

Abstract
Inhaled anesthetics have been shown to increase the aggregation of amyloid beta in vitro through the stabilization of intermediate toxic oligomers, which are thought to contribute to neurocognitive dysfunction in Alzheimer's disease. Inhaled anesthetics may escalate cognitive dysfunction through enhancement of these intermediate oligomer concentrations. We intermittently exposed 12-month-old Tg2576 transgenic mice and nontransgenic littermates to isoflurane and halothane for 5 days. Cognitive function was measured before and after anesthetic exposures using the Morris Water Maze; amyloid beta plaque burden and caspase-3 mediated apoptosis were quantified by immunohistochemistry. At 12 months of age, anesthetic exposure did not further enhance cognitive decline in the transgenic mice. Immunohistochemistry, however, revealed that the halothane-exposed Tg2576 mice had more amyloidopathy than the isoflurane treated mice or the nonexposed transgenic mice. Isoflurane exposure impaired cognitive function in the nontransgenic mice, implying an alternative pathway for neurodegeneration. These findings indicate that inhaled anesthetics influence cognition and amyloidogenesis, but that the mechanistic relationship remains unclear.
Keywords: Anesthesia, Isoflurane, Halothane, Transgenic mice, Neurodegenerative disease, Morris Water Maze, Immunohistochemistry, Caspase-3, Amyloid, Cognitive dysfunction, Learning, Memory, Inhaled anesthetics, Alzheimer's
1. Introduction
Every year, more than 100 million people undergo surgery worldwide, most of which is carried out under general anesthesia, and most of these with an inhaled anesthetic, such as isoflurane, halothane, or sevoflurane. Today, because of the improved safety of anesthesia through the use of advanced monitoring technology and training, increasing numbers of the elderly are safely enduring general anesthesia; even though the mechanism of action and the full measure of possible side effects of the inhaled anesthetics have yet to be elucidated. These drugs clearly influence cognition, at least temporarily, in that the patient is made unconscious, unaware, insensate and amnesic for the duration of the surgery, and briefly thereafter. However, there is growing concern that inhaled anesthetics might affect a patient's cognitive abilities beyond the perioperative period, even permanently, and especially at the extremes of age.
Current evidence suggests that some inhaled anesthetics are capable of causing apoptosis, neuronal damage and durable cognitive dysfunction (Culley et al., 2003, 2004a,b; Jevtovic-Todorovic et al., 2003; Kvolik et al., 2005; Loop et al., 2005; Muravchick and Smith, 1995; Wei et al., 2005; Xie et al., 2006; Xie and Tanzi, 2006; Yon et al., 2005). Potential mechanisms are varied but a recent study shows that anesthetics can enhance protein misfolding and aggregation. Thus, anesthetic-induced neuronal injury could follow similar pathways as the neurodegenerative disorders, such as Alzheimer's or Parkinson's disease (Eckenhoff et al., 2004; Ghirlanda et al., 2004; Xie et al., 2006). Interestingly, the age of Alzheimer's disease onset has been associated with previous surgery at odds ratios of 1.2–1.7 in three different studies (Bohnen et al., 1994; Gasparini et al., 2002; Lee et al., 2005), which, while the studies themselves were not large enough to show statistical significance, certainly constitutes a worrisome trend.
Although the mechanism of neurodegeneration in Alzheimer's disease is still unclear, the leading theory implicates the accumulation and aggregation of amyloid-β peptides into a variety of soluble oligomers, ranging in size from 4 to 20 monomers, instead of the insoluble fibrils found in plaque (Billings et al., 2005; Catalano et al., 2006; Chromy et al., 2003; Kayed et al., 2003; Klein et al., 2004; Lesne et al., 2006; Petkova et al., 2005; Selkoe, 2004). These small toxic oligomers may then initiate a second sequence, associated with the hyperphosphorylation of tau followed by aggregation into neurofibrillary tangles, another hallmark of Alzheimer's neuropathogenesis in humans (Avila, 2006; Binder et al., 2005; Churcher, 2006; Gotz et al., 2001; Lee and Trojanowski, 2006; Oddo et al., 2006). Nevertheless, it remains unclear how amyloid-β aggregation, neurotoxicity and/or tauopathy occurs.
Inhaled anesthetics are mostly small haloalkanes or haloethers that preferentially bind to hydrophobic cavities in proteins (Eckenhoff, 2001; Eckenhoff and Johansson, 1997; Liu et al., 2005). Although the amyloid-β monomer is too small to contain cavities, the oligomers theoretically could possess such features at their interpeptide interfaces. If so, and if of appropriate size, inhaled anesthetics should bind and stabilize oligomers, enhancing their rate of formation. In fact, we previously demonstrated that isoflurane and halothane do increase aggregation of amyloid beta proteins and induce cytotoxicity (Eckenhoff et al., 2004). A follow up study reported that the population of small oligomers was enhanced by anesthetics (Carnini et al., in press). In addition to aggregation, other investigators have reported that anesthetics alter APP processing, increasing amyloid-β levels and apoptosis in H4 human neuroglioma cell lines after clinically relevant isoflurane exposures (Xie et al., 2006).
To determine whether these in vitro effects have an in vivo correlate, we subjected Alzheimer Tg2576 mice, and their nontransgenic littermates to isoflurane and halothane at 12 months of age and studied subsequent behavior and changes in brain amyloid beta plaque burden and caspase-3 mediated apoptosis. The Tg2576 transgenic mouse, harboring the human APP-Swedish mutation, produces ever-increasing amyloid-β concentrations, leading to age-dependent memory deficits by 9–10 months of age (Hsiao et al., 1996).
2. Methods
2.1. Animals
Animal protocols were approved as required by the University of Pennsylvania Institutional Animal Care and Use Committees and all mice were treated in strict accordance to APS/NIH guidelines. Eleven to twelve-month-old female Tg2576 mice and nontransgenic wild-type littermates, bred by mating Tg2576 males with C57B6/SJL F1 females, were used in this study. The APP-Swedish mutation inserted into the mice is a human APP gene with a double mutation at 670 (Lys → Asn) and 671 (Met → Leu) that creates a transgenic hemizygote characterized by a significant increase in amyloid-β levels, with the appearance of amyloid-β plaque at 11–13 months and altered behavior at 9–10 months of age (Hsiao et al., 1996). All mice were bred in our colony at the University of Pennsylvania. Initial breeding pairs were a gift from Dr. Virginia Lee, University of Pennsylvania. The mice were genotyped from tail biopsies by means of proteinase K digest, DNA extraction and PCR probe.
2.2. Anesthetic exposures
Female Tg2576 mice and wild-type littermates were anesthetized with either humidified 0.8 MAC halothane (2-bromo-2-chloro-1,1,1-trifluoroethane) (0.8–1%) or isoflurane (1-chloro-2,2,2-trifluoroethyl difluoromethyl ether) (0.9–1%) in 30% oxygen, balanced by N2. Both groups were exposed to anesthetics for 120 min per day for 5 days, for a total of five-exposures. The exposures were carried out in a monitored chamber in a fume hood. The mice breathed spontaneously without intubation or other support while being warmed with a heating pad. Anesthetic gas phase concentrations were verified by IR spectroscopy. Daily body weights and temperatures were recorded for each animal at the end of the exposures and no changes were detected. Control Tg2576 and nontransgenic littermates were exposed to humidified 30% O2 balanced by N2 for 120 min in similar chambers concurrently. After the exposures, the animals were returned to their cages, and in all cases, recovery was rapid and complete within 10 min.
2.3. Behavioral testing
2.3.1. Morris Water Maze
Before and after anesthetic exposures, behavioral testing of the Tg2576 mice and wild-type littermates was performed using the Morris Water Maze. A schematic of the experimental paradigm is shown in Fig. 1 and explained in detail below. A round, fiberglass pool, 1.5m in diameter, was filled with water to a height of 1.5 cm above the top of the movable clear 15 cm diameter platform. Water was maintained at 20 °C and opacified with titanium dioxide throughout all training and testing. Mice were tracked with a video camera mounted above the pool that fed to a computer running IMAQ PCI-1407 software (National Instruments, Austin TX; www.ni.com). After every trial, each mouse was placed in a holding cage, under a heat lamp until dry (~1–2 min), before returning to its regular cage. The time between trials was at least 30–45 min. Body temperatures were maintained by active heating and protracted intervals between trials. Mice were omitted from the study if they were consistent poor performers in the pre-exposure visual cued or reference memory tests. Poor performance was defined as failure to find the cued platform, excessive floating or thigmotaxis. In all, 8% nontransgenic and 12.5% transgenic mice were eliminated from the study.
Fig. 1.

Schematic time-line of the experimental paradigm. RMT= reference memory testing.
2.3.2. Pre-exposure cued training
All mice were trained, 4 trials per day for 5 days, to swim to the platform, which was marked with a vertical flag (Fig. 1, days 1–5). They were allotted 60 s to find the platform upon which they sat for 10–15 s before being removed from the pool. If an animal did not find the platform within 60 s, it was gently guided to the platform and allowed to remain there for 10–15 s. The platform was placed in the target quadrant for all trials but the starting points were random for each mouse. A white curtain surrounded the pool to prevent confounding visual cues.
2.3.3. Pre-exposure reference memory testing
Two days after the end of cued training, pre-exposure reference memory testing (RMT) was performed for 5 days (Fig. 1, days 8–12). The white curtain and platform flag were removed and fixed external visual cues were provided to enable reference memory. The platform was placed in the target quadrant for all trials but the starting points were random for each mouse. In order to reach criterion, the mice were required to reach the platform within 60 s and were given 4 trials per day. After 60 s, the mice were gently guided to the platform and required to remain for 10–15 s. The time to reach the platform and the swim speed were recorded for each trial.
2.3.4. Pre-exposure probe test
After all mice completed the last reference memory trial on the 5th day, a probe test was performed to determine memory retention (Fig. 1, day 12). For this trial, the platform was removed from the water maze and the movements of the mice were recorded for 60 s. The time spent in each quadrant was measured.
2.3.5. Post-exposure reference memory and probe testing
After the anesthetic exposures, animals were allowed to recover for 2 days to assure all acute cognitive effects attributable to the anesthetics per se had dissipated. The mice were given 1 day of reference memory testing and a probe test (Fig. 1, day 22). This consisted of 4 reference memory trials, as performed prior to the anesthetic exposures, where the hidden platform was in the target quadrant, the starting points for the mice were random and the same visual cues were present. The time to reach the platform and the swim speed were recorded. After all mice completed the 4th RMT trial, a probe test was given as described above.
2.3.6. Post-exposure spatial memory testing
The day after the post-exposure reference memory/probe testing, the mice began 10 days of spatial memory testing (Fig. 1, days 23–32). For these trials, the criterion for advancing to a new position was reaching the platform position in less than 15 s for 3 consecutive trials. If criterion was not reached within 8 trials, then the mouse would begin the next day anew with the same platform location and 8 trials per day until criterion was reached or through day 10. For these trials, the time to reach the platform, the number of trials, and the swim speed were recorded.
2.4. Immunohistochemistry
One to four days after the last day of behavioral testing, all animals were briefly anesthetized with halothane, perfused via the left ventricle with ice cold 0.1 M PBS with simultaneous exsanguination from the right atrium. The brains were quickly removed, placed on ice, and hemisected in the sagittal plane. The time required from loss of consciousness to brain removal was routinely less than 5 min. One half of the brain was fixed in 4% paraformaldyhyde overnight at 4 °C and then processed for paraffin embedding. Serial coronal 8–10 μm sections were cut and at least three slides for each animal were subjected to amyloid beta or caspase-3 immunohistochemical techniques using DAB or immunofluorescence detection, respectively. Briefly, the sections were dewaxed, rehydrated through a series of graded alcohols, and washed in distilled water followed by PBS. Antigen retrieval was performed by incubation in 0.1 M PBS for 20 min in a steamer at 95 °C. The sections were then incubated in 3% hydrogen peroxide in 0.1 M PBS for 5 min to quench endogenous peroxidase activity and then washed. For amyloid beta staining, the Vectastain MOM kit (Vector Labs) was utilized. Nonspecific antibody binding was inhibited by incubating the sections in MOM Mouse Ig Blocking Reagent for 1 h. Next, slides were incubated with the primary antibody to amyloid beta at 1:50 (Vector Labs). Following the primary antibody incubation, sections were washed in PBS, incubated in biotinylated anti-mouse IgG (1:250) for 10 min, washed again and incubated with ABC reagent. Finally, slides were developed with DAB for approximately 6 min. The sections were counter-stained with hematoxylin and viewed and quantitated with an Olympus IX70 Microscope equipped with a SensiCam SVGA High-Speed Cooled Digital Camera (Cooke Corp., Auburn Hills, MI www.cookecorp.com) and IPLab Suite v3.6 Imaging Processing and Analysis software (Biovision Technologies, Exton, PA www.BioVis.com). To obtain an accurate quantification, nonselective background staining was subtracted from the amyloid beta positive slides by comparing them to nontransgenic control slides. The mean density of plaques and the total area occupied by plaques in the cortex, hippocampus and dentate gyrus was determined from at least six sections per animal using a customized program developed for us by Biovision Technologies (Exton, PA www.BioVis.com). For caspase-3 staining, slides were incubated with blocking buffer for 1 h, washed, and then incubated at 4 °C overnight with anti-activated caspase-3 (Cell Signaling) at 1:100. After washing the next day, sections were incubated in a dark chamber with IgG-FITC (Santa Cruz) at 1:200 for 1 h at room temperature followed by counterstain with propidium iodide at 1:3000 for 30 min. Finally, sections were wet mounted and viewed immediately using appropriate filters on the microscope described previously. The density of caspase-positive cells from at least nine sections per animal was calculated. The regions were photographed and caspase-positive cells were counted by eye per unit area.
2.5. Statistics
One-way ANOVA followed by Student–Newman–Keuls post hoc tests was used to analyze the water maze escape latency, swim speed and probe trial quadrant data. The percent to reach criterion data was empirically modeled using least-squares regression and then compared using F-tests. Finally, the density of amyloid plaques and caspase-positive cells and the percent area occupied by the plaques per unit area in either the wild-type or transgenic anesthetic exposed groups were compared to their respective controls and tested using the one-way Analysis of Variance (ANOVA) with the post hoc Dunnett's Multiple Comparisons Test.
3. Results
3.1. Reference memory testing
Prior to the anesthetic exposures, the Tg2576 (Tg) mice took longer to reach the platform than the nontransgenic wild-type (Wt) mice. After exposure to the inhaled anesthetics, halothane (Hal) or isoflurane (Iso), all mice including controls took longer to reach the platform, with the transgenic mice taking even longer than wild-type. There were no changes in the escape latencies of any group before and after the exposures that were attributable to anesthetic exposure (Fig. 2) (Tg-Hal, n = 12; Tg-Iso, n = 5; Tg-Con, n = 12; Wt-Hal, n = 14; Wt-Iso, n = 7; Wt-Con, n = 24).
Fig. 2.

Escape latencies from the reference memory testing for transgenic (A) and wild-type mice (B). No changes were found in the escape latencies of any group before and after the exposures. The x-axis, days, corresponds to the testing paradigm in Fig. 1 for pre-exposures (days 8–12) and post-exposure (day 22). Black squares and solid line, controls; open triangles and dashed line, halothane-exposed; black circles and small dashed lines, isoflurane-exposed (Tg-Hal, n = 12; Tg-Iso, n = 5; Tg-Con, n = 12; Wt-Hal, n = 14; Wt-Iso, n = 7; Wt-Con, n = 24).
3.2. Spatial memory testing
After the anesthetic exposures, significantly fewer Tg2576 mice reached criterion than Wt mice for both the exposed and control groups (p < 0.0001) (Fig. 3A). No anesthetic effect could be discerned in the transgenic mice (Fig. 3B). In the Wt animals (Fig. 3C), both controls and those exposed to halothane performed at about the same level. However, significantly fewer isoflurane exposed Wt mice reached criterion than unexposed control Wt mice (p < 0.0001) (Tg-Con, n = 14; Tg-Hal, n = 12; Tg-Iso, n = 5; Wt-Con, n = 25; Wt-Hal, n = 14; Wt-Iso, n = 11). Exposures to anesthetics had no detectable motor effect as reflected by the average swim speed during the spatial memory testing trials (Fig. 4).
Fig. 3.

The percent of transgenic and wild-type mice that reached criterion in the spatial memory testing after anesthetic exposures. The x-axis represents the number of different hidden platform locations reached by the mice during the 10 days of testing. (A) Control mice: wild-type (solid squares, solid line) (n = 25) compared to transgenic (open squares, solid line) (n = 14) (p < 0.0001). (B) Transgenic mice: controls (open squares, solid line) (n = 14), isoflurane exposed (open circles, dashed line) (n=5) and halothane-exposed (open triangles, dotted line) (n = 12) (p > 0.05). No significant difference was detected between any transgenic group. (C) Wild-type mice: controls (solid squares, solid line) (n = 25), isoflurane exposed (solid circles, dashed line) (n = 11) and halothane-exposed (solid triangles, dotted line) (n = 14). Performance in the isoflurane group was significantly different from control, but not in the halothane group.
Fig. 4.
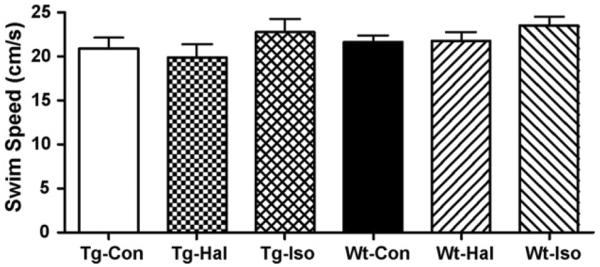
The average swim speeds of transgenic (Tg) and wild-type (Wt) mice during the spatial memory testing after exposures to halothane (Hal) or isoflurane (Iso). No significant differences in swim speed were observed (p = 0.4037) (Tg-Con, n = 14; Tg-Hal, n = 12; Tg-Iso, n = 5; Wt-Con, n = 25; Wt-Hal, n = 14; Wt-Iso, n = 11).
3.3. Memory retention testing
Probe trails were performed before and after anesthetic exposure. The Tg2576 mice demonstrated memory of the platform quadrant pre-exposure when comparing target quadrant percent time versus other quadrants (Con, p < 0.001; Hal, p < 0.01; Iso, p < 0.05) (Fig. 5A). This memory ability continued in the halothane group after exposure (p < 0.05, n = 6), but not in the isoflurane group (p > 0.05, n = 5). However, since the “post-exposure” control group also showed no preference for the target quadrant (p > 0.05, n = 10), we cannot attribute the decrement in memory in the isoflurane group to the drug. Wild-type mice spent significantly more time searching the target quadrant for the platform than the other quadrants pre-exposure (Fig. 5B) (Con, p < 0.05; Hal, p < 0.05; Iso, p < 0.05). This memory ability continued after anesthetic exposures in the control and halothane groups, but not in the isoflurane exposed group (Con, p < 0.01, n = 10; Hal, p < 0.05, n = 6; Iso, p > 0.05, n = 7), consistent with the spatial memory task results in the Wt animals. This result is further confirmed by comparing the time spent in the target quadrant with the mean of the time spent in the other three quadrants both pre-exposure (Con, p < 0.01; Hal, p < 0.001; Iso, p < 0.01) and post-exposure (Con, p < 0.001; Hal p < 0.001; Iso, p > 0.05), demonstrating a lack of search polarity and an impairment in memory in the isoflurane exposed wild-type mice.
Fig. 5.

The percent of total time that mice spent in each quadrant during pre- and post-exposure probe tests for controls (black), halothane (grey) and isoflurane (white) exposed mice. (A) Transgenic mice, prior to anesthetic exposure, spent more time in the target quadrant than the other quadrants. However, after the exposures, only the halothane group spent significantly more time in the target quadrant. (B) Wild-type mice significantly preferred the target over adjacent or opposite quadrants both before and after anesthetic exposures, except for the post-exposure isoflurane group (*, p < 0.05 target vs. opposite and adjacent quadrants; +, p < 0.05 target vs. opposite quadrant only) (Tar = target, Adj = adjacent, Opp = opposite) (Tg-Con, n = 10; Tg-Hal, n = 6; Tg-Iso, n = 5; Wt-Con, n = 10; Wt-Hal, n = 6; Wt-Iso, n = 7).
3.4. Immunohistochemistry
Significant numbers of plaque were found in the transgenic mice at 12 months of age (Fig. 6B) and, as expected, no plaques were found in the nontransgenic wild-type mice (Fig. 6A). The density of plaques in the cortex, CA1 region and the dentate gyrus of the hippocampus combined (Fig. 7A), was significantly elevated in the halothane-exposed transgenic animals (p < 0.001) compared to controls, while no difference was found in the isoflurane-exposed mice. The percent area occupied by amyloid beta plaques was not significantly different in either anesthetic group (Fig. 7B) (Con, n = 13; Hal, n = 8; Iso, n = 5), although a trend for being increased in the halothane group was apparent.
Fig. 6.

Micrographs of a wild-type mouse brain (A) and transgenic brain (B). Variously sized amyloid plaques were found in the cortex, CA1 region and dentate gyrus in the transgenic brain at 12 months of age (B). Scale bar = 180 μm.
Fig. 7.

The plaque load in the Tg2576 mouse cortex, CA1 and dentate gyrus combined. (A) The density of plaques in controls and anesthetic exposedmice. The halothane-exposed Tg2576 mice had a significantly increased plaque burden compared to control Tg2576 mice, p < 0.05. (B) The percent area occupied by plaque normalized to area. No significant differences were detected in the anesthetic exposed groups (Con, n = 13; Hal, n = 8; Iso, n = 5).
Caspase-3 positive cells were found in both wild-type and transgenic anesthetic exposed and control mice (Fig. 8). The density of caspse-3 positive cells in the three regions combined was significantly greater in the transgenic control mice than the wild-type mice (p < 0.02). However, the anesthetic effect on apoptosis was limited to nonsignificant positive trends in the exposed transgenic and wild-type mice as compared to their respective controls (Fig. 9) (Wt: Con, n = 12; Hal, n = 6; Iso, n = 6; Tg: Con, n = 9; Hal, n = 7; Iso, n = 5).
Fig. 8.
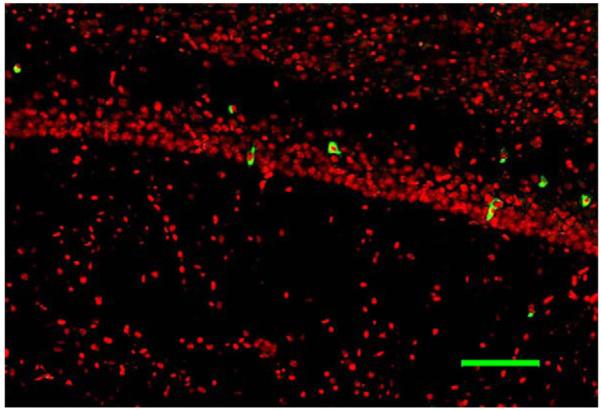
Caspase-3 immunohistochemistry of the CA1 region of the hippocampus in a transgenic mouse exposed to isoflurane. Caspase-3 positive cells are green. Scale bar = 50μm.
Fig. 9.

The density of caspase-3 positive cells in the cortex, CA1 and dentate gyrus combined. Overall, the density of caspase-positive cells is significantly greater in all transgenic mice than wild-type mice (p < 0.02), but an anesthetic effect could not be demonstrated (Wt-Con, n = 12; Wt-Hal n = 6; Wt- Iso n = 6; Tg-Con, n = 9; Tg-Hal, n = 7; Tg-Iso, n = 5).
4. Discussion
In this study, we have demonstrated that moderate exposures to inhaled anesthetics can produce subtle but durable alterations in the aging rodent central nervous system. Consistent with our protein-based hypothesis, we find that halothane enhances plaque deposition in the Tg2576 mouse model, but that isoflurane does not. On the other hand, in wild-type animals, isoflurane causes a decrease in cognitive performance whereas halothane does not. These findings emphasize that these drugs are not only capable of producing sustained alterations in the CNS, but that they may do so via different mechanisms. Whether these effects persist longer than a few weeks is not yet clear.
Our previous work showed that isoflurane and halothane enhance oligomerization and cytotoxicity of amyloid-β in vitro by means of a reduction in solubility of the amyloid-β monomer, in part through a preference for binding and stabilizing the intermediate oligomers (Carnini et al., in press; Eckenhoff et al., 2004). Halothane was more potent than isoflurane, and the effect was small at clinical concentrations. Thus, the predicted effect of anesthetic exposure in an animal model that expresses human amyloid-β is an enhancement of oligomer formation and subsequent plaque deposition, especially in those exposed to halothane. Our results are consistent in that halothane enhanced plaque load and there was a nonsignificant trend for isoflurane to do the same. Interestingly, the plaque data suggests that the size distribution shifts to smaller plaques in the halothane group (a larger increase in number than area), the expected result if a recent perturbation had increased the available oligomer. In fact, it is striking that this increase was detectable only 2 weeks after anesthetic exposure.
Despite these modest effects on plaque deposition, neither anesthetic produced a detectable alteration in the cognitive performance of the Tg2576 mouse on the Morris Water Maze. Although puzzling, the relationship between plaque burden and neurodegeneration or cognitive performance is known to be weak in this mouse model (Westerman et al., 2002). A lack of anesthetic effect on behavior might also indicate an already advanced degree of neuronal or synaptic injury at the time of exposure. This is supported by the fact that at 12 months of age, the transgenic mice demonstrated significant cognitive impairments compared to their wild-type littermate controls. Thus, a small incremental effect of anesthetic-enhanced oligomerization may have been difficult to detect. The clinical correlate of this study might be those patients with a diagnosis of Alzheimer disease at the time of surgery (quite common). Thus, these studies may warrant repeating at an earlier time point in this model, and/or in another mouse model, to more closely represent the group of patients with mild cognitive impairment at the time of surgery, a much larger cohort than those with established Alzheimer disease.
Interestingly, cognitive performance was clearly influenced by isoflurane in the wild-type littermate control animals. Two features suggest that this effect is unrelated to our protein oligomerization hypothesis. First, wild-type animals do not develop plaque or, indeed, detectable levels of insoluble amyloid-β. Second, although there might be other potentially neurotoxic proteins susceptible to anesthetic-induced oligomerization, the opposite rank order to what we observed in vitro (halothane > isoflurane) renders this unlikely. Further, isoflurane has been previously demonstrated to be neurotoxic in both very young and aged wild-type rats (Culley et al., 2003, 2004a,b; Jevtovic-Todorovic et al., 2003; Yon et al., 2005). At least one potential mechanism for isoflurane-induced neurotoxicity is calcium dysregulation (Wei et al., 2005), whereby the anesthetic induces endoplasmic reticulum calcium depletion, triggering apoptosis. Halothane is less potent at inducing calcium depletion than isoflurane, in agreement with our results in the wild-type animals. The fact that an isoflurane effect could not be detected in the Tg2576 model is further support for our hypothesis that incremental effects of potential neurotoxic agents become difficult to detect in already established compromise.
Thus, these results confirm that anesthetics may have effects on the CNS that outlast the period of anesthesia itself. Whether these effects are reversible is not yet clear, but reversibility is likely to depend on the degree of reserve or plasticity. Thus, durable effects might be undetectable in younger animals without compromise (or genetic vulnerability), but are predicted to become apparent in animals with a degree of compromise, such as early neurodegenerative disease, head trauma or advanced age. This is consistent with recent clinical reports of post-operative cognitive dysfunction, and the fact that age appears to be the primary risk factor (Abildstrom et al., 2000; Ancelin et al., 2001; Bedford, 1955; Davidsson et al., 2002; Johnson et al., 2002; Moller et al., 1998). It is too early to alter clinical practice based on these data, but the ability of volatile anesthetics to produce changes in cognition and aggregation of proteins suggests the need for closer examination of this issue, particularly in the elderly, and the need to investigate new anesthetic compounds.
Acknowledgements
This work was supported by the Austin Lamont Endowment and the T32 GM 7612 to the Department of Anesthesiology and Critical Care, University of Pennsylvania. The authors would like to thank Angelina Sylvestro for tissue sectioning and technical assistance, Gia Guo for genotyping of the transgenic mice, and the Center for Neurodegenerative Disorders at the University of Pennsylvania for technical assistance.
Footnotes
Conflicts of interests All authors state that there are no actual or potential conflicts of interests.
References
- Abildstrom H, Rasmussen LS, Rentowl P, Hanning CD, Rasmussen H, Kristensen PA, Moller JT. Cognitive dysfunction 1–2 years after non-cardiac surgery in the elderly. ISPOCD group. International Study of Post-Operative Cognitive Dysfunction. Acta Anaesthesiol. Scand. 2000;44(10):1246–1251. doi: 10.1034/j.1399-6576.2000.441010.x. [DOI] [PubMed] [Google Scholar]
- Ancelin ML, de Roquefeuil G, Ledesert B, Bonnel F, Cheminal JC, Ritchie K. Exposure to anaesthetic agents, cognitive functioning and depressive symptomatology in the elderly. Br. J. Psychiatry. 2001;178:360–366. doi: 10.1192/bjp.178.4.360. [DOI] [PubMed] [Google Scholar]
- Avila J. Tau phosphorylation and aggregation in Alzheimer's disease pathology. FEBS Lett. 2006;580(12):2922–2927. doi: 10.1016/j.febslet.2006.02.067. [DOI] [PubMed] [Google Scholar]
- Bedford PD. Adverse cerebral effects of anesthesia on old people. Lancet. 1955;ii:259–263. doi: 10.1016/s0140-6736(55)92689-1. [DOI] [PubMed] [Google Scholar]
- Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45(5):675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Binder LI, Guillozet-Bongaarts AL, Garcia-Sierra F, Berry RW. Tau, tangles, and Alzheimer's disease. Biochim. Biophys. Acta. 2005;1739(2–3):216–223. doi: 10.1016/j.bbadis.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Bohnen N, Warner MA, Kokmen E, Beard CM, Kurland LT. Alzheimer's disease and cumulative exposure to anesthesia: a case-control study. J. Am. Geriatr. Soc. 1994;42(2):198–201. doi: 10.1111/j.1532-5415.1994.tb04952.x. [DOI] [PubMed] [Google Scholar]
- Carnini A, Lear JD, Eckenhoff RG. Inhaled anesthetic modulation of amyloid beta1-40 assembly and growth. Curr. Alzheimer Res. 2007 doi: 10.2174/156720507781077278. in press. [DOI] [PubMed] [Google Scholar]
- Catalano SM, Dodson EC, Henze DA, Joyce JG, Krafft GA, Kinney GG. The role of amyloid-beta derived diffusible ligands (ADDLs) in Alzheimer's disease. Curr. Top. Med. Chem. 2006;6(6):597–608. doi: 10.2174/156802606776743066. [DOI] [PubMed] [Google Scholar]
- Chromy BA, Nowak RJ, Lambert MP, Viola KL, Chang L, Velasco PT, Jones BW, Fernandez SJ, Lacor PN, Horowitz P, Finch CE, Krafft GA, Klein WL. Self-assembly of Abeta (1–42) into globular neurotoxins. Biochemistry. 2003;42(44):12749–12760. doi: 10.1021/bi030029q. [DOI] [PubMed] [Google Scholar]
- Churcher I. Tau therapeutic strategies for the treatment of Alzheimer's disease. Curr. Top. Med. Chem. 2006;6(6):579–595. doi: 10.2174/156802606776743057. [DOI] [PubMed] [Google Scholar]
- Culley DJ, Baxter M, Yukhananov R, Crosby G. The memory effects of general anesthesia persist for weeks in young and aged rats. Anesth. Analg. 2003;96(4):1004–1009. doi: 10.1213/01.ANE.0000052712.67573.12. [DOI] [PubMed] [Google Scholar]
- Culley DJ, Baxter MG, Crosby CA, Yukhananov R, Crosby G. Impaired acquisition of spatial memory 2 weeks after isoflurane and isoflurane-nitrous oxide anesthesia in aged rats. Anesth. Analg. 2004a;99(5):1393–1397. doi: 10.1213/01.ANE.0000135408.14319.CC. [DOI] [PubMed] [Google Scholar]
- Culley DJ, Baxter MG, Yukhananov R, Crosby G. Long-term impairment of acquisition of a spatial memory task following isoflurane-nitrous oxide anesthesia in rats. Anesthesiology. 2004b;100(2):309–314. doi: 10.1097/00000542-200402000-00020. [DOI] [PubMed] [Google Scholar]
- Davidsson P, Westman-Brinkmalm A, Nilsson CL, Lindbjer M, Paulson L, Andreasen N, Sjogren M, Blennow K. Proteome analysis of cerebrospinal fluid proteins in Alzheimer patients. Neuroreport. 2002;13(5):611–615. doi: 10.1097/00001756-200204160-00015. [DOI] [PubMed] [Google Scholar]
- Eckenhoff RG. Promiscuous ligands and attractive cavities: How do the inhaled anesthetics work? Mol. Interv. 2001;1:258–268. [PubMed] [Google Scholar]
- Eckenhoff RG, Johansson JS. Molecular interactions between inhaled anesthetics and proteins. Pharmacol. Rev. 1997;49(4):343–367. [PubMed] [Google Scholar]
- Eckenhoff RG, Johansson JS, Wei H, Carnini A, Kang B, Wei W, Pidikiti R, Keller JM, Eckenhoff MF. Inhaled anesthetic enhancement of amyloid-beta oligomerization and cytotoxicity. Anesthesiology. 2004;101(3):703–709. doi: 10.1097/00000542-200409000-00019. [DOI] [PubMed] [Google Scholar]
- Gasparini M, Vanacore N, Schiaffini C, Brusa L, Panella M, Talarico G, Bruno G, Meco G, Lenzi GL. A case-control study on Alzheimer's disease and exposure to anesthesia. Neurol. Sci. 2002;23(1):11–14. doi: 10.1007/s100720200017. [DOI] [PubMed] [Google Scholar]
- Ghirlanda G, Hilcove SA, Pidikiti R, Johansson JS, Lear JD, DeGrado WF, Eckenhoff RG. Volatile anesthetic modulation of oligomerization equilibria in a hexameric model peptide. FEBS Lett. 2004;578(1–2):140–144. doi: 10.1016/j.febslet.2004.10.087. [DOI] [PubMed] [Google Scholar]
- Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293(5534):1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274(5284):99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Jevtovic-Todorovic V, Hartman RE, Izumi Y, Benshoff ND, Dikranian K, Zorumski CF, Olney JW, Wozniak DF. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J. Neurosci. 2003;23(3):876–882. doi: 10.1523/JNEUROSCI.23-03-00876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T, Monk T, Rasmussen LS, Abildstrom H, Houx P, Korttila K, Kuipers HM, Hanning CD, Siersma VD, Kristensen D, Canet J, Ibanaz MT, Moller JT. Postoperative cognitive dysfunction in middle-aged patients. Anesthesiology. 2002;96(6):1351–1357. doi: 10.1097/00000542-200206000-00014. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300(5618):486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Klein WL, Stine WB, Jr., Teplow DB. Small assemblies of unmodified amyloid beta-protein are the proximate neurotoxin in Alzheimer's disease. Neurobiol. Aging. 2004;25(5):569–580. doi: 10.1016/j.neurobiolaging.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Kvolik S, Glavas-Obrovac L, Bares V, Karner I. Effects of inhalation anesthetics halothane, sevoflurane, and isoflurane on human cell lines. Life Sci. 2005;77(19):2369–2383. doi: 10.1016/j.lfs.2004.12.052. [DOI] [PubMed] [Google Scholar]
- Lee TA, Wolozin B, Weiss KB, Bednar MM. Assessment of the emergence of Alzheimer's disease following coronary artery bypass graft surgery or percutaneous transluminal coronary angioplasty. J. Alzheimers Dis. 2005;7(4):319–324. doi: 10.3233/jad-2005-7408. [DOI] [PubMed] [Google Scholar]
- Lee VM, Trojanowski JQ. Progress from Alzheimer's tangles to pathological tau points towards more effective therapies now. J. Alzheimers Dis. 2006;9(3 Suppl):257–262. doi: 10.3233/jad-2006-9s328. [DOI] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440(7082):352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Liu R, Loll PJ, Eckenhoff RG. Structural basis for high-affinity volatile anesthetic binding in a natural 4-helix bundle protein. FASEB J. 2005;19(6):567–576. doi: 10.1096/fj.04-3171com. [DOI] [PubMed] [Google Scholar]
- Loop T, Dovi-Akue D, Frick M, Roesslein M, Egger L, Humar M, Hoetzel A, Schmidt R, Borner C, Pahl HL, Geiger KK, Pannen BH. Volatile anesthetics induce caspase-dependent, mitochondria-mediated apoptosis in human T lymphocytes in vitro. Anesthesiology. 2005;102(6):1147–1157. doi: 10.1097/00000542-200506000-00014. [DOI] [PubMed] [Google Scholar]
- Moller JT, Cluitmans P, Rasmussen LS, Houx P, Rasmussen H, Canet J, Rabbitt P, Jolles J, Larsen K, Hanning CD, Langeron O, Johnson T, Lauven PM, Kristensen PA, Biedler A, van Beem H, Fraidakis O, Silverstein JH, Beneken JE, Gravenstein JS. Long-term postoperative cognitive dysfunction in the elderly ISPOCD1 study. ISPOCD investigators. International Study of Post-Operative Cognitive Dysfunction. Lancet. 1998;351(9106):857–861. doi: 10.1016/s0140-6736(97)07382-0. erratum appears in Lancet 1998 June 6;351(9117):1742. [DOI] [PubMed] [Google Scholar]
- Muravchick S, Smith DS. Parkinsonian symptoms during emergence from general anesthesia. Anesthesiology. 1995;82(1):305–307. doi: 10.1097/00000542-199501000-00039. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, LaFerla FM. Temporal profile of Abeta oligomerization in an in vivo model of Alzheimer's disease: a link between Abeta and tau pathology. J. Biol. Chem. 2006;281(3):1599–1604. doi: 10.1074/jbc.M507892200. [DOI] [PubMed] [Google Scholar]
- Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer's beta-amyloid fibrils. Science. 2005;307(5707):262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Cell biology of protein misfolding: the examples of Alzheimer's and Parkinson's diseases. Nat. Cell Biol. 2004;6(11):1054–1061. doi: 10.1038/ncb1104-1054. [DOI] [PubMed] [Google Scholar]
- Wei H, Kang B, Wei W, Liang G, Meng QC, Li Y, Eckenhoff RG. Isoflurane and sevoflurane affect cell survival and BCL-2/BAX ratio differently. Brain Res. 2005;1037(1–2):139–147. doi: 10.1016/j.brainres.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin SG, Ashe KH. The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer's disease. J. Neurosci. 2002;22(5):1858–1867. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Dong Y, Maeda U, Alfille P, Culley DJ, Crosby G, Tanzi RE. The common inhalation anesthetic isoflurane induces apoptosis and increases amyloid beta protein levels. Anesthesiology. 2006;104(5):988–994. doi: 10.1097/00000542-200605000-00015. [DOI] [PubMed] [Google Scholar]
- Xie Z, Tanzi RE. Alzheimer's disease and post-operative cognitive dysfunction. Exp. Gerontol. 2006;41(4):346–359. doi: 10.1016/j.exger.2006.01.014. [DOI] [PubMed] [Google Scholar]
- Yon JH, Daniel-Johnson J, Carter LB, Jevtovic-Todorovic V. Anesthesia induces neuronal cell death in the developing rat brain via the intrinsic and extrinsic apoptotic pathways. Neuroscience. 2005;135(3):815–827. doi: 10.1016/j.neuroscience.2005.03.064. [DOI] [PubMed] [Google Scholar]