

Abstract
Disruptions in folate-mediated one-carbon metabolism (FOCM) are associated with risk for several pathologies including developmental anomalies such as neural tube defects and congenital heart defects, diseases of aging including cognitive decline, neurodegeneration and epithelial cancers, and hematopoietic disorders including megaloblastic anemia. However, the causal pathways and mechanisms that underlie these pathologies remain unresolved. Because folate-dependent anabolic pathways are tightly interconnected and best described as a metabolic network, the identification of causal pathways and associated mechanisms of pathophysiology remains a major challenge in identifying the contribution of individual pathways to disease phenotypes. Investigations of genetic mouse models and human inborn errors of metabolism enable a more precise dissection of the pathways that constitute the FOCM network and enable elucidation of causal pathways associated with NTDs. In this overview, we summarize recent evidence that the enzyme MTHFD1 plays an essential role in FOCM in humans and in mice, and that it determines the partitioning of folate-activated one carbon units between the folate-dependent de novo thymidylate and homocysteine remethylation pathways through its regulated nuclear localization. We demonstrate that impairments in MTHFD1 activity compromise both homocysteine remethylation and de novo thymidylate biosynthesis, and provide evidence that MTHFD1-associated disruptions in de novo thymidylate biosynthesis lead to genome instability that may underlie folate-associated immunodeficiency and birth defects.
Keywords: MTHFD1, SHMT, TYMS, DHFR, thymidylate, folate, lamin, DNA replication, multi-enzyme complex
I. Overview of folate-mediated one-carbon metabolism
Tetrahydrofolates (THF) are a family of cofactors that carry and chemically-activate one-carbon units for the de novo synthesis of guanosine, adenosine and thymidine nucleotides, and for the remethylation of homocysteine to methionine (Figure 1).1 Folate-dependent pathways are compartmentalized in the mitochondria, cytosol and nucleus, and each compartment is associated with a particular metabolic function.2 These folate-dependent pathways are tightly interconnected within the cell and communicate across the compartments, and thereby function as a metabolic network, as opposed to independent autonomously regulated pathways (Figure 1). Folate cofactors are also compartmentalized and do not readily exchange across compartments. Folate-dependent pathways are interconnected across compartments though the exchange of metabolic substrates, including serine, glycine and formate (Figure 1). 11
Figure 1.

Folate-Mediated One-Carbon Metabolism. One-carbon metabolism is required for the de novo synthesis of purines and thymidylate, and for the remethylation of homocysteine to methionine. The de novo thymidylate pathway is SUMOylated and translocates to the nucleus during S-phase. Mitochondria generate formate from the amino acids serine and glycine. THF, tetrahydrofolate; AdoMet, S-adenosylmethionine; MTHFD, Methylenetetrahydrofolate Dehydrogenase; MTR, Methionine Synthase; MTHFR, Methylenetetrahydrofolate Reductase; SHMT1, Cytoplasmic Serine Hydroxymethyltransferase; SHMT2α, Serine Hydroxymethyltransferase 2α TYMS, Thymidylate Synthase; DHFR, Dihydrofolate Reductase; GSC, Glycine Cleavage System.
In mitochondria, folate cofactors function in: 1) the de novo thymidylate synthesis for mitochondrial DNA replication, which involves the enzymes serine hydroxymethyltransferase (SHMT2), thymidylate synthase (TYMS), and dihydrofolate reductase like 1 (DHFRL1)3; 2) for the N-formylation of Met-tRNA for the initiation of mitochondrial protein synthesis, and 3) for the generation of formate from the catabolism of the amino acids serine, glycine, dimethylglycine and sarcosine by the enzymes SHMT2, sarcosine dehydrogenase, dimethylglycine dehydrogenase, methylenetetrahydrofolate dehydrogenase 2, methylenetetrahydrofolate dehydrogenase like-2 and methylenetetrahydrofolate dehydrogenase like-1.
In the cytosol, formate is an important source of one-carbons for FOCM. Mitochondrial-derived formate translocates to the cytoplasm where it is essential for the functioning of folate metabolism in the cytosol and the nucleus. Formate is a primary source of one-carbons for the de novo synthesis of purines and for the remethylation of homocysteine to methionine, catalyzed by the vitamin B12-dependent enzyme methionine synthase (MTR). Methionine can be converted to S-adenosylmethionine (AdoMet) by AdoMet synthetase. AdoMet is a cofactor for numerous methylation reactions including the methylation of DNA, RNA, proteins, neurotransmitters, phospholipids and numerous metabolites.
Nuclear folate metabolism involves the conversion of uridylate to thymidylate through reductive methylation. In this reaction, thymidylate synthase (TYMS) transfers-while simultaneously reducing- the one-carbon group from 5, 10-methylenetetrahydrofolate to deoxyuridine monophosphate, yielding thymidylate and dihydrofolate. 5, 10-methylenetetrahydrofolate can be produced either by the activity of serine hydroxymethyltransferase isozymes SHMT1 and SHMT2α4 from serine and tetrahydrofolate or by the activity of methylenetetrahydrofolate dehydrogenase 1 (MTHFD1) from formate and tetrahydrofolate5,6. Metabolic labeling studies in MCF-7 cells measured relative contribution of one-carbon group donors to thymidylate synthesis: serine contributes about 30% whereas formate contributes about 70% of one-carbon groups used by TYMS7. To regenerate tetrahydrofolate from dihydrofolate, cells use the activity of dihydrofolate reductase (DHFR).
In mammalian cells, the enzymes of thymidylate biosynthesis pathway are SUMOylated (covalently linked to the Small Ubiquitin-like MOdifier) during S-phase of the cell cycle and following DNA damage 5,8,9. This enables nuclear translocation of these enzymes, where they form a physical complex with nuclear lamin proteins and other enzymes of the DNA replication machinery (Figure 2).5 SHMT1 and SHMT2α are key enzymes in the complex, as they were shown to serve as scaffold proteins that tether the entire enzymatic complex to the nuclear lamina at sites of replication.5 Impairments in de novo thymidylate synthesis result in uracil misincorporation into DNA, which leads to single- and double-strand breaks during base-excision DNA repair.10
Figure 2.
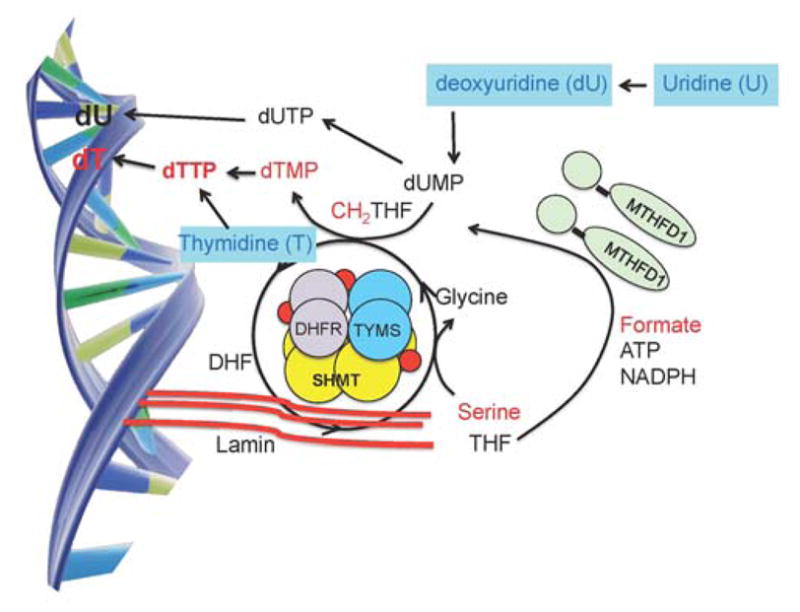
The de novo thymidylate synthesis pathway as a nuclear multienzyme complex at sites of DNA replication. THF, tetrahydrofolate; DHF, dihydrofolate; MTHFD1, Methylenetetrahydrofolate Dehydrogenase; SHMT1, Cytoplasmic Serine Hydroxymethyltransferase; TYMS, Thymidylate Synthase; DHFR, Dihydrofolate Reductase; dUMP, deoxyuridine monophosphate; dUTP, deoxyuridine triphosphate; dTMP, thymidine monophosphate; dTTP, thymidine triphosphate.
II. Partitioning of folate cofactors between the de novo thymidylate synthesis and homocysteine remethylation pathways
Folate-dependent pathways are regulated by the availability of one-carbon activated folate cofactors. This occurs because the total cellular concentration of folate-dependent enzymes exceeds the total cellular concentration of folate (for review see12), thus requiring regulation of the partitioning of folate among the pathways that constitute the folate-dependent one-carbon (FOCM) network. This competition for folate cofactor is most pronounced for the partitioning of methylenetetrahydrofolate between the de novo thymidylate cycle and the homocysteine remethylation cycle (Figure 1).7 Methylenetetrahydrofolate is used directly in the conversion of dUMP to dTMP by thymidylate synthase, or alternatively can be reduced to 5-methyltetrahydrofolate by the enzyme MTHFR, a reaction that commits the folate cofactor to the homocysteine remethylation pathway because this reaction is essentially irreversible in vivo (Figure 1). Mathematical modeling, using ordinary differential equations and the kinetic properties of methylenetetrahydrofolate reductase and thymidylate synthase, was employed to describe the competition for methylenetetrahydrofolate.13 Under conditions of limited folate availability or folate deficiency, experimental evidence shows that de novo thymidylate biosynthesis has priority over homocysteine remethylation.14
More recent studies indicate that the competition between MTHFR and TYMS enzymes for folate cofactors is achieved not through kinetic competition but rather through partitioning in distinct cellular compartments.6 De novo thymidylate synthesis occurs in the nucleus during S-phase 4, whereas MTHFR and the homocysteine remethylation cycle function in the cytosol. The competition for folate cofactors between the homocysteine remethylation and the de novo thymidylate synthesis pathway is explained by the partitioning of folate cofactors and the enzyme MTHFD1 between the nucleus and cytosol. Folate-deficient animals and human cells in culture were shown to enrich both folate cofactors and the MTHFD1 enzyme in the nucleus, at the expense of levels in the cytosol.6 MTHFD1 is the primary source of methylenetetrahydrofolate generation, and therefore the nuclear enrichment of this enzyme and folate cofactors ensures the functioning of de novo thymidylate synthesis, but at the expense of homocysteine remethylation in the cytosol.6
III. Mouse models of impaired de novo thymidylate biosynthesis
Loss-of-function mouse models have been studied to understand the physiological consequences of impaired de novo thymidylate synthesis. An N-ethyl-N-nitrosourea (ENU) mutagenesis screen yielded a T > A transversion in the TYMS codon encoding amino acid 106, causing an asparagine to lysine substitution in the folate binding site of the enzyme. This mouse model displays an early post-implantation embryonic lethal phenotype. Embryos homozygous for this mutation die due to defective gastrulation. Mice heterozygous for this mutation exhibit ~2.6 fold elevated TYMS protein in the liver compared to wild type mice.15 This elevated TYMS expression is likely due to enhanced translation, as TYMS protein autoregulates its expression by binding its mRNA through its catalytic active site and inhibiting its translation.16
The Shmt1 gene has also been disrupted in the mouse genome. Mice with a homozygous deletion of the Shmt1 gene are viable and fertile but exhibit depressed rates of de novo thymidylate synthesis and elevated levels of uracil in nuclear DNA.17 The viability of these mice is accounted for by functional redundancy with SHMT2α encoded in an alternative transcript of the Shmt2 gene, whose primary transcript expresses the mitochondrial SHMT isozyme SHMT2.4 The Shmt1+/− mice are more susceptible to intestinal tumors when crossed to the ApcMin/+ mouse model compared to wild-type mice18, and embryos exhibit sporadic neural tube defects when the dams are placed on a folate-deficient diet.19,20 Interestingly, neural tube defects associated with embryonic Shmt1 disruption can be rescued with maternal deoxyuridine supplementation, which stimulates rates of thymidylate synthesis.21
Homozygous disruption of Mthfd1 in mice through a gene-trap (gt) is early embryonic lethal, consistent with MTHFD1 serving as a primary source of one-carbon-activated folate cofactors for purine, thymidylate and methionine synthesis.22 Heterozygous Mthfd1gt/+ mice exhibit lower hepatic AdoMet levels, which are expected because MTHFD1-derived methylenetetrahydrofolate is a source of one-carbons for cellular methylation reactions. Unexpectedly, Mthfd1gt/+ mice were shown to have decreased levels of uracil in nuclear DNA, indicating enhanced rates of de novo thymidylate synthesis.22 This observation was later explained by the enrichment of MTHFD1 in the nucleus at the expense of MTHFD1 levels in the cytosol when total cellular MTHFD1 is limiting, indicating that MTHFD1 preferentially supports de novo thymidylate biosynthesis at the expense of homocysteine remethylation.6,23,24 During embryonic development, maternal Mthfd1gt/+ genotype impairs fetal growth compared to wild-type dams, but neither maternal nor embryonic Mthfd1 genotype influences risk for neural tube defects when dams are fed a folate- and choline-deficient diet.25
These mouse models demonstrate the multiple feedback mechanisms that protect de novo thymidylate biosynthesis in the nucleus. This includes the robust upregulation of TYMS expression at the level of translation, enrichment of MTHFD1 protein in the nucleus when Mthfd1 expression is limiting, and enrichment of folate cofactors in the nucleus during folate deficiency.
IV. Human MTHFD1 mutations
A novel inborn error of metabolism was recently described resulting from loss of MTHFD1 function due to deleterious mutations in both the paternal and maternal MTHFD1 alleles. 23,26,27 The patient presented with severe combined immunodeficiency (SCID), megaloblastic anemia and neurologic abnormalities. Fibroblasts from the patient exhibited decreased flux of formate into methionine and dTMP by 90% and 50%, respectively, with elevated uracil in DNA, lower rates of de novo dTMP synthesis and increased salvage pathway dTMP generation relative to control fibroblasts. As seen in the mouse models, MTHFD1 was enriched in the nucleus in the patient fibroblasts at the expense of MTHFD1 levels in the cytosol. Patient fibroblasts exhibited increased DNA damage (double-stranded DNA breaks) compare to control fibroblasts. These results provide strong evidence for the role of MTHFD1 in nuclear de novo dTMP biosynthesis, and connect impaired MTHFD1 specific activity to both megaloblastic anemia and SCID.
V. Conclusions
The identification of causal pathways and associated mechanisms of folate-associated pathophysiology remains a major challenge. Because folate-dependent anabolic pathways are tightly interconnected and best described as a metabolic network, identifying the contribution of individual pathways to disease phenotypes requires relevant model systems that capture all of the system dynamics. These dynamics include metabolic subcellular compartmentation and its regulation, as well as feed back loops that maintain the functioning of the network and metabolic priority among interconnected pathways. Recent evidence suggests that the enzyme MTHFD1 plays an essential role in FOCM in humans and in mice, and that it determines the flux of folate-activated one carbon units between the folate-dependent de novo thymidylate and homocysteine remethylation pathways through the regulation of its nuclear localization. Furthermore, investigations of genetic mouse models and human inborn errors of metabolism are enabling a more precise dissection of the pathways that constitute the FOCM network and enable elucidation of causal pathways associated with folate pathologies including neural tube defects. Evidence to date suggests that folate-dependent de novo thymidylate synthesis plays an important role in folate-associated pathologies, and that the FOCM network is configured to protect this pathway at the expense of homocysteine remethylation.
Highlights.
The mechanisms for folate-associated pathologies remain unresolved.
MTHFD1 nuclear localization determines folate one-carbon unit partitioning.
MTHFD1 may underlie folate-associated immunodeficiency and birth defects.
Acknowledgments
This work was supported by NIH grant DK58144 and HD059120 to PJS.
Abbreviations used are
- FOCM
folate-mediated one-carbon metabolism
- MTHFD1
methylenetetrahydrofolate dehydrogenase 1
- SHMT1
cytoplasmic serine hydroxymethyltransferase
- TYMS
thymidylate synthase
- DHFR
dihydrofolate reductase
- DHF
dihydrofolate
- THF
tetrahydrofolate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
VI. Literature Cited
- 1.Fox JT, Stover PJ. Folate-mediated one-carbon metabolism. Vitamins and hormones. 2008;79:1–44. doi: 10.1016/S0083-6729(08)00401-9. [DOI] [PubMed] [Google Scholar]
- 2.Tibbetts AS, Appling DR. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annual review of nutrition. 2010;30:57–81. doi: 10.1146/annurev.nutr.012809.104810. [DOI] [PubMed] [Google Scholar]
- 3.Anderson DD, Quintero CM, Stover PJ. Identification of a de novo thymidylate biosynthesis pathway in mammalian mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:15163–8. doi: 10.1073/pnas.1103623108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson DD, Stover PJ. SHMT1 and SHMT2 are functionally redundant in nuclear de novo thymidylate biosynthesis. PloS one. 2009;4:e5839. doi: 10.1371/journal.pone.0005839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson DD, Woeller CF, Chiang EP, Shane B, Stover PJ. Serine hydroxymethyltransferase anchors de novo thymidylate synthesis pathway to nuclear lamina for DNA synthesis. The Journal of biological chemistry. 2012;287:7051–62. doi: 10.1074/jbc.M111.333120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Field MS, et al. Nuclear Enrichment of Folate Cofactors and Methylenetetrahydrofolate Dehydrogenase 1 (MTHFD1) Protect de Novo Thymidylate Biosynthesis during Folate Deficiency. J Biol Chem. 2014;289:29642–50. doi: 10.1074/jbc.M114.599589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herbig K, et al. Cytoplasmic serine hydroxymethyltransferase mediates competition between folate-dependent deoxyribonucleotide and S-adenosylmethionine biosyntheses. The Journal of biological chemistry. 2002;277:38381–9. doi: 10.1074/jbc.M205000200. [DOI] [PubMed] [Google Scholar]
- 8.Woeller CF, Anderson DD, Szebenyi DM, Stover PJ. Evidence for small ubiquitin-like modifier-dependent nuclear import of the thymidylate biosynthesis pathway. The Journal of biological chemistry. 2007;282:17623–31. doi: 10.1074/jbc.M702526200. [DOI] [PubMed] [Google Scholar]
- 9.Anderson DD, Eom JY, Stover PJ. Competition between sumoylation and ubiquitination of serine hydroxymethyltransferase 1 determines its nuclear localization and its accumulation in the nucleus. The Journal of biological chemistry. 2012;287:4790–9. doi: 10.1074/jbc.M111.302174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blount BC, et al. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: implications for cancer and neuronal damage. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:3290–5. doi: 10.1073/pnas.94.7.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stover PJ, Field MS. Trafficking of intracellular folates. Advances in nutrition. 2011;2:325–31. doi: 10.3945/an.111.000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suh JR, Herbig AK, Stover PJ. New perspectives on folate catabolism. Annu Rev Nutr. 2001;21:255–82. doi: 10.1146/annurev.nutr.21.1.255. [DOI] [PubMed] [Google Scholar]
- 13.Green JM, MacKenzie RE, Matthews RG. Substrate flux through methylenetetrahydrofolate dehydrogenase: predicted effects of the concentration of methylenetetrahydrofolate on its partitioning into pathways leading to nucleotide biosynthesis or methionine regeneration. Biochemistry. 1988;27:8014–22. doi: 10.1021/bi00421a007. [DOI] [PubMed] [Google Scholar]
- 14.Scotti M, Stella L, Shearer EJ, Stover PJ. Modeling cellular compartmentation in one-carbon metabolism. Wiley interdisciplinary reviews Systems biology and medicine. 2013;5:343–65. doi: 10.1002/wsbm.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ching YH, et al. High resolution mapping and positional cloning of ENU-induced mutations in the Rw region of mouse chromosome 5. BMC genetics. 2010;11:106. doi: 10.1186/1471-2156-11-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chu E, et al. Autoregulation of human thymidylate synthase messenger RNA translation by thymidylate synthase. Proc Natl Acad Sci U S A. 1991;88:8977–81. doi: 10.1073/pnas.88.20.8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacFarlane AJ, et al. Cytoplasmic serine hydroxymethyltransferase regulates the metabolic partitioning of methylenetetrahydrofolate but is not essential in mice. The Journal of biological chemistry. 2008;283:25846–53. doi: 10.1074/jbc.M802671200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Macfarlane AJ, Perry CA, McEntee MF, Lin DM, Stover PJ. Shmt1 heterozygosity impairs folate-dependent thymidylate synthesis capacity and modifies risk of Apc(min)-mediated intestinal cancer risk. Cancer research. 2011;71:2098–107. doi: 10.1158/0008-5472.CAN-10-1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beaudin AE, et al. Dietary folate, but not choline, modifies neural tube defect risk in Shmt1 knockout mice. The American journal of clinical nutrition. 2012;95:109–14. doi: 10.3945/ajcn.111.020305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beaudin AE, et al. Shmt1 and de novo thymidylate biosynthesis underlie folate-responsive neural tube defects in mice. The American journal of clinical nutrition. 2011;93:789–98. doi: 10.3945/ajcn.110.002766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martiniova L, Field MS, Finkelstein JL, Perry CA, Stover PJ. Maternal dietary uridine causes, and deoxyuridine prevents, neural tube closure defects in a mouse model of folate-responsive neural tube defects. American Journal of clinical nutrition. 2015 doi: 10.3945/ajcn.114.097279. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacFarlane AJ, et al. Mthfd1 is an essential gene in mice and alters biomarkers of impaired one-carbon metabolism. The Journal of biological chemistry. 2009;284:1533–9. doi: 10.1074/jbc.M808281200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Field MS, Kamynina E, Watkins D, Rosenblatt DS, Stover PJ. Human mutations in methylenetetrahydrofolate dehydrogenase 1 impair nuclear de novo thymidylate biosynthesis. Proc Natl Acad Sci U S A. 2015;112:400–5. doi: 10.1073/pnas.1414555112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Field MS, et al. Reduced MTHFD1 activity in male mice perturbs folate- and choline-dependent one-carbon metabolism as well as transsulfuration. The Journal of nutrition. 2013;143:41–5. doi: 10.3945/jn.112.169821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beaudin AE, Perry CA, Stabler SP, Allen RH, Stover PJ. Maternal Mthfd1 disruption impairs fetal growth but does not cause neural tube defects in mice. The American journal of clinical nutrition. 2012;95:882–91. doi: 10.3945/ajcn.111.030783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watkins D, et al. Novel inborn error of folate metabolism: identification by exome capture and sequencing of mutations in the MTHFD1 gene in a single proband. J Med Genet. 2011;48:590–2. doi: 10.1136/jmedgenet-2011-100286. [DOI] [PubMed] [Google Scholar]
- 27.Keller MD, et al. Severe combined immunodeficiency resulting from mutations in MTHFD1. Pediatrics. 2013;131:e629–34. doi: 10.1542/peds.2012-0899. [DOI] [PubMed] [Google Scholar]