

Abstract
In this article, we address the problem of modeling generic features of structurally but not textually related DNA motifs, that is, motifs whose consensus sequences are entirely different but nevertheless share “metasequence features” reflecting similarities in the DNA-binding domains of their associated protein recognizers. We present MotifPrototyper, a profile Bayesian model that can capture structural properties typical of particular families of motifs. Each family corresponds to transcription regulatory proteins with similar types of structural signatures in their DNA-binding domains. We show how to train MotifPrototypers from biologically identified motifs categorized according to the TRANSFAC categorization of transcription factors and present empirical results of motif classification, motif parameter estimation, and de novo motif detection by using the learned profile models.
Keywords: mixture model, Dirichlet density, hidden Markov model, classification, semi-unsupervised learning
All motifs are not created equal.
Michael Eisen
Transcription regulation is mediated primarily by combinatorial interactions between protein regulators called transcription factors (TFs), and their corresponding cis-regulatory recognition sites on the noncoding genomic sequences, often referred to as DNA motifs. In general, the motif that is recognized by any DNA-binding protein is not a unique sequence. Rather, the sites of recognition are a set of similar sequences that are somewhat complementary in structure to their corresponding TFs within a certain degree of variability tolerance (1). As Michael Eisen (personal communication) has pointed out, great potential exists for improving motif recognition by modeling and exploiting such structural regularities. In addition to biologically functional motifs, complex genomes also contain nonspecific binding sites (nonsites) that can interact with a protein but do not fall into its set of specific recognition sequences and other recurring patterns not recognizable by any TF despite their enriched occurrences. The sequence variabilities among the set of instances of each motif (corresponding to a unique TF) and the possible ambiguities between true motif sites and nonsites at the sequence level make it difficult to identify biologically plausible motif patterns during de novo motif detection from long and complex genome sequences and to infer the function of identified motifs in silico.
For the gene regulatory system to work properly, a TF must display much higher binding affinities to its own recognition sites than to nonsite DNA. This correspondence suggests possible regularities in the DNA motif structure that match the structural signatures in the DNA-binding domains of their corresponding TFs. Can these regularities hidden in the true DNA motif patterns be exploited to improve sensitivity and specificity during motif discovery?
A commonly used representation for motifs in extant motif-finding algorithms is the position weight matrix (PWM), which records the relative frequency (or a related score) of each potential DNA nucleotide at the positions of a motif (2, 3). Statistically, a PWM defines a product multinomial (PM) model for the observed instances of a motif, which inherently assumes that the nucleotide contents of positions within the motif are independent of each other. Thus, a PWM only models independent statistical variations with respect to a consensus pattern of a motif, but it ignores potential couplings between positions inside the motif. This limitation often weakens the ability of a PWM to discern genuine instances of a motif from a very complex background that may harbor random recurring patterns because of the low signal/noise ratios reflected in the likelihood-based scores computed from the PM model.
A recent article by Barash et al. (4) proposed a family of more sophisticated representations to capture richer characteristics of motifs. These representations are based on probabilistic graphical models (also referred to as Bayesian networks for the cases of directed acyclic models), a formalism that captures probabilistic dependencies among random variables in complex domains by using graph-theoretic representations with associated probabilistic semantics (5, 6). Barash et al. (4) suggested that a mixture of PM models can capture potential multimodalities of the biophysical mechanism underlying the protein-DNA recognition between a TF and its target motif sites. They further proposed a tree-based Bayesian network capable of capturing pairwise dependencies of nucleotide contents between nonadjacent positions within the motif. A natural combination of the above two models leads to a more expressive model, a mixture of trees, which captures more complex dependency characteristics of motifs. In a series of experiments with simulated and real data, Barash et al. (4) showed that these more expressive motif models lead to better likelihood scores for motifs and can improve the sensitivity and specificity of motif detection in yeast regulatory sequences under a simple scenario of motif occurrence (i.e., at most one motif per sequence).
In principle, it is possible to construct even more expressive models for motifs by systematically exploiting the power of graphical models, although fitting more complex models reliably demands more training data. Thus, striking the right balance between expressiveness and complexity remains an open research problem in motif modeling.
This progress notwithstanding, it should be clear that all extant motif models are essentially motif-specific and are intended to generalize only to different instances of the same motif. An important issue that remains little addressed is how to build models that can generalize over different motifs that are somewhat related (for instance, belonging to a family of regulatory sites that are targets of TFs bearing the same class of binding domains) even though they do not share apparent commonality in consensus sequences. This issue is important in computational motif analysis because,
often, we want to roughly predict the biological property of an in silico identified motif pattern (e.g., to what kind of TFs it is likely to bind) to reduce the search space of experimental verification;
we may need to introduce some generic but biologically meaningful bias during de novo motif detection so that we can distinguish a biologically plausible binding site (i.e., specifically recognizable by some TF) from a trivial recurring pattern (e.g., microsatellites);
we may also want to restrict attention to a particular class of proteins in performing tasks, such as, “find a regulatory site that potentially binds to type X TF,” or “find co-occurring regulatory sites that can be recognized by type X and type Y TFs, respectively.”
These tasks are important in inferring gene regulatory networks from genomic sequences, possibly in conjunction with relevant expression information.
In this article, we address the problem of modeling generic features of structurally but not textually related DNA motifs, that is, motifs whose consensus sequences are entirely different but nevertheless share “metasequence features,” reflecting similarities in the DNA-binding domains of their associated protein recognizers. We present MotifPrototyper, a profile hidden Markov–Dirichlet multinomial (HMDM) model, which can capture regularities of nucleotide-distribution prototypes and site-conservation couplings typical of each particular family of motifs that corresponds to TFs with similar types of structural signatures in their DNA-binding domains. Central to our framework is the idea of formulating a profile motif model as a family-specific, structured Bayesian prior model for the PWMs of motifs belonging to the family being modeled, thereby relating these motif patterns at the metasequence level. We developed the theoretical framework of the HMDM model in an earlier technical article (7). In this article, we show how to learn family-specific profile HMDMs, or MotifPrototypers, from biologically identified motifs categorized in standard biological databases; how the model can be used as a classifier for aligned multiple instances of motifs; and, most importantly, how a mixture model built on top of multiple profile models can facilitate a Bayesian estimation of the PWM of a novel motif. The Bayesian estimation approach connects biologically identified motifs in the database to previously unknown motifs in a statistically consistent way (which is not possible under the single-motif-based representations described previously) and turns de novo motif detection, a task conventionally cast as an unsupervised learning problem, into a semiunsupervised learning problem that makes substantial use of existing biological knowledge.
Categorization of Motifs Based on Biological Classification of DNA-Binding Proteins
Unlike proteins or genes, which usually have a one-to-one correspondence to monomer sequences and hence are directly comparable based on sequence similarity, a DNA motif is a collective object referring to a set of similar short DNA substrings that can be recognized by a specific protein transcription factor. Different motifs are characterized by differences in consensus, stochasticity, and the number of occurrences. Since each motif usually corresponds to a profile of gapless, multiple-aligned instances rather than a single sequence as for genes and proteins, comparisons based on sequence similarity for different motif patterns are not as straightforward as for genes or proteins.
From a biological point of view, perhaps the most informative way of categorizing DNA motifs is according to the regularities of the DNA-binding domains of their corresponding transcription factors. Advances in structural biology have provided an extensive categorization of the biophysical structures of DNA-binding proteins. The most recent update of the TRANSFAC database (www.gene-regulation.com) (8) lists 4,219 entries, many of which are homologous proteins from different species but nevertheless indicative of the vast number of transcription factors now known that regulate gene expression. The TRANSFAC categorization of TFs (Table 2, which is published as supporting information on the PNAS web site.) provides a good indication of the types of binding mechanisms involved in motif-TF recognition. (For briefness, we refer to the supporting information, which provides detailed methods in the Supporting Text, as well as Table 2 and Figs. 7 and 8, which are published on the PNAS web site.) For concreteness, the following is a brief summary of the structural regularities of four of the major classes of DNA-binding proteins, paraphrasing ref. 9. Due to the correspondence between a TF and a DNA motif, the TF categorization strongly suggests possible features in the structure of motif sequences that are intrinsic to a family of motifs corresponding to a specific class of TFs.
The leucine zipper signature (Fig. 1A) under the superclass of basic domain is an important feature of many eukaryotic regulatory proteins. The hallmark of leucine zipper proteins is the presence of leucine at every seventh position in a stretch of 35 residues. This regularity suggests the presence of a zipper-like α-helical coiled coil bringing together a pair of DNA-binding modules to bind two adjacent DNA sequences. Leucine zippers can couple identical or nonidentical chains, suggesting homodimeric or heterodimeric signature in the recognition site.
Fig. 1.

DNA-binding domains in TFs. (A) Leucine zipper. (B) Zinc fingers. (C) Helix–turn–helix. (D) Beta scaffold.
The zinc finger domain (Fig. 1B) is also common in eukaryotic TFs and regulates gene expression by binding to extended DNA sequences. A zinc finger grips a specific region of DNA, binds to the major groove of DNA, and wraps part of the way around the double helix. Each finger makes contact with a short stretch of the DNA, and residues from the amino-terminal part of the α-helix form hydrogen bonds with the exposed bases in the major groove. Zinc-finger DNA-binding proteins are highly versatile and can have various numbers of zinc fingers in the binding domain. Arrays of zinc fingers are well suited for combinatorial recognition of DNA sequences.
The helix–turn–helix domain (Fig. 1C) contains two α-helices separated by 34 Å, the pitch of a DNA double helix. Molecular modeling studies showed that these two helices would fit into two successive major grooves. This domain, common in bacterial DNA-binding proteins, such as the bacteriophage λ Cro protein, also occurs in the eukaryotic homeobox proteins controlling development in insects and vertebrates.
The beta-scaffold factors (Fig. 1D) are somewhat unusual in that they bind to the minor groove of DNA. The binding domain is globular rather than elongated, suggesting an extensive contact between the DNA sequence and the protein binding domain.
These class-specific protein-binding mechanisms suggest the existence of features that are characteristic of different families of DNA motifs and shared by different motifs in the same family. It is evident that the positions within the motifs are not necessarily uniformly conserved, nor are the conserved positions randomly distributed. Since only a subset of the positions inside the motif are directly involved in protein binding, the degree of conservation of positions inside the motif is likely to be spatially dependent, and such dependencies may be typical for each motif family corresponding to a TF class due to structural complementarity between motifs and the corresponding TFs. It is also possible that due to different degrees of variability tolerance for different TF classes, each family of motifs may require a different selection of prototypes for the distributions of possible nucleotides at the positions within the motifs. Note that such regularities are less likely to be preserved in a nonfunctional recurring pattern, thus they also provide important clues to distinguishing genuine from false motif patterns during de novo motif finding. Fig. 2 provides two examples for the so-called conservation-coupling property of the position dependencies in functional motifs. On the left-hand side are two genuine motifs from two different families. On the right are artificial patterns resulting from a column permutation of the original motifs. Although the two patterns will receive the same likelihood score under conventional PWM representations, clearly the patterns on the left are biologically more plausible because of the complementarity of their patterns of conserved positions to the structures of their binding proteins. Again, it is important to remember that the conservation-coupling property and nucleotide-distribution prototypes are only associated with the generic biophysical properties of a motif family, but not with any specific consensus sequence of a single motif; thus, we call them metasequence features.
Fig. 2.

Conservation coupling of a zinc-finger motif gal4 and a helix–loop–helix motif pho4. Since typical conservation couplings are often reflected in the “contour shape” (e.g., U or bell shape) of the motif logo (a graphical display of the spatial pattern of information content over all sites), we can understand this property as a “shape bias.”
Bayesian Profile Models for Motif Families
Our goal is to build a statistical model to capture the generic properties of a motif family so that it can generalize to novel motifs belonging to the same family. In the following text, we develop such a model using a hierarchical Bayesian approach.
The column of nucleotides at each position in a motif can be modeled by a position-specific multinomial distribution (PSMD). A multinomial distribution over K symbols can be viewed a point in a regular (K – 1)-dimensional simplex; the probabilities of the symbols are the distances from the point to the faces of the simplex (an example of a 2D simplex is shown in Fig. 3A). A Dirichlet distribution is a particular type of distribution over the simplex, hence, a distribution over the multinomial distributions. Each specific Dirichlet is characterized by a vector of K parameters. It can impose a bias toward a particular type of PSMD in terms of how strongly it is conserved and to what nucleotide it is conserved. For example, in Fig. 3A, the center of probability mass is near the center of the simplex, meaning that the multinomial distributions that define a near uniform probability of all possible nucleotides will have a higher prior probability. But for a Dirichlet density whose center of mass is close to a corner associated with a particular nucleotide, say, “A” (Fig. 3B), the multinomial distributions with high frequencies for A have high prior probabilities. Therefore, we can regard a Dirichlet distribution as a “prototype” of the PSMDs of motifs.
Fig. 3.

Dirichlet densities over a 3-nt simplex.
We propose a generative model that generates a multialignment A containing M instances of a motif of length L, in the following way (as illustrated in Fig. 4). (i) We sample a sequence of states 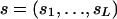 from a first-order Markov chain with initial distribution π and transition matrix B. The states in this sequence can be viewed as prototype indicators for the columns (positions) of the motif. Associated with each state is a corresponding Dirichlet distribution specified by the value of the state. For example, if sl = i, then column l is associated with a Dirichlet distribution parameterized by
from a first-order Markov chain with initial distribution π and transition matrix B. The states in this sequence can be viewed as prototype indicators for the columns (positions) of the motif. Associated with each state is a corresponding Dirichlet distribution specified by the value of the state. For example, if sl = i, then column l is associated with a Dirichlet distribution parameterized by 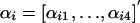 . (ii) For each
. (ii) For each 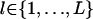 , sample a multinomial distribution θl according to p(θ|αsl), the probability defined by the Dirichlet component αsl. (iii) All the nucleotides in column l are generated iid according to the multinomial distribution parameterized by θl.
, sample a multinomial distribution θl according to p(θ|αsl), the probability defined by the Dirichlet component αsl. (iii) All the nucleotides in column l are generated iid according to the multinomial distribution parameterized by θl.
Fig. 4.

The graphical model representation of a MotifPrototyper. Empty circles represent random variables associated with a single motif and the boxes are plates representing iid replicates (i.e., M observed instances of the motif). Black arrows denote dependencies between the variables. Parameters of the MotifPrototyper are represented by the center-dotted circles, and the round-cornered box over the α parameter denotes I sets of Dirichlet parameters.
Thus, the complete likelihood of a motif alignment AM×L characterized by a nucleotide-count matrix h is:
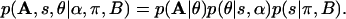 |
[1] |
Technically, such a model, which we refer to as a MotifPrototyper, is a HMDM model (7, 10). It defines a structured prior for the PWM of a motif. Formal development of the HMDM model and mathematical details of Bayesian inference using this model can be found in an earlier technical article (7) and hence are omitted here for simplicity. With the availability of a categorization for motifs, each family of motifs can be associated with a family-specific profile HMDM model that imposes PSMD prototypes and positional dependencies unique to this family.
What do we gain from a MotifPrototyper? First, a MotifPrototyper introduces prior information about the joint distribution of the nucleotide distribution in different positions of a motif of the corresponding family and gives high probabilities to those commonly found distributions possibly compatible with the degree of variability tolerance intrinsic to the class of TFs corresponding to the motif family. Under a MotifPrototyper, a posteriori, each PSMD in a motif follows a family-specific mixture of multiple Dirichlet distributions, which blends the different prototypes that might dictate the nucleotide distribution at that position. Furthermore, a MotifPrototyper stochastically imposes family-specific spatial dependencies for different columns within a motif. As Fig. 4 makes clear, a MotifPrototyper is not a simple hidden Markov model (HMM) for sequence data. In an HMM the transitions would be between the emission models (i.e., multinomials) themselves, and the output at each step would be a single monomer in the sequence. In MotifPrototyper, the transitions are between different prior components for the emission models, and the direct output of this HMM is the parameter vector of a generative model, which will be sampled multiple times at each position to generate iid instances. This approach is especially useful when we have prior knowledge about motif properties, such as conservation-coupling or other positional dependencies.
Second, rather than using a maximum likelihood (ML) approach to estimate the PWM, which considers only the relative frequency of nucleotides but is indifferent to the actual number of instances observed, MotifPrototyper facilitates a Bayesian estimation of the PWM under a family-specific prior, thus taking into consideration the actual number of observations available for PWM estimation along with the biological prior. It is possible with only a few instances to obtain a robust estimation of the nucleotide frequency at each position of a motif.
Note that a MotifPrototyper defines a family-specific structured prior for the PWMs without committing to any specific consensus motif sequence.
Training a MotifPrototyper. Given biologically identified instances of motifs of a particular family, we can compile a multiple-alignment for each motif and write down the joint likelihood of the training data under a single-profile model (i.e., a MotifPrototyper) by marginalizing the PWMs (i.e., θ's) and the hidden Markov states (i.e., s) of each motif in Eq. 1. This likelihood is a function of the model parameters. Thus, we can compute the empirical Bayesian estimation of the model parameters by maximizing the likelihood over each parameter by using a quasi-Newton procedure (11). The result is a set of parameters intrinsic to the training data.
Note that this training process also involves a model selection issue of how many Dirichlet components should be used. As in any statistical model, a balance must be struck between the complexity of the model and the data available to estimate the parameters of the model. Empirically, we found that eight components appear to be a robust choice and also provide good interpretability.
Classifying Motifs. Identifying that a motif belongs to a family and relating it to other members of the family often allows inference about its functions. Given multiple profile models, each corresponding to a distinct motif family, we can compute the conditional likelihood of a set of aligned instances of an unlabeled motif under each profile model by integrating out the hidden variables (i.e., θ and s) in each resulting complete likelihood function. The posterior probability of each possible assignment of class membership to the motif under test is proportional to the magnitude of the conditional likelihood multiplied by the prior probabilities of the respective motif families (which can be computed from the empirical frequency of each motif family) (see supporting information).
Thus, we can estimate the family membership by a maximum a posteriori scheme. It is noteworthy that, here, we are classifying a set of aligned instances of a motif as a whole rather than a single sequence substring as in a standard classification task, such as, predicting the function or structure of a protein based on its amino acid sequence (12, 13).
Bayesian Estimation of PWM and Semiunsupervised de Novo Motif Detection. Given a set of aligned instances of a motif, if we know the family membership of this motif, we can directly compute the posterior distribution of its PWM, using the family-specific MotifPrototyper as a prior according to the Bayesian rule. The Bayesian estimation of a PWM is defined as the expectation of the PWM with respect to this posterior. If the family membership is not known a priori (i.e., we do not prespecify what family of motif to look for, but allow the motif to come from any family), then we can simply assume that the PWM admits a mixture of profile models (see supporting information).
In de novo motif detection where locations of motif instances are not known, the motif matrix A is an unobserved random variable. We can iterate between predicting motif locations based on the current Bayesian estimation of the motif PWM and updating the Bayesian estimation based on newly predicted motif instances. It can be proved that such a procedure is guaranteed to converge to a locally optimal solution (14). But unlike the standard EM algorithm for estimating a PWM, since we can compute the Bayesian estimation based on a trained profile motif prior, we essentially turn de novo motif detection from an originally unsupervised learning problem into a semiunsupervised learning problem that can make use of biological training data without committing to any particular consensus motif pattern.
It is straightforward to generalize our current formulation of the MotifPrototyper model to family-specific prior distributions of more sophisticated motif representations, such as trees or mixture of trees (4) by slightly reparameterizing the MotifPrototyper model. The training procedure and the usage for classification and de novo motif detection require little modification.
Experiments
In this section, we present results of learning MotifPrototyper models from categorized families of motifs and demonstrate applications of the learned MotifPrototypers with three experiments, each addressing a typical issue of interest in in silico motif analysis. (i) Given instances of a (computationally) identified motif, assign the motif to a motif family that corresponds to a particular class of transcription factors. (ii) Provide a Bayesian estimation of PWM that be more informative than a ML estimation. (iii) Improve de novo motif detection by casting the problem as a semisupervised learning task that makes use of biological prior knowledge incorporated in the family-specific MotifPrototypers (with a small-scale demonstration).
Parameter Estimation. The TRANSFAC database (version 6.0) contains 336-nt count matrices of aligned motif sequences. These matrices summarize a significant portion of the biologically identified transcription regulatory motifs reported in the literature and are well categorized and curated (although the original aligned sequences corresponding to the count matrices are not provided). We used 271 of the matrices as training data, each derived from at least 10 recognition sites of a TF in one of the four well represented superclasses (Table 2), to compute the empirical Bayesian estimations of the parameters of four profile Bayesian models of motif families.
We performed 50 random restarts for the quasi-Newton algorithm for parameter estimation and picked the solutions corresponding to the highest log likelihood achieved at convergence (Fig. 7 illustrates the parameters of the four resulting profile models pictorially). We have not attempted to interpret the numerical representations of each profile model in terms of their biological implications, but it is possible to read off some interesting high-level biological characteristics therefrom (see supporting information). In this article we refrain from such elaborations but simply maintain that MotifPrototyper is a formal mathematical abstraction of the metasequence properties intrinsic to a motif profile represented by the training examples.
To evaluate the training quality of our profile models, we define the training error as the percentage of misclassification of the superclass identities of the training motif matrices using profile models learned from the full training set. Our training errors ranged from 10% to 28%, with the beta-scaffold MotifPrototyper having the best fit (basic domain, 16.8%; zinc finger, 17.3%; helix–turn–helix, 27.6%; and beta-scaffold, 10%). Given that motif family is a rather loose definition based on TF superclasses, and that each superclass still has very diverse and ambiguous internal structures, these training errors indicate that family-specific regularities can be captured reasonably well by MotifPrototyper.
Motif Classification. To examine the generalizability of MotifPrototyper to newly encountered motif patterns, we performed a 10-fold cross-validation test for motif classification (see supporting information). The performances over each family of motifs are summarized in Table 1. We present classification error rates for both the entire data set and the slashed data set that contains only the major motif subclasses (i.e., those with at least 10 different motifs, see Table 2 for details of the class hierarchy) under each superclass. Not surprisingly, performance on the data set with only major subclasses is significantly better, suggesting that the minor classes in each superclass are possibly more ambiguous and less typical with respect to the overall characteristics of the superclass. In fact, some minor classes were unanimously assigned to a different superclass by our classifier; for example, all six members of class 1.6 (bHSH) and all seven members of class 3.4 (heat-shock factors) are assigned to superclass 4 (beta-scaffold), whereas all five members of class 4.7 (HMG) are assigned to superclass 3 (helix–turn–helix). Whether such inconsistencies reflect a deficiency of our classifier or possible true biological ambiguity of these motif patterns is an interesting problem to be investigated further.
Table 1. Motif classification with MotifPrototyper.
| CV error | Basic domain | Zinc finger | Heliz—turn—helix | Beta scaffold |
|---|---|---|---|---|
| Whole set | 0.256 | 0.423 | 0.443 | 0.403 |
| Major classes | 0.217 | 0.373 | 0.379 | 0.178 |
CV, cross-validation
To our knowledge, there has been no algorithm that classifies aligned sets of motif instances as a collective object based on metasequence features shared within motif families. The closest counterpart in sequence analysis is the profile HMM (pHMM) for protein classification (15), but pHMM is based on the assumption that proteins of the same family share sequence-level similarities, and the objects classified are single sequences. Thus, no direct comparison can be made between pHMM and MotifPrototyper. Nevertheless, we note that although pHMM is based on much more stringent features at the sequence level and aimed at a relatively simpler task of evaluating single sequences, typical performance of pHMM is ≈20–50% for short polypeptides (i.e., <100 aa) (12, 13), similar to the performance of motif classification using MotifPrototyper. Thus, we believe that MotifPrototyper exhibits a reasonable performance given that the labeling of motif family membership is more ambiguous than that of single protein sequences, the metasequence features we use are far less stringent than sequence similarities, and motif patterns are much shorter than polypeptides.
PWM Estimation and Motif Scoring. A major application of MotifPrototyper is to serve as an informative prior for Bayesian estimation of the PWM from a set of aligned instances of a novel motif. Since in a realistic de novo motif detection scenario, we have to evaluate many substrings corresponding to either a true motif, or random patterns in the background, we expect that the Bayesian estimation of PWM resulted from a mixture of MotifPrototypers provides a more reliable discriminability than the ML estimation between true motifs and background sequences. We demonstrate this ability by comparing the likelihood of a true motif substring with the likelihoods of background substrings, all scored under the estimated PWM of the motif (see supporting information).
As evident from Fig. 5, the discriminability of the Bayesian estimation of the PWM, measured by the log likelihood odds (of motif vs. background substrings), is indeed better than that of the ML estimation for most of the motifs we tested. A more detailed analysis (supporting information) further reveals that, in cases where only a small number of instances are available for estimation, mixture of profile models still leads to a good estimation that generalizes well to new instances and results in high log likelihood odds, whereas the ML estimation does not generalize as well.
Fig. 5.

A comparison of Bayesian and ML estimations of the PWM. Each point represents a motif being tested, and the x coordinate (respectively, y coordinate) represents the log likelihood odds due to the ML (respectively, Bayesian) estimation.
These results give strong support to the claim that, in many cases, a MotifPrototyper-based approach can significantly improve the sensitivity and specificity for novel motifs and provide a robust estimation of their PWM under few observations. These are very useful properties for de novo motif detection in complex genomic sequences.
De Novo Motif Discovery. Finally, we present a comparison of the (mixture of) profile Bayesian motif model, MotifPrototyper, with the conventional PM model for de novo motif detection, using semirealistic test data of which the ground truth (i.e., full annotation of motif types and locations) is known for evaluating the prediction results (see supporting information).
We tested on sequences each containing a single “authentic” motif instance and contaminated by artificial “decoy” patterns (e.g., the permuted patterns in Fig. 2). This scenario frees us from modeling the global distribution of motif occurrences, as needed for more complex sequences (compare the LOGOS model, ref. 10) and therefore demonstrates the influence of different models for motif patterns on de novo detection.
As shown in Fig. 6, MotifPrototyper significantly outperforms PM [i.e., with >20% margin in “hit-rate” (supporting information)] on 11 of the 28 motifs and is comparable with PM (within ±10% difference) for the remaining 17 motifs. Overall, MotifPrototyper correctly identifies 50% or more of the motif instances for 16 of the 28 motifs, whereas the PM model achieves a 50% hit rate for only 8 of the 28 motifs. Note that MotifPrototyper is fully autonomous and requires no user specification of which particular profile motif model to use. If we are willing to introduce a manual postprocessing step, in which we use each of the four profile motif models described before separately for de novo motif finding, and generate four sets of motif predictions instead of one (as of MotifPrototyper) for visual inspection, it is possible to obtain even better predictions (Fig. 6, ♦).
Fig. 6.

Median hit rates of de novo detection of yeast motifs with MotifPrototyper (□), PM (○), and the best outcome of four single-profile-based predictions with MotifPrototyper (♦). Motifs are listed along the x axis, ordered by the hit rates of MotifPrototyper for each motif.
The ability to provide multiple candidate solutions, each corresponding to a specific TF category, manifests a key advantage of the profile motif model. It allows a user to capture different types of prior knowledge about motif structures and bias motif prediction toward a particular metasequence structure in a well controlled way. A human observer given a visual presentation of the most likely motifs suggested by different profile motif models could easily pick out the best one from these candidates, whereas PM can yield only a single most likely answer.
Conclusion
We have presented MotifPrototyper, a novel profile Bayesian motif model that captures generic metasequence features shared by motifs corresponding to common transcription factor superclasses. It is a probabilistic graphical model that captures the positional dependencies and nucleotide distribution prototypes typical to each motif family, and it defines a prior distribution of the positional weight matrices of motifs for each family. We demonstrated how MotifPrototyper can be trained from biologically identified motif examples and its applications for motif classification, Bayesian estimation of PWM, and de novo motif detection.
To the best of our knowledge, all extant motif models are intended to be motif-specific, emphasizing the ability to characterize sequence-level features unique to a particular motif pattern. Thus, when one defines a model in such a way for a novel motif not biologically characterized before, one needs to solve a completely unsupervised learning problem to identify the possible instances and fit the motif parameters simultaneously. Under this unsupervised framework, there is little explicit connection between the novel motif to be estimated from the unannotated sequences and the rich collection of biologically identified motifs recorded in various databases. It is reasonable to expect that the fruitful biological investigations of gene regulatory mechanisms and the resulting large number of known motifs could contribute more information to the unraveling of novel motifs. MotifPrototyper represents an initial foray into the development of a new framework that turns de novo motif detection into a semiunsupervised learning problem. It provides more control during the search of novel motif patterns by making use of prior knowledge implied in the known motifs, helps to improve sensitivity to biologically plausible motifs, and potentially reduces spurious solutions often occurring in an pure unsupervised setting.
It is possible to build a stronger motif classifier by using discriminative approaches, such as neural networks or support vector machines, and we are currently pursuing this direction. But since the goal of this article is not merely to build a classifier but to develop a model that can be easily integrated into a more general architecture for de novo motif detection, we feel that a generative framework, especially by means of a Bayesian prior model, provides the desired generalizability and flexibility for such tasks. As discussed in ref. 10, a graphical model formalism of the motif detection problem allows a modular combination of heterogeneous submodels, each addressing a particular component of the overall problem, i.e., the local structure of a motif pattern, the global organization of motif instances and motif modules, and the distribution of background sequences, thereby enabling a complex modeling and inference problem to be handled in a divide-and-conquer fashion. The design of MotifPrototyper aligns with this principle and can be used as the “local” submodel under the LOGOS framework (10).
In should also be clear that the main aim of this article is to demonstrate the profile Bayesian model as a modeling approach to capture metasequence motif features. To make the presentation simple and focused, in this article we did not intend to present working software that performs motif discovery in real complex sequences, which also requires appropriate modeling of other aspects of gene regulatory sequences, such as genomic distribution of motif locations. This issue should be addressed with another probabilistic model and de novo motif detection in metazoan genomes using a joint model should be investigated.
Supplementary Material
Abbreviations: TF, transcription factors; PWM, position weight matrix; PM, product multinomial; HMDM, hidden Markov–Dirichlet multinomial; PSMD, position-specific multinomial distribution; HMM, hidden Markov model; pHMM, profile HMM; ML, maximum likelihood.
References
- 1.Stormo, G. D & Fields, D. S. (1998) Trends Biochem. Sci. 23, 109–113. [DOI] [PubMed] [Google Scholar]
- 2.Lawrence, C & Reilly, A. (1990) Proteins 7, 41–51. [DOI] [PubMed] [Google Scholar]
- 3.Bailey, T. L & Elkan, C. (1994) in Proceedings of the 2nd International Conference on Intelligent Systems for Molecular Biology (AAAI Press, Menlo Park, CA), pp. 28–36.
- 4.Barash, Y, Elidan, G, Friedman, N. & Kaplan, T. (2003) in Proceedings of the 7th International Conference on Research in Computational Molecular Biology (ACM Press, New York), pp. 28–37.
- 5.Cowell, R. G., Dawid, A. P., Lauritzen, S. L. & Spiegelhalter, D. J. (1999) Probabilistic Networks and Expert Systems (Springer, New York).
- 6.Pearl, J. (1988) Probabilistic Reasoning in Intelligent System: Networks of Plausible Inference (Morgan Kaufmann, San Francisco).
- 7.Xing, E. P., Jordan, M. I., Karp, R. M. & Russell, S. (2003) in Advances in Neural Information Processing Systems 15, eds. Becker, S., Thrun, S. & Obermayer, K. (MIT Press, Cambridge, MA).
- 8.Wingender, E., Chen, X., Hehl, R., Karas, H., Liebich, I., Matys, V., Meinhardt, T., Pruss, M., Reuter, I. & Schacherer, F. (2000) Nucleic Acids Res. 28, 316–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stryer, L. (1995) Biochemistry (Freeman, New York), 4th Ed.
- 10.Xing, E. P., Wu, W., Jordan, M. I. & Karp, R. M. (2004) J. Bioinformatics Comput. Biol. 2, 127–154. [DOI] [PubMed] [Google Scholar]
- 11.Sjölander, K., Karplus, K., Brown, M., Hughey, R., Krogh, A., Mian, I. & Haussler, D. (1996) Comput. Appl. Biosci. 12, 327–345. [DOI] [PubMed] [Google Scholar]
- 12.Karchin, R., Karplus, K. & Haussler, D. (2002) Bioinformatics 18, 147–159. [DOI] [PubMed] [Google Scholar]
- 13.Moriyama, E. N & Kim, J. (2003) Proceedings of the 23rd Stadler Genetics Symposium (Plenum, New York). Available at http://bioinfolab.unl.edu/emlab/index.html. Accessed June 25, 2004.
- 14.Xing, E. P., Jordan, M. I. & Russell, S. (2003) in Uncertainty in Artificial Intelligence, eds. Kjaerulff, U. & Meek, C. (Morgan Kaufmann, San Francisco), Vol. 19, pp. 583–591. [Google Scholar]
- 15.Krogh, A., Brown, M., Mian, I., Sjölander, K. & Haussler, D. (1994) J. Mol. Biol. 235, 1501–1531. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.