

Abstract
Purpose
Although limited understanding exists for the presence of specific genetic mutations and aberrantly methylated genes in pancreatobiliary intraductal papillary mucinous neoplasms (IPMNs), the fundamental understanding of the dynamics of methylation expansion across CpG dinucleotides in specific gene promoters during carcinogenesis remains unexplored. Expansion of DNA methylation in some gene promoter regions, such as EFEMP1, one of the fibulin family, with tumor progression has been reported in several malignancies. We hypothesized that DNA hypermethylation in EFEMP1 promoter would expand with the tumor grade of IPMN.
Methods
A sample of 65 IPMNs and 30 normal pancreatic tissues was analyzed. IPMNs were divided into the following three subsets according to pathological findings: 31 with low-grade dysplasia (low grade), 11 with high-grade dysplasia (high grade), and 23 with associated invasive carcinoma (invasive Ca). Mutations in the KRAS or GNAS genes were analyzed by Sanger sequencing, and methylation status of two discrete regions within the EFEMP1 promoter, namely region 1 and region 2, was analyzed by bisulfite sequencing and fluorescent high-sensitive assay for bisulfite DNA (Hi-SA). Expression status of EFEMP1 was investigated by immunohistochemistry (IHC).
Results
KRAS mutations were detected in 39, 55, and 70 % of low-grade, high-grade, and invasive Ca, respectively. GNAS mutations were observed in 32, 55, and 22 % of low-grade, high-grade, and invasive Ca, respectively. The methylation of individual regions (region 1 or 2) in the EFEMP1 promoter was observed in 84, 91, and 87 % of low-grade, high-grade, and invasive Ca, respectively. However, simultaneous methylation of both regions (extensive methylation) was exclusively detected in 35 % of invasive Ca (p = 0.001) and five of eight IPMNs (63 %) with extensive methylation, whereas 20 of 57 (35.1 %) tumors of unmethylation or partial methylation of the EFEMP1 promoter region showed weak staining EFEMP1 in extracellular matrix (p = 0.422). In addition, extensive EFEMP1 methylation was particularly present in malignant tumors without GNAS mutations and associated with disease-free survival of patients with IPMNs (p < 0.0001).
Conclusions
Extensive methylation of the EFEMP1 gene promoter can discriminate invasive from benign IPMNs with superior accuracy owing to their stepwise accumulation of tumor progression.
Electronic supplementary material
The online version of this article (doi:10.1007/s00432-016-2164-x) contains supplementary material, which is available to authorized users.
Keywords: Mucinous neoplasms, Methylation, Epigenetics, EFEMP1, Invasive carcinoma, Dysplasia
Introduction
Pancreatic intraductal papillary mucinous neoplasms (IPMNs) are precursor lesions characterized by an atypical degree of intraductal proliferation of neoplastic mucinous cells arising in the pancreatic duct (Das et al. 2013; Matthaei et al. 2012; Tanaka et al. 2012; Wasif et al. 2010). Histologically, IPMNs may progress from low-grade to high-grade dysplasia and finally to an invasive carcinoma (Das et al. 2013; Farrell and Brugge 2002; Salvia et al. 2004), canonical to the adenoma-carcinoma sequence in colorectal cancer and pancreatic ductal adenocarcinoma (PDAC) (Wasif et al. 2010). While IPMNs with invasive carcinoma have a poor 5-year survival ratio of 33–43 %, patients with resected IPMNs without any invasive cancer features generally have a better 5-year survival ratio of 77–94 % (Chari et al. 2002; Das et al. 2013; Farrell and Brugge 2002; Maire et al. 2002; Raimondo et al. 2002; Sohn et al. 2004), highlighting the importance of efficient diagnosis of IPMNs with invasive carcinoma. Therefore, preoperative identification of dysplastic IPMNs is challenging even with a multimodality approach including radiographic imaging, endoscopic ultrasound-guided fine-needle aspiration (EUSFNA), cytological examination, and tumor markers (Schoedel et al. 2006). This problem has prompted the development of other analytical tools, including genomic biomarkers that can predict IPMNs at a high risk of developing dysplasia with malignant potential (Schoedel et al. 2006).
Applying molecular techniques to evaluate surgical and cytological specimens is evolving in conjunction with our understanding of the IPMN molecular makeup (Schoedel et al. 2006). Studies aimed at characterizing genetic profiles in IPMNs have identified activating mutations of KRAS and GNAS oncogenes and inactivating mutations in RNF43, CDKN2A/p16, and TP53 tumor suppressor genes (Amato et al. 2014; Cooper et al. 2013; Dal Molin et al. 2013; Furukawa et al. 2011; Kanda et al. 2013; Komatsu et al. 2014; Schonleben et al. 2007, 2008; Sessa et al. 1994). Collectively, these studies underscored the importance of genetic alterations in IPMN progression; however, the prevalence of such genetic events generally occurs at lower frequencies than in PDAC (Adsay 2002; Cooper et al. 2013; Kanda et al. 2013; Sato and Goggins 2006).
Similar to other cancers, epigenetic alterations, such as promoter hypermethylation of tumor suppressor genes, are considered a critical process in IPMN development (Sato and Goggins 2006). Results suggest a gradual expansion of methylation across CpG islands in MGMT, RASSF2, and SFRP2 promoters during colorectal cancer progression and highlighted their potential role as biomarkers for diagnosis and disease prediction for specific cancer types (Nagasaka et al. 2008, 2009; Takeda et al. 2011). In this study, we evaluated the methylation status of the epidermal growth factor-containing fibulin-like extracellular matrix protein 1 gene (EFEMP1, alternative annotation is FIbulin3), a member of the fibulin family of extracellular matrix (ECM) proteins. EFEMP1 is involved in malignant transformation through modulation of cell proliferation, angiogenesis, and invasion in a tissue-dependent manner (Kobayashi et al. 2007; Sadr-Nabavi et al. 2009; Wang et al. 2010, 2012; Yang et al. 2013; Yue et al. 2007; Zhu et al. 2014), and alterations in this gene expression have often been linked to aberrant DNA methylation (Nomoto et al. 2010; Sadr-Nabavi et al. 2009; Wang et al. 2010, 2012; Yang et al. 2013; Yue et al. 2007; Zhu et al. 2014). Moreover, recent studies have reported an association between a reduction in protein expression and EFEMP1 methylation expansion in breast and lung cancers using immunohistochemistry and sequencing approaches (Chen et al. 2014; Sadr-Nabavi et al. 2009). For these reasons, the presence or absence of methylation and gradual expansion of specific gene promoter methylation may help diagnose and differentiate invasive carcinoma from normal adjacent tissues and dysplastic lesions.
To systematically test this hypothesis, we first analyzed mutations in the KRAS and GNAS genes to confirm the genetic background of IPMNs. Next, to determine whether extensive methylation of candidate genes may serve as a predictive alteration for malignant IPMNs, we performed a comprehensive investigation of the methylation status of EFEMP1 promoter and examined EFEMP1 expression status by IHC.
Materials and methods
Samples and tumor classifications
Nine tissues from non-necrotic areas of PDAC were frozen immediately at −80 °C, and DNA was extracted from the tissues using QIAamp DNA mini kits (Qiagen, Valencia, CA, USA). DNAs of 21 tissues from non-necrotic areas of PDAC were macrodissected from formalin-fixed, paraffin-embedded (FFPE) specimens. DNAs of IPMNs were macrodissected from 65 patients who underwent surgical resection and who were pathologically diagnosed with IPMNs or invasive IPMNs. All samples were collected from the Okayama University Hospital, Okayama, Japan, between January 2001 and December 2012. Institutional review board approval was granted by the Ethics Committee of the Okayama University, and written informed consent was obtained from all patients to use their tissues for research. The medical records of the patients were retrospectively explored and matched with clinical and pathological data. We defined IPMN classification based on the International Consensus Guidelines from 2012 as follows: MD-IPMNs were characterized by segmental or diffused dilation of the main pancreatic duct by >5 mm in the absence of other causes of obstruction; BD-IPMNs comprised pancreatic cysts of >5 mm in diameter communicating with the main pancreatic duct; finally, mixed-type lesions were those lesions that simultaneously met the criteria of both MD-IPMNs and BD-IPMNs (Tanaka et al. 2012). In addition, we used a revised terminology to classify IPMNs. Formally, IPMNs are classified according to World Health Organization classification based on pathological findings as follows: IPMNs with low-grade dysplasia, intermediate-grade dysplasia, high-grade dysplasia (carcinoma in situ) and associated invasive carcinoma. Based on the revised classification criteria proposed in 2015, IPMNs with intermediate-grade dysplasia are combined together with the ones with low-grade dysplasia and are now called low-grade IMPNs. Therefore, we classified IPMNs according to this revised nomenclature.
IPMN patients’ characteristics
Using the criteria mentioned above, our sample of 65 IPMNs was classified as follows: 31 (48 %), 11 (17 %), and 23 (35 %) IPMNs were classified as low- or intermediate-grade dysplasia (low grade), high-grade dysplasia (high grade), and invasive Ca, respectively. To clarify the clinicopathological features of IPMNs, statistical analyses were performed between low-grade and high-grade and invasive Ca (Supplementary Table 1). Within the group of 23 invasive Ca, only one case showed a distant metastasis in the liver at surgical resection (and hence classified as stage IV); the remainder of the invasive Ca cases was categorized as stage I (10 of 23; 44 %) and stage II (12 of 23; 52 %, Supplementary Table 2). Disease-free survival (DFS) and overall survival (OS) of IPMNs were estimated according to their clinicopathological characteristics (Supplementary Fig. 1). The median time of the follow-up period of 65 IPMNs after surgical resection was 48 months (range 8–108 months). The lesions categorized as invasive Ca were also classified by the tumor node metastasis classification system (Adsay et al. 2010; Kim et al. 2008).
Direct sequencing of KRAS and GNAS mutations in IPMNS tissues
KRAS mutations in codons 12 and 13 were determined by the method previously described (Nagasaka et al. 2008, 2009; Takeda et al. 2011). GNAS mutations in codon 201 were analyzed by direct sequencing using GNAS primers (Supplementary Fig. 2 and Supplementary Table 3).
Analysis of DNA methylation
DNA was subjected to sodium bisulfite modification using the EZ DNA Methylation Kit (ZYMO Research, Irvine, CA). As shown in Fig. 1, methylation status of the EFEMP1 gene promoter was studied at various regions in previous studies (Kobayashi et al. 2007; Nomoto et al. 2010; Sadr-Nabavi et al. 2009; Wang et al. 2010, 2012; Yang et al. 2013; Yue et al. 2007; Zhu et al. 2014). In this study, we searched CTCF biding sites by CTCFBSDB 2.0 (http://insulatordb.uthsc.edu/) in promoter region of the EFEMP1 gene and found a CTCF biding site ‘TGACATCTGTTGGG,’ called as the EMBL_M1 motifs (Schmidt et al. 2012). CTCF, also known as CCCTC-binding factor, is a transcription factor involved in many cellular processes, including transcriptional regulation, insulator activity, V(D)J recombination, and regulation of chromatin architecture (Chaumeil and Skok 2012; Phillips and Corces 2009). Interestingly, this biding site was between the two regions which were analyzed by previous studies as mentioned above. For the reason, we divided the EFEMP1 gene promoter into two regions (regions 1 and 2, Fig. 1), which were located as dividing line on the CTCF biding site, and analyzed by a fluorescence high-sensitive assay (Hi-SA) using bisulfite-modified DNA template as previously described (Nagasaka et al. 2009). The sense and antisense nonspecific primers and internal methylation-specific primers with enhanced sensitivity for polymerase chain reaction (PCR) amplification have been described previously (Nagasaka et al. 2009) and are shown in Supplementary Table 3. PCR products digested with HhaI (New England BioLabs, Massachusetts, USA) were loaded simultaneously onto an ABI 310R or 31000 Genetic Analyzer (Applied Biosystems, California, USA). Signals from individual PCR products were distinguished by the unique fluorescent PCR signal from each target and their fragment length, and the data were analyzed using GeneMapper software version 4.0 (Applied Biosystems, California, USA). In this study, the percentages of methylated HhaI sites were calculated by determining the ratios between the HhaI-cleaved PCR product and the total amount of PCR product in each loci, and methylation positivity was defined as a proportion of >1.0 % of methylated HhaI sites. Direct sequencing of the two regions in the EFEMP1 promoter was performed by PCR products obtained from bisulfite DNA extracted from normal pancreatic tissues and IPMNs. Primer sequences are shown in Supplementary Table 3.
Fig. 1.
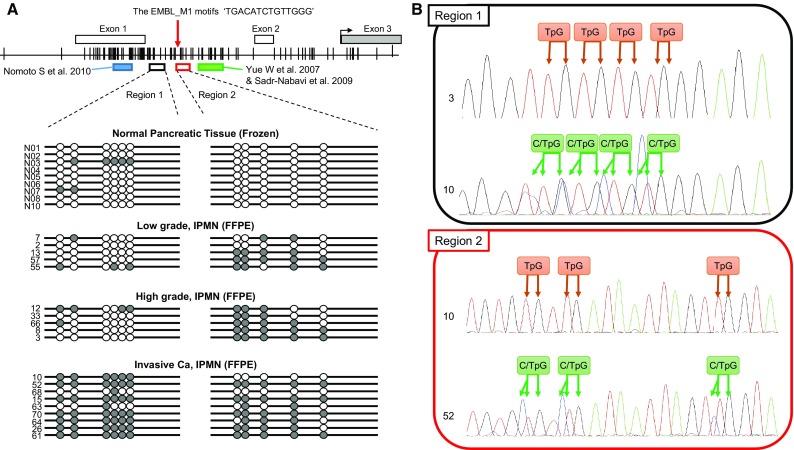
Bisulfite sequencing of the discrete EFEMP1 gene promoter regions. a Schematic representation of the location of discrete EFEMP1 gene promoter regions and the result of bisulfite sequencing. The white and gray boxes denote untranslated and translated exon in the EFEMP1 gene, respectively. The red allow indicates the location of the EMBL_M1 motifs ‘TGACATCTGTTGGG’, a candidate of CTCF biding site. The black arrow indicates the transcriptional starting site. The blue box indicates the regions of which methylation status was analyzed by Nomoto et al. (2010). The green box indicates the regions of which methylation status was analyzed by Yue et al. (2007) and Sadr-Nabavi et al. (2009). Vertical lines indicate CpG sites; white circles represent unmethylated CpGs; and gray circles represent methylated and unmethylated CpGs observed by bisulfite direct sequencing. b Examples of bisulfite sequencing in region 1 and 2. Each CpG was categorized as unmethylated or methylated CpG. TpG denotes the CpG site consisting of unmethylated CpG only. C/TpG denotes the CpG site consisting of both methylated (CpG) and unmethylated CpGs (TpG)
Immunohistochemical analysis
EFEMP1 localization was performed by immunohistochemical (IHC). A sample of 65 IPMNs was available for IHC staining for EFEMP1 protein expression analysis. Staining was carried out manually with FFPE tissues. Thin (5 µm) sections of representative blocks were deparaffinized and dehydrated using gradient solvents. Following antigen retrieval in the citrate buffer (pH 6.0), endogenous peroxidase was blocked with 3 % H2O2. Thereafter, slides were incubated overnight in the presence of a purified mouse anti-human EFEMP1 monoclonal antibody (sc-33722, Santa Cruz, Dallas, TX, USA; dilution 1:250). Further incubation was carried out with a secondary antibody and the avidin–biotin–peroxidase complex (Vector Laboratories, Burlingame, CA, USA) and then incubated with biotinyl-tyramide followed by streptavidin-peroxidase. Diaminobenzidine was used as a chromogen and hematoxylin as a nuclear counterstain.
EFEMP1 was detectable in normal pancreatic tissue; weak staining was detected in islets of Langerhans, whereas intense staining was observed in the peripheral nerve fiber. IHC results were interpreted by pathologists blinded to the corresponding clinicopathological data. The expression status of EFEMP1 was evaluated by an immunoreactive score (IRS), which was calculated by scoring of the percentage of positive cells and their expression intensities. The percentage of positive ECM staining was rated as described previously and as follows: 1 = 0–10 %, 2 = 11–50 %, 3 = 51–80 %, and 4 = 81–100 % (Sadr-Nabavi et al. 2009). Staining intensity was scored as follows: 1 = weak, 2 = moderate, and 3 = intensive. All IPMNs were categorized into four subsets by IRS score as follows: 0–1 = no staining, 2–3 = weak staining, 4–8 = moderate staining, and 9–12 = strong staining (Remmele and Stegner 1987).
Statistical analyses
All statistical analyses were performed using EZR (Saitama Medical Center, Jichi Medical University), which is a graphical user interface for R (The R Foundation for Statistical Computing, version 2.13.0). First, methylation levels were analyzed as continuous variables. Next, the methylation status was analyzed as a categorical variable (positive, methylation level >1.0 %; negative, methylation level ≤1.0 %), as described previously. Each IPMN specimen was given a numerical score so as to reflect the number of methylated loci. Categorical variables were compared by Fisher’s exact test. Differences between continuous variables were determined using the Mann–Whitney U test or the Kruskal–Wallis test. Multiple comparisons were performed using the Steel–Dwass test. OS was calculated from the date of surgical resection to the date of death due to IPMNS or last follow-up for censored patients. DFS was calculated from the date of surgical resection to the date of the first documentation of local, regional, or distant relapse, appearance of a second primary lesion by computed tomography and/or magnetic resonance imaging routinely performed per 6 months. OS and DFS were univariately estimate with the Kaplan–Meier method. All p values reported were calculated by two-sided tests, and values <0.05 were considered statistically significant.
Results
KRAS and GNAS mutations in IPMNs
To evaluate the genetic background in our current cohort, we analyzed mutations in KRAS and GNAS genes as shown in Supplementary Figure 2. KRAS mutations were detected in 33 IPMNs (51 %), and the spectrum of relative frequencies of individual mutations was 5 (15 % of KRAS mutants), 12 (36 %), and 16 (49 %) for G12R, G12D, and G12V mutations, respectively (Supplementary Tables 1 and 2). Meanwhile, 19 IPMNs (29 %) harbored GNAS mutations, and the following mutations were primarily found in codon 201: R201H, 5 IPMNs (26 %); R201C, 13 IPMNs (68 %); and R201S, 1 IPMNs (5 %) (Table 2). One R201S was a novel mutation, which had not been previously described for IPMNs. KRAS mutation frequency tended to increase with IPMN grade: 12 of 31 were low grade (39 %), 6 of 11 were high grade (55 %), and 16 of 23 were invasive Ca (70 %, p = 0.084; low-grade vs. high-grade/invasive Ca). In contrast, GNAS mutations were constantly observed, with 10 of 32 (32 %) in low-grade and 12 of 33 (36 %) in high-grade and invasive Ca. Concurrent KRAS and GNAS mutations were observed in 11 IPMNs (17 %), KRAS mutation were present only in 22 IPMNs (34 %), GNAS mutations only in 8 IPMNs (12 %), and wild-type status of both genes was observed in 24 IPMNs (37 %, Supplementary Fig. 2C).
Table 2.
Methylation status of EFEMP1and Genetic profiles of KRAS and GNAS in this cohort
| Sample no. | Gender | Age | Duct involvement | KRAS | GNAS | EFEMP | |
|---|---|---|---|---|---|---|---|
| Region 1 | Region 2 | ||||||
| Low grade | |||||||
| 14 | M | 71 | MD | WT | WT | U | U |
| 44 | M | 54 | Mixed | WT | WT | U | U |
| 47 | M | 58 | MD | WT | WT | U | U |
| 46 | M | 63 | BD | WT | R201C | U | U |
| 7 | F | 66 | BD | WT | WT | U | U |
| 22 | F | 63 | Mixed | WT | WT | U | M |
| 50 | M | 68 | Mixed | WT | WT | U | M |
| 54 | F | 76 | BD | WT | WT | U | M |
| 60 | M | 66 | Mixed | WT | WT | U | M |
| 65 | M | 63 | MD | WT | WT | U | M |
| 39 | M | 64 | BD | WT | R201C | U | M |
| 42 | M | 59 | MD | G12V | WT | M | U |
| 56 | M | 65 | BD | G12V | R201C | U | M |
| 17 | F | 71 | BD | WT | WT | U | M |
| 21 | M | 67 | BD | WT | WT | U | M |
| 31 | M | 78 | MD | WT | WT | U | M |
| 35 | F | 57 | BD | WT | WT | U | M |
| 36 | F | 62 | BD | WT | WT | U | M |
| 2 | M | 61 | Mixed | WT | WT | U | M |
| 58 | M | 60 | BD | WT | R201H | U | M |
| 13 | F | 74 | MD | G12R | WT | U | M |
| 30 | M | 73 | Mixed | G12D | WT | U | M |
| 57 | M | 74 | Mixed | G12R | WT | U | M |
| 67 | M | 81 | BD | G12D | WT | U | M |
| 55 | F | 71 | MD | G12V | R201C | U | M |
| 41 | M | 50 | BD | G12D | WT | U | M |
| 1 | F | 65 | Mixed | G12V | R201H | U | M |
| 69 | M | 71 | Mixed | G12D | R201S | U | M |
| 18 | M | 68 | BD | WT | R201C | U | M |
| 25 | M | 69 | Mixed | G12D | R201C | U | M |
| 9 | M | 73 | Mixed | G12V | R201C | U | M |
| High grade | |||||||
| 12 | M | 81 | MD | WT | WT | U | M |
| 16 | M | 72 | MD | WT | WT | U | M |
| 33 | F | 70 | Mixed | G12D | WT | U | M |
| 43 | M | 75 | Mixed | G12V | R201H | U | M |
| 66 | M | 79 | Mixed | WT | R201C | U | M |
| 19 | F | 67 | Mixed | G12R | WT | U | M |
| 38 | M | 66 | Mixed | G12D | R201H | U | U |
| 8 | M | 73 | Mixed | WT | WT | U | M |
| 3 | M | 70 | MD | G12V | WT | U | M |
| 34 | F | 59 | BD | WT | WT | U | M |
| 53 | M | 72 | MD | WT | R201C | U | M |
| Invasive Ca | |||||||
| 48 | F | 66 | Mixed | WT | WT | U | M |
| 49 | M | 67 | MD | WT | R201C | U | U |
| 10 | M | 73 | MD | G12V | WT | M | U |
| 37 | F | 59 | Unknown | G12V | WT | U | M |
| 4 | F | 73 | MD | G12V | WT | U | M |
| 59 | M | 66 | Mixed | G12V | WT | U | U |
| 6 | F | 82 | MD | G12D | R201C | U | U |
| 20 | M | 74 | MD | G12D | R201H | U | M |
| 32 | M | 56 | BD | WT | WT | U | M |
| 11 | M | 74 | MD | WT | R201C | U | M |
| 52 | M | 51 | Mixed | G12R | WT | M | M |
| 68 | M | 78 | MD | WT | WT | M | M |
| 15 | M | 67 | MD | G12D | WT | M | M |
| 24 | F | 72 | BD | G12V | WT | U | M |
| 51 | M | 57 | MD | G12D | WT | U | M |
| 62 | F | 81 | BD | G12V | R201C | U | M |
| 63 | M | 75 | BD | WT | WT | M | M |
| 70 | M | 57 | Mixed | WT | WT | M | M |
| 5 | F | 74 | MD | G12V | WT | U | M |
| 40 | M | 39 | Mixed | G12V | WT | M | M |
| 64 | M | 63 | BD | G12D | WT | M | M |
| 26 | F | 79 | MD | G12V | WT | M | M |
| 61 | M | 88 | Mixed | G12R | WT | M | M |
M methylation, U unmethylation, MD, MD-IPMN; BD, BD-IPMN; Mixed, Mixed-IPMN
We assessed DFS and OS according to KRAS or GNAS mutation status. Although KRAS mutations were more frequently observed in invasive Ca, there was no difference in outcome between KRAS mutants and wild type (Supplementary Fig. 2E and F). In contrast, GNAS mutations were less frequently observed in invasive Ca compared with low and high grade, and there was no difference in outcome between GNAS mutants and wild type (Supplementary Fig. 2G and H).
Methylation profiles of EFEMP1 promoter in IPMNs
We investigated the methylation status of discrete regions in the EFEMP1 promoter in 65 IPMNs and 30 normal pancreatic tissues obtained from PDAC patients. The location of the EFEMP1 gene and a panel of representative bisulfite sequencing and fluorescent Hi-SA results are depicted in Figs. 1 and 2a, respectively. Methylation status in the discrete regions obtained from fluorescent Hi-SA was analyzed as the categorical variable. Figure 2b presents methylation frequency in each discrete region according to pathological features, and Table 1 presents the correlation between the methylation status of EFEMP1 and the clinical and pathological features of IPMNs. The region-1 methylation was frequently observed in invasive Ca (p = 0.0016), whereas the region-2 methylation was commonly observed in IPMN (>80 % of IPMNs) but less frequently in normal pancreatic tissues (<20 %). Another interesting feature of IPMNs with extensive EFEMP1 methylation was histologic subtype, in which extensive EFEMP1 methylation was frequently observed in pancreatobiliary type lesions (4 of 6; 67 %, p = 0.027).
Fig. 2.
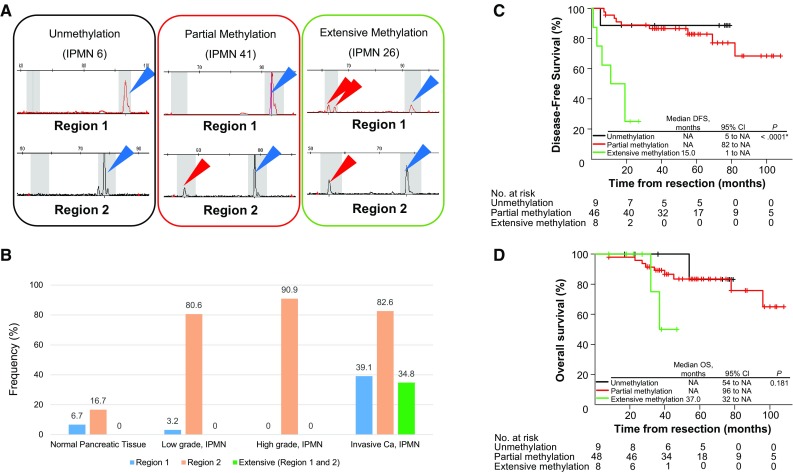
Methylation analysis of EFEMP1. a Results of EFEMP1 methylation by fluorescent Hi-SA. Blue arrows represent PCR fragments with non-methylation in HhaI sites. Red arrows represent methylated PCR fragments cleaved by HhaI. b Frequencies of EFEMP1 methylation according to pathological findings. Kaplan–Meier survival curves for disease-free survival (c), excluding a patient with remaining cancer at the resected margin, and overall survival (d) according to EFEMP1 promoter methylation status
Table 1.
Correlation between characteristics of patients and EFEMP1 methylation status
| All no. (%) | Methylation status of EFEMP1—no (%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Unmethylation n = 9 (%) | Partial methylation n = 48 (%) | Extensive methylation n = 8 (%) | p value | Region 1 unmethylation n = 55 (%) | Region 1 methylation n = 10 (%) | p value | Region 2 unmethylation n = 11 (%) | Region 2 methylation n = 54 (%) | p value | ||
| Age (years) | |||||||||||
| Median (range) | 68 (39–88) | 66 (54–82) | 70 (39–81) | 71 (51–88) | 0.482a | 63 (39–82) | 70 (51–88) | 0.716c | 68 (54–82) | 70 (39–88) | 0.234c |
| Gender | |||||||||||
| Female | 21 (32) | 2 (22) | 17 (35) | 2 (25) | 0.753b | 19 (35) | 2 (20) | 0.479d | 2 (18) | 19 (35) | 0.48d |
| Male | 44 (68) | 7 (78) | 31 (65) | 6 (75) | 36 (76) | 8 (80) | 9 (82) | 35 (65) | |||
| Location | |||||||||||
| Head, neck and uncinate | 34 (52) | 6 (67) | 24 (50) | 4 (50) | 0.344b | 29 (53) | 5 (50) | 1d | 7 (64) | 27 (50) | 0.182d |
| Body and tail | 29 (45) | 2 (22) | 23 (48) | 4 (50) | 24 (44) | 5 (50) | 3 (27) | 26 (48) | |||
| Whole pancreas | 2 (3) | 1 (11) | 1 (2) | 0 (0) | 2 (4) | 0 (0) | 1 (9) | 1 (2) | |||
| Duct involvement* | |||||||||||
| MD-IPMN | 21 (33) | 4 (44) | 14 (30) | 3 (38) | 0.896b | 16 (30) | 5 (50) | 0.473d | 6 (55) | 15 (28) | 0.261d |
| BD-IPMN | 20 (31) | 3 (33) | 15 (32) | 2 (25) | 18 (33) | 2 (20) | 3 (27) | 17 (32) | |||
| Mixed type | 23 (36) | 2 (22) | 18 (38) | 3 (38 | 20 (37) | 3 (30) | 2 (18) | 21 (40) | |||
| Cystic size** (cm) | |||||||||||
| Median size (range) | 2.7 (0.5–7.5) | 2.6 (0.5–6.7) | 2.7 (0.5–7.5) | 3.9 (0.5–6.1) | 0.872a | 2.7 (0.5–7.5) | 2.7 (0.5–6.1) | 0.958c | 1.7 (0.5–6.7) | 2.9 (0.5–7.5) | 0.517c |
| <3.0 cm | 35 (54) | 5 (56) | 27 (56) | 3 (38) | 0.657b | 30 (55) | 5 (50) | 1d | 7 (64) | 28 (52) | 0.526d |
| >3.0 cm | 30 (46) | 4 (44) | 21 (44) | 5 (63) | 25 (46) | 5 (50) | 4 (36) | 26 (48) | |||
| Main duct diameter (cm) | |||||||||||
| Median size (range) | 0.7 (0.2–7.1) | 0.6 (0.2–6.7) | 0.7 (0.2–7.1) | 0.6 (0.2–4.0) | 0.822a | 2.7 (0.5–7.5) | 2.7 (0.5–6.2) | 0.82d | 0.7 (0.2–6.7) | 0.7 (0.2–7.1) | 0.441d |
| <1.0 cm | 45 (69) | 6 (67) | 33 (69) | 6 (75) | 1b | 39 (71) | 6 (60) | 0.482b | 6 (54) | 39 (72) | 0.292b |
| >1.0 cm | 20 (31) | 3 (33) | 15 (31) | 2 (25) | 16 (29) | 4 (40) | 5 (46) | 15 (28) | |||
| Presence of mural nodule | |||||||||||
| Yes | 38 (59) | 5 (56) | 25 (52) | 8 (100) | 0.032 a | 29 (53) | 9 (90) | 0.037 a | 6 (55) | 32 (59) | 1a |
| No | 27 (42) | 4 (44) | 23 (48) | 0 (0) | 26 (47) | 1 (10) | 5 (46) | 22 (41) | |||
| Histologic subtype | |||||||||||
| Gastric | 33 (51) | 6 (67) | 25 (52) | 2 (25) | 0.027 a | 31 (56) | 2 (20) | 0.006 a | 6 (55) | 27 (50) | 0.756a |
| Intestinal | 25 (39) | 3 (33) | 20 (42) | 2 (25) | 21 (38) | 4 (40) | 5 (46) | 20 (37) | |||
| Pancreatobillary | 6 (9) | 0 (0) | 2 (4) | 4 (50) | 2 (4) | 4 (40) | 0 (0) | 6 (11) | |||
| Oncocytic | 1 (2) | 0 (0) | 1 (2) | 0 (0) | 1 (2) | 0 (0) | 0 (0) | 1 (2) | |||
| Atypical grade | |||||||||||
| Low grade | 31 (48) | 5 (56) | 26 (54) | 0 (0) | 0.0016 b | 30 (55) | 1 (10) | 0.00044 d | 6 (55) | 25 (46) | 0.908d |
| High grade | 11 (17) | 1 (11) | 11 (21) | 0 (0) | 11 (20) | 0 (0) | 1 (9) | 10 (19) | |||
| Invasive Ca | 23 (35) | 3 (33) | 12 (25) | 8 (100) | 14 (26) | 9 (90) | 4 (36) | 19 (35) | |||
| KRAS | |||||||||||
| Mutant | 33 (51) | 3 (33) | 25 (52) | 5 (63) | 0.471b | 26 (47) | 7 (70) | 0.303d | 5 (46) | 28 (52) | 0.751d |
| Wild type | 32 (49) | 6 (67) | 23 (48) | 3 (38) | 29 (53) | 3 (30) | 6 (55) | 26 (48) | |||
| GNAS | |||||||||||
| Mutant | 19 (29) | 4 (44) | 15 (31) | 0 (0) | 0.102b | 19 (35) | 0 (0) | 0.028 d | 4 (36) | 15 (28) | 0.718d |
| Wild type | 46 (71) | 5 (56) | 33 (69) | 8 (100) | 36 (66) | 10 (100) | 7 (64) | 39 (72) | |||
| Combined mutational type | |||||||||||
| Mutant in the both genes | 11 (17) | 2 (22) | 9 (19) | 0 (0) | 0.364b | 11 (20) | 0 (0) | 0.058d | 2 (18) | 9 (17) | 0.899d |
| KRAS mutation alone | 22 (34) | 1 (11) | 16 (33) | 5 (63) | 15 (27) | 7 (70) | 3 (27) | 19 (35) | |||
| GNAS mutation alone | 8 (12) | 2 (22) | 6 (13) | 0 (0) | 8 (15) | 0 (0) | 2 (18) | 6 (11) | |||
| Wild type in the both genes | 24 (37) | 4 (44) | 17 (35) | 3 (38) | 21 (38) | 3 (30) | 4 (36) | 20 (37) | |||
Low grade Low-grade dysplasia, high grade high-grade dysplasia, invasive Ca invasive carcinoma, MD-IPMN main duct type IPMN, BD-IPMN branch duct type IPMN
* One unknown case was excluded
** Cystic size reprsented the largest one measured by CT or MRI
a p value were calculated among unmethylation, partial methylation and extensive methylation by Kruskal–Wallis test
b p value were calculated among unmethylation, partial methylation and extensive methylation by Fisher exact test
c p value were calculated between unmethylation and methylation of region 1 or 2 within EFEMP1 promoter by Mann–Whitney U test
d p value were calculated between unmethylation and methylation of region 1 or 2 within EFEMP1 promoter by Fisher exact test
With respect to KRAS/GNAS mutation status, interestingly, IPMNs with methylation of the region-1 never harbored GNAS mutations (0 of 10; 0 %, p = 0.028), while 7 of 10 IPMNs with methylation of the region-1 harbored KRAS mutations (70 %). While methylation of the region-2 promoter was frequently observed (54 of 65 IPMNs [83 %]), no associations were observed among the frequencies of region-2 methylation and any of the clinicopathological factors.
Next, we evaluated correlations between the methylation status of EFEMP1 promoter and the clinicopathological features of invasive Ca (Supplementary Table 3). However, methylation status had no association with any of the features explored in invasive Ca.
Finally, we also assessed DFS and OS according to methylation status in the discrete EFEMP1 promoter regions. As extensive EFEMP1 methylation was a specific feature of invasive Ca, IMPNs with extensive EFEMP1 methylation showed a poor prognosis compared with IMPNs without extensive EFEMP1 methylation (Fig. 2c, d).
Association of EFEMP1 promoter methylation and protein expression
We investigated EFEMP1 protein expression in 65 IPMN tissues. Representative examples of IHC staining results are shown in Fig. 3a–c. Using these criteria, no IPMNs were categorized as having no staining, whereas 25, 38, and 2 IPMNs were deemed to have weak, moderate, and strong staining, respectively. Although five of eight IPMNs (63 %) showed weak staining and extensive methylation of the EFEMP1 promoter region, no significant differences in the frequencies of EFEMP1 protein expression were observed in IPMNs exhibiting partial methylation or non-methylation in the EFEMP1 promoter (Fig. 3d).
Fig. 3.
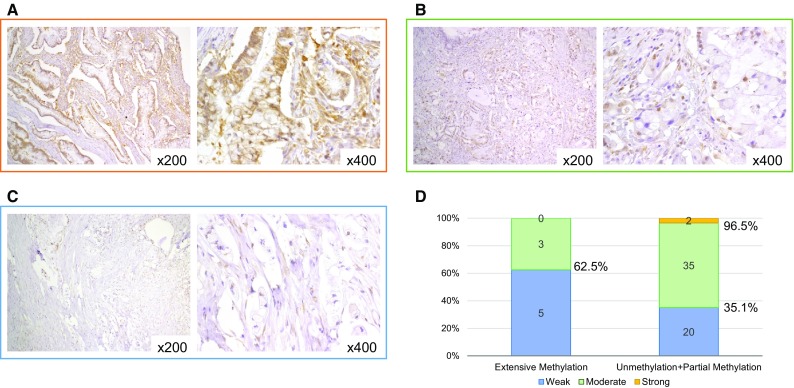
Expression analysis of EFEMP1. IHC staining of EFEMP1 in intraductal papillary mucinous neoplasms with strong staining (a), moderate staining (b), and weak staining (c). Association between EFEMP1 methylation status and IHC staining (d). EFEMP1, epidermal growth factor-containing fibulin-like extracellular matrix protein 1; IHC immunohistochemical; NA not available
Discussion
We have shown for the first time the biological significance of methylation in discrete promoter regions of EFEMP1 gene in tissue specimens obtained from patients with pancreatobiliary IPMNs, playing an important functional role in malignant transformation by modulating cell proliferation, angiogenesis, and invasion in a tissue-dependent context (Kobayashi et al. 2007); it is a common target of promoter hypermethylation in various tumors (Nomoto et al. 2010; Sadr-Nabavi et al. 2009; Wang et al. 2010, 2012; Yang et al. 2013; Yue et al. 2007; Zhu et al. 2014). Aberrant hypermethylation of the EFEMP1 promoter region is a potential biomarker. Sadr et al. reported an association between reduction in protein expression and EFEMP1 methylation expansion in breast cancer using sequencing approaches and IHC (Sadr-Nabavi et al. 2009). In this study, we also examined the association between EFEMP1 expression in ECM by IHC and EFEMP1 methylation profiles. Unfortunately, although 63 % of IPMNs with extensive methylation of the EFEMP1 promoter region showed weak staining in ECM, no significant differences in the frequencies of EFEMP1 protein expression were observed in IPMNs exhibiting partial methylation or non-methylation in the EFEMP1 promoter.
Considering the importance of EFEMP1, we hypothesized that gradual expansion of methylation across its promoter during the development of IPMN serves as a biomarker for distinguishing malignant IPMNs from the non-malignant ones. To systematically test this hypothesis, we first analyzed mutations in the KRAS and GNAS oncogenes to confirm the genetic background of IPMNs. Usually, the methylation pattern in a gene promoter is considered either entirely methylated or non-methylated. However, as demonstrated by bisulfite sequencing, individual CpG residues within the EFEMP1 promoter were not equally methylated. In this study, region-2 of the EFEMP1 promoter was more frequently methylated than region-1, and the aberrant methylation tended to spread from region-2 toward region-1 with IPMN progression. More importantly, none of the normal pancreatic tissues, low- or high-grade IMPNs showed extensive methylation in the EFEMP1 promoter. These characteristics of the EFEMP1 methylation pattern in IPMN carcinogenesis appeared similar to those of MGMT, SFRP2, and RASSF2 in the adenoma-carcinoma sequence of colorectal cancer (Nagasaka et al. 2008, 2009; Takeda et al. 2011). Therefore, the presence or absence of methylation and the gradual expansion of methylation of specific gene promoters may help diagnose and differentiate invasive carcinoma from normal adjacent tissues and dysplastic lesions. This fundamental concept of stepwise expansion of DNA methylation across specific gene promoters during the neoplastic progression of IPMNs remains unexplored and is still an active area of investigation.
KRAS and GNAS mutations, the most common genetic mutations observed in IPMNs, occurred in the early stages of disease progression. KRAS and GNAS mutations were also found at codon 12 (a G12D, G12V, or G12R) and codon 201 (an R201H or R201C), respectively. These genetic features are in line with those previously reported in other studies (Amato et al. 2014; Furukawa et al. 2011; Sadr-Nabavi et al. 2009; Wu et al. 2011a, b). This agreement indicates that KRAS mutations at codon 12 or GNAS mutations at codon 201 could play a key role as the driver of carcinogenesis, providing a selective advantage in tumor formation associated with these IPMNs (Parmigiani et al. 2009). However, the frequencies of both genetic mutations, especially GNAS mutations, were different among genetic mutational analyses. In the current study, GNAS mutations were observed with similar frequencies in low-grade and invasive Ca. However, Furukawa et al. (2011) reported that GNAS mutations were more common in low-grade lesions. In contrast, Amato et al. and other authors showed that GNAS mutation frequency tended to increase with tumor progression (Amato et al. 2014; Wu et al. 2011a, b). These differences in observations might partly be due to smaller sample size or variations in the detection technologies used in the studies.
To our knowledge, no studies have been published so far demonstrating a correlation between genetic and epigenetic alterations in IPMNs although several previous reports have revealed that there are distinct patterns of genetic mutations in IPMN subtypes (Amato et al. 2014; Chadwick et al. 2009; Cooper et al. 2013; Dal Molin et al. 2013; Fritz et al. 2009; Komatsu et al. 2014; Mino-Kenudson et al. 2011; Mohri et al. 2012). In this study, extensive methylation of EFEMP1 was likely to occur in IPMNs without GNAS mutations (Table 2). This result might reflect the biological behavior of the GNAS gene pathway, which is less aggressive than that of the KRAS gene, and further investigation is needed to evaluate the roles of KRAS and GNAS mutations in IPMN carcinogenesis.
Our study has several limitations. One is the sample size. Another is that, although we examined the correlation between methylation status of our analyzed regions in the EFEMP promoter and its expression status by IHC, there was no strong correlation between them; hence, further investigation is needed. Beyond the limitations, we demonstrated that the extensive methylation in the EFEMP1 promoter could be a useful predictive marker for invasive IPMNs and could serve as a possible means to noninvasively screen for invasive IPMNs using DNA obtained from EUSFNA, pancreatic juice, and fecal samples.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Kaplan–Meier survival curves for DFS and OS according to clinicopathological characteristics. Kaplan–Meier survival curves for DFS (A, C, and E) excluding a patient with remaining cancer at the resected margin and OS (B, D, and F) according to atypical grade, invasive carcinoma by stage I and II, and invasive carcinoma by histological type, respectively. DFS, disease-free survival; NA, not available; OS, overall survival (PPTX 91 kb)
KRAS and GNAS mutations in IPMNs. (A) and (B) demonstrate the mutations occurring in KRAS codon 12 and GNAS codon 201, respectively. (C) Venn diagram of IPMNs with KRAS mutations, GNAS mutations, and concurrent KRAS and GNAS mutations. (D) Population of IPMNs with mutations in both genes, KRAS mutation alone, GNAS mutation alone, and wild type of both genes. Kaplan–Meier survival curves for disease-free survival (E and G), excluding a patient with remaining cancer at the resected margin, and overall survival (F and H) according to KRAS and GNAS mutation status, respectively. IPMN, intraductal papillary mucinous neoplasm; NA, not available (PPTX 126 kb)
Acknowledgments
The authors would like to thank Mr. Toru Nakai, Mrs. Tae Yamanishi, and Mr. Akihiro Nyuya for technical assistance and Enago (www.enago.jp) for English language editing.
Authors’ contributions
KY extracted DNA, performed methylation and genetic analyses, and drafted the manuscript. TN assisted with data interpretation, designed the project, secured the funding, and drafted the manuscript. YU and TY provided patient samples and clinicopathological data. TT and HY performed IHC and pathological investigations. KK and FT assisted with KRAS and GNAS mutation analysis and provided clinical information. TomF, YM, and SK assisted with IHC staining and summarized clinicopathological data. AG assisted with data interpretation and revised the manuscript. TosF provided patient samples and clinicopathological data, assisted with data interpretation, and revised the manuscript. All authors have read and approved the final manuscript.
Compliance with ethical standards
Conflict of interest
All the authors declare that they have no conflict of interest.
Funding
This study was funded by KAKENHI (20590572, 25860409, 26462016, and 15H03034).
Ethical approval
All procedures were performed in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
References
- Adsay NV. Intraductal papillary mucinous neoplasms of the pancreas: pathology and molecular genetics. J Gastrointest Surg. 2002;6:656–659. doi: 10.1016/S1091-255X(02)00057-4. [DOI] [PubMed] [Google Scholar]
- Adsay NV, Fukushima N, Furuawa T et al (2010) Intraductal neoplasm of the pancreas. In: Bosman FT, Carneiro F, Hruban RH, Theise ND (eds) WHO classification of tumors of digestive system. WHO press, Lyon, pp 304–313
- Amato E, et al. Targeted next-generation sequencing of cancer genes dissects the molecular profiles of intraductal papillary neoplasms of the pancreas. J Pathol. 2014;233:217–227. doi: 10.1002/path.4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick B, Willmore-Payne C, Tripp S, Layfield LJ, Hirschowitz S, Holden J. Histologic, immunohistochemical, and molecular classification of 52 IPMNs of the pancreas. Appl Immunohistochem Mol Morphol. 2009;17:31–39. doi: 10.1097/PAI.0b013e31817c02c6. [DOI] [PubMed] [Google Scholar]
- Chari ST, et al. Study of recurrence after surgical resection of intraductal papillary mucinous neoplasm of the pancreas. Gastroenterology. 2002;123:1500–1507. doi: 10.1053/gast.2002.36552. [DOI] [PubMed] [Google Scholar]
- Chaumeil J, Skok JA. The role of CTCF in regulating V(D)J recombination. Curr Opin Immunol. 2012;24:153–159. doi: 10.1016/j.coi.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Meng J, Yue W, Yu J, Yang J, Yao Z, Zhang L. Fibulin-3 suppresses Wnt/beta-catenin signaling and lung cancer invasion. Carcinogenesis. 2014;35:1707–1716. doi: 10.1093/carcin/bgu023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper CL, O’Toole SA, Kench JG. Classification, morphology and molecular pathology of premalignant lesions of the pancreas. Pathology. 2013;45:286–304. doi: 10.1097/PAT.0b013e32835f2205. [DOI] [PubMed] [Google Scholar]
- Dal Molin M, et al. Clinicopathological correlates of activating GNAS mutations in intraductal papillary mucinous neoplasm (IPMN) of the pancreas. Ann Surg Oncol. 2013;20:3802–3808. doi: 10.1245/s10434-013-3096-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das KK, et al. mAb Das-1 is specific for high-risk and malignant intraductal papillary mucinous neoplasm (IPMN) Gut. 2013;63:1626–1634. doi: 10.1136/gutjnl-2013-306219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell JJ, Brugge WR. Intraductal papillary mucinous tumor of the pancreas. Gastrointest Endosc. 2002;55:701–714. doi: 10.1067/mge.2002.123641. [DOI] [PubMed] [Google Scholar]
- Fritz S, et al. Global genomic analysis of intraductal papillary mucinous neoplasms of the pancreas reveals significant molecular differences compared to ductal adenocarcinoma. Ann Surg. 2009;249:440–447. doi: 10.1097/SLA.0b013e31819a6e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa T, et al. Whole-exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Sci Rep. 2011;1:161. doi: 10.1038/srep00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda M, et al. Mutant TP53 in duodenal samples of pancreatic juice from patients with pancreatic cancer or high-grade dysplasia. Clin Gastroenterol Hepatol. 2013;11(719–730):e5. doi: 10.1016/j.cgh.2012.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SC, et al. Intraductal papillary mucinous neoplasm of the pancreas: clinical characteristics and treatment outcomes of 118 consecutive patients from a single center. J Hepatobiliary Pancreat Surg. 2008;15:183–188. doi: 10.1007/s00534-007-1231-8. [DOI] [PubMed] [Google Scholar]
- Kobayashi N, et al. A comparative analysis of the fibulin protein family. Biochemical characterization, binding interactions, and tissue localization. J Biol Chem. 2007;282:11805–11816. doi: 10.1074/jbc.M611029200. [DOI] [PubMed] [Google Scholar]
- Komatsu H, et al. A GNAS mutation found in pancreatic intraductal papillary mucinous neoplasms induces drastic alterations of gene expression profiles with upregulation of mucin genes. PLoS One. 2014;9:e87875. doi: 10.1371/journal.pone.0087875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maire F, et al. Prognosis of malignant intraductal papillary mucinous tumours of the pancreas after surgical resection. Comparison with pancreatic ductal adenocarcinoma. Gut. 2002;51:717–722. doi: 10.1136/gut.51.5.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthaei H, et al. Clinicopathological characteristics and molecular analyses of multifocal intraductal papillary mucinous neoplasms of the pancreas. Ann Surg. 2012;255:326–333. doi: 10.1097/SLA.0b013e3182378a18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mino-Kenudson M, et al. Prognosis of invasive intraductal papillary mucinous neoplasm depends on histological and precursor epithelial subtypes. Gut. 2011;60:1712–1720. doi: 10.1136/gut.2010.232272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohri D, et al. Different subtypes of intraductal papillary mucinous neoplasm in the pancreas have distinct pathways to pancreatic cancer progression. J Gastroenterol. 2012;47:203–213. doi: 10.1007/s00535-011-0482-y. [DOI] [PubMed] [Google Scholar]
- Nagasaka T, et al. Methylation pattern of the O6-methylguanine-DNA methyltransferase gene in colon during progressive colorectal tumorigenesis. Int J Cancer. 2008;122:2429–2436. doi: 10.1002/ijc.23398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasaka T, et al. Analysis of fecal DNA methylation to detect gastrointestinal neoplasia. J Natl Cancer Inst. 2009;101:1244–1258. doi: 10.1093/jnci/djp265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomoto S, et al. Epidermal growth factor-containing fibulin-like extracellular matrix protein 1, EFEMP1, a novel tumor-suppressor gene detected in hepatocellular carcinoma using double combination array analysis. Ann Surg Oncol. 2010;17:923–932. doi: 10.1245/s10434-009-0790-0. [DOI] [PubMed] [Google Scholar]
- Parmigiani G, Boca S, Lin J, Kinzler KW, Velculescu V, Vogelstein B. Design and analysis issues in genome-wide somatic mutation studies of cancer. Genomics. 2009;93:17–21. doi: 10.1016/j.ygeno.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137:1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondo M, Tachibana I, Urrutia R, Burgart LJ, DiMagno EP. Invasive cancer and survival of intraductal papillary mucinous tumors of the pancreas. Am J Gastroenterol. 2002;97:2553–2558. doi: 10.1111/j.1572-0241.2002.06022.x. [DOI] [PubMed] [Google Scholar]
- Remmele W, Stegner HE. Recommendation for uniform definition of an immunoreactive score (IRS) for immunohistochemical estrogen receptor detection (ER-ICA) in breast cancer tissue. Pathologe. 1987;8:138–140. [PubMed] [Google Scholar]
- Sadr-Nabavi A, et al. Decreased expression of angiogenesis antagonist EFEMP1 in sporadic breast cancer is caused by aberrant promoter methylation and points to an impact of EFEMP1 as molecular biomarker. Int J Cancer. 2009;124:1727–1735. doi: 10.1002/ijc.24108. [DOI] [PubMed] [Google Scholar]
- Salvia R, et al. Main-duct intraductal papillary mucinous neoplasms of the pancreas: clinical predictors of malignancy and long-term survival following resection. Ann Surg. 2004;239:678–685. doi: 10.1097/01.sla.0000124386.54496.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Goggins M. Epigenetic alterations in intraductal papillary mucinous neoplasms of the pancreas. J Hepatobiliary Pancreat Surg. 2006;13:280–285. doi: 10.1007/s00534-005-1056-2. [DOI] [PubMed] [Google Scholar]
- Schmidt D, et al. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell. 2012;148:335–348. doi: 10.1016/j.cell.2011.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoedel KE, Finkelstein SD, Ohori NP. K-Ras and microsatellite marker analysis of fine-needle aspirates from intraductal papillary mucinous neoplasms of the pancreas. Diagn Cytopathol. 2006;34:605–608. doi: 10.1002/dc.20511. [DOI] [PubMed] [Google Scholar]
- Schonleben F, et al. BRAF and KRAS gene mutations in intraductal papillary mucinous neoplasm/carcinoma (IPMN/IPMC) of the pancreas. Cancer Lett. 2007;249:242–248. doi: 10.1016/j.canlet.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonleben F, Qiu W, Remotti HE, Hohenberger W, Su GH. PIK3CA, KRAS, and BRAF mutations in intraductal papillary mucinous neoplasm/carcinoma (IPMN/C) of the pancreas. Langenbecks Arch Surg. 2008;393:289–296. doi: 10.1007/s00423-008-0285-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa F, et al. Intraductal papillary-mucinous tumours represent a distinct group of pancreatic neoplasms: an investigation of tumour cell differentiation and K-ras, p53 and c-erbB-2 abnormalities in 26 patients. Virchows Archiv Int J Pathol. 1994;425:357–367. doi: 10.1007/BF00189573. [DOI] [PubMed] [Google Scholar]
- Sohn TA, Yeo CJ, Cameron JL, Hruban RH, Fukushima N, Campbell KA, Lillemoe KD. Intraductal papillary mucinous neoplasms of the pancreas: an updated experience. Ann Surg. 2004;239:788–797. doi: 10.1097/01.sla.0000128306.90650.aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda M, et al. Expansion of CpG methylation in the SFRP2 promoter region during colorectal tumorigenesis. Acta Med Okayama. 2011;65:169–177. doi: 10.18926/AMO/46628. [DOI] [PubMed] [Google Scholar]
- Tanaka M, et al. International consensus guidelines 2012 for the management of IPMN and MCN of the pancreas. Pancreatology. 2012;12:183–197. doi: 10.1016/j.pan.2012.04.004. [DOI] [PubMed] [Google Scholar]
- Wang R, Zhang YW, Chen LB. Aberrant promoter methylation of FBLN-3 gene and clinicopathological significance in non-small cell lung carcinoma. Lung Cancer. 2010;69:239–244. doi: 10.1016/j.lungcan.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Wang Z, Yuan X, Jiao N, Zhu H, Zhang Y, Tong J. CDH13 and FLBN3 gene methylation are associated with poor prognosis in colorectal cancer. Pathol Oncol Res POR. 2012;18:263–270. doi: 10.1007/s12253-011-9437-0. [DOI] [PubMed] [Google Scholar]
- Wasif N, Bentrem DJ, Farrell JJ, Ko CY, Hines OJ, Reber HA, Tomlinson JS. Invasive intraductal papillary mucinous neoplasm versus sporadic pancreatic adenocarcinoma: a stage-matched comparison of outcomes. Cancer. 2010;116:3369–3377. doi: 10.1002/cncr.25070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc Natl Acad Sci USA. 2011;108:21188–21193. doi: 10.1073/pnas.1118046108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med. 2011;3:92ra66. doi: 10.1126/scitranslmed.3002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, et al. Epigenetic inactivation of EFEMP1 is associated with tumor suppressive function in endometrial carcinoma. PLoS One. 2013;8:e67458. doi: 10.1371/journal.pone.0067458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue W, et al. Frequent inactivation of RAMP2, EFEMP1 and Dutt1 in lung cancer by promoter hypermethylation. Clin Cancer Res Off J Am Assoc Cancer Res. 2007;13:4336–4344. doi: 10.1158/1078-0432.CCR-07-0015. [DOI] [PubMed] [Google Scholar]
- Zhu XJ, Liu J, Xu XY, Zhang CD, Dai DQ. Novel tumor-suppressor gene epidermal growth factor-containing fibulin-like extracellular matrix protein 1 is epigenetically silenced and associated with invasion and metastasis in human gastric cancer. Mol Med Rep. 2014;9:2283–2292. doi: 10.3892/mmr.2014.2135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Kaplan–Meier survival curves for DFS and OS according to clinicopathological characteristics. Kaplan–Meier survival curves for DFS (A, C, and E) excluding a patient with remaining cancer at the resected margin and OS (B, D, and F) according to atypical grade, invasive carcinoma by stage I and II, and invasive carcinoma by histological type, respectively. DFS, disease-free survival; NA, not available; OS, overall survival (PPTX 91 kb)
KRAS and GNAS mutations in IPMNs. (A) and (B) demonstrate the mutations occurring in KRAS codon 12 and GNAS codon 201, respectively. (C) Venn diagram of IPMNs with KRAS mutations, GNAS mutations, and concurrent KRAS and GNAS mutations. (D) Population of IPMNs with mutations in both genes, KRAS mutation alone, GNAS mutation alone, and wild type of both genes. Kaplan–Meier survival curves for disease-free survival (E and G), excluding a patient with remaining cancer at the resected margin, and overall survival (F and H) according to KRAS and GNAS mutation status, respectively. IPMN, intraductal papillary mucinous neoplasm; NA, not available (PPTX 126 kb)