism and its role in pharyngitis. Molecular microbiology. 2008;69:436–452. doi: 10.1111/j.1365-2958.2008.06290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick GM. A short history of SHELX. Acta crystallographica Section A, Foundations of crystallography. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Siebold C, Flükiger K, Beutler R, Erni B. Carbohydrate transporters of the bacterial phosphoenolpyruvate: sugar phosphotransferase system (PTS) FEBS letters. 2001;504:104–111. doi: 10.1016/s0014-5793(01)02705-3. [DOI] [PubMed] [Google Scholar]
- Sugiyama JE, Mahmoodian S, Jacobson GR. Membrane topology analysis of Escherichia coli mannitol permease by using a nested-deletion method to create mtlA-phoA fusions. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:9603–9607. doi: 10.1073/pnas.88.21.9603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervoort EB, Bultema JB, Schuurman-Wolters GK, Geertsma ER, Broos J, Poolman B. The first cytoplasmic loop of the mannitol permease from Escherichia coli is accessible for sulfhydryl reagents from the periplasmic side of the membrane. Journal of molecular biology. 2005;346:733–743. doi: 10.1016/j.jmb.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated structure solution with autoSHARP. Methods in molecular biology. 2007;364:215–230. doi: 10.1385/1-59745-266-1:215. [DOI] [PubMed] [Google Scholar]
- Webb AJ, Homer KA, Hosie AH. A phosphoenolpyruvate-dependent phosphotransferase system is the principal maltose transporter in Streptococcus mutans. Journal of bacteriology. 2007;189:3322–3327. doi: 10.1128/JB.01633-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu EL, Cheng X, Jo S, Rui H, Song KC, Davila-Contreras EM, Qi Y, Lee J, Monje-Galvan V, Venable RM, et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. Journal of computational chemistry. 2014;35:1997–2004. doi: 10.1002/jcc.23702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagur-Kroll S, Amster-Choder O. Dynamic membrane topology of the Escherichia coli beta-glucoside transporter BglF. The Journal of biological chemistry. 2005;280:19306–19318. doi: 10.1074/jbc.M410896200. [DOI] [PubMed] [Google Scholar]
- Yagur-Kroll S, Ido A, Amster-Choder O. Spatial arrangement of the beta-glucoside transporter from Escherichia coli. Journal of bacteriology. 2009;191:3086–3094. doi: 10.1128/JB.01037-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Levin EJ, Pan Y, McCoy JG, Sharma R, Kloss B, Bruni R, Quick M, Zhou M. Structural basis of the alternating-access mechanism in a bile acid transporter. Nature. 2014;505:569–573. doi: 10.1038/nature12811. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.