

Abstract
Mutations of the CLN3 gene lead to juvenile neuronal ceroid lipofuscinosis (JNCL), an autosomal recessive lysosomal storage disorder that causes progressive neurodegeneration in children and adolescents. There is evidence of immune system involvement in pathology that has been only minimally investigated. We characterized bone marrow stem cell-derived antigen presenting cells (APCs), peritoneal macrophages, and leukocytes from spleen and blood, harvested from the Cln3−/− mouse model of JNCL. We detected dramatically elevated CD11c surface levels and increased total CD11c protein in Cln3−/− cell samples compared to wild type. This phenotype was specific to APCs and also to a loss of CLN3, as surface levels did not differ from wild type in other leukocyte subtypes nor in cells from two other NCL mouse models. Subcellularly, CD11c was localized to lipid rafts, indicating that perturbation of surface levels is attributable to derangement of raft dynamics, which has previously been shown in Cln3 mutant cells. Interrogation of APC function revealed that Cln3−/− cells have increased adhesiveness to CD11c ligands as well as an abnormal secretory pattern that closely mimics what has been previously reported for Cln3 mutant microglia. Our results show that CLN3 deficiency alters APCs, which can be a major contributor to the autoimmune response in JNCL.
Keywords: Batten disease, JNCL, CLN3, CD11c, integrin
1. Introduction
Juvenile neuronal ceroid lipofuscinosis (JNCL), or Batten disease, is an autosomal recessive lysosomal storage disorder that causes progressive neurodegeneration in children and adolescents. It is caused by mutation of the CLN3 gene, which encodes a protein product of the same name (CLN3). Patients present around age 5, typically with visual deterioration, followed by blindness, behavioral, cognitive, and psychiatric disturbances, progressive motor dysfunction, seizures, and eventually death in the third decade of life (1). The signs and symptoms of disease come from the extensive loss of neurons seen in multiple brain regions, the magnitude of which correlates with widespread proliferation and activation of glial cells (2–4). The pathogenesis of JNCL is not well-characterized, however, nor is the role of the CLN3 protein. While CLN3 has been extensively implicated in intracellular trafficking, potentially involving the endosomal/lysosomal system and Golgi transport, its function remains incompletely understood [reviewed by Cotman and Staropoli (2012)] (5).
There is evidence of an autoimmune component in JNCL. Patients and Cln3−/− mice produce IgG autoantibodies reactive to brain proteins (6–9). While there is no direct evidence that autoantibodies are harmful in JNCL, IgG deposition can be appreciated in multiple brain regions in human autopsy and genetic mouse model specimens (9). Moreover, administration of immunosuppressant drugs ameliorates the characteristic motor phenotype of Cln3−/− mice and reduces glial activation (10). These discoveries have led to a clinical trial of mycophenolate mofetil as a treatment for JNCL, which is currently in progress.
Immune cell abnormalities at the cellular level have been only minimally investigated; although several lines of evidence suggest there is potential for cellular dysfunction. Microglia are the resident mononuclear phagocytes of the central nervous system (CNS). Cln3 mutant microglia, which show abnormal secretion of pro-inflammatory cytokines, appear to contribute directly to neuronal loss (11, 12). Macrophages (Mφ) have a secretory defect that has not been well-defined, and they accumulate particularly large amounts of lysosomal storage material (11–13). Also suggestive is the reliance of Mφ and the other primary antigen presenting cell (APC), the dendritic cell (DC), rely heavily on processes in which CLN3 and its interacting partners and associated regulatory proteins are implicated. Shwachman-Bodian-Diamond Syndrome protein (SBDS), myosin IIb, and Cdc42 variably regulate endocytosis of extracellular antigens and subsequent trafficking through the endosomal/lysosomal system, polarization, migration, and chemotaxis, and are needed for normal hematopoiesis (14–27). Of particular interest is the possibility of leukocyte dysfunction in JNCL via perturbation of surface protein homeostasis; surface protein abnormalities have been described in CLN3-deficient cells, including a reduction of lipid raft membrane microdomain associated proteins, inappropriate Na+/K+ ATPase subunit composition, and decreased surface expression of rho GTPase activating protein ARHGAP21 (24, 28–30).
Immune cells rely on the proper repertoire of surface proteins to mediate interactions with other cells and the environment, with altered levels affecting leukocyte function in a variety of autoimmune diseases (31–38). The β2-integrins are a leukocyte specific family of adhesion molecules comprised of a common CD18 beta subunit that binds to one of four alpha subunits—CD11a, CD11b, CD11c, and CD11d. While all heteromers function primarily in adhesion-based processes, CD11b and CD11c have additional roles in phagocytosis and inflammatory signaling [reviewed by Mazzone and Ricevuti (1995) and Tan (2012)] (39, 40). CD11c is of particular interest as an overall increase in CD11c is observed in chronic inflammation and in autoimmune diseases associated with autoantibody production (i.e. multiple sclerosis and rheumatoid arthritis) (31, 41–47). It serves as the main dendritic cell marker and is also found on microglia, subsets of Mφ and neutrophils, and on limited numbers of natural killer cells and lymphocytes (46, 48–51).
With this in mind, we hypothesized that CLN3 deficiency might be associated with an aberrant phenotype in APCs, potentially through alteration of cell surface proteins and subsequent perturbation of downstream function. We characterized surface proteins as well as function for APCs from wild type mice and the Cln3−/− mouse model of JNCL, which recapitulates several neuropathological and behavioral phenotypes of human JNCL patients (52). We looked at DCs and Mφ derived from culture of bone marrow stem cells—BM-DCs and BM-Mφ, respectively—as well as elicited peritoneal Mφ and a wider variety of leukocytes harvested from blood and spleen. We also set out to better characterize potential secretory defects in APCs to the extent this has previously been done in microglia. We believe that a better understanding of the mechanisms underlying potential immune dysfunction in JNCL will further the development of more targeted immunotherapies and also help elucidate further the function of CLN3.
2. Materials and Methods
2.1. Mice
All experiments used the Cln3−/− (Cln3Δex1-6) mouse model of juvenile Batten disease inbred on the C57BL/6J background (52). Ppt1−/−and Tpp1−/− mice, mouse models of infantile and late infantile Batten disease, respectively, were assessed for CD11c in the comparative NCL mouse model study only (53, 54). Control animals were the C57BL/6J strain. Mice were male and aged 70 to 120 days as younger mice lack a fully developed immune system that includes impaired function of both lymphocytes and APCs [see Adkins, Leclerc et al. (2004) for review] (55). Injections and euthanasia procedures were in accordance with the NIH Guide for the Care and Use of Laboratory Animals (56) and approved by the institutional animal care and use committee for Sanford Research, Sioux Falls, South Dakota.
2.2. Bone marrow cultured cells
Bone marrow stem cells were cultured as previously described in RPMI 1640 medium with 2.05 mM L-glutamine supplemented with 1 μg/ml amphotericin B, 50 IU/ml penicillin, 50 μg/ml streptomycin, 10% heat-inactivated fetal bovine serum (FBS), and 20 ng/ml granulocyte macrophage-colony stimulating factor (GM-CSF) (Life Technologies, Carlsbad, CA) (57). For cells cultured beyond 8 days, GM-CSF was reduced to 10 ng/ml. Where indicated, cells were stimulated on day 8 of culture with addition of toll-like receptor (TLR) ligands lipopolysaccharide (LPS) (100 ng/ml) (Sigma-Aldrich, St. Louis, MO) or poly I:C (PIC) (50 μg/ml) (Sigma-Aldrich). Cells were harvested on days 10 to 12 of culture by fraction — non-adherent and lightly adherent cells comprised the dendritic cell fraction, while firmly adhered cells dislodged with a cell scraper comprised the Mφ fraction. Mixed bone marrow cultured cell (BMCC) populations included both adherent and non-adherent fractions. Previous research has demonstrated the fractions are distinct populations, although phenotypes overlap somewhat (57–59). Our own experiments consistently showed nearly 100% CD11b positivity and slightly less CD11c positivity in all bone marrow populations (some data not shown). A previous study indicates that few neutrophils should be present (57), consistent with our observation based on morphology.
2.3. Elicited peritoneal Mφ
Peritoneal Mφ were elicited with Bio-Gel P100 and harvested as previously described (60). Cells were seeded into 6-well tissue culture plates, 2.5 × 106 cells per well, with some activated by priming with 10 ng/ml interferon-γ (IFN-γ) (Novus Biologicals, Littleton, CO) for 6 hours followed by addition of 100 ng/ml LPS or 50 μg/ml PIC (LIγ=IFN-γ + LPS; PIγ=IFN-γ + PIC). Cells were harvested 48 to 96 hours later by first vacuuming off any non-adherent cells (including any residual, non-apoptosed B1 lymphocytes) and then detaching adherent Mφ by incubating in 6 mM ethylenediaminetetraacetic acid (EDTA) in phosphate buffered saline (PBS) at 37° C for 10 minutes.
2.4. Splenocytes
Mice were euthanized and spleens excised and macerated with a 5 ml syringe plunger in 10-cm dishes with PBS. Cell suspensions were transferred to 50 ml conical tubes, avoiding larger debris, and washed with PBS. Red blood cells were lysed by resuspending the cell pellet in 9 ml of molecular grade water. After 25 seconds, 1 ml of 10× PBS was added to the tube followed by 30 ml of 1× PBS. The suspension was filtered through a 40 μm cell strainer and then washed again with PBS.
2.5. Peripheral blood cells
Syringes (1 ml) were prepared by flushing with heparinized saline (1000 U/ml), leaving a residual of 10 μl. Mice were euthanized, the chest cavity opened, and blood withdrawn from the right ventricle. Samples were refrigerated at 4° C for 2 to 4 hours followed by pelleting of cells by centrifuging for 10 minutes at 200 x g. Red blood cells were lysed and the cells filtered and washed as described above.
2.6. Gene expression
Day 10 and 11 wild type or Cln3−/− mixed BMCCs, with or without activation by LPS for 24 hours or 48 hours, were harvested as described above, washed three times with PBS, and the cell pellet snap frozen on dry ice. Total RNA was isolated with the Maxwell 16 LEV simplyRNA Cells Kit (Promega, Madison, WI) on a Maxwell 16 Instrument (AS2000) (Promega), according to the manufacturer’s instructions. Quantification and assessment of RNA integrity was performed on an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Relative integrity numbers (RIN) for all samples were >7.0. Reverse transcription to cDNA and real-time quantitative PCR (qPCR) were performed as previously described (61). TaqMan hydrolysis assays (Life Technologies) included the following: Cln3 (Mm00487021_m1), Itgam (Mm00434455_m1 ), Itgal (Mm00801807_m1), Itgax (Mm00498698_m1 ), Itgb2(Mm00434513_m1), B2m (Mm00437762_m1), Gapdh (Mm99999915_g1), Gusb (Mm00446962_g1) and Hgprt (Mm01318741_g1). The latter four served as housekeeping genes. There were at least 2 biological replicates per group, with four technical replicates per biological replicate. Cln3 expression was assessed in wild type cells only.
Light Cycler 480 software version 1.5 (Roche Diagnostics, Indianapolis, IN) was used to analyze raw data with the second derivative method (Cq). Cq values for all samples had a standard deviation of < 0.11. No template controls indicated no nucleic acid contamination. All assays had PCR efficiencies of 0.90 to 0.99, determined with calibration curves and calculated with the following equation: PCR efficiency = 10−1/slope – 1. Expression levels were normalized to housekeeping genes before calculating relative expression of target genes with REST-MCS software. Cln3 expression was calculated as relative expression levels compared to 24 hour unstimulated samples. Statistical significance was determined with 2-way ANOVA to look at differences by day of culture and length of time of LPS stimulation. For assessment of integrin subunit gene expression, relative expression levels were calculated for each time point as compared to unstimulated wild type samples. Two-way ANOVA assessed factors genotype and stimulation (unstimulated vs. LPS stimulated). Post-hoc pairwise comparisons for all experiments are reported for Fisher’s least squares difference (LSD). Statistics were generated with SYSTAT 13 v13.1 (SYSTAT Software, Inc. San Jose, CA). Graphs were made with GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA).
2.7. Fluorochrome-conjugated antibody staining
Cell populations were stained with fluorochrome-conjugated primary antibodies to proteins of interest in PBS/0.5% BSA, followed by live/dead staining with Fixable Viability Dye eFluor 780 (eBioscience). After washing, cells were read immediately on the flow cytometer or fixed with IC Fixation Buffer (eBioscience) according to the manufacturer’s instructions before reading. Cells were assessed with a Guava easyCyte 8HT Flow Cytometer (EMD-Millipore, Billerica, MA) or a BD Accuri C6 (BD Biosciences, San Jose, CA). Data were analyzed with FlowJo v7.6.5 (Tree Star, Inc., Ashland, OR) using compensation matrices generated from single stain controls and/or compensation beads (BD CompBead anti-rat/hamster Ig, κ, BD Biosciences). Outcome measures included the percent of cells in the sample gating positively for specific markers (i.e. %CD19) and the intensity of fluorescence signal of positive cells (median fluorescence intensity, MFI). Statistics were calculated with SYSTAT 13 v13.1 (SYSTAT Software, Inc). The Student’s t-test was used to compare means. Dunn-Sidak corrected P values were used to reduce the risk of type I errors where appropriate (62). Two-way analysis of variance (ANOVA) was used to look at effects of several factors and their interaction. With multiple ANOVAs within the same experiment, uncorrected P values were reported but the alpha level was adjusted according to the Bonferroni method (alpha level = 0.05/# of tests) (63). We performed post-hoc testing with Fisher’s LSD and maintained adjusted alpha levels when determining significance.
Antibodies used included the following: phycoerythrin (PE)-conjugated Armenian hamster anti-mouse CD11c (clone N417, #12-0114, eBioscience, San Diego, CA), Alexa Fluor 488-conjugated Armenian hamster anti-mouse CD11c (clone N417, #53-0114, eBioscience), allophycocyanin (APhC)-conjugated Armenian hamster anti-mouse CD11c (clone N417, #17-0114, eBioscience), APhC-conjugated rat IgG2a K anti-mouse CD11a (clone M17/4, #17-0111, eBioscience), Alexa Fluor 488-conjugated rat IgG2b K anti-mouse CD11b (clone M1/70, # RM2820, Life Technologies), fluorescein isothiocyanate (FITC)-conjugated rat IgG2a K anti-mouse CD18 (clone M18/2, #11-0181, eBioscience), eFluor 660-conjugated rat IgG2a K anti-mouse F4/80 (clone BM8, #50-0141, eBioscience), FITC-conjugated rat IgG2a K anti-mouse CD19 (clone eBio1D3, #11-0193, eBioscience), FITC-conjugated rat IgG2a K anti-mouse CD4 (clone GK1.5, #11-0041, eBioscience), peridinin-chlorophyll protein (PerCP)-Cy5.5-conjugated rat IgG2a K anti-mouse CD8a (clone 53-6.7, #45-0081, eBioscience), APhC-conjugated Armenian hamster anti-mouse CD3 (clone 145-2C11, #17-0031, eBioscience), eFluor 660-conjugated rat IgG2a K anti-mouse B220/CD45R (clone RA3-6B2, #50-0452, eBioscience), PerCP-Cy5.5-conjugated rat IgG2a K anti-mouse CD14 (clone Sa2-8, #45-0141, eBioscience), and FITC-conjugated rat anti-mouse MHCII (I-A/I-E) (clone M5/114.15.2, #11-5321, eBioscience).
2.8. CD11c comparative NCL mouse model study
Two experiments were conducted. In the first, day 10 unstimulated mixed BMCCs cultivated from Cln3−/− and Ppt1−/− mouse models and wild type mice were compared for mean CD11c MFI with one-way ANOVA, with post-hoc Fisher’s LSD for pairwise comparisons. In the second, day 10 unstimulated mixed BMCCs from the Tpp1−/− mouse model and wild type mice were compared for mean CD11c MFI using a Student’s t-test. The graph represents both experiments, showing fold expression relative to wild type, with wild type set at 1, calculated with the following formula (mean CD11c MFI mouse model/ mean CD11c MFI wild type).
2.9. Fluorescence-based immunocytochemistry
Cells from BM-Mφ, BM-DCs, and peritoneal Mφ, were stimulated as described above with LIγ or LPS, respectively, and incubated on coverslips at 37° C for 24 to 36 hours. Cells were fixed to coverslips with 4% paraformaldehyde in Dulbecco’s Phosphate Buffered Saline (DPBS) with calcium and magnesium for 10 minutes. Prior to staining, coverslips were blocked with blocking buffer (5% goat serum, 1% BSA, and .01% sodium azide in PBS) for 1 hour at room temperature. Colocalization studies, with the exception of lipid raft labeling (i.e. cholera toxin B and anti-caveolin-1 antibody) required intracellular staining such that blocking buffer was supplemented with 0.5% Triton-X 100.
Primary antibodies were diluted to appropriate concentrations in blocking buffer, 1:200 for all except CD11b (1:500): purified Armenian hamster anti-mouse CD11c, clone N417, 14-0114 (eBioscience), rat anti-mouse CD11b, clone M1/70, ab8878 (AbCam, Cambridge, MA), mouse anti-mouse GM-130, clone 25, BDB610822 (BD Biosciences), rabbit anti-mouse EEA-1, clone C45B10, 3288S (Cell Signaling, Danvers, MA), rabbit anti-mouse Rab11, clone D4F5, 5589S (Cell Signaling), rabbit anti-mouse EPS-15, clone D3K8R, 12460S (Cell Signaling), mouse anti-mouse caveolin-1, clone 125, 610406 (BD Biosciences), and FITC conjugated Cholera toxin B (CTB), C1655 (Sigma-Aldrich). Coverslips were incubated with the primary antibodies overnight at 4° C. Coverslips then were washed three times with PBS and incubated for 1 to 2 hours at room temperature with fluorophore-conjugated secondary antibodies (2 μg/ ml, with 0.5% Triton-X 100 for intracellular staining), which included DyLight 488-conjugated goat anti-Armenian hamster IgG, 405503 (Biolegend, San Diego, CA), AlexaFluor 594-conjugated chicken anti-rat IgG, A21471 (Life technologies), AlexaFluor 647-labeled goat anti-mouse IgG, A21235 (Life technologies), AlexaFluor 488-conjugated donkey anti-rabbit IgG, A21206 (Life technologies), and DyLight™ 649-labeled goat anti-Armenian hamster IgG, 405505 (Biolegend).
Coverslips were mounted onto slides with 10% Mowiol mounting medium [Mowiol 4-88 (Sigma-Aldrich), glycerol, water, 0.2M Tris HCl] and allowed to dry overnight at room temperature before storage at 4°C. Control coverslips contained wild type or Cln3−/− cells pooled from multiple individuals and were incubated with secondary antibody only. Coverslips were imaged with the Nikon A1si confocal microscope (Nikon Inc., Melville, NY). The plane capturing the largest area of cell surface was selected for analysis of surface staining, while multiple planes (i.e. z-stacks) were visualized for intracellular staining. All z-stacks were captured at 0.5 μm depth intervals. NIS-Elements AR software (Nikon Inc.) was used to characterize surface staining and colocalization and prepare images.
Surface staining of integrin subunits was assessed by auto-selecting ROIs corresponding to whole cells for all images associated with each coverslip. For control coverslips, cells were auto-selected where possible or random ROIs were drawn manually. The ROI mean intensities were averaged for each image. The average ROIs for all images were then averaged per coverslip for final values. Final values were compared between wild type and Cln3−/− cells by stimulation condition and surface protein with the Student’s t-test. A one-tailed t-test was used to assess CD11c surface staining as previous data indicated CD11c would be higher in Cln3−/− cells. Two-tailed t-tests were used for CD11b and CD18. Interactions between genotype and stimulation (i.e. LPS or LIγ) could not be assessed as optimal imaging required that laser settings be changed between stimulated and unstimulated samples.
For colocalization studies, the colocalization tool was used to generate Pearson’s correlation coefficients, which are insensitive to differences in channel intensity (64). Values can range from −1.0 to 1.0, with ≥ 0.5 indicating significant colocalization (65). The average of individuals produced the average Pearson’s R by group. Where Pearson’s R for both genotypes indicated significant colocalization, differences between Cln3−/− and wild type cells were calculated with a Student’s t-test for mean correlation coefficients.
Statistics were generated with SYSTAT® 13 v13.1 (SYSTAT Software, Inc.) with additional graphing using GraphPad Prism 5 (GraphPad Software, Inc.). Image formatting was performed in Adobe Photoshop CC (Adobe Systems Incorporated).
2.10. Western blotting
Unstimulated and LPS-stimulated day 12 BMCCs were harvested by fraction. Cells were resuspended in 150 μl denaturing cell lysis buffer [50 mM Tris-HCl, 250 mM NaCl, 5 mM EDTA, 10 mM NaF, 0.5 mM Na3VO4, 50 μM ZnCL2, 0.1% SDS, 1mM DTT, HALT protease inhibitor single-use cocktail (Thermo Fisher Scientific), Phosphatase Inhibitor Cocktail A and Phosphatase Inhibitor Cocktail B (Santa Cruz Biotechnology, Dallas, TX)], vortexed for 20 seconds in 1 second pulses and then placed on dry ice to freeze for increased protein yield. After freezing, tubes were thawed and immediately placed on ice to rest for 30 minutes. Tubes then were centrifuged at 4°C for 10 minutes at 14,000 x g and the supernatant stored at −80°C until needed. Protein concentrations were quantified using the Pearce 660nm Protein Assay according to the manufacturer’s instructions (Thermo Fisher Scientific), and read on a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA).
Total protein lysate was run on 10% Mini-PROTEAN TGX Precast Gels (Bio-Rad), 55 μg protein/well for 10-well gels (LPS-stimulated samples) and 35 μg protein/well for 12-well gels (unstimulated samples), alongside the Fisher BioReagents EZ-Run Prestained Rec Protein Ladder (Thermo Fisher Scientific). Proteins were transferred to Immobilon-P PVDF transfer membrane (EMD-Millipore), blocked for 1 hour in 5% non-fat dry milk in Tris-buffered saline supplemented with 0.1% Tween-20 (TBST), and then incubated overnight at 4°C in primary antibody against CD11c (rat IgG2B CD11c Mab clone 435421, #MAB6950, R&D Systems, Minneapolis, MN) 2 μg/ml, on a rotating platform. After washing with TBST, membranes were incubated with secondary antibody for 1.5 hours (ECL horseradish peroxidase-conjugated sheep anti-rat IgG, #NA935, GE Healthcare, Piscataway, NY) at 1:5000 dilution in 5% milk. After washing, bands were developed with Luminata Forte Western HRP Substrate (EMD-Millipore) for 4 minutes. Imaging was performed on a UVP Biospectrum 800 (UVP, LLC, Upland, CA).
After imaging, antibody was removed with Restore PLUS Western Blot Stripping Buffer (Thermo Fisher Scientific). Membranes then were probed for GM130, with processing as described above. Anti-GM130 primary antibody (mouse IgG1 GM130 clone 25, #BDB610822, BD Biosciences) was diluted 1:200, and anti-mouse secondary diluted 1:2000 (ECL horseradish peroxidase conjugated sheep anti-mouse IgG, #NA931, GE Healthcare). Immunoreactive bands were quantified with densitometry in Image J (NIH, Bethesda, MD). Values used for analysis were derived by dividing the integrated density of the band for the protein of interest by the integrated density of the band for the loading control for each lane (integrated density protein of interest/ integrated density loading control) and then averaging this value within each genotype and treatment group. Samples were compared within gels only. Graphs were created with GraphPad Prism 5 (GraphPad Software, Inc).
2.11. Adhesion assays
Wild type and Cln3−/− elicited peritoneal Mφ and day 10 BMCCs by fraction were harvested as described above. Black, opaque 96-well polystyrene plates were prepared by incubating overnight with 100 μl of ligand per well — 100 μg/ml fibrinogen in saline, 0.2 μg/ml poly-D-lysine (PDL) in water, or 1% BSA in PBS. After incubation, wells were washed twice with PBS and then incubated for 1 hour with PBS/1% BSA to block nonspecific protein binding to the polystyrene surface. A volume containing 30,000 cells in RPMI supplemented with 1% BSA was added to each well. Four to five technical replicates were plated per individual for each ligand. Four to five negative wells not seeded with cells were included for each ligand. Plates were incubated for 30 minutes at 37°C. Hoechst 33342 (BD Pharmingen, San Jose, CA) in PBS (1μg/ml final concentration) was added to all wells and plates incubated for 10 minutes at 37°C. The number of cells per well was assessed by reading the intensity of Hoechst staining by well on a SpectraMax M5 plate reader (Molecular Devices) (9 reads; excitation: 350 nm, emission: 461 nm). Results were calculated for each ligand by subtracting the average of negative wells from the values of the experimental wells. The net experimental well fluorescence values were averaged for all technical replicates by individual to produce the total adhesion values. Statistics were generated with SYSTAT 13 v13.1 (SYSTAT Software, Inc.), with graphs created with GraphPad Prism 5 (GraphPad Software, Inc). The Student’s t-test was used to compare mean total adhesion by genotype. P values were Dunn-Sidak adjusted to control for type I errors (62).
2.12. Cytokine/chemokine secretion
Day 10 BMCCs were plated in triplicate per individual (n = 5, wild type; n = 5, Cln3−/−), 1 × 105 cells per well, in two 96-well v-bottom plates in RPMI with or without LPS (final concentration 100 ng/ml). Plates were incubated for 24 hours at 37°C. The medium from each triplicate was pooled in 1.5 ml tubes and stored at −80°C until needed. Cytokines were assessed using the MILLIPLEX MAP Mouse Cytokine/ Chemokine Luminex kit (EMD-Millipore; kit# MPXMCYTO-70K) according to the manufacturer’s instructions. Samples were diluted with RPMI (1:3 for unstimulated cells, 1:5 for LPS-stimulated cells) for analytes at concentrations above the maximum detectable level. Plates were read on Luminex 100/200 System (Luminex Corporation, Austin, TX). Analyte concentrations were derived with xPONENT software (Luminex Corporation), which compares the average intensity of secondary antibody per bead by well for technical replicates to the average intensity of secondary antibody per bead by well for the prepared standard. Where analytes were undetectable, the value for that sample was set to zero. Data were analyzed with SYSTAT 13 v13.1 (SYSTAT Software, Inc.). Analytes were assessed with a Student’s t-test. Multivariate testing was not possible due to multi-colinearity among dependent variables. Genotypic differences in cytokine secretion were calculated by cell fraction and LPS stimulation (groups: BM-Mφ, no LPS; BM-DCs, no LPS; BM-Mφ, LPS; BM-DCs, LPS). Within each group, P values were Dunn-Sidak adjusted, with adjusted values requiring an alpha level of 0.017, to account for each sample being included in analyses from 3 groups (62).
3. Results
3.1. Cln3 gene expression in bone marrow cultured cells (BMCCs)
We first investigated expression of Cln3 in wild type cells to confirm that BMCCs are a relevant model for studying the effects of absent or mutated CLN3. Cln3 transcript levels were assessed with qPCR in unstimulated, mixed BMCCs or after stimulation with lipopolysaccharide (LPS) for 24 or 48 hours. Cln3 was expressed by cells in all groups. Main effects were significant for both day [F(1,4) = 30.505, P = .005] and LPS stimulation [F(1,4) = 31.455, P = .005]. Graphing of the interaction effect [F(1,4) = 31.761, P = .005] revealed that Cln3 expression was significantly increased 24 hours after LPS activation compared to all other groups, which did not differ from each other (Fig. 1).
Fig. 1. Relative gene expression of Cln3 in wild type mixed bone marrow cultured cells with or without stimulation with lipopolysaccharide.

Wild type mixed bone marrow cultured cells (BMCCs) were harvested on days 10 and 11 of culture, with or without 24 or 48 hours of stimulation with lipopolysaccharide (LPS), respectively. Cln3 transcript levels were assessed with real-time quantitative PCR and normalized to housekeeping genes. Gene expression is reported relative to day 10 unstimulated samples. LPS stimulation for 24 h significantly increased Cln3 expression. P values are derived from post-hoc pairwise comparisons using Fisher’s LSD. Columns and bars represent mean ± S.E.M. (n=2, all groups).
3.2. Characterization of surface proteins on APCs
We characterized surface levels of the β2-integrin alpha subunits (CD11a, CD11b, and CD11c) and the common beta subunit (CD18) in wild type and Cln3−/− BM-DCs, BM-Mφ, and peritoneal Mφ, with or without prior stimulation with toll-like receptor (TLR) ligands. Surface proteins first were assessed with flow cytometry. Preliminary testing indicated that endogenous fluorescence was not a confounding factor at the voltage used for fluorescence-conjugated antibody detection (data not shown). Dramatically increased CD11c surface levels and a higher percentage of CD11c+ cells were detected in Cln3−/− samples compared to wild type samples for all cell types, (Fig. 2, supplementary Table 1). Stimulation with TLR ligands decreased CD11c surface expression in both wild type and Cln3−/− cells; however, levels were reduced to very low to undetectable levels in wild type cells, while LPS-stimulated Cln3−/− cells maintained surface levels greater or equivalent to those on unstimulated wild type cells (Fig. 2A, C and E, supplementary Fig. 1, supplementary Table 1). In turn, fewer wild type cells were CD11c positive, while the rate of CD11c positive Cln3−/− cells remained unchanged (Fig. 2B and F, supplementary Table 1). Follow-up studies with fluorescence-based immunocytochemistry confirmed that Cln3−/− BM-DCs, BM-Mφ, and peritoneal Mφ have markedly elevated surface expression of CD11c (Fig. 3, supplementary Fig. 2 and 3). Assessment of control coverslips stained with secondary antibody only indicated that neither non-specific staining nor endogenous fluorescence was a confounding factor in analyses (Fig. 3A).
Fig. 2. Flow cytometric analysis of CD11c surface expression in wild type and Cln3−/− bone marrow macrophage, bone marrow dendritic cell, and elicited peritoneal macrophage samples, unstimulated or stimulated with TLR ligands.

Cln3−/− and wild type bone marrow dendritic cells (BM-DCs), bone marrow macrophages (BM-Mφ), and peritoneal macrophages (Mφ) staining negatively for Fixable Viability Dye eFluor 780 were assessed for expression of CD11c with flow cytometry, with or without 24 hours of stimulation by toll-like receptor (TLR) ligands lipopolysaccharide (LPS) or poly I:C (PIC). Mφ were pre-primed with IFN-γ (LIγ or Piγ). (A, C, E) CD11c surface level was characterized by the median fluorescence intensity (MFI). (B, D, F) The percentage of CD11c positive cells was characterized by positive gating compared to isotype controls. Statistical significance was determined by 2-way ANOVA with genotype and stimulation as factors, with P values shown derived from post-hoc pairwise comparisons using Fisher’s LSD. Columns and bars represent mean ± S.E.M. (n=3, all groups for peritoneal Mφ; n=5, all groups for BM-DC and BM-Mφ except PIC-stimulated Cln3−/− BM-Mφ, n=4).
Fig. 3. Fluorescence-based immunocytochemical analysis of CD11c surface expression on Cln3−/− and wild type bone marrow macrophages (BM-Mφ), bone marrow dendritic cells (BM-DCs), and peritoneal macrophages (Mφ), with or without 24 hours of stimulation by lipopolysaccharide (LPS).

To detect surface expression cells were immunofluorescently labeled for CD11c and CD11b without membrane permeabilization and imaged with confocal microscopy using a 60× objective. Mφ were pre-primed with IFN-γ (LIγ or Piγ). Control coverslips were stained with secondary antibody (2°) only. The average mean fluorescence signal intensity for individual cells was compared by genotype, with statistical significance determined with the one-tailed Student’s t test and Dunn-Sidak adjusted P values shown (A). Columns and bars represent mean ± S.E.M. (n=3 for unstimulated wild type and Cln3−/− BM-Mφ and BM-DCs; n=2 for LPS-stimulated wild type and Cln3−/− BM-Mφ and BM-DCs and all Mφ groups). Representative confocal images show surface expression of CD11c and CD11b on wild type and Cln3−/− BM-Mφ, with and without LPS stimulation (B). Merged images compare distribution of CD11c and CD11b on the cell surface. Scale bar = 50 microns.
To determine if the CD11c phenotype was specific to the Cln3−/− JNCL mouse model, levels of CD11c on unstimulated mixed BMCCs from two additional NCL mouse models were assessed with flow cytometry. CD11c surface expression, quantified as median fluorescence intensity (MFI), did not significantly differ from wild type samples when compared to the infantile NCL model, Ppt1−/− mouse [mean(SD): Ppt1−/−, 433.30(34.05); wild type, 393.00(23.27)]; or the late infantile NCL model, Tpp1−/−, mouse [mean(SD): Tpp1, 74.13(1.012); wild type, 71.65(9.405)]. Graphing shows relative CD11c surface expression normalized to wild type with fold expression of Cln3−/− samples (Fig. 4). As the phenotype does not extend to other NCL mouse models, these results indicate that increased CD11c found on the surface of Cln3−/− cells is not a general feature of NCL disease and likely is more specifically related to the loss of CLN3 function.
Fig. 4. Comparative study of CD11c surface expression across NCL mouse models assessed with flow cytometry.
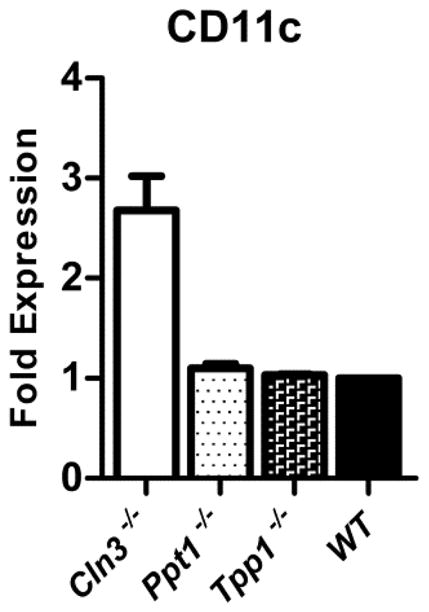
Mixed bone marrow cultured cells from wild type mice and three mouse models of neuronal ceroid lipofuscinoses (NCLs) staining negatively for Fixable Viability Dye eFluor 780 were assessed for CD11c surface expression as determined by median fluorescence intensity (MFI) with flow cytometry. Models identified are infantile NCL (Ppt1−/−, n=4), late infantile NCL (Tpp1−/−, n=3), and juvenile NCL (Cln3−/−, n=9). Results are shown as the ratio of CD11c MFI for each mouse model relative to wild type (n=2), set at 1. Columns and bars represent mean ± S.E.M.
We also discovered a phenotype for CD11b. Surface levels were reduced on Cln3−/− BM-DCs, BM-Mφ, and peritoneal Mφ, compared to wild type cells as assessed with flow cytometry (supplementary Fig. 4, supplementary Table 1). Genotypic differences, however, were more subtle than those reported for CD11c. Fluorescence-based immunocytochemical staining showed reduced surface CD11b on Cln3−/− cells only in LPS-stimulated samples (Fig. 3B, supplementary Fig. 2, 3 and 5). Microscopy might not have been sensitive enough to detect the subtle differences in surface levels on unstimulated samples.
No consistent effects of genotype or any interaction effects were detected for surface levels of CD11a, CD18, or other proteins (F4/80, CD86, CD14, CD40, MHCI, CD43, GR-1, MMR) (data not shown). Notably, we report no significant genotypic differences in MHCII surface levels [LPS-stimulated mixed BMCCs, mean(SD): wild type, 290.250(36.918), Cln3−/−, 318.400(17.126)].
3.3. Characterization of immune cells in spleen and blood
We next investigated whether the CD11c and CD11b phenotypes extended to in vivo APCs harvested from blood and spleen. CD11c was increased on Cln3−/− mononuclear phagocytes — CD14+ monocytes (CD14+ non-granulocytes) in the blood and F4/80+ Mφ in the spleen (F4/80+ non-granulocytes) (Table 1, supplementary Fig. 6A, D). In spleen, CD11c on F4/80+ Mφ segregated into high and low expressing populations, with genotypic differences detected in the CD11clow population only (Table 1). No genotypic differences in surface levels of integrins were detected on lymphocytes, neutrophils (GR-1hi non-lymphocytes with high side scatter), or splenic DCs (data not shown). We did not specifically identify DC subtypes in blood as most CD11c+ cells are pDCs, DC precursors, or monocytes, and a high degree of surface marker redundancy made definitive identification of these groups difficult with our technical capabilities (66–68).
Table 1.
Genotypic differences in CD11c on Cln3−/− and wild type leukocytes from spleen and blood samples as assessed by flow cytometry.
| Variable | Wild type | Cln3−/− | |||
|---|---|---|---|---|---|
|
| |||||
| Mean | SD | Mean | SD | P value | |
| Spleen | |||||
| CD11chi MFI on F480+ macrophages | 76160.00 | 1864.94 | 72000.000 | 2815.138 | 0.073 |
| 0 | 0 | ||||
| CD11clow MFI on F480+ macrophages | 19320.00 | 248.998 | 21020.000 | 426.615 | <.0001 |
| 0 | |||||
| CD11chi MFI | 53280.00 | 6488.20 | 55680.000 | 3343.900 | 0.929 |
| 0 | 0 | ||||
| CD11clow MFI | 11080.00 | 258.800 | 11860.000 | 391.200 | 0.023 |
| 0 | |||||
| %CD11chi of CD11c+ | 14.480 | 3.238 | 21.620 | 2.759 | 0.033 |
| %CD11clow of CD11c+ | 85.500 | 3.160 | 78.300 | 2.723 | 0.033 |
| Blood | |||||
| CD11c MFI on CD14+ monocytes | 11600.00 | 374.166 | 15366.667 | 2361.920 | 0.029 |
| 0 | |||||
| CD11chi MFI | 10340.00 | 364.692 | 14250.000 | 2375.500 | 0.050 |
| 0 | |||||
| CD11clow MFI | 3035.400 | 51.432 | 2760.167 | 124.857 | 0.013 |
| %CD11chi of CD11c+ | 50.340 | 2.606 | 57.217 | 2.460 | 0.007 |
| %CD11clow of CD11c+ | 49.700 | 2.639 | 42.783 | 2.431 | 0.007 |
Significant results in bold. MFI = median fluorescence intensity. SD = standard deviation.
Mean defined as average MFI or average percent of positively gated cells within each genotype.
We additionally characterized CD11c on non-granulocytic leukocytes more generally, again noting a natural division between CD11clow and CD11chi cells. Out of CD11c+ cells, a significantly greater percentage were CD11chi in Cln3−/−samples compared to wild type samples in both spleen and blood (Table 1). This would suggest a higher prevalence of leukocyte subtypes known to be CD11chi in Cln3−/− samples, or that levels of CD11c were elevated on subtypes typically characterized as CD11clow such that they were included in the CD11chi population. In blood, CD11c was significantly increased on CD11chi Cln3−/− cells compared to wild type cells (Table 1, supplementary Fig. 6C). A more general increase in CD11c surface levels was also found for CD11clow cells in Cln3−/− spleen samples (supplementary Fig. 6E). Disparate findings for blood and spleen were not unexpected as their leukocyte composition differs. Genotypic differences were not detected in other cell surface proteins (CD3, CD4, CD8, CD11b, CD19, CD69, CD86, B220, F4/80, GR-1) (data not shown). Altogether these data indicate that cells of Mφ lineage have higher levels of CD11c and a greater percentage of cells with significantly elevated surface CD11c are present in Cln3−/− mice in vivo compared to wild type mice.
3.4. Mechanism of increased CD11c surface expression
Leukocytes contain CD11c in two compartments—the surface compartment and an intracellular pool ready for mobilization (69, 70). As CLN3-deficient cells experience a variety of trafficking defects, we hypothesized the increased surface CD11c could stem from abnormal distribution whereby cells might traffic all synthesized protein to the surface, failing to maintain intracellular stores. CD11c total protein levels assessed by Western blotting, however, were increased in Cln3−/− BM-DCs and BM-Mφ compared to wild type cells in both unstimulated and LPS-stimulated conditions (Fig. 5), indicating that CD11c expression was increased overall in Cln3−/− samples and not merely redistributed to the surface compartment.
Fig. 5. CD11c total protein levels are significantly elevated in unstimulated and LPS-stimulated Cln3−/− bone marrow macrophages and bone marrow dendritic cells compared to their wild type counterparts.

Total protein levels of CD11c were assessed in Cln3−/− and wild type (WT) bone marrow cultured cells with Western blot. Day 12 bone marrow macrophages (BM-Mφ) and bone marrow dendritic cells (BM-DCs), were harvested, unstimulated or 48 hours after addition of lipopolysaccharide (LPS). Total protein lysate was interrogated for levels of CD11c protein (top band in B, C, and D) with GM130 as the loading control (bottom double band in B, C, and D). Densitometric analysis of CD11c protein expression is shown (A). Band intensities were quantified by densitometry and the ratio of integrated densities of CD11c to GM130 was calculated. Columns and bars represent mean ± S.E.M. For unstimulated samples, statistical significance was determined by 2-way ANOVA with genotype and fraction as factors, with reported P values for post-hoc Fisher’s LSD (n=2, all groups). For LPS-stimulated samples, statistical significance was determined with the one-tailed Student’s t test (n= 3 wild type BM-Mφ; n=2 Cln3−/− BM-Mφ; n=2 wild type BM-DCs; n=3 Cln3−/− BM-DCs). Representative Western blots are shown (B–D).
A greater total amount of CD11c protein in Cln3−/− cells may result from overproduction of protein or a failure of degradation. To explore potential changes in production, we interrogated the first point of regulation for protein synthesis, transcription, by determining the relative expression levels of integrin subunit genes for CD11c (Itgax) and CD11b (Itgam) with qPCR. LPS stimulation significantly reduced transcript levels at both the 24 hour and 48 hour time points in Cln3−/− and wild type mixed BMCCs (Table 2). This concurs with our finding of decreased β2-integrin surface levels on BMCCs after addition of TLR ligands. No genotypic differences in CD11c transcript levels were detected, suggesting that increased levels of CD11c on Cln3−/− cells do not stem from increased expression of Itgax and that the cause is further downstream (Table 2). Interestingly, gene expression assays determined that the CD11b phenotype likely is attributable to reduced Itgam transcripts in Cln3−/− cells compared to wild type cells (Table 2, supplementary Fig. 7).
Table 2.
Two-way ANOVA tables assessing effects of genotype and lipopolysaccharide stimulation on β2-integrin subunit gene expression in Cln3−/− and wild type mixed bone marrow cultured cells.
| Subunit, gene | F-ratio | df | P value |
|---|---|---|---|
| CD11c, itgax | |||
| 24 hours | |||
| genotype | 0.005 | 1,6 | 0.945 |
| stimulation | 39.395 | 1,6 | 0.001 |
| genotype x stimulation | 2.012 | 1,6 | 0.206 |
| 48 hours | |||
| genotype | 0.478 | 1,6 | 0.515 |
| stimulation | 196.333 | 1,6 | <.0001 |
| genotype x stimulation | 2.251 | 1,6 | 0.184 |
| CD11b, itgam | |||
| 24 hours | |||
| genotype | 2201.588 | 1,6 | <.0001 |
| stimulation | 253.009 | 1,6 | <.0001 |
| genotype x stimulation | 128.628 | 1,6 | <.0001 |
| 48 hours | |||
| genotype | 628.928 | 1,6 | <.0001 |
| stimulation | 284.380 | 1,6 | <.0001 |
| genotype x stimulation | 174.628 | 1,6 | <.0001 |
Significant results in bold. df = degrees of freedom. Genotype = wild type vs. Cln3−/−. F-ratio = mean square of the factor/ mean square of error. The larger the F-ratio value, the greater the likelihood that the difference between the means is due to something other than chance alone.
To further characterize potential points of CD11c dysregulation, we investigated the cellular localization of CD11c in bone marrow cultured cells with immunolabeling of CD11c and proteins known to reside in specific surface and intracellular domains. We also looked at whether localization differed between Cln3−/− and wild type cells. CD11c did not show significant colocalization with GM130, Rab11, EEA-1, or EPS15, indicating that very little protein is present in the Golgi, recycling pathway, early endosomes, or clathrin-coated pits, respectively (see supplementary Table 2). CD11c colocalized with lipid raft markers in unstimulated and stimulated cells, identified with cholera toxin B (CTB), which binds GM1 gangliosides, or with an antibody against raft protein caveolin-1 (71–73). The average Pearson’s correlation coefficient for wild type and Cln3−/− cells met the threshold for positive colocalization, reflected by an overlap of signals in imaging (Fig. 6). Correlation coefficients did not differ by genotype for unstimulated cells stained with CTB [mean(SD): wild type, 0.532(0.021), Cln3−/−, 0.587(0.080), P = .448] or stimulated cells stained with the anti-caveolin-1 antibody [mean(SD): wild type, 0.713(0.034), Cln3−/−, 0.745(0.043), P = .412].
Fig. 6. Localization of CD11c to lipid rafts in unstimulated or LPS-stimulated Cln3−/− and wild type bone marrow cultured cells assessed by fluorescence-based immunocytochemistry.

Cln3−/− and wild type bone marrow cultured cells (BMCCs) were assessed for surface CD11c and lipid raft markers with fluorescence-based immunocytochemistry without membrane permeabilization, and imaged with confocal microscopy using a 100× objective lens. Representative images are shown. Unstimulated mixed BMCCs were labeled with an anti-CD11c antibody (green fluorescence, A, D) and lipid raft binding cholera toxin B (CTB; red fluorescence, B, E). LPS-stimulated BMCCs were labeled with antibodies against CD11c shown in green (G, J) and lipid raft protein caveolin-1 shown in red (H, K). Yellow in the merged images indicates colocalization. Scale bar = 10 microns.
3.5. Adhesion assays
As CD11c functions primarily as an adhesion molecule, we speculated that increased surface levels might enhance the adhesive properties of Cln3−/− cells. An analogous situation is found in Down’s syndrome with overexpression of the CD18 β2 subunit gene (ITG2B, chromosome 21), which upregulates surface CD11a (LFA-1) on patient lymphoblast cell lines, increasing adhesiveness (74). We assessed the ability of BM-DCs and BM-Mφ to adhere to several different ligands of varying specificity for β2-integrins. Fibrinogen is a well-characterized ligand for both CD11b and CD11c. Coating with bovine serum albumin (BSA) often is used to block non-specific binding to polystyrene plates. Our own preliminary experiments, however, indicated that CD11c might mediate binding to BSA-coated plates, as illustrated by a reduction in adhesion in the presence of blocking antibodies (supplementary Fig. 8). Control wells were coated with poly-D-lysine (PDL), a positively charged lysine polymer that non-specifically binds negatively-charged ions on the cell membrane. No difference in adhesion to PDL was detected in BM-DCs (P = .717) or BM-Mφ (P = .627), indicating that results are not attributable to an error in the number of cells aliquoted per well. In BM-Mφ samples, adhesion was significantly higher to BSA and fibrinogen for Cln3−/− cells compared to wild type with no difference in adhesion to PDL (Fig. 7). Adhesion to BSA also was significantly higher for Cln3−/− BM-DCs compared to wild type cells, but no differences were detected for adhesion to fibrinogen (P = .362) (Fig. 7). This indicates that ligand binding potentially mediated by CD11c is greater in Cln3−/− samples, suggesting that increased CD11c might enhance adhesive properties.
Fig. 7. Significantly increased adhesion of Cln3−/− compared to wild type bone marrow macrophages and bone marrow dendritic cells.
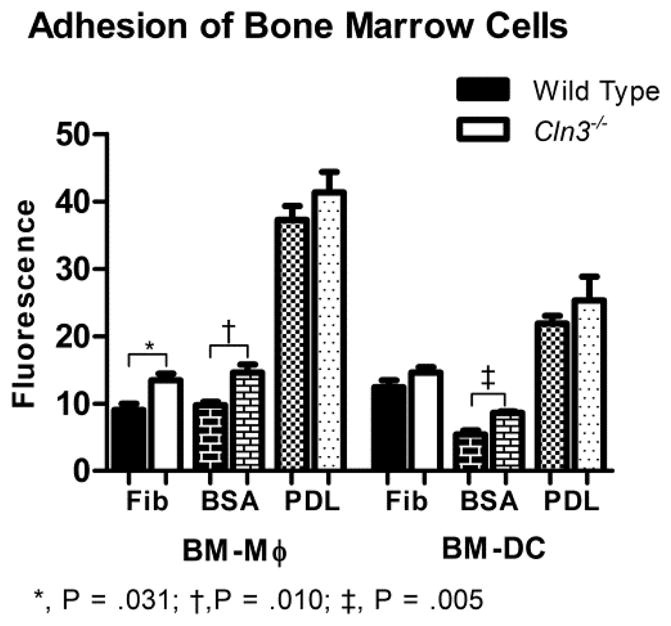
Day 10 bone marrow macrophages (BM-Mφ) and bone marrow dendritic cells (BM-DCs) from Cln3−/− and wild type mice were assessed for adhesion to polystyrene plates coated with bovine serum albumin (BSA), fibrinogen (Fib), or poly-D-lysine (PDL). Wells were loaded with 30,000 cells per sample, with five technical replicates, incubated for 30 minutes, and then stained with Hoechst 33342 solution. The outcome variable, fluorescence, characterized the intensity of Hoechst staining measured with a fluorescence plate reader and is proportional to the number of fluorescently labeled cells adhered to ligand. Columns and bars represent mean ± S.E.M (n=6 for all wild type groups; n=5 for all Cln3−/− groups). Means for Cln3−/− and wild type cells were compared with the Student’s t-test. Reported P values were Dunn-Sidak corrected.
3.6. Cytokine/chemokine secretion
We next characterized secretion of cytokines and chemokines by BM-Mφ and BM-DCs. Twenty-four hour culture medium of unstimulated and LPS-stimulated Cln3−/− and wild type cells was assessed for concentrations of cytokines and chemokines. Without stimulation, no significant genotypic differences in analyte concentration were detected for BM-Mφ or BM-DCs (supplementary Tables 3, 4). When BM-Mφ were stimulated with LPS, levels of IFN-γ, IL-1β, IL-17, IL-12p70, and IL-9, were significantly decreased in medium collected from Cln3−/− cells compared to wild type medium (Table 3), a complement of proteins similar to what was previously reported for Cln3 mutant microglia (12). No genotypic differences in cytokine secretion were present in LPS-stimulated BM-DCs (supplementary Table 5). These results indicate that broad defects in secretory function likely extend to APCs of Mφ lineage.
Table 3.
Statistical comparison of mean analyte concentrations in 24 hour medium from Cln3−/− and wild type bone marrow macrophages stimulated with lipopolysaccharide as assessed with Luminex assay.
| Analyte | Wild Type | Cln3−/− | P value | ||
|---|---|---|---|---|---|
|
| |||||
| Concentration (pg/ml) | Concentration (pg/ml) | ||||
|
| |||||
| Mean | SD | Mean | SD | ||
| G-CSF | 846.600 | 808.141 | 1592.000 | 972.391 | 0.994 |
| IFN-γ | 12.078 | 1.507 | 7.074 | 0.984 | 0.005 |
| IL-1α | 1130.400 | 103.106 | 786.000 | 112.927 | 0.020* |
| IL-1β | 2474.800 | 475.035 | 931.800 | 88.220 | 0.002 |
| IL-2 | 10.322 | 4.300 | 5.080 | 1.914 | 0.534 |
| IL-5 | 25.310 | 4.097 | 24.158 | 1.976 | 1.000 |
| IL-6 | 2268.010 | 1963.369 | 2689.000 | 2045.084 | 1.000 |
| IL-7 | 3.796 | 1.425 | 1.836 | 0.463 | 0.321 |
| IL-10 | 1720.400 | 352.180 | 974.000 | 113.475 | 0.039 |
| IL-13 | 13.270 | 18.994 | 71.830 | 68.706 | 0.888 |
| IL-15 | 173.000 | 24.607 | 130.000 | 9.000 | 0.119 |
| IL-17 | 4.382 | 0.575 | 1.444 | 0.243 | <.0001 |
| IP-10 | 269.630 | 261.456 | 84.990 | 73.487 | 0.974 |
| KC | 8011.600 | 372.679 | 6174.600 | 1500.536 | 0.444 |
| MCP-1 | 1186.800 | 132.006 | 1512.000 | 298.268 | 0.687 |
| MIP-1α | 5408.800 | 878.988 | 4969.600 | 154.165 | 0.999 |
| RANTES | 2559.400 | 1974.671 | 945.200 | 337.517 | 0.901 |
| TNF-α | 1824.000 | 505.860 | 1443.800 | 668.982 | 1.000 |
| IL-12p70 | 78.174 | 5.338 | 61.392 | 4.937 | 0.017 |
| IL-9 | 243.000 | 41.641 | 134.800 | 13.349 | 0.011 |
not significant after type I correction. Significant results in bold. P values are Dunn-Sidak adjusted. n=5 per genotype. SD = standard deviation. Mean defined as average analyte concentration (pg/ml).
4. Discussion
This is the first study to describe an abnormal phenotype in CLN3-deficient peripheral immune cells. Previous research has demonstrated reactive lymphocyte infiltration of the CNS in the NCLs in response to brain insult (9, 75). Our discovery, however, is in accordance with recent reports that, beyond the reactive gliosis observed in the NCL brain, microglia appear to be functionally impaired. Cln3 mutant microglia manifest secretory defects and influence the survival and morphology of wild type neurons in culture (11, 12). These studies and ours emphasize the need to characterize dysfunction of non-neuronal cell types to adequately understand JNCL pathology.
Our main finding is a robust increase in surface and total protein levels of the β2-integrin family adhesion molecule CD11c in Cln3−/− BM-Mφ, BM-DCs, and peritoneal Mφ. That the phenotype did not extend to other NCL mouse models indicates it is not a general feature of NCL disease and more likely is related to the loss of CLN3 function. In addition, the phenotype largely spared other cell surface proteins, suggesting perturbation of a CD11c specific regulatory pathway. We speculate that this could be the pathway involved in reduction of surface CD11c, possibly via the same mechanisms mediating downregulation after LPS stimulation. This is supported by our data showing that stimulation increases Cln3 expression in wild type cells, while CD11c surface downregulation is notably impaired in Cln3−/−APCs. The LPS stimulation-associated pathways have not been defined in healthy cells.
We did not fully characterize the specific mechanism underlying abnormal CD11c levels in the context of CLN3 deficiency; however, our results suggest involvement of lipid rafts. In healthy cells, lipid raft membrane microdomains stabilize cell surface proteins, facilitating appropriate regulation of intracellular signal transduction pathways (76). That we localized CD11c to rafts is compelling given that CLN3 also has been localized to rafts, and, in cells with mutated or absent CLN3, rafts are smaller and less stable, with altered surface levels of some raft proteins and impaired caveolae-mediated endocytosis (28, 29, 77). Dysregulation of rafts generally could lead to abnormal CD11c surface levels, or CLN3 might play a more direct role in CD11c trafficking. That we did not detect genotypic differences in CD11a lends support to the latter as CD11a has been localized to rafts in other studies (78).
An increase in the total amount of CD11c protein indicates an increase in synthesis or reduced degradation and not merely redistribution. Increased synthesis was not supported at the level of transcription as no genotypic differences in CD11c mRNA expression was detected. Blocking synthesis of nascent protein with the ribosome translocation inhibitor cycloheximide seems not to have a marked effect on CD11c surface levels on mixed BMCCs of either genotype (unpublished results), indicating that synthesis of new subunits does not play a major role in regulating CD11c surface levels. That we did not find any CD11c in the Golgi supports this notion, which was not unexpected as integrins tend to be recycled (79). It is thus likely that increased CD11c in Cln3−/− cells is attributable to defects in degradative or recycling pathways. Nonetheless, CD11c did not significantly colocalize to early endosomes, or recycyling endosomes. Degradation would seem a promising candidate to mediate CD11c perturbation given the broadly characterized lysosomal dysfunction (29, 80–83). One would then expect altered expression of MHCII surface molecules, which are heavily regulated by the lysosome, but genotypic difference in MHCII surface levels were not detected. We hope that future studies tracking the life cycle of CD11c in wild type and Cln3−/− mice with pulse chase experiments will clarify the point of dysregulation.
Beyond simple surface upregulation, the CD11c phenotype of Cln3−/− cells is associated with increased adhesion compared to wild type cells. This concurs with a previous report that CD11c enhances the adhesive properties of macrophages to more efficiently localize them to atherosclerotic plaques, while CD11c-KO mouse monocytes are less able to form strong adhesive interactions with the endothelium in vivo (47). We do have preliminary data indicating that 3D migration of LPS-stimulated Cln3−/− BM-DCs is impaired, while no differences were detected in unstimulated BM-DCs (unpublished data). This could indicate that functional consequences are most critical if CD11c remains on the surface when normally it would be completely absent (i.e. wild type LPS-stimulated cells have little to no CD11c, but Cln3−/− LPS-stimulated cells maintain significant surface levels). Follow-up studies are needed to replicate findings and extend investigation to Mφ.
Our data show that Cln3−/− BM-Mφ have reduced cytokine secretion compared to wild type cells after LPS stimulation, which might suggest that CLN3-deficient cells are actually less inflammatory. Cln3 mutant microglia also show reduced cytokine secretion after LPS stimulation (12). Endogenous inflammatory stimuli (C6 ceramide and neuronal lysate), however, induces an exaggerated cytokine secretion response by Cln3 mutant microglia (12). We speculate that, similarly to Cln3 mutant microglia Cln3−/−, BM-Mφ may increase cytokine secretion with exposure to appropriate endogenous stimuli in vivo, inciting systemic inflammation. We will need to conduct further testing of BM-Mφ and peritoneal Mφ using C6 ceramide and neuronal lysate to determine if activity is dichotomous as in microglia. We also will need to explore the mechanism underlying abnormal secretion and if it is directly related to Cln3 mutation.
Our in vivo studies demonstrated a greater percentage of cells with significantly elevated surface CD11c in Cln3−/− mice compared to wild type mice. It is possible that this increase confers a pro-inflammatory state on affected mice as studies have demonstrated an association between increased cell surface levels of CD11c and inflammation and autoimmunity. CD11c is upregulated on APCs in the inflammatory processes of metabolic syndrome and polycystic ovarian syndrome, while CD11c-KO mice have attenuated adipose tissue inflammation and insulin resistance (41–45, 47, 84). High numbers of CD11chi NK cells secrete pro-inflammatory cytokines and predict relapse in MS patients (31, 46, 85). Monocytes and neutrophils isolated from rheumatoid arthritis patients also have increased CD11c, with elevated levels suggesting a pro-inflammatory phenotype (37, 86). The mechanism underlying the association of elevated CD11c with inflammation is not known.
There is mounting evidence that the immune system is abnormal in many neurodegenerative lysosomal storage diseases. Patients with Fabry disease have increased incidence of a range of autoimmune disorders, while immune cell dysfunction of various types has been reported in models of Gaucher disease, mucopolysaccharidosis I, and Niemann Pick type C, to name a few (87–91). Potentially important is the predisposition to infection of children with Gaucher disease (92). It has long been known that peripheral inflammation exacerbates neurodegeneration, suggesting that a simple inflammatory state, whether through primary immune dysfunction or infection, could accelerate disease (93). The use of anti-inflammatories in mouse and cell models of lysosomal storage disorders have supported this notion, although there is no evidence this extends to patients (94–96). Currently, there is no indication that children with NCL are at risk for infection outside that conferred by immobility; however, this has not been systematically studied. Use of the anti-inflammatory Flupirtine has conferred little to no benefit in children with NCL (97), making it more likely that the beneficial phenotypic changes in the Cln3−/− mouse model after immunosuppression did not stem purely from reduced inflammation. Trials of immunosuppression in JNCL patients are still underway and should help determine if the immune system’s role is robust enough to warrant directed therapy.
This study lays the foundation for further understanding of the role of APCs in the pathophysiology of JNCL. While much is still unknown, we speculate that CLN3 deficiency could facilitate an environment where APCs with increased adhesive properties have prolonged contact with the cells they activate in the setting of increased concentrations of pro-inflammatory cytokines, priming the body for autoimmunity. Activated lymphocytes then migrate into the CNS and destroy neural tissue. Determining the pathological significance of APC subtypes could potentially lead to immunotherapy with monoclonal antibodies that specifically target the offending cells. In addition, as the function of CLN3 remains elusive, our discovery is additionally useful in establishing a consistent, measurable CLN3-deficient phenotype in an easily obtainable cell population. This is an important tool that can be used to investigate the cellular function of CLN3, as well as to test the efficacy of novel pharmaceutical treatments in cellular assays.
Supplementary Material
Highlights.
Cln3−/− antigen presenting cells were interrogated for phenotypic differences.
CD11c surface levels are elevated on Cln3−/− antigen presenting cells.
CD11c localizes to lipid rafts known to be abnormal in CLN3-deficient cells.
Cytokine secretion is altered in LPS-stimulated Cln3−/− bone marrow macrophages.
LPS-stimulated Cln3−/− antigen presenting cells have increased adhesive properties.
Acknowledgments
We thank Satoshi Nagata, PhD, and Tomoko Ise, MD, PhD, of the Sanford Research Flow Cytometry Core for the use of their equipment and extensive technical assistance. We also thank Jake Miller for performing mRNA isolation and real time quantitative PCR. This work was supported by the National Institutes of Health (NIH) R01 NS044310 and the Luke and Rachel Batten Foundation.
Abbreviations
- APCs
antigen presenting cells
- APhC
allophycocyanin
- BM
bone marrow
- BMCCs
bone marrow cultured cells
- BM-DCs
bone marrow dendritic cells
- BM-Mφ
bone marrow macrophages
- BSA
bovine serum albumin
- CHX
cycloheximide
- CTB
cholera toxin B
- DCs
dendritic cells
- DPBS
Dulbecco’s phosphate buffered saline
- EDTA
ethylenediaminetetraacetic acid
- FBS
fetal bovine serum
- GM-CSF
granulocyte macrophage-colony stimulating factor
- IFN-γ
interferon-γ
- JNCL
juvenile neuronal ceroid lipofuscinosis
- LPS
lipopolysaccharide
- LSD
least squares difference
- LUTs
lookup tables
- MFI
median fluorescence intensity
- Mφ
macrophages
- NCL
neuronal ceroid lipofuscinosis
- PBS
phosphate buffered saline
- PDL
poly-D-lysine
- PE
phycoerythrin
- PerCP
peridinin-chlorophyll protein
- PIC
poly I:C
- ROI
region of interest
- SBDS
Shwachman-Bodian-Diamond Syndrome protein
- TLR
toll-like receptor
- wild type
WT
Footnotes
Competing Interest
The authors have no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Goebel HH, Wisniewski KE. Current state of clinical and morphological features in human NCL. Brain pathology. 2004;14:61–69. doi: 10.1111/j.1750-3639.2004.tb00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pontikis CC, Cella CV, Parihar N, Lim MJ, Chakrabarti S, Mitchison HM, Mobley WC, Rezaie P, Pearce DA, Cooper JD. Late onset neurodegeneration in the Cln3− mouse model of juvenile neuronal ceroid lipofuscinosis is preceded by low level glial activation. Brain research. 2004;1023:231–242. doi: 10.1016/j.brainres.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 3.Tyynela J, Cooper JD, Khan MN, Shemilts SJ, Haltia M. Hippocampal pathology in the human neuronal ceroid-lipofuscinoses: distinct patterns of storage deposition, neurodegeneration and glial activation. Brain pathology. 2004;14:349–357. doi: 10.1111/j.1750-3639.2004.tb00077.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weimer JM, Benedict JW, Getty AL, Pontikis CC, Lim MJ, Cooper JD, Pearce DA. Cerebellar defects in a mouse model of juvenile neuronal ceroid lipofuscinosis. Brain research. 2009;1266:93–107. doi: 10.1016/j.brainres.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cotman SL, Staropoli JF. The juvenile Batten disease protein, CLN3, and its role in regulating anterograde and retrograde post-Golgi trafficking. Clinical lipidology. 2012;7:79–91. doi: 10.2217/clp.11.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castaneda JA, Pearce DA. Identification of alpha-fetoprotein as an autoantigen in juvenile Batten disease. Neurobiology of disease. 2008;29:92–102. doi: 10.1016/j.nbd.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 7.Chattopadhyay S, Ito M, Cooper JD, Brooks AI, Curran TM, Powers JM, Pearce DA. An autoantibody inhibitory to glutamic acid decarboxylase in the neurodegenerative disorder Batten disease. Human molecular genetics. 2002;11:1421–1431. doi: 10.1093/hmg/11.12.1421. [DOI] [PubMed] [Google Scholar]
- 8.Chattopadhyay S, Kriscenski-Perry E, Wenger DA, Pearce DA. An autoantibody to GAD65 in sera of patients with juvenile neuronal ceroid lipofuscinoses. Neurology. 2002;59:1816–1817. doi: 10.1212/01.wnl.0000041913.97883.8b. [DOI] [PubMed] [Google Scholar]
- 9.Lim MJ, Alexander N, Benedict JW, Chattopadhyay S, Shemilt SJ, Guerin CJ, Cooper JD, Pearce DA. IgG entry and deposition are components of the neuroimmune response in Batten disease. Neurobiology of disease. 2007;25:239–251. doi: 10.1016/j.nbd.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 10.Seehafer SS, Ramirez-Montealegre D, Wong AM, Chan CH, Castaneda J, Horak M, Ahmadi SM, Lim MJ, Cooper JD, Pearce DA. Immunosuppression alters disease severity in juvenile Batten disease mice. Journal of neuroimmunology. 2011;230:169–172. doi: 10.1016/j.jneuroim.2010.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lange JPL, Anderson G, Dihanich S, Rezaie P, Mitchison H, Williams B, Cooper JD. Glia impair neuron health in juvenile neuronal ceroid lipofuscinosis. Glia. 2013;61:S78. [Google Scholar]
- 12.Xiong J, Kielian T. Microglia in juvenile neuronal ceroid lipofuscinosis are primed toward a pro-inflammatory phenotype. Journal of neurochemistry. 2013;127:245–258. doi: 10.1111/jnc.12385. [DOI] [PubMed] [Google Scholar]
- 13.Staropoli JF, Haliw L, Biswas S, Garrett L, Holter SM, Becker L, Skosyrski S, Da Silva-Buttkus P, Calzada-Wack J, Neff F, et al. Large-scale phenotyping of an accurate genetic mouse model of JNCL identifies novel early pathology outside the central nervous system. PloS one. 2012;7:e38310. doi: 10.1371/journal.pone.0038310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Araki N, Hatae T, Furukawa A, Swanson JA. Phosphoinositide-3-kinase-independent contractile activities associated with Fcgamma-receptor-mediated phagocytosis and macropinocytosis in macrophages. Journal of cell science. 2003;116:247–257. doi: 10.1242/jcs.00235. [DOI] [PubMed] [Google Scholar]
- 15.Benedict JW, Sommers CA, Pearce DA. Progressive oxidative damage in the central nervous system of a murine model for juvenile Batten disease. Journal of neuroscience research. 2007;85:2882–2891. doi: 10.1002/jnr.21416. [DOI] [PubMed] [Google Scholar]
- 16.Bhuwania R, Cornfine S, Fang Z, Kruger M, Luna EJ, Linder S. Supervillin couples myosin-dependent contractility to podosomes and enables their turnover. Journal of cell science. 2012;125:2300–2314. doi: 10.1242/jcs.100032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burroughs L, Woolfrey A, Shimamura A. Shwachman-Diamond syndrome: a review of the clinical presentation, molecular pathogenesis, diagnosis, and treatment. Hematology/oncology clinics of North America. 2009;23:233–248. doi: 10.1016/j.hoc.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Getty AL, Benedict JW, Pearce DA. A novel interaction of CLN3 with nonmuscle myosin-IIB and defects in cell motility of Cln3(−) cells. Experimental cell research. 2011;317:51–69. doi: 10.1016/j.yexcr.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harada Y, Tanaka Y, Terasawa M, Pieczyk M, Habiro K, Katakai T, Hanawa-Suetsugu K, Kukimoto-Niino M, Nishizaki T, Shirouzu M, et al. DOCK8 is a Cdc42 activator critical for interstitial dendritic cell migration during immune responses. Blood. 2012;119:4451–4461. doi: 10.1182/blood-2012-01-407098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harris KP, Tepass U. Cdc42 and vesicle trafficking in polarized cells. Traffic. 2010;11:1272–1279. doi: 10.1111/j.1600-0854.2010.01102.x. [DOI] [PubMed] [Google Scholar]
- 21.Koivusalo M, Welch C, Hayashi H, Scott CC, Kim M, Alexander T, Touret N, Hahn KM, Grinstein S. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. The Journal of cell biology. 2010;188:547–563. doi: 10.1083/jcb.200908086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krits I, Wysolmerski RB, Holliday LS, Lee BS. Differential localization of myosin II isoforms in resting and activated osteoclasts. Calcified tissue international. 2002;71:530–538. doi: 10.1007/s00223-001-1112-0. [DOI] [PubMed] [Google Scholar]
- 23.Lammermann T, Renkawitz J, Wu X, Hirsch K, Brakebusch C, Sixt M. Cdc42-dependent leading edge coordination is essential for interstitial dendritic cell migration. Blood. 2009;113:5703–5710. doi: 10.1182/blood-2008-11-191882. [DOI] [PubMed] [Google Scholar]
- 24.Schultz ML, Tecedor L, Stein CS, Stamnes MA, Davidson BL. CLN3 deficient cells display defects in the ARF1-Cdc42 pathway and actin-dependent events. PloS one. 2014;9:e96647. doi: 10.1371/journal.pone.0096647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stepanovic V, Wessels D, Goldman FD, Geiger J, Soll DR. The chemotaxis defect of Shwachman-Diamond Syndrome leukocytes. Cell motility and the cytoskeleton. 2004;57:158–174. doi: 10.1002/cm.10164. [DOI] [PubMed] [Google Scholar]
- 26.Vitiello SP, Benedict JW, Padilla-Lopez S, Pearce DA. Interaction between Sdo1p and Btn1p in the Saccharomyces cerevisiae model for Batten disease. Human molecular genetics. 2010;19:931–942. doi: 10.1093/hmg/ddp560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang L, Wang L, Kalfa TA, Cancelas JA, Shang X, Pushkaran S, Mo J, Williams DA, Zheng Y. Cdc42 critically regulates the balance between myelopoiesis and erythropoiesis. Blood. 2007;110:3853–3861. doi: 10.1182/blood-2007-03-079582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rusyn E, Mousallem T, Persaud-Sawin DA, Miller S, Boustany RM. CLN3p impacts galactosylceramide transport, raft morphology, and lipid content. Pediatric research. 2008;63:625–631. doi: 10.1203/PDR.0b013e31816fdc17. [DOI] [PubMed] [Google Scholar]
- 29.Tecedor L, Stein CS, Schultz ML, Farwanah H, Sandhoff K, Davidson BL. CLN3 loss disturbs membrane microdomain properties and protein transport in brain endothelial cells. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:18065–18079. doi: 10.1523/JNEUROSCI.0498-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uusi-Rauva K, Luiro K, Tanhuanpaa K, Kopra O, Martin-Vasallo P, Kyttala A, Jalanko A. Novel interactions of CLN3 protein link Batten disease to dysregulation of fodrin-Na+, K+ ATPase complex. Experimental cell research. 2008;314:2895–2905. doi: 10.1016/j.yexcr.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 31.Aranami T, Miyake S, Yamamura T. Differential expression of CD11c by peripheral blood NK cells reflects temporal activity of multiple sclerosis. Journal of immunology. 2006;177:5659–5667. doi: 10.4049/jimmunol.177.8.5659. [DOI] [PubMed] [Google Scholar]
- 32.Berkun Y, Verbovetski I, Ben-Ami A, Paran D, Caspi D, Krispin A, Trahtemberg U, Gill O, Naparstek Y, Mevorach D. Altered dendritic cells with tolerizing phenotype in patients with systemic lupus erythematosus. European journal of immunology. 2008;38:2896–2904. doi: 10.1002/eji.200838342. [DOI] [PubMed] [Google Scholar]
- 33.Catalan D, Aravena O, Sabugo F, Wurmann P, Soto L, Kalergis AM, Cuchacovich M, Aguillon JC Millenium Nucleus on, I. and Immunotherapy, P.F. B cells from rheumatoid arthritis patients show important alterations in the expression of CD86 and FcgammaRIIb, which are modulated by anti-tumor necrosis factor therapy. Arthritis research & therapy. 2010;12:R68. doi: 10.1186/ar2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Folzenlogen D, Hofer MF, Leung DY, Freed JH, Newell MK. Analysis of CD80 and CD86 expression on peripheral blood B lymphocytes reveals increased expression of CD86 in lupus patients. Clinical immunology and immunopathology. 1997;83:199–204. doi: 10.1006/clin.1997.4353. [DOI] [PubMed] [Google Scholar]
- 35.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 36.Mackay M, Stanevsky A, Wang T, Aranow C, Li M, Koenig S, Ravetch JV, Diamond B. Selective dysregulation of the FcgammaIIB receptor on memory B cells in SLE. The Journal of experimental medicine. 2006;203:2157–2164. doi: 10.1084/jem.20051503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mera S, Magnusson M, Tarkowski A, Bokarewa M. Extracellular survivin up-regulates adhesion molecules on the surface of leukocytes changing their reactivity pattern. Journal of leukocyte biology. 2008;83:149–155. doi: 10.1189/jlb.0507287. [DOI] [PubMed] [Google Scholar]
- 38.Zhu J, Liu X, Xie C, Yan M, Yu Y, Sobel ES, Wakeland EK, Mohan C. T cell hyperactivity in lupus as a consequence of hyperstimulatory antigen-presenting cells. The Journal of clinical investigation. 2005;115:1869–1878. doi: 10.1172/JCI23049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mazzone A, Ricevuti G. Leukocyte CD11/CD18 integrins: biological and clinical relevance. Haematologica. 1995;80:161–175. [PubMed] [Google Scholar]
- 40.Tan SM. The leucocyte beta2 (CD18) integrins: the structure, functional regulation and signalling properties. Bioscience reports. 2012;32:241–269. doi: 10.1042/BSR20110101. [DOI] [PubMed] [Google Scholar]
- 41.Brake DK, Smith EO, Mersmann H, Smith CW, Robker RL. ICAM-1 expression in adipose tissue: effects of diet-induced obesity in mice. American journal of physiology. Cell physiology. 2006;291:C1232–1239. doi: 10.1152/ajpcell.00008.2006. [DOI] [PubMed] [Google Scholar]
- 42.Gonzalez F, Rote NS, Minium J, Kirwan JP. Increased activation of nuclear factor kappaB triggers inflammation and insulin resistance in polycystic ovary syndrome. The Journal of clinical endocrinology and metabolism. 2006;91:1508–1512. doi: 10.1210/jc.2005-2327. [DOI] [PubMed] [Google Scholar]
- 43.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. The Journal of clinical investigation. 2007;117:175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nguyen MT, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, Liu-Bryan R, Glass CK, Neels JG, Olefsky JM. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. The Journal of biological chemistry. 2007;282:35279–35292. doi: 10.1074/jbc.M706762200. [DOI] [PubMed] [Google Scholar]
- 45.Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell metabolism. 2008;8:301–309. doi: 10.1016/j.cmet.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pillarisetty VG, Katz SC, Bleier JI, Shah AB, Dematteo RP. Natural killer dendritic cells have both antigen presenting and lytic function and in response to CpG produce IFN-gamma via autocrine IL-12. Journal of immunology. 2005;174:2612–2618. doi: 10.4049/jimmunol.174.5.2612. [DOI] [PubMed] [Google Scholar]
- 47.Wu H, Gower RM, Wang H, Perrard XY, Ma R, Bullard DC, Burns AR, Paul A, Smith CW, Simon SI, et al. Functional role of CD11c+ monocytes in atherogenesis associated with hypercholesterolemia. Circulation. 2009;119:2708–2717. doi: 10.1161/CIRCULATIONAHA.108.823740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Akiyama H, McGeer PL. Brain microglia constitutively express beta-2 integrins. Journal of neuroimmunology. 1990;30:81–93. doi: 10.1016/0165-5728(90)90055-r. [DOI] [PubMed] [Google Scholar]
- 49.Beyer M, Wang H, Peters N, Doths S, Koerner-Rettberg C, Openshaw PJ, Schwarze J. The beta2 integrin CD11c distinguishes a subset of cytotoxic pulmonary T cells with potent antiviral effects in vitro and in vivo. Respiratory research. 2005;6:70. doi: 10.1186/1465-9921-6-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, Marrack P. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c(+) B-cell population is important for the development of autoimmunity. Blood. 2011;118:1305–1315. doi: 10.1182/blood-2011-01-331462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walzog B, Weinmann P, Jeblonski F, Scharffetter-Kochanek K, Bommert K, Gaehtgens P. A role for beta(2) integrins (CD11/CD18) in the regulation of cytokine gene expression of polymorphonuclear neutrophils during the inflammatory response. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 1999;13:1855–1865. doi: 10.1096/fasebj.13.13.1855. [DOI] [PubMed] [Google Scholar]
- 52.Mitchison HM, Bernard DJ, Greene ND, Cooper JD, Junaid MA, Pullarkat RK, de Vos N, Breuning MH, Owens JW, Mobley WC, et al. Targeted disruption of the Cln3 gene provides a mouse model for Batten disease. The Batten Mouse Model Consortium [corrected] Neurobiology of disease. 1999;6:321–334. doi: 10.1006/nbdi.1999.0267. [DOI] [PubMed] [Google Scholar]
- 53.Gupta P, Soyombo AA, Atashband A, Wisniewski KE, Shelton JM, Richardson JA, Hammer RE, Hofmann SL. Disruption of PPT1 or PPT2 causes neuronal ceroid lipofuscinosis in knockout mice. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:13566–13571. doi: 10.1073/pnas.251485198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sleat DE, Wiseman JA, El-Banna M, Kim KH, Mao Q, Price S, Macauley SL, Sidman RL, Shen MM, Zhao Q, et al. A mouse model of classical late-infantile neuronal ceroid lipofuscinosis based on targeted disruption of the CLN2 gene results in a loss of tripeptidyl-peptidase I activity and progressive neurodegeneration. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:9117–9126. doi: 10.1523/JNEUROSCI.2729-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adkins B, Leclerc C, Marshall-Clarke S. Neonatal adaptive immunity comes of age. Nature reviews. Immunology. 2004;4:553–564. doi: 10.1038/nri1394. [DOI] [PubMed] [Google Scholar]
- 56.Academies, N.R.C.o.t.N. Guide for the Care and Use of Laboratory Animals. The National Academies Press; Washington: 2011. [PubMed] [Google Scholar]
- 57.Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. Journal of immunological methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 58.Campisano S, Mac Keon S, Gazzaniga S, Ruiz MS, Traian MD, Mordoh J, Wainstok R. Anti-melanoma vaccinal capacity of CD11c-positive and -negative cell populations present in GM-CSF cultures derived from murine bone marrow precursors. Vaccine. 2013;31:354–361. doi: 10.1016/j.vaccine.2012.10.114. [DOI] [PubMed] [Google Scholar]
- 59.Inaba K, Inaba M, Deguchi M, Hagi K, Yasumizu R, Ikehara S, Muramatsu S, Steinman RM. Granulocytes, macrophages, and dendritic cells arise from a common major histocompatibility complex class II-negative progenitor in mouse bone marrow. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:3038–3042. doi: 10.1073/pnas.90.7.3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. In: Coligan John E, et al., editors. Current protocols in immunology / edited. Unit 14 11. Chapter 14. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miller JN, Chan CH, Pearce DA. The role of nonsense-mediated decay in neuronal ceroid lipofuscinosis. Human molecular genetics. 2013;22:2723–2734. doi: 10.1093/hmg/ddt120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sidak ZK. Rectangular confidence regions for the means of multivariate normal distributions. J Am Stat Assoc. 1967;62:626–633. [Google Scholar]
- 63.Dunn OJ. Multiple comparisons among means. J Am Stat Assoc. 1961;56:52–64. [Google Scholar]
- 64.Adler J, Parmryd I. Quantifying colocalization by correlation: the Pearson correlation coefficient is superior to the Mander’s overlap coefficient. Cytometry. Part A : the journal of the International Society for Analytical Cytology. 2010;77:733–742. doi: 10.1002/cyto.a.20896. [DOI] [PubMed] [Google Scholar]
- 65.Zinchuk V, Grossenbacher-Zinchuk O. Quantitative colocalization analysis of confocal fluorescence microscopy images. In: Bonifacino Juan S, et al., editors. Current protocols in cell biology / editorial board. Unit4 19. Chapter 4. 2011. [DOI] [PubMed] [Google Scholar]
- 66.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 67.O’Keeffe M, Hochrein H, Vremec D, Scott B, Hertzog P, Tatarczuch L, Shortman K. Dendritic cell precursor populations of mouse blood: identification of the murine homologues of human blood plasmacytoid pre-DC2 and CD11c+ DC1 precursors. Blood. 2003;101:1453–1459. doi: 10.1182/blood-2002-03-0974. [DOI] [PubMed] [Google Scholar]
- 68.Sunderkotter C, Nikolic T, Dillon MJ, Van Rooijen N, Stehling M, Drevets DA, Leenen PJ. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. Journal of immunology. 2004;172:4410–4417. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 69.Kiley SC, Parker PJ. Differential localization of protein kinase C isozymes in U937 cells: evidence for distinct isozyme functions during monocyte differentiation. Journal of cell science. 1995;108(Pt 3):1003–1016. doi: 10.1242/jcs.108.3.1003. [DOI] [PubMed] [Google Scholar]
- 70.Miller LJ, Bainton DF, Borregaard N, Springer TA. Stimulated mobilization of monocyte Mac-1 and p150,95 adhesion proteins from an intracellular vesicular compartment to the cell surface. The Journal of clinical investigation. 1987;80:535–544. doi: 10.1172/JCI113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Heyningen SV. Cholera toxin: interaction of subunits with ganglioside GM1. Science. 1974;183:656–657. doi: 10.1126/science.183.4125.656. [DOI] [PubMed] [Google Scholar]
- 72.Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR, Anderson RG. Caveolin, a protein component of caveolae membrane coats. Cell. 1992;68:673–682. doi: 10.1016/0092-8674(92)90143-z. [DOI] [PubMed] [Google Scholar]
- 73.Skocaj M, Bakrac B, Krizaj I, Macek P, Anderluh G, Sepcic K. The sensing of membrane microdomains based on pore-forming toxins. Current medicinal chemistry. 2013;20:491–501. doi: 10.2174/0929867311320040002. [DOI] [PubMed] [Google Scholar]
- 74.Taylor GM, Haigh H, Williams A, D’Souza SW, Harris R. Down’s syndrome lymphoid cell lines exhibit increased adhesion due to the over-expression of lymphocyte function-associated antigen (LFA-1) Immunology. 1988;64:451–456. [PMC free article] [PubMed] [Google Scholar]
- 75.Groh J, Kuhl TG, Ip CW, Nelvagal HR, Sri S, Duckett S, Mirza M, Langmann T, Cooper JD, Martini R. Immune cells perturb axons and impair neuronal survival in a mouse model of infantile neuronal ceroid lipofuscinosis. Brain : a journal of neurology. 2013;136:1083–1101. doi: 10.1093/brain/awt020. [DOI] [PubMed] [Google Scholar]
- 76.Simons K, Toomre D. Lipid rafts and signal transduction. Nature reviews. Molecular cell biology. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 77.Persaud-Sawin DA, McNamara JO, 2nd, Rylova S, Vandongen A, Boustany RM. A galactosylceramide binding domain is involved in trafficking of CLN3 from Golgi to rafts via recycling endosomes. Pediatric research. 2004;56:449–463. doi: 10.1203/01.PDR.0000136152.54638.95. [DOI] [PubMed] [Google Scholar]
- 78.Fabbri M, Di Meglio S, Gagliani MC, Consonni E, Molteni R, Bender JR, Tacchetti C, Pardi R. Dynamic partitioning into lipid rafts controls the endoexocytic cycle of the alphaL/beta2 integrin, LFA-1, during leukocyte chemotaxis. Molecular biology of the cell. 2005;16:5793–5803. doi: 10.1091/mbc.E05-05-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bridgewater RE, Norman JC, Caswell PT. Integrin trafficking at a glance. Journal of cell science. 2012;125:3695–3701. doi: 10.1242/jcs.095810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fossale E, Wolf P, Espinola JA, Lubicz-Nawrocka T, Teed AM, Gao H, Rigamonti D, Cattaneo E, MacDonald ME, Cotman SL. Membrane trafficking and mitochondrial abnormalities precede subunit c deposition in a cerebellar cell model of juvenile neuronal ceroid lipofuscinosis. BMC neuroscience. 2004;5:57. doi: 10.1186/1471-2202-5-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Golabek AA, Kida E, Walus M, Kaczmarski W, Michalewski M, Wisniewski KE. CLN3 protein regulates lysosomal pH and alters intracellular processing of Alzheimer’s amyloid-beta protein precursor and cathepsin D in human cells. Molecular genetics and metabolism. 2000;70:203–213. doi: 10.1006/mgme.2000.3006. [DOI] [PubMed] [Google Scholar]
- 82.Holopainen JM, Saarikoski J, Kinnunen PK, Jarvela I. Elevated lysosomal pH in neuronal ceroid lipofuscinoses (NCLs) European journal of biochemistry / FEBS. 2001;268:5851–5856. doi: 10.1046/j.0014-2956.2001.02530.x. [DOI] [PubMed] [Google Scholar]
- 83.Padilla-Lopez S, Pearce DA. Saccharomyces cerevisiae lacking Btn1p modulate vacuolar ATPase activity to regulate pH imbalance in the vacuole. The Journal of biological chemistry. 2006;281:10273–10280. doi: 10.1074/jbc.M510625200. [DOI] [PubMed] [Google Scholar]
- 84.Wu H, Perrard XD, Wang Q, Perrard JL, Polsani VR, Jones PH, Smith CW, Ballantyne CM. CD11c expression in adipose tissue and blood and its role in diet-induced obesity. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:186–192. doi: 10.1161/ATVBAHA.109.198044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Homann D, Jahreis A, Wolfe T, Hughes A, Coon B, van Stipdonk MJ, Prilliman KR, Schoenberger SP, von Herrath MG. CD40L blockade prevents autoimmune diabetes by induction of bitypic NK/DC regulatory cells. Immunity. 2002;16:403–415. doi: 10.1016/s1074-7613(02)00290-x. [DOI] [PubMed] [Google Scholar]
- 86.Stuhlmuller B, Haupl T, Hernandez MM, Grutzkau A, Kuban RJ, Tandon N, Voss JW, Salfeld J, Kinne RW, Burmester GR. CD11c as a transcriptional biomarker to predict response to anti-TNF monotherapy with adalimumab in patients with rheumatoid arthritis. Clinical pharmacology and therapeutics. 2010;87:311–321. doi: 10.1038/clpt.2009.244. [DOI] [PubMed] [Google Scholar]
- 87.Archer LD, Langford-Smith KJ, Critchley WR, Bigger BW, Fildes JE. Characterisation of the T cell and dendritic cell repertoire in a murine model of mucopolysaccharidosis I (MPS I) Journal of inherited metabolic disease. 2013;36:257–262. doi: 10.1007/s10545-012-9508-8. [DOI] [PubMed] [Google Scholar]
- 88.Liu J, Halene S, Yang M, Iqbal J, Yang R, Mehal WZ, Chuang WL, Jain D, Yuen T, Sun L, et al. Gaucher disease gene GBA functions in immune regulation. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:10018–10023. doi: 10.1073/pnas.1200941109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rahman P, Gladman DD, Wither J, Silver MD. Coexistence of Fabry’s disease and systemic lupus erythematosus. Clinical and experimental rheumatology. 1998;16:475–478. [PubMed] [Google Scholar]
- 90.Speak AO, Te Vruchte D, Davis LC, Morgan AJ, Smith DA, Yanjanin NM, Simmons L, Hartung R, Runz H, Mengel E, et al. Altered distribution and function of natural killer cells in murine and human Niemann-Pick disease type C1. Blood. 2014;123:51–60. doi: 10.1182/blood-2013-03-488692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Whybra C, Schwarting A, Kriegsmann J, Gal A, Mengel E, Kampmann C, Baehner F, Schaefer E, Beck M. IgA nephropathy in two adolescent sisters heterozygous for Fabry disease. Pediatric nephrology. 2006;21:1251–1256. doi: 10.1007/s00467-006-0176-5. [DOI] [PubMed] [Google Scholar]
- 92.Marodi L, Kaposzta R, Toth J, Laszlo A. Impaired microbicidal capacity of mononuclear phagocytes from patients with type I Gaucher disease: partial correction by enzyme replacement therapy. Blood. 1995;86:4645–4649. [PubMed] [Google Scholar]
- 93.Perry VH, Newman TA, Cunningham C. The impact of systemic infection on the progression of neurodegenerative disease. Nature reviews. Neuroscience. 2003;4:103–112. doi: 10.1038/nrn1032. [DOI] [PubMed] [Google Scholar]
- 94.Dhar S, Bitting RL, Rylova SN, Jansen PJ, Lockhart E, Koeberl DD, Amalfitano A, Boustany RM. Flupirtine blocks apoptosis in batten patient lymphoblasts and in human postmitotic CLN3- and CLN2-deficient neurons. Annals of neurology. 2002;51:448–466. doi: 10.1002/ana.10143. [DOI] [PubMed] [Google Scholar]
- 95.Smith D, Wallom KL, Williams IM, Jeyakumar M, Platt FM. Beneficial effects of anti-inflammatory therapy in a mouse model of Niemann-Pick disease type C1. Neurobiology of disease. 2009;36:242–251. doi: 10.1016/j.nbd.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 96.Williams IM, Wallom KL, Smith DA, Al Eisa N, Smith C, Platt FM. Improved neuroprotection using miglustat, curcumin and ibuprofen as a triple combination therapy in Niemann-Pick disease type C1 mice. Neurobiology of disease. 2014;67:9–17. doi: 10.1016/j.nbd.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 97.Cialone J, Augustine EF, Newhouse N, Adams H, Vierhile A, Marshall FJ, de Blieck EA, Kwon J, Rothberg PG, Mink JW. Parent-reported benefits of flupirtine in juvenile neuronal ceroid lipofuscinosis (Batten disease; CLN3) are not supported by quantitative data. Journal of inherited metabolic disease. 2011;34:1075–1081. doi: 10.1007/s10545-011-9346-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.