

Abstract
Impairment of brain-glucose uptake and brain-copper regulation occurs in Alzheimer’s disease (AD). Here we sought to further elucidate the processes that cause neurodegeneration in AD by measuring levels of metabolites and metals in brain regions that undergo different degrees of damage. We employed mass spectrometry (MS) to measure metabolites and metals in seven post-mortem brain regions of nine AD patients and nine controls, and plasma-glucose and plasma-copper levels in an ante-mortem case-control study. Glucose, sorbitol and fructose were markedly elevated in all AD brain regions, whereas copper was correspondingly deficient throughout (all P < 0.0001). In the ante-mortem case-control study, by contrast, plasma-glucose and plasma-copper levels did not differ between patients and controls. There were pervasive defects in regulation of glucose and copper in AD brain but no evidence for corresponding systemic abnormalities in plasma. Elevation of brain glucose and deficient brain copper potentially contribute to the pathogenesis of neurodegeneration in AD.
“Alzheimer’s disease” (AD) is the predominant cause of ageing-related dementia1 and refers to a constellation of cognitive and behavioural changes that are typical for patients who have substantial amounts of its hallmark pathological lesions in their brain2. AD may occur through interplay of environmental, genetic, and metabolic factors3,4, but some brain regions are impacted more severely than others, consistent with operation of a specific disease mechanism5,6. The pathophysiological process of AD is thought to begin many years before the diagnosis of AD-related dementia7,8, and sporadic AD accounts for the vast majority of cases2. AD is a non-communicable disease which displays several pathogenic mechanisms in brain, including: Aβ-amyloid deposition9; neurofibrillary tangles comprising tau protein2,8; impaired cerebral glucose metabolism (‘hypometabolism’) with insulin resistance and defective carbohydrate regulation8,10; oxidative stress and inflammation4,11; cerebrovascular amyloid angiopathy (CAA)12; enhanced advanced glycation end-product (AGE) formation9; and defective copper regulation13,14. Risk of sporadic AD has also been linked to presence of type-2 diabetes (T2D) in epidemiological studies15,16.
Glucose uptake17,18 and regional cerebral perfusion6 are impaired in AD, particularly in severely-damaged brain regions5,19, and progressive reductions in glucose-uptake may occur years before clinically-evident dementia19: however, the molecular basis for these defects is uncertain. Impaired brain-glucose regulation has been tentatively attributed to a functional defect of glucose transporter (GLUT)-mediated glucose uptake20, but neuronal loss may be responsible for some of this effect, and there is ongoing argument concerning the contribution of this mechanism to defective brain-glucose uptake and neurodegeneration.
Here we analyzed post-mortem tissue from seven brain regions of patients with AD and controls in whom clinical dementia had been excluded and who had no ante-mortem evidence for neurodegeneration or T2D. Diabetes was excluded in this post-mortem study on the basis of a negative history and lack of any biochemical evidence of hyperglycemia but not by systematic ante-mortem glucose-tolerance testing: it is therefore possible, albeit unlikely, that one or more patients had undiagnosed mild T2D or prediabetes. Elevated glucose levels have previously been linked to altered tissue-copper regulation in the context of diabetes21. Furthermore, in AD, some brain regions are more severely damaged than others22,23. Therefore, we measured tissue levels of metabolites and trace metals24,25 in seven human brain-regions in patients and controls matched for age and gender: three of these undergo severe neuronal damage in AD (hippocampus, entorhinal cortex and middle temporal gyrus); three are less affected (cingulate gyrus, sensory cortex and motor cortex); and one (cerebellum) is relatively spared5,6,22. We also measured levels of brain metabolites and copper in (i) post-mortem whole-brain tissue of diabetic and control rats, to probe potential effects of post-mortem delay (PMD) on metabolite levels and contrast effects of AD with those of diabetes in an animal model; and (ii) metabolites and metal levels in brains from a triple-transgenic mutant model of familial AD (fAD) and controls26. Finally, we measured fasting-plasma glucose (FPG), hemoglobin A1c (HbA1c), and fasting-plasma copper in AD-patients and controls from a clinical case-control study, to ascertain whether patients might show evidence of elevated systemic glucose consistent with diabetes or lesser degrees of hyperglycemia, or signs of impaired systemic copper metabolism.
Results
Post-mortem study of AD brain: group characteristics
All participants in this study were from New Zealand and were matched for age, gender and PMD (Tables 1 and 2). Median (range) brain-weight was 1062 (831–1355) g in cases and 1260 (1094–1461; P = 0.005) in controls (Tables 1 and 2): the ~16% decline in brain-weight in AD is consistent with histological severity22. The case-control matching and short PMD support the quality of these data (Tables 1 and 2). Cases were diagnosed with AD by clinical27, CERAD28 and Braak criteria22: patients had late-onset/sporadic AD and none had findings consistent with early-onset AD, fAD, or T2D.
Table 1. Case-control study of post-mortem human brain in Alzheimer’s disease: group characteristics.
| Variable | Control | Alzheimer’s disease |
|---|---|---|
| Number | 9 | 9 |
| Age | 70.1 (6.7) | 70.3 (7.1) |
| Male sex, n (%) | 5 (55.6) | 5 (55.6) |
| PMD (h) | 9 (5.5–13.0) | 7 (4.0–12.0) |
| Brain-weight (g) | 1260 (1094–1461) | 1062 (831–1355)* |
| Plaques, n (%) | 1 (11) | 9 (100)** |
| Tangles, n (%) | 1 (11) | 9 (100)** |
Values are: age, mean (SD); PMD and brain-weights, median (range): *P = 0.005, **P < 0.0001 compared with corresponding Control value; all other differences are non-significant.
Table 2. Individual patient characteristics.
| No | Code | Group | Age/Sex | Ante-mortem assessment of brain disease/mental state | Cause of death | Braak Stage (Amyloid load) | PMD (h) | Brain Wt (g) |
|---|---|---|---|---|---|---|---|---|
| 1 | H155 | Control | 61/M | No brain disease or dementia | Ischemic heart disease | 0 (0) | 7.0 | 1258 |
| 2 | H121 | Control | 64/F | No brain disease or dementia | Pulmonary embolism | 0 (0) | 5.5 | 1260 |
| 3 | H132 | Control | 63/F | No brain disease or dementia | Ruptured aorta | 0 (0) | 12.0 | 1280 |
| 4 | H122 | Control | 72/F | No brain disease or dementia | Emphysema | 0 (0) | 9.0 | 1230 |
| 5 | H204 | Control | 66/M | No brain disease or dementia | Ischemic heart disease | 0 (0) | 9.0 | 1461 |
| 6 | H241 | Control | 76/F | No brain disease or dementia | Metastatic carcinoma | II (A3, B1, C1) | 12.0 | 1094 |
| 7 | H164 | Control | 73/M | No brain disease or dementia | Ischemic heart disease | 0 (0) | 13.0 | 1315 |
| 8 | H123 | Control | 78/M | No brain disease or dementia | Ruptured aortic aneurysm | 0 (0) | 7.5 | 1260 |
| 9 | H150 | Control | 78/M | No brain disease or dementia | Ruptured MI | 0 (0) | 12.0 | 1416 |
| 10 | AZ42 | AD | 60/M | Alzheimer’s dementia | Alzheimer’s disease | VI (3/3) | 7.0 | 1020 |
| 11 | AZ71 | AD | 62/F | Alzheimer’s dementia | Alzheimer’s disease | VI (3/3) | 6.0 | 831 |
| 12 | AZ48 | AD | 63/F | Alzheimer’s dementia | Bronchopneumonia | VI (2/3) | 7.0 | 1080 |
| 13 | AZ72 | AD | 70/F | Alzheimer’s dementia | Lung cancer | V (3/3) | 7.0 | 1044 |
| 14 | AZ90 | AD | 73/M | Alzheimer’s dementia | GI hemorrhage | IV (3/3) | 4.0 | 1287 |
| 15 | AZ96 | AD | 74/F | Alzheimer’s dementia | Metastatic cancer | V (3/3) | 8.5 | 1062 |
| 16 | AZ39 | AD | 74/M | Alzheimer’s dementia | Pseudomonas bacteremia | VI (2/3) | 12.0 | 1355 |
| 17 | AZ80 | AD | 77/M | Alzheimer’s dementia | Myocardial infarction | VI (3/3) | 4.5 | 1180 |
| 18 | AZ38 | AD | 80/M | Alzheimer’s dementia | Bronchopneumonia/ pulmonary oedema | V (3/3) | 5.5 | 1039 |
Abbreviations: GI, gastrointestinal; MI, myocardial infarction; PMD, post-mortem delay; wt, weight. Cause of death was determined by post-mortem examination, and brain pathology and Braak Stage were determined by specialist neuropathological examination. All AD cases had ‘Age-Related Plaque’ scores of “C”. Causes of death were the primary causes listed on the death certificate. Patient H241 had post-mortem signs consistent with AD and was therefore diagnosed with preclinical disease: the corresponding data have been retained in the main analysis presented in the manuscript, and removed from the control group for the secondary analysis. Control patient H241 (preclinical AD) was described as A3, B1, C1 using the ‘ABC’ criteria for AD neuropathologic change that incorporates histopathological assessments of Aβ deposits (A), staging of neurofibrillary tangles (B), and scoring of neuritic plaques (C)2.
Glucose levels in AD brain
Levels of glucose were elevated in all brain regions of AD-patients (Table 3, Figs 1, 2, 3). Mean (±95% CI) elevations in glucose varied from 5.2-fold (1.4–18.6) in cerebellum to 16.4-fold (5.2–51.2) in middle temporal gyrus (Figs 1 and 2a). There was evidence for differences in glucose elevations between different brain regions in AD where levels tended to be higher in more severely affected regions such as the middle temporal gyrus (Table 3; Figs 1 and 2a). Elevated glucose levels can damage proteins and lipids through chemical attack via the aldehyde group, forming AGEs in diabetic tissues9,29. Our data indicate that potential exists for similar glucose-driven damage mechanisms to occur in AD brain, consistent with co-localization there of AGEs with Aβ-containing plaque9. Variation in PMD did not affect glucose values here (data not shown).
Table 3. Study of post-mortem human brain: relative fold-changes in glucose, sorbitol and fructose in seven brain regions from nine Alzheimer’s cases and nine controls.
| Metabolite | Brain-region | Estimate | Lower Bound | Upper Bound | P-value | BH FDR Q-value |
|---|---|---|---|---|---|---|
| Glucose | Cerebellum | 5.2 | 1.4 | 18.6 | 0.015 | 0.015 |
| Entorhinal cortex | 7.4 | 2.4 | 23.2 | 0.0028 | 0.0041 | |
| Cingulate gyrus | 8.0 | 2.4 | 26.1 | 0.0023 | 0.0037 | |
| Hippocampus | 8.9 | 2.9 | 27.4 | 0.0016 | 0.0030 | |
| Sensory cortex | 9.2 | 3.0 | 28.8 | 0.0014 | 0.0029 | |
| Motor cortex | 11.8 | 3.7 | 37.7 | 0.0006 | 0.0019 | |
| Middle temporal gyrus | 16.4 | 5.2 | 51.2 | 0.0002 | 0.0019 | |
| Sorbitol | Cerebellum | 3.7 | 1.6 | 8.6 | 0.0036 | 0.0044 |
| Entorhinal cortex | 4.3 | 2.0 | 9.1 | 0.0006 | 0.0019 | |
| Cingulate gyrus | 3.1 | 1.5 | 6.5 | 0.0048 | 0.0053 | |
| Hippocampus | 4.1 | 1.9 | 8.5 | 0.0009 | 0.0021 | |
| Sensory cortex | 4.1 | 1.9 | 8.6 | 0.0009 | 0.0021 | |
| Motor cortex | 3.0 | 1.4 | 6.6 | 0.0090 | 0.0094 | |
| Middle temporal gyrus | 3.3 | 1.5 | 6.9 | 0.0039 | 0.0046 | |
| Fructose | Cerebellum | 5.3 | 2.0 | 14.4 | 0.0018 | 0.0031 |
| Entorhinal cortex | 5.7 | 2.4 | 13.5 | 0.0004 | 0.0019 | |
| Cingulate gyrus | 3.9 | 1.7 | 9.2 | 0.0030 | 0.0041 | |
| Hippocampus | 5.5 | 2.3 | 12.9 | 0.0005 | 0.0019 | |
| Sensory cortex | 5.4 | 2.4 | 12.4 | 0.0004 | 0.0019 | |
| Motor cortex | 4.2 | 1.7 | 10.2 | 0.0032 | 0.0041 | |
| Middle temporal gyrus | 5.7 | 2.6 | 12.9 | 0.0003 | 0.0019 |
Estimates and their lower and upper bounds were derived by Bayesian modelling. This table incorporates values from n = 9 patients with clinical diagnoses of AD and n = 9 matched asymptomatic controls. Abbreviation: ‘BH FDR Q-value’ is the Benjamini-Hochberg False- Discovery Rate-adjusted P-value.
Figure 1. Log2-transformed concentrations of glucose, sorbitol and fructose in post-mortem tissue from seven brain regions of nine patients with AD and nine controls.

Individual values are shown for each region. At the right-hand side are detailed the identity, sex, age, brain-weight, and PMD of each participant, along with the histopathological diagnosis. One control (Case 15: F76, brain wt 1094 g; black squares) with higher metabolite concentrations than other controls had preclinical AD (Braak Stage II).
Figure 2.

Probability density distributions of fold-changes from controls to cases in glucose, sorbitol and fructose measured in brain tissue from (a) each of seven brain regions from patients with AD (n = 9) and controls (n = 9), and (b) whole brain homogenates from diabetic (n = 7) and control (n = 7) rats. Distributions were derived by Bayesian modeling and illustrate the plausible ranges (posterior distributions) of inferred fold changes for each region and metabolite. This analysis complements the results of a mixed-effects model assessment of the fold-change between AD cases and controls for measurements of metabolites in each brain region. Posterior means (solid lines) and 95% credible intervals (dashed lines) are shown, along with the ‘no fold change’ value (solid black line). (b) Shown are results of an equivalent simulation of data from a metabolomic study of ex-vivo whole-brain tissue from diabetic rats and controls. Data were log2-transformed before analysis.
Figure 3. Regional brain glucose (upper panel) and brain copper (μmol/dry kg; lower panel) levels in patients with AD (n = 7–9/region; red) and controls (n = 7–9; black).
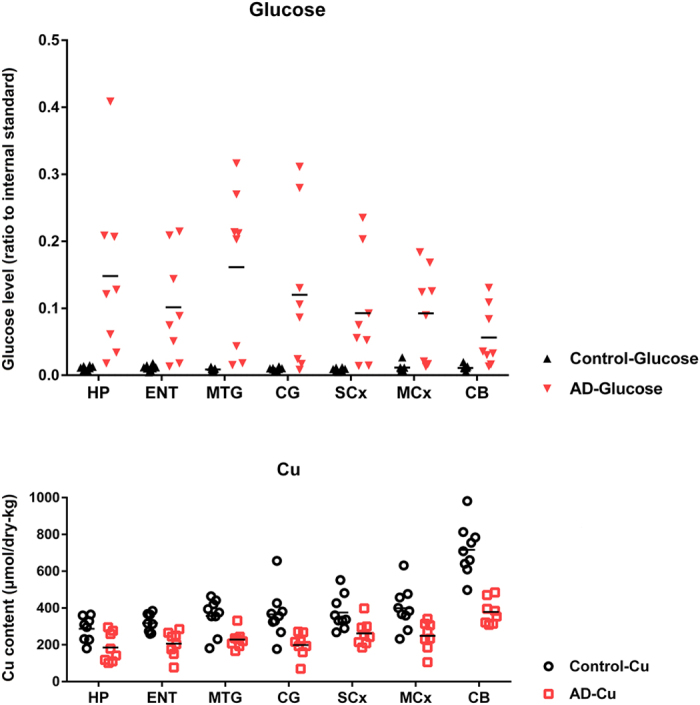
Overall mean ( ± 95% CI; Confidence Interval) wet-weight/dry-weight ratio was 5.6 (5.4–5.8). Overall mean brain-glucose levels were higher (P = 9.7 × 10−13) and brain copper values lower (P = 1.6 × 10−8) in cases than controls. Mean brain copper in AD trended lower in every region: CB (P < 0.0001); MCx (P = 0.021); SCx (P = 0.027); CG (P = 0.0079); MTG (P = 0.013); ENT (P = 0.0046); and HP (P = 0.070); and there was modest evidence that overall brain-copper and brain-glucose values were inversely correlated (P = 0.021) in AD-patients but not controls. Abbreviations: CB, cerebellum; CG, cingulate gyrus; ENT, entorhinal cortex; HP, hippocampus; MCx, motor cortex; MTG, middle temporal gyrus; SCx, sensory cortex.
Sorbitol levels in AD brain
Sorbitol, formed from glucose, is the first metabolite in the polyol pathway, which usually accounts for a few percent at most of glucose utilization under normal conditions30. Here, sorbitol was elevated in all AD brain regions: mean (±95% CI) fold-elevations ranged from 3.0 (1.4–6.6) in motor cortex to 4.3 (2.0–9.1) in entorhinal cortex (Figs 1 and 2; Table 3). Variation in sorbitol levels was less than for glucose and there was no evidence for inter-regional differences, possibly reflecting sorbitol formation from glucose as the rate-limiting step in the polyol pathway. Variation in PMD did not affect sorbitol levels.
Fructose levels in AD brain
Fructose is the second metabolite in the polyol pathway30. Fructose may be even more reactive in AGE-mediated damage mechanisms than glucose, and levels of the two are similar in AD brain. Here, brain-fructose levels were elevated in all cases, consistent with the findings for glucose and sorbitol (Figs 1 and 2a; Table 3). Elevations in brain fructose showed less variation than glucose, consistent with sorbitol: fold-elevations were between 3.9 (1.7–9.2) in cingulate gyrus and 5.7 (2.6–12.9) in middle temporal gyrus. Inter-regional differences were not significant and variation in PMD did not affect fructose levels.
Inter-regional comparisons of metabolite levels in human brain
Mean elevations in glucose fold-change, 9.5 (3.0–30.4) were significantly higher across all regions in AD brain than those for sorbitol and fructose (P = 0.0002). Values for sorbitol, 3.6 (1.7–7.8), and fructose, 5.1 (2.1–12.2) did not differ significantly across regions. Mean values of all three metabolites were substantively elevated in all AD brain regions (Figs 1 and 2): these elevations were widespread in AD, consistent with a global defect.
Post-mortem brain metabolites in a patient with preclinical AD
Elevated brain levels of glucose, sorbitol and fructose were present in one control patient, a 76 year-old female (Code H241) who had no ante-mortem clinical evidence for brain disease or dementia, but had preclinical AD characterized by low brain-weight (1,094 g) and positive post-mortem histology (Braak Stage II; ABC score2 of A3, B1, C1; black squares in Fig. 1) consistent with “AD neuropathologic change”2. This finding indicates that substantive disturbance of brain glucose and polyol pathway regulation may antedate the onset of clinical dementia, possibly by a considerable period.
Copper levels in AD brain
Mean (±95% CI) brain-copper levels summed across all seven regions were 256 (232–279) μmol/dry kg in AD and 406 (363–449) μmol/dry kg in controls (P = 1.6 × 10−8): there was thus an overall decrease of ~40% in brain-copper levels in AD. Mean brain-copper levels tended to be lower in AD than controls in all brain regions examined (Fig. 3). In addition, there was moderate evidence for a trend in copper levels to be inversely proportional to tissue-glucose values in AD-patients (P = 0.021; restricted iterative generalized least squares31) but not in controls.
Levels of tissue metabolites and copper in rodent brain
We did not have access to post-mortem brain from diabetic patients with dementia suitable for this study. Instead, we measured levels of metabolites and trace metals in whole brains from male rats with streptozotocin-induced diabetes (a model of severe T1D)21 and whole mouse brains from a model of AD (13–14-month-old male triple-transgenic mice)26, by applying the same methods we used for analysis of human brain. Diabetic-rat brain displayed marked elevations in glucose, sorbitol, and fructose, similar to those in human AD brain (Suppl. Table S1; Fig. 2b; all P < 0.0001). Thus, elevations in post-mortem human AD brain tissue were similar to those in rat-brain snap-frozen within ~1 minute after death. By contrast, there was no significant difference in brain-copper levels between diabetic and control rats: mean (±95% CI) copper values were 209.0 (189.8–228.1) μmol/dry-kg in diabetic rats and 188.4 (147.1–229.8) μmol/dry-kg in controls.
In the triple-transgenic model of AD26, brain-glucose levels were below the limits of detection (~1–5 μmol/kg) in both experimental groups; brain-fructose levels were lower in transgenic mice than controls (P = 0.0005); sorbitol levels trended lower in transgenic mice but differences were not significant; and copper values did not differ between transgenic mice and controls (data not shown).
Case-control study of plasma glucose and plasma-copper levels in patients with AD
Participants in this case-control study were matched for gender and age (see Suppl. Table S2). Consistent with expectation, measured cognitive function scores (MMSE) in the AD group were significantly lower than those of controls whereas the ApoE4 allele was more prevalent. FPG and serum HbA1c levels were equivalent between groups so there was no evidence for elevated rates of undiagnosed T2D, impaired glucose tolerance (IGT) or impaired fasting glucose (IFG) in these British patients with late-onset AD. Fasting plasma-copper levels did not differ significantly between the AD group and controls (see Suppl. Table S2).
Discussion
Glucose is the obligatory energy-generating substrate in the brain32, and impaired brain-glucose uptake and utilization is a key metabolic defect in AD33: this phenomenon is generally worse in brain regions with greater histological3,34, functional19 and molecular4 evidence of damage, consistent with the operation of a specific disease process17,19. Transport of glucose across the plasma membrane of mammalian cells is the first rate-limiting step for glucose metabolism and is mediated by facilitative GLUT proteins35. One potential explanation for decreased brain-glucose uptake could be that of impaired GLUT activity, leading to consequent cerebral ‘hypometabolism’. Consistently, lowered levels of GLUT1, the main glucose transporter in the blood-brain barrier (BBB), have been reported in several studies of AD20,36,37,38,39; many GLUT-related changes in AD pathogenesis may occur before the onset of neuronal dysfunction40. A reduction of GLUTs at the BBB that occurs before the onset of the main pathophysiological changes and symptoms of AD could contribute to pathogenesis39. However, direct measurements of glucose concentrations in AD brain tissue have hitherto been lacking.
Unexpectedly, we found here that glucose levels in AD were markedly elevated rather than decreased in all seven brain regions of these patients with late-onset AD: therefore, diminished cerebral GLUT activity is unlikely to play an initiating role in decreased glucose uptake in AD. Rather, these data indicate that lower brain-GLUT/glucose-uptake rates in AD probably occur in response to elevated brain-glucose levels; consistently, there is substantive evidence that elevated cellular glucose can mediate down-regulation of GLUT1 expression and function41,42. Elevated brain-glucose levels may therefore antedate the occurrence of GLUT down-regulation at the BBB in the pathogenesis of AD. The presence of elevated brain-glucose levels in AD accords with impaired intracellular glucose utilization through glycolysis and the tricarboxylic acid (TCA) cycle: consistently, there is substantive evidence for defects in AD of TCA-related mitochondrial enzymes including the pyruvate dehydrogenase complex, the α-ketoglutarate dehydrogenase complex, and cytochrome c oxidase/complex IV43.
Here, marked elevations in levels of sorbitol and fructose were also present in all AD brain regions. These metabolites are produced from glucose via the polyol pathway in neurons and astrocytes44. The proportion of cortical oxidative metabolism attributable to astrocytes, ~30%, roughly corresponds to their volume fraction, indicating that astrocytes and neurons have similar fuel oxidation rates45, so both cell-types could contribute to this effect, in proportions depending on the relative distribution of polyol-pathway intermediates in the two.
Brain-glucose levels were lower in cerebellum than other regions whereas elevations in sorbitol and fructose were similar in all regions studied (Fig. 2). Glucose levels trended higher in regions more prone to tissue-damage in AD, consistent with insufficient clearance via glycolysis with consequent up-regulation of the alternative polyol pathway29. The human brain comprises similar numbers of neurons and non-neuronal cells46: both make substantive contributions to functional in vivo indices of energy metabolism47, although non-neuronal cells have more prominent gluconeogenesis and glycogenesis48,49. It will therefore be important to determine whether the elevation of glucose and polyol-pathway intermediates in AD is caused by metabolic defects in neurons, glia, or both.
Our measurements of FPG and HbA1c50 in this group of patients with AD provide no evidence for increased prevalence of undiagnosed T2D, IGT or IFG (see Suppl. Table S2): therefore, it is unlikely that dementia in these patients occurred as a result of systemic disorders of glucose metabolism such as diabetes or IGT. Rather, our findings indicate that defective glucose metabolism in AD may occur only in a restricted location, the brain. Our data in brain tissue from AD patients and rats with severe diabetes were similar with respect to their glucose, sorbitol and fructose content. Therefore, the brain in patients with AD presents an abnormality of glucose utilization closely resembling that in the brain (the current study) and peripheral nervous tissue (sciatic nerve, dorsal root ganglia, and trigeminal ganglia) of the rats with severe diabetes51.
Our finding of elevated blood glucose, sorbitol and fructose in a patient with Braak Stage-II disease without clinical evidence of dementia is potentially significant, and suggests that metabolic disturbances in AD- brain may occur relatively early in disease progression. It was recently reported that acute hyperglycemia in young AD-model mice had increased Aβ production in the interstitial fluid, which was augmented in aged AD-mice with marked Aβ plaque pathology, identifying a mechanism by which systemic glucose elevation could damage brain cells52. However, in light of our current findings of marked elevations of brain glucose, sorbitol and fructose levels in AD without systemic elevation of glucose, direct toxicity caused by isolated intra-cerebral metabolite elevations could provide an alternative mechanism for neurodegeneration in AD.
How might elevated brain levels of glucose, sorbitol and fructose cause neurodegeneration? One potential mechanism is that glucose (an aldehyde) and fructose (a ketone) can attack macromolecules through their reactive functional groups leading to AGE formation, as occurs in the diabetic complications29. Tissues in AD brain are known to be modified by N-epsilon-carboxymethyllysine (CML) formation9,53, which coordinates divalent copper9,13,54, localizing it to affected regions and thereby enhancing localized pro-oxidant stress55,56; consistent with this mechanism, copper homeostasis is evidently impaired in AD (summarized in)13. These mechanisms may well make similar contributions to AGE formation and downstream pathogenesis in AD.
Brain glucose uptake in AD is lower in severely affected regions and occurs early in disease development18. For example, using 2-[18F]fluoro-2-deoxy-D-glucose positron-emission tomography (FDG-PET), it has been shown that there is diminished glucose uptake in the hippocampus, parietotemporal cortex and/or posterior cingulate cortex in: (i) individuals who are at genetic risk for fAD57,58; (ii) individuals with a history of AD59; as well as (iii) those who have mild or no cognitive impairment but eventually go on to develop AD19. The relevance of studies in fAD to those of sporadic AD are uncertain; there is thus an identified need for similar metabolomic and metallomic studies in suitable cohorts of fAD patients. Elevated intracellular glucose diminishes the trans-membrane gradient that drives facilitative glucose uptake via glucose transporters, so these findings in fAD are not inconsistent with the current results: however, levels of brain glucose and polyol pathway intermediates in fAD are currently unknown. The findings in (iii) indicate that impaired cerebral glucose uptake presages the development of (presumably sporadic) AD: these findings are consistent with elevated brain glucose as a key early aspect of pathogenesis.
It has also been shown that, before neuronal dysfunction develops, mice heterozygous for GLUT1 (Slc2a1) at the BBB develop cerebral blood flow perfusion deficits and BBB breakdown that leads to vascular-mediated neurodegeneration40: the relevance of this study in engineered mice to our findings in human brain is uncertain, given that these authors did not report brain-glucose levels. Our own findings show that adult triple-mutant mice did not develop elevated brain glucose, and that patterns of sorbitol and fructose levels differed markedly from those in patients with AD; we therefore conclude that these mice may not reflect disturbances of glucose and the polyol pathway that occur in humans with Alzheimer’s dementia, although the proviso remains that the disease in these mice is highly region-specific and may not have generated the widespread dysregulation present in patients. Further studies in this and other transgenic models of AD may be indicated. Here we employed male transgenic mice, to avoid the increased metabolic variability which can occur in female mice and is attributable to effects of the estrus cycle, and in light of the similar prevalence of AD in male and female patients. Study of only male transgenic mice may have affected our results since Dr LaFerla, who developed this model, has recently reported that male triple-transgenic mice may not exhibit the phenotypic traits originally described (see: https://www.jax.org/strain/004807). Prevalence of AD is similar in male and female patients, so the potential sexual dimorphism in this model may represent another difference between it and the human disease process. Further studies in this and other transgenic models of AD are therefore indicated in order to ascertain whether any of the available models might demonstrate defective glucose and polyol pathway regulation similar to that present in AD brain.
In glycolysis, hexokinase and phosphofructokinase catalyse reactions where ATP is the phosphate-group donor. Current results could be consistent with inefficient glucose utilization caused by impaired glycolysis and consequent elevation in polyol-pathway metabolites; consistent with this interpretation, for example, hippocampal lactate was elevated by 2.6-fold in cases compared with controls; fold-values (case-control ratios) measured in other brain regions were as follows: EC, 0.5; MTG, 1.3; SCx, 3.4; MCx, 7.3; CG, 1.7; CB, 0.6; thus values in SCx and MCx showed apparent upward trends, although these were not significant, possibly due to the limited numbers in each group. Further study of lactate levels in larger numbers of cases and controls will be required to confirm and extend these data. Elevation of brain glucose and polyol-pathway intermediates might be explained by impaired ATP supply leading to diminished glycolysis, suppressing these ATP-requiring phosphorylation reactions, and could ultimately result from impaired TCA cycle function60. Additionally, in the physiological cerebral milieu, glycogen provides a metabolically-active storage form of glucose in astrocytes61,62. Therefore, it is possible that some of the elevated free glucose we measured in AD brains could have been derived from increased rates of glycogen breakdown via glycogenolysis; alternatively, elevated glucose could mediate increased deposition of brain glycogen or other forms of polymerized glucose63 such as metabolically-inactive, high-molecular-weight amylose that may be formed at the expense of glycogen in AD brains, thereby decreasing the pool of metabolically-accessible stored glucose63.
Elevated free glucose promotes glycation of macromolecules leading to formation of CML. Here, our finding of elevated brain glucose in AD provides a molecular explanation for previously-reported elevations of CML in AD brain9. PET-imaging studies in AD, where the rate-constant k2 is ~normal64, argue against substantively-elevated cytoplasmic free glucose available for retrograde transport into the extracellular space. PET findings are not inconsistent with the current findings, however, with respect to large elevations in tissue-glucose since it binds to macromolecules in a form releasable upon chemical extraction. For example, formation of Schiff-base (aldimine) linkages to proteins is the first step in a reversible pathway leading to CML formation65, reconciling published imaging results with elevated brain-glucose levels; additionally, fructose binds to proteins through an equivalent mechanism66.
Cells require a substantial, uninterrupted copper supply to maintain effective levels of metabolic-fuel utilization and anti-oxidant defense67. Copper for support of fuel utilization is required particularly by the copper-enzymes cytochrome oxidase I (COI) and COII (both inner-membrane components of mitochondrial respiratory-chain complex IV), and for anti-oxidant defense by copper-enzymes superoxide dismutase 1 (SOD1, intracellular) and SOD3 (extracellular)21,68. Deficient enzyme-bound copper caused by defective cell-copper uptake impairs enzyme function that can cause metabolic sequelae including severe tissue damage21,69. The brain has particularly large copper requirements compared with most other organs, since it is very metabolically active70 and generates large amounts of reactive oxygen species, particularly superoxide anion that requires clearance by SOD1 and SOD3. In this context, our current findings of severely-elevated glucose coupled with markedly-deficient copper levels in AD brain are significant, particularly given that elevated CML levels are also widespread in AD brain9. CML directly links elevated tissue-glucose with low tissue-copper, since increased tissue-glucose levels drive CML-modification of collagen71, which inhibits cell-copper uptake by suppressing cell-membrane copper transport via copper transporter 121. A similar process occurs in diabetic heart, where cardiac copper is deficient along with hyperglycemia and elevated coronary CML21, causing myocardial-copper deficiency that mirrors the current findings in AD brain and causes defective copper supply to SOD1 and cytochrome c oxidase/complex IV13,21. There is substantive evidence that defective tissue-copper uptake is triggered by coordination of bioactive divalent copper to CML in collagen54. These findings provide a new and potentially reversible mechanism for the causation of brain damage in AD. Whether this mechanism can be targeted by pharmacological intervention in AD remains to be determined.
This study has limitations. There is evidence that brain from significant numbers of aged but cognitively unimpaired patients may display elevated Braak stages, not infrequently up to III-IV72,73, whereas by contrast, our group of 9 controls showed scores of 0-II. In several studies, Braak scores in the range of 0-II have been reported to occur in more than half of all cognitively-normal elderly persons73,74,75, so our sample may well be consistent with derivation from a similar population. Knopman et al. concluded that “the majority of individuals who are cognitively normal near the time of their death have minimal amounts of tau-positive neuritic pathology (that is, Braak stage <IV)”73. Cases at the more severe end of the pathologic spectrum (Braak stages III-IV) lacking Aβ plaques have been reportedly reported in 2–10% of brains in large autopsy series that included community-based sampling75,76,77. Thus it is not entirely clear whether our control group is representative of the status of aged patients without dementia: larger follow-up studies will be required in clinic-based and community-based cohorts to avoid selection bias77 and thereby to better characterise the status of the polyol pathway in aged patients with and without varying degrees of cognitive impairment.
Despite the limited numbers (n = 9 cases and n = 9 controls), our findings in seven brain regions are statistically robust; however, they require replication in other cohorts of AD patients, and also extension to subjects with preclinical AD, mild cognitive impairment (MCI), and fAD syndromes. Data from the single patient with preclinical AD reported here provide compelling, albeit preliminary data that similar metabolic perturbations might occur in other patients with preclinical disease, but this remains to be verified in suitable larger studies. Equivalent post-mortem metabolomic studies in preclinical AD, MCI, and fAD will require studies of suitable brain tissue from cohorts of patients with these conditions, which are likely to be challenging, and lie beyond the scope of the current study. We did not have the opportunity of performing ante-mortem assessments by MMSE or CDR scoring in the post-mortem study: this represents a limitation that will only be addressed once suitable brain tissue from post-mortem cohorts of patients who have previously been characterized by MMSE/CDR scoring are analyzed by suitable metabolomic methods. The current study does, however, open the field up to consideration of the roles played by elevations in brain glucose and polyol-pathway metabolites and associated copper deficiency in the pathogenesis of AD.
In conclusion, we have shown here that free glucose is markedly elevated in AD brain, particularly in vulnerable regions where elevated glucose may well be linked to copper deficiency and tissue-damage. Brain-sorbitol and brain-fructose levels are also elevated in AD brain, consistent with increased polyol-pathway flux, potentially caused by defective glucose utilization via glycolysis and/or the TCA-cycle. A further, mechanistically-related finding is that pan-cerebral brain-copper deficiency accompanies these glucose and polyol-pathway defects: elevated tissue-CML levels provide a molecular linkage between elevated brain glucose and copper deficiency. Impaired neuronal glucose utilization could play a major role in tissue-damage in AD, possibly via defective mitochondrial metabolism caused by deficient copper supply to COI and COII, leading to impaired function of cytochrome c oxidase78. These findings provide a clear molecular linkage between mechanisms of tissue damage in AD and T2D. The process by which elevated brain glucose is linked to defective cell-copper uptake and intracellular transport to key copper proteins such as COI, COII, SOD1 and SOD3, may well provide a new target for diagnostic imaging or therapeutic intervention.
Methods
Ethics
All experiments were performed in accordance with relevant guidelines and regulations as stated below: the case-control study of post-mortem human brain was approved by the University of Auckland Human Participants Ethics Committee with informed consent from all families. The observational case-control study was performed under a protocol approved by the Leeds Teaching Hospitals NHS Trust Research Ethics Committee with informed consent from participants or families of patients.
The studies in the streptozotocin-induced rat model of diabetes and the triple-transgenic mouse model of AD were performed under UK Home Office Licenses, according to protocols approved by the University of Manchester Animal Welfare and Ethical Review Body (AWERB), and were consistent with the Animals (Scientific Procedures) Act 1986 and the ARRIVE guidelines.
Human brains: acquisition and analysis
Human brains were obtained from the New Zealand Neurological Foundation Human Brain Bank, University of Auckland79. Tissue quality was confirmed by demonstration of mRNA integrity (data not shown). Each brain was dissected under the supervision of neuroanatomists (JX, SP, HJW and RLMF), who accurately identified each region. Ante-mortem MMSE and CDR (Clinical Dementia Rating) examinations were not performed on patients enrolled in the study of post-mortem human brain tissue. Tissue was analyzed for metabolites (~50–800 Da) and copper: regions studied were entorhinal cortex, hippocampus, middle temporal gyrus, cingulate gyrus, sensory cortex, motor cortex, and cerebellum: grey matter from each region was sampled as shown (Suppl. Fig. S1). Aliquots of 50 ± 5 mg were dissected from each region and stored at −80 °C until analysis, and were otherwise treated as previously described4. Patients had ante-mortem evidence of clinical dementia, whereas controls did not. Controls were selected by matching for age, sex and PMD (Tables 1 and 2). A consultant neuropathologist diagnosed or excluded AD by applying the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) criteria28, and determined the neuropathological severity by assigning the Braak stage22 and amyloid load by applying the 2013 consensus National Institute on Aging–Alzheimer’s Association guidelines2 (Tables 1 and 2).
Data from one control participant with Braak Stage II disease
One control patient had neuropathological findings consistent with AD (Braak Stage II; Table 2) and was therefore diagnosed with preclinical disease: this finding is consistent with the known frequency of asymptomatic AD in similarly-aged groups in the study population80. To assess the impact of this patient’s data on the overall outcome, the main statistical analysis was performed twice, first by retaining the corresponding patient’s data in the control group (Table 3) and then by performing a second analysis using the same methods but excluding this patient’s data only: the two analyses yielded equivalent results so this patient’s data have been retained in the control group, despite the presence of some of the metabolic changes observed in diagnosed AD cases.
Observational case-control study
Patients with memory disorder and control participants without cognitive impairment were recruited81 respectively through memory clinics in Leeds and Dewsbury (England), and the Leeds Family Health Services Authority day hospitals and elderly medicine outpatient clinics in the Leeds area. All were of European Caucasian background and gave written informed consent (consent from relatives of the AD-patients was provided, where appropriate). Diagnosis of probable AD was made in accordance with international diagnostic criteria (National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer’s Disease and Related Disorders Association Work Group: NINCDS-ADRDA)82. All participants underwent a standardised clinical evaluation: medical history, fasting plasma glucose (FPG) and HbA1c, and cognitive function assessment by MMSE. AD samples were refined by excluding the few patients with diagnosed T1D or T2D, including those on insulin, or others with significant co-morbidities. Samples were then selected from the whole-study population for whom required measurements (FPG, HbA1c) were available83. The resulting 42 AD-patients were then age- and gender-matched to 43 corresponding controls.
Analysis of rat brain
We employed a validated model of diabetes21 using 6-7 week-old male Wistar rats (n = 14), with body-weights of 220–250 g, maintained under a 12:12-h light:dark cycle and housed at 22 ± 2 °C and 60% humidity, with ad libitum access to standard rat chow (Special Diets Services, Dietex International, Essex, UK) and fresh water. Diabetes was induced (n = 7) by intraperitoneal injection of streptozotocin (55 mg/kg; Sigma Aldrich, Gillingham, UK) and diagnosed by blood-glucose levels of >15 mmol/l on two consecutive measurements21; non-diabetic control animals (n = 7) were age-matched littermates who received a single intraperitoneal injection of sodium-citrate buffer (vehicle) only. Eleven weeks after induction, we euthanized rats by terminal isoflurane anesthesia, excised brains, and stored them at −80 °C until analysis. Whole brains were homogenized and sampled to yield 50 ± 5 mg (wet-weight) aliquots. Equivalent methods were used for analysis of mouse-brain tissue.
Transgenic mice: methods
Male triple transgenic (3xTgAD) mice expressing mutant PS1M146V, APPSwe, TauP301L, and control non-transgenic (non-Tg, 129/C57BL6) mice, were originally supplied by Frank LaFerla (Irvine, CA, USA)26 and an in-house colony established at the University of Manchester. All mice were kept in standard housing conditions (humidity 50–60%, temperature 21 ± 1 °C, 12:12 hour light-dark cycle) and given ad libitum access to standard rodent chow (Special Diets Services). At 13–14 months of age, non-Tg (n = 10) and 3xTgAD (n = 10) were terminally anesthetized with 3-5% isoflurane (30% O2 and 70% N2O) and then transcardially perfused with 0.9% saline. The brains were quickly removed, and one hemisphere was snap-frozen on dry ice and stored at −80 °C until analysis.
Analytical methods
More detailed descriptions are given in the Supplementary Materials. Briefly, metabolite levels were compared between cases and controls by gas chromatography mass spectrometry (GC-MS)-based metabolomics in wet-tissue24, copper was measured in dry-tissue by inductively-coupled-plasma mass spectrometry (ICP-MS)25, and statistical analysis was performed as described in the Supplementary Materials. P-values of < 0.05 were considered significant.
Additional Information
How to cite this article: Xu, J. et al. Elevation of brain glucose and polyol-pathway intermediates with accompanying brain-copper deficiency in patients with Alzheimer’s disease: metabolic basis for dementia. Sci. Rep. 6, 27524; doi: 10.1038/srep27524 (2016).
Supplementary Material
Acknowledgments
We thank Cynthia Tse and Michael Anderson for their assistance with the management of these studies. We acknowledge Professor Sir John Scott for his encouragement, moral support, and helpful discussions with us. We thank the families of patients who supported this research through their donation of brains to the New Zealand Neurological Foundation Human Brain Bank. This work was supported by the Endocore Research Trust (60147); the Maurice and Phyllis Paykel Trust (3627036; and 3709342); the Health Research Council of New Zealand (3338701); the Ministry of Business, Innovation & Employment, New Zealand (MBIE; UOAX0815); the Maurice Wilkins Centre for Molecular Biodiscovery (Tertiary Education Commission 9431-48507; and Doctoral Scholarship funding to Jingshu Xu); the University of Auckland (Doctoral Student funding to Jingshu Xu - JXU058 PReSS); Lottery Health New Zealand (3626585; 3702766); the Oakley Mental Health Research Foundation (3456030; 3627092; 3701339; 3703253; 3702870); the New Zealand Neurological Foundation; the Medical Research Council (UK, MR/L010445/1 and MR/L011093/1); Alzheimer’s Research UK (ARUK-PPG2014B-7); the Guangdong High-end Foreign Expert Fund; the University of Manchester, the CMFT, and the Northwest Regional Development Agency through a combined programme grant to CADET; and was facilitated by the Manchester Biomedical Research Centre and the Greater Manchester Comprehensive Local Research Network. This manuscript is dedicated to the memory of Professor Sir P John Scott in recognition of his moral courage, leadership and support of justice, profound humanity, and outstanding care and concern for all people.
Footnotes
Author Contributions J.X. and P.B. designed and performed research, analyzed and interpreted data, and wrote the manuscript; S.J.C. and S.P. performed research, analyzed data and revised the manuscript; S.Mc.H., K.A.H., N.K., H.J.W., H.L., S.Z., W.L., C.T., B.J.S., M.A.C., J.R.-A. and C.B.L. performed research and revised the manuscript; R.L.M.F. led development of the New Zealand Neurological Foundation Human Brain Bank, supervised brain collection and dissection, and revised the manuscript; K.H. and D.W. interpreted data and revised the manuscript; K.A.B.K., E.R.L.C.V. and N.M.H. designed and performed research, and wrote and revised the manuscript; R.D.U. designed and supervised research, interpreted data and revised the manuscript; A.W.D. designed and developed the statistical models, analyzed data and wrote the manuscript; G.J.S.C. conceived, designed and supervised research, analyzed and interpreted data, wrote the manuscript, and bears overall responsibility for the integrity of the study and the manuscript.
References
- Ferri C. P. et al. Global prevalence of dementia: a Delphi consensus study. Lancet 366, 2112–2117 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montine T. J. et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 123, 1–11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson M. P. & Magnus T. Ageing and neuronal vulnerability. Nat Neurosci 7, 278–294 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönberger S. J., Edgar P. F., Kydd R., Faull R. L. M. & Cooper G. J. S. Proteomic analysis of the brain in Alzheimer’s disease: molecular phenotype of a complex disease process. Proteomics 1, 1519–1528 (2001). [DOI] [PubMed] [Google Scholar]
- Jobst K. A. et al. Rapidly progressing atrophy of medial temporal lobe in Alzheimer’s disease. Lancet 343, 829–830 (1994). [DOI] [PubMed] [Google Scholar]
- Bradley K. M. et al. Cerebral perfusion SPET correlated with Braak pathological stage in Alzheimer’s disease. Brain 125, 1772–1781 (2002). [DOI] [PubMed] [Google Scholar]
- Sperling R. A. et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 280–292 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman R. J. et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med 367, 795–804 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki N. et al. Advanced glycation end products in Alzheimer’s disease and other neurodegenerative diseases. Am J Pathol 153, 1149–1155 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot K. et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest 122, 1–23 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka M. T. et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14, 388–405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll J. A., Yamada M., Frackowiak J., Mazur-Kolecka B. & Weller R. O. Cerebral amyloid angiopathy plays a direct role in the pathogenesis of Alzheimer’s disease. Pro-CAA position statement. Neurobiol Aging 25, 589–597 discussion 603–604 (2004). [DOI] [PubMed] [Google Scholar]
- Cooper G. J. S. Therapeutic potential of copper chelation with triethylenetetramine in managing diabetes and Alzheimer’s disease. Drugs 71, 1281–1320 (2011). [DOI] [PubMed] [Google Scholar]
- Bush A. I. The metallobiology of Alzheimer’s disease. Trends Neurosci 26, 207–214, doi: S0166223603000675 [pii] (2003). [DOI] [PubMed] [Google Scholar]
- Huang C.-C. et al. Diabetes mellitus and the risk of Alzheimer’s disease: a nationwide population-based study. PLOS ONE 9, e87095 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchsinger J. A., Tang M. X., Stern Y., Shea S. & Mayeux R. Diabetes mellitus and risk of Alzheimer’s disease and dementia with stroke in a multiethnic cohort. Am J Epidemiol 154, 635–641 (2001). [DOI] [PubMed] [Google Scholar]
- Arnáiz E. et al. Impaired cerebral glucose metabolism and cognitive functioning predict deterioration in mild cognitive impairment. Neuroreport 12, 851–855 (2001). [DOI] [PubMed] [Google Scholar]
- Anchisi D. et al. Heterogeneity of brain glucose metabolism in mild cognitive impairment and clinical progression to Alzheimer disease. Arch Neurol 62, 1728–1733 (2005). [DOI] [PubMed] [Google Scholar]
- Mosconi L. et al. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur J Nucl Med Mol Imaging 36, 811–822 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson I. A., Chundu K., Davies-Hill T., Honer W. G. & Davies P. Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann Neurol 35, 546–551 (1994). [DOI] [PubMed] [Google Scholar]
- Zhang S. et al. Diabetic cardiomyopathy is associated with defective myocellular copper regulation and both defects are rectified by divalent copper chelation. Cardiovasc Diabetol 13, 100 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H. & Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82, 239–259 (1991). [DOI] [PubMed] [Google Scholar]
- Lüth H.-J. et al. Age- and stage-dependent accumulation of advanced glycation end products in intracellular deposits in normal and Alzheimer’s disease brains. Cerebral Cortex 15, 211–220 (2006). [DOI] [PubMed] [Google Scholar]
- Begley P. et al. Development and performance of a gas chromatography-time-of-flight mass spectrometry analysis for large-scale nontargeted metabolomic studies of human serum. Anal Chem 81, 7038–7046 (2009). [DOI] [PubMed] [Google Scholar]
- Church S. J. et al. Deficient copper concentrations in dried-defatted hepatic tissue from ob/ob mice: A potential model for study of defective copper regulation in metabolic liver disease. Biochem Biophys Res Commun 460, 549–554, doi: 10.1016/j.bbrc.2015.03.067 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S. et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421 (2003). [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. DSM-5. 5th edn, (American Psychiatric Publishing, 2013). [Google Scholar]
- Mirra S. S. et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurol 41, 479–486 (1991). [DOI] [PubMed] [Google Scholar]
- Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes 54, 1615–1625 (2005). [DOI] [PubMed] [Google Scholar]
- Gabbay K. H. Hyperglycemia, polyol metabolism, and complications of diabetes mellitus. Annu Rev Med 26, 521–536 (1975). [DOI] [PubMed] [Google Scholar]
- Goldstein H. Multilevel mixed linear model analysis using iterative generalized least squares. Biometrika 78, 43–56 (1986). [Google Scholar]
- Gruetter R. et al. Direct measurement of brain glucose concentrations in humans by 13C NMR spectroscopy. Proc Natl Acad Sci USA 89, 1109–1112 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetelat G. et al. Direct voxel-based comparison between grey matter hypometabolism and atrophy in Alzheimer’s disease. Brain 131, 60–71 (2008). [DOI] [PubMed] [Google Scholar]
- Blennow K., de Leon M. J. & Zetterberg H. Alzheimer’s disease. Lancet 368, 387–403 (2006). [DOI] [PubMed] [Google Scholar]
- Macheda M. L., Rogers S. & Best J. D. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol 202, 654–662 (2005). [DOI] [PubMed] [Google Scholar]
- Horwood N. & Davies D. C. Imunolabelling of hippocampal microvessel glucose transporter protein is reduced in Alzheimer’s disease. Virchows Arch 425, 69–72 (1994). [DOI] [PubMed] [Google Scholar]
- Kalaria R. N. & Harik S. I. Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in Alzheimer disease. J Neurochem 53, 1083–1088 (1989). [DOI] [PubMed] [Google Scholar]
- Mooradian A. D., Chung H. C. & Shah G. N. GLUT-1 expression in the cerebra of patients with Alzheimer’s disease. Neurobiol Aging 18, 469–474 (1997). [DOI] [PubMed] [Google Scholar]
- Patching S. G. Glucose transporters at the blood-brain barrier: function, regulation and gateways for drug delivery. Mol Neurobiol PMID: 26801191 (2016). [DOI] [PubMed] [Google Scholar]
- Winkler E. A. et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat Neurosci 18, 521–530 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klip A., Tsakiridis T., Marette A. & Ortiz P. A. Regulation of expression of glucose transporters by glucose: a review of studies in vivo and in cell culture. FASEB J 8, 43–53 (1994). [DOI] [PubMed] [Google Scholar]
- Wertheimer E., Sasson S., Cerasi E. & Ben-neriah Y. The ubiquitous glucose transporter GLUT-1 belongs to the glucose-regulated protein family of stress-inducible proteins. Proc Natl Acad Sci USA 88, 2525–2529 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson G. E., Sheu K.-F. R. & Blass J. P. Abnormalities of mitochondrial enzymes in ALzheimer disease. J Neural Transm 105, 855–870 (1998). [DOI] [PubMed] [Google Scholar]
- Bergbauer K. et al. Studies on fructose metabolism in cultured astroglial cells and control hepatocytes: lack of fructokinase activity and immunoreactivity in astrocytes. Dev Neurosci 18, 371–379 (1996). [DOI] [PubMed] [Google Scholar]
- Hertz L., Peng L. & Dienel G. A. Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J Cereb Blood Flow Metab 27, 219–249 (2007). [DOI] [PubMed] [Google Scholar]
- Azevedo F. C. et al. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol 513, 532–541 (2009). [DOI] [PubMed] [Google Scholar]
- Lebon V. et al. Astroglial contribution to brain energy metabolism in humans revealed by 13C nuclear magnetic resonance spectroscopy: elucidation of the dominant pathway for neurotransmitter glutamate repletion and measurement of astrocytic oxidative metabolism. J Neurosci 22, 1523–1153 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dringen R., Schmoll D., Cesar M. & Hamprecht B. Incorporation of radioactivity from [14C]lactate into the glycogen of cultured mouse astroglial cells. Evidence for gluconeogenesis in brain cells. Biol Chem Hoppe Seyler 374, 343–347 (1993). [DOI] [PubMed] [Google Scholar]
- Dringen R., Gebhardt R. & Hamprecht B. Glycogen in astrocytes: possible function as lactate supply for neighboring cells. Brain Res 623, 208–214 (1993). [DOI] [PubMed] [Google Scholar]
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 36 Suppl 1, S67–S74 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman O. J. et al. Metabolic dysfunction is restricted to the sciatic nerve in experimental diabetic neuropathy. Diabetes 65, 228–238 (2016). [DOI] [PubMed] [Google Scholar]
- Macauley S. L. et al. Hyperglycemia modulates extracellular amyloid-β concentrations and neuronal activity in vivo. J Clin Invest 125, 2463–2467, doi: 10.1172/JCI79742 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M. X. et al. The advanced glycation end product, Ne-(carboxymethyl)lysine, is a product of both lipid peroxidation and glycoxidation reactions. J Biol Chem 271, 9982–9986 (1996). [DOI] [PubMed] [Google Scholar]
- Kamalov M. et al. Physicochemical studies on the copper(II) binding by glycated collagen telopeptides. Org Biomol Chem 13, 3058–3063 (2015). [DOI] [PubMed] [Google Scholar]
- Sultana R. et al. Proteomics analysis of the Alzheimer’s disease hippocampal proteome. J Alzheimers Dis 11, 153–164 (2007). [DOI] [PubMed] [Google Scholar]
- Martinez A., Portero-Otin M., Pamplona R. & Ferrer I. Protein targets of oxidative damage in human neurodegenerative diseases with abnormal protein aggregates. Brain Pathol 20, 281–297 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele R. et al. Differential effect of APOE genotype on amyloid load and glucose metabolism in AD dementia. Neurol 80, 359–265 (2013). [DOI] [PubMed] [Google Scholar]
- Protas H. D. et al. Posterior cingulate glucose metabolism, hippocampal glucose metabolism, and hippocampal volume in cognitively normal, late-middle-aged persons at 3 levels of genetic risk for Alzheimer disease. JAMA Neurol 70, 320–325 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L. et al. Amyloid and metabolic positron emission tomography imaging of cognitively normal adults with Alzheimer’s parents. Neurobiol Aging 34, 22–34 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai K. et al. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci 21, 3017–3023 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti P. J., Sorg O. & Martin J.-L. In Astrocytes: Pharmacology and Function (ed Murphy S.) 243–265 (Academic Press, 1993). [Google Scholar]
- Brown A. M. & Ransom B. R. Astrocyte glycogen and brain energy metabolism. Glia 55, 1263–1271 (2007). [DOI] [PubMed] [Google Scholar]
- Huang L., Hillingsworth R. I. & Castellani R. Accumulation of high-molecular-weight amylose in Alzheimer’s disease brains. Glycobiol 14, 409–416 (2004). [DOI] [PubMed] [Google Scholar]
- Piert M., Koeppe R. A., Giordani B., Berent S. & Kuhl D. E. Diminished glucose transport and phosphorylation in Alzheimer’s disease determined by dynamic FDG-PET. J Nucl Med 37, 201–208 (1996). [PubMed] [Google Scholar]
- Venkatraman J., Aggarwal K. & Balaram P. Helical peptide models for protein glycation: proximity effects in catalysis of the Amadori rearrangement. Chem Biol 8, 611–625 (2001). [DOI] [PubMed] [Google Scholar]
- McPherson J. D., Shilton B. H. & Walton D. J. Role of fructose in glycation and cross-linking of proteins. Biochemistry 27, 1901–1907 (1988). [DOI] [PubMed] [Google Scholar]
- Medeiros D. M., Davidson J. & Jenkins J. A unified perspective on copper deficiency and cardiomyopathy. Exp Biol Med (Maywood) 203, 262–273 (1993). [DOI] [PubMed] [Google Scholar]
- Cooper G. J. S. Selective divalent copper chelation for the treatment of diabetes mellitus. Curr Med Chem 19, 2828–2860 (2012). [DOI] [PubMed] [Google Scholar]
- Medeiros D. M. & Wildman R. E. C. Newer findings on a unified perspective of copper restriction and cardiomyopathy. Exp Biol Med (Maywood) 215, 299–313 (1997). [DOI] [PubMed] [Google Scholar]
- Raichle M. E. & Gusnard D. A. Appraising the brain’s energy budget. Proc Natl Acad Sci USA 99, 10237–10239 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brings S. et al. Diabetes-induced alterations in tissue collagen and carboxymethyllysine in rat kidneys: association with increased collagen-degrading proteinases and amelioration by Cu(II)-selective chelation. Biochim Biophys Acta Mol Basis Dis 1852, 1610–1618 (2015). [DOI] [PubMed] [Google Scholar]
- Kovacs G. G. et al. Non-Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community-based autopsy series. Acta Neuropathol 126, 365–384 (2013). [DOI] [PubMed] [Google Scholar]
- Knopman D. et al. Neuropathology of cognitively normal elderly. J Neuropathol Exp Neurol 62, 1087–1095 (2003). [DOI] [PubMed] [Google Scholar]
- Wharton S. B. et al. Epidemiological pathology of Tau in the ageing brain: application of staging for neuropil threads (BrainNet Europe protocol) to the MRC cognitive function and ageing brain study. Acta Neuropathol Commun 4, 11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crary J. et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 128, 755–766 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson P. T. et al. Brains with medial temporal lobe neurofibrillary tangles but no neuritic amyloid plaques are a diagnostic dilemma but may have pathogenetic aspects distinct from Alzheimer disease. J Neuropathol Exp Neurol 68, 774–784 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider J. A., Aggarwal N. T., Barnes L., Boyle P. & Bennett D. A. The neuropathology of older persons with and without dementia from community versus clinic cohorts. J Alzheimers Dis 18, 691–701 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros D. M. & Jennings D. B. Role of copper in mitochondrial biogenesis via interaction with ATP synthase and cytochrome c oxidase. J Bioenerg Biomemb 34, 389–395 (2002). [DOI] [PubMed] [Google Scholar]
- Waldvogel H. J. et al. The collection and processing of human brain tissue for research. Cell Tissue Bank 9, 169–179 (2008). [DOI] [PubMed] [Google Scholar]
- Skoog I. Detection of preclinical Alzheimer’s disease. N Engl J Med 343, 502–503 (2000). [DOI] [PubMed] [Google Scholar]
- Vardy E. R. et al. Plasma angiotensin-converting enzyme in Alzheimer’s disease. J Alzheimers Dis 16, 609–618 (2009). [DOI] [PubMed] [Google Scholar]
- McKhann G. et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurol 34, 939–944 (1984). [DOI] [PubMed] [Google Scholar]
- Vardy E. R. et al. Increased circulating insulin-like growth factor-1 in late-onset Alzheimer’s disease. J Alzheimers Dis 12, 285–290 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.