

Abstract
Purpose of review
This review will examine advances in our understanding of the role kidneys play in HDL metabolism and the effect on levels, composition, and function of HDL particles.
Recent findings
Components of the HDL particles can cross the glomerular filtration barrier. Some of these components, including apolipoproteins and enzymes involved in lipid metabolism, are taken up by the proximal tubule and degraded, modified, salvaged/returned to the circulation, or lost in the urine. Injury of the glomerular capillaries or tubules can affect these intrarenal processes and modify HDL. Changes in the plasma and urine levels of HDL may be novel markers of kidney damage and/or mechanism(s) of kidney disease.
Summary
The kidneys have a significant role in metabolism of individual HDL components, which in turn modulate HDL levels, composition and functionality of HDL particles. These intrarenal effects may be useful markers of kidney damage and have consequences on kidney-related perturbations in HDL.
Keywords: HDL, ApoA-I, kidney, chronic kidney disease, cardiovascular disease
Introduction
Excessive tissue accumulation of circulating lipoproteins such as low density lipoprotein (LDL) causes atherosclerotic cardiovascular disease (CVD). By contrast, HDL has cardioprotective effects through elimination of excess cholesterol from peripheral tissues via the liver and gut. HDL levels, and more recently functionality, are believed to underlie these beneficial effects. Thus, processes that determine synthesis, but especially metabolism, of HDL regulate the circulating levels, composition and function of this lipoprotein. Liver, skeletal muscles, adipose tissue and certain cells such as macrophages are considered to be the primary sites of lipoprotein metabolism. Traditionally, the kidneys have not been regarded as important regulators of lipid and lipoprotein metabolism because the glomerular filtration barrier prevents passage of all but the smallest size molecules. However, recent studies have made it clear that HDL is not synthesized as an intact particle but assembled in the circulation from its constituent components including lipids, apolipoproteins, transfer proteins, and enzymes. Critically, metabolism of HDL also does not proceed as removal of the whole intact particle and instead represents metabolism of its individual components such as apolipoprotein A-I, apoE, and apoL. HDL’s other cargo includes proteins involved in lipid metabolism such as cholesteryl ester transfer protein (CETP), lecithin cholesteryl ester acyltransferase (LCAT), phospholipid transfer protein (PLTP), which are also metabolized by the kidneys.
Kidney dysfunction causes abnormalities in lipids and lipoproteins, the extent and character of which depend on the degree of kidney impairment, underlying etiology, and whether proteinuria, especially nephrotic syndrome, is present. This review will focus on the concept of a significant role for kidneys in HDL homeostasis, underscoring the previously underappreciated role for kidneys in metabolism of individual HDL components, which control levels, composition, and functionality, which in turn have consequences on kidney-related perturbations in HDL.
Renal handling of HDL: biosynthesis and metabolism
Biogenesis of HDL begins with hepatic, and to a lesser extent, intestinal production of ApoA-I. Once released into the circulation, assembly and maturation of HDL particles proceeds with lipidation of ApoA-I with cholesterol and phospholipids and interaction with the ATP-binding cassette class A (ABCA1) transporter on various cells, especially macrophages, adipocytes, skin fibroblasts and skeletal muscle cells to form nascent preβ-HDL. This is followed by a critical interaction with the enzyme, LCAT, and formation of cholesteryl ester-rich spherical HDL that circulates in the plasma. HDL acquires additional lipids from other circulating lipoproteins, including VLDL and LDL, and also from circulating albumin. Following this remodeling, HDL is catabolized by plasma proteins and through interactions with hepatic receptors that recycle several of its components. Thus, scavenger receptor class BI (SRBI) binds cholesterol ester-rich HDL2 particles, which undergo hydrolysis by hepatic lipase that detaches ApoA-I, returning it back into the circulation. This hepatic catabolism has long been believed to be the primary pathway of clearance of HDL-associated lipids while endocytic receptors in the liver degrade HDL-associated apolipoproteins. Conspicuously, this classic overview of HDL homeostasis did not include the kidneys. The idea that kidneys do not participate in HDL metabolism is based on limitations imposed by the filtration barrier in the glomerular capillaries that prevent passage of molecules >60–100kD. Although HDL is the smallest and densest of the circulating lipoproteins, all mature spherical HDL subclasses (HDL3, HDL2), exceed this mass. Nonetheless, HDL particles are heterogeneous macromolecules that carry various species of lipids and proteins, and thus, there now is greater appreciation for a kidney role in modulating filtration, reabsorption, degradation, and urinary loss of some components and cargo of HDL particles.
Apolipoprotein A-I, the most abundant protein in HDL, constitutes the nidus around which the particle is assembled. In animals, 30–70% of injected radiolabeled human ApoA-I is cleared by the kidneys [1]. In humans, ApoA-I as well as pre-β HDL are filtered by the glomerular capillaries. This is not unexpected since the molecular weight of ApoA-I (28kD) and pre-β HDL (60–85kD for pre-β), are similar to or smaller than albumin (66.5kD) and thus are predicted to cross the normal glomerular filtration barrier. The discoidal shape of pre-β HDL may also enhance its ability to cross the filtration barrier by changing the particle’s orientation. Currently, there are no studies examining the potential effect of charge on these particles’ glomerular permeability. In preliminary studies, we examined the key role of podocytes in maintaining the normal filtration barrier and its relationship to ApoA-I excretion [2]. Nphs1-hCD25 transgenic mice (Nep25) express the human CD25 receptor on podocytes, which can be selectively injured by injection of immunotoxin, LMB2. Low dose LMB2 caused albuminuria and edema by 2 weeks that recovered by 6 weeks. This exposure led to rare podocyte detachment, diffuse foot process effacement and reduced synaptopodin expression, indicating podocyte injury. Urinary ApoA-I increased >10-fold. Interestingly, urinary ApoA-I remained very elevated (>7-fold) at week 6, despite normalization in urinary albumin excretion. High dose LMB2 caused more severe podocyte injury with greater podocyte detachment, exposure of the GBM, followed by segmental glomerulosclerosis and reduced GFR by 6 weeks. In these mice, urinary ApoA-I increased 1400-fold compared to baseline. The results illustrate parallelism between disruption of the glomerular filtration barrier and urinary excretion of both albumin and ApoA-I. The data also make the noteworthy point that urinary loss of ApoA-I continues in the face of more subtle disruption in the glomerular filtration barrier, even in the absence of albuminuria. Thus, urinary ApoA-I may be a very sensitive marker of disruption in the glomerular filtration barrier.
Once it crosses the glomerular filtration barrier, ApoA-I is taken up by the cubilin-megalin-amnionless complex in the proximal tubule where it undergoes endocytosis and degradation [3]. Cubilin deficiency and proximal tubular reabsorption failure due to Fanconi syndrome increase urinary excretion of ApoA-I [4]. Cubilin-deficient mice have reduced proximal tubule uptake and increased urinary loss of ApoA-I as well as albumin, attended by significant decrease in plasma levels of ApoA-I and HDL3 along with hypoalbuminemia [5]. Interestingly, the larger, more mature HDL2 subclass was not detected in the urine and the plasma level was not reduced compared to mice with intact renal cubilin. The results support the idea that some HDL subclasses and certain components carried by all HDL particles can indeed cross the glomerular filtration barrier. Moreover, although proximal tubule cubilin uptake followed by endocytosis and lysosomal degradation of ApoA-I is well recognized, the results illustrate an additional important metabolic pathway. Thus, ApoA-I can be filtered, taken up, degraded or salvaged by the proximal tubule, and delivered back to the circulation, thereby regulating the plasma level of ApoA-I and HDL. The different possibilities for renal handling of ApoA-I are illustrated in Figure 1. Currently, the relative effects of the cubilin-megalin complex or specific lipoprotein transporters including ABCA1, ABCG1 or SRBI on these pathways remain uncertain. In preliminary studies, we examined the contribution of proximal and distal nephrons to ApoA-I handling [2]. We generated two tubular injury models: diphtheria toxin (DT) transgenic mouse, which express the human DT receptor in proximal tubular epithelial cells and in which DT injection causes acute tubular injury (ATI), and the folic acid injury model, where injected folic acid forms crystals in the distal nephron with distal tubular necrosis. Compared to baseline, mice with DT-induced proximal tubular injury had increased urinary KIM-1, a marker of ATI, accompanied by doubling in urinary excretion of ApoA-I. By contrast, folic acid caused greater increase in urinary NGAL compared to KIM-1, but did not affect urinary ApoA-I or albuminuria. Interestingly, in DT mice, proximal tubule expression of cubilin was reduced and ApoA-I localized to the apical side. By contrast, in the LMB2 podocyte injury model noted above, we saw increased cubilin expression without a change in ApoA-I localization. High dose LMB2 not only increased cubilin expression but translocated ApoA-I to the cytoplasm of tubular cells and peritubular capillaries. Examination of the transporters for ApoA-I revealed that ABCA1, but not G1 or SRBI, translocated from the apical membrane to cytoplasm of glomerular parietal and proximal tubular epithelial cells. Taken together, these data indicate that renal handling of ApoA-I is determined by the glomerular filtration barrier and proximal, but not distal, tubules through mechanisms that involve cubilin and ABCA1.
Figure 1.
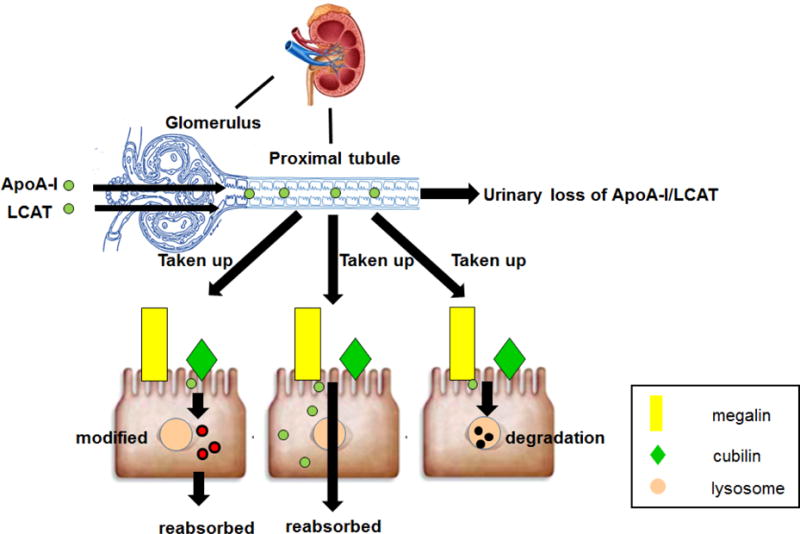
Renal handling of HDL components. ApoA-I, pre β-HDL and LCAT can be filtered by the glomerular capillaries and taken up by proximal tubular epithelial cells through the cubilin-megalin-amnionless complex or other transporters. ApoA-I can undergo endocytosis and degradation, or direct transcytosis, or transcytosis after modification. The salvaged ApoA-I enters the interstitium, and finally is delivered back to the circulation.
The demonstration that ApoA-I can cross the glomerular filtration barrier has additional implications that even in the absence of glomerular injury, conditions that impede lipidation and maturation of ApoA-I or encourage its dissociation from the mature HDL, permit it to be filtered. Once in the ultrafiltrate, ApoA-I can be taken up, degraded, salvaged, modified, or lost in the urine. For example, individuals with LCAT deficiency have impaired maturation of preβ-HDL, and their low levels of HDL reflect increased renal catabolism of the immature, small HDL particles [6]. This is relevant since the molecular weight of LCAT (63kD) predicts its ability to cross the glomerular filtration barrier and be lost in the urine. Indeed, reduced concentration and activity of LCAT have been documented in puromycin-induced nephrotic rats, which were specifically linked to urinary losses of the enzyme [7]. Acquired LCAT deficiency has been suggested to decrease plasma HDL in CKD stage 3 and 4 patients [8]. The study did not specifically address to what extent urinary loss contributed to the reduced plasma LCAT levels. Experimental studies show that inactivation of Abca1 increases plasma removal and renal degradation of lipid-free ApoA-I. Similarly, individuals with Tangier’s disease, due to ABCA1 gene mutations, have renal hypercatabolism of HDL, which results in low plasma Apo-AI and HDL [9]. Overall these observations indicate that certain apolipoproteins and enzymes on HDL particles pass through the glomerulus and can be reabsorbed, metabolized, or excreted in the urine. Interestingly, mass spectrometry of urine from patients with fibrotic kidney disease revealed that an HDL component, PLTP, along with cadherin and macrophage mannose receptor C1, is significantly increased [10]. Complimentary experimental studies showed that urinary exertion of PLTP paralleled increased gene and protein expression in tubules of kidneys from several animal models and biopsy specimens, suggesting PLTP may be a novel biomarker of kidney fibrosis. Whether PLTP also has a mechanistic role in progressive kidney damage is currently unknown.
It is important to emphasize that common glomerulopathies that disrupt the glomerular filtration barrier and tubular injuries that disrupt tubular handling of HDL proteins could permit filtration and urinary losses of greater quantity and greater variety of HDL particles, apolipoproteins, and enzymes. This would enhance the impact of kidney-originating disruption in HDL beneficial effects. Consistent with this possibility are reports of increased fractional catabolic rate of ApoA-I and small HDL3 in patients with moderate glomerular impairment [11]. Even in the absence of reduced GFR, proteinuria per se can reduce HDL levels [12]. In normoalbuminuric type 1 diabetics, lower levels of plasma HDL and HDL3 were independently associated with albuminuria, even after adjusting for glycemic control and other risk factors [13]. Notably, proteinuric patients have a larger HDL particle size distribution [14]. In addition to urinary losses of HDL components contributing to low levels of circulating HDL, kidneys also modulate HDL composition. Experimental proteinuria causes HDL enrichment in harmful epoxides, diols, hydroxyeicosatetraenoic acids and hydroxyoctadecadienoic acids [15]. Patients with type 2 diabetes have methylglyoxal modification of their HDL that causes particle re-structuring, which decreases the stability and plasma half-life [16]. This increased plasma clearance reflects greater shedding of lipoproteins, yielding smaller HDL particles that are more easily catabolized by the liver and kidneys. These considerations are relevant since proteinuria is a strong risk factor for cardiovascular disease, and progressive kidney damage, even in the absence of GFR changes [17, 18]. Overall, kidney handling of HDL components can affect assembly, maturation, and circulating levels of HDL particles. Disruption in the glomerular filtration barrier reflected by proteinuria and/or tubular injury may signal changes in the kidney’s role in the homeostasis of ApoA-I and other proteins, enzymes and cargo on HDL particles which would in turn affect levels, composition, and function of HDL.
Consequences of abnormal levels, composition, and function of HDL
Kidney impairment alters the composition and functionality of HDL. Biochemical and mass spectrometry analyses have documented a number of changes in the protein and lipid moieties of HDL particles; however, no clear footprint for CKD-impaired HDL exists. On the other hand, there is considerable evidence that CKD impairs many HDL functions. We and others have shown that patients with pre-dialysis CKD, ESRD receiving hemodialysis or peritoneal dialysis, patients following kidney transplantation have HDL with compromised capacity to elicit cholesterol efflux, which measures the ability of HDL to accept cholesterol from cells [19–24]. The HDL is also less effective in anti-inflammatory, anti-oxidative, antithrombotic actions and has suppressed capacity to protect the endothelium [25]. Among the potentially beneficial functions of HDL, the cholesterol efflux capacity is considered most important. This property is consistent with the concept that impaired HDL function contributes to the pathogenesis and risk for CVD in the non-CKD populations [26–28]. In the CKD population, despite the substantial number of reports showing reduced cholesterol efflux capacity, there is currently little data on HDL functionality predicting CVD or acute events. A prospective study in renal transplant recipients examined whether cholesterol efflux capacity can predict future cardiovascular mortality, all-cause mortality and graft failure [29]. Decreased cholesterol efflux capacity was associated with all-cause mortality after adjustment for recipient age and gender, but not if also adjusted for ApoA-I, HDL-C, and creatinine clearance. On the other hand, there was a strong inverse association between efflux capacity and graft failure. The results make several important points. First, HDL modulates distinct processes, and impairment in one function does not necessarily cause functional deficiencies in other tissues. Thus, HDL protection against progressive loss of kidney graft function occurs through mechanisms that are distinct from those in extra-renal cells and tissues. These may include modulation in the number and type of infiltrating inflammatory and immune cells as well as modulation of resident cells including maintenance of podocyte slit-diaphragm integrity and effects on autophagy. The link between impaired cholesterol efflux and graft failure reiterates the postulated relationship between HDL and kidney damage.
Epidemiologic studies have reported that low HDL is a significant risk factor for developing renal dysfunction in apparently healthy individuals and in community-based populations [30]. Moreover, in patients with existing CKD (stage 2–3), those with low HDL-C had worsening of kidney function, an increased rate of creatinine doubling and earlier entry into dialysis, independent of other risk factors including diabetes and hypertension. However, HDL-C did not predict decline in renal function [31, 32]. Since kidneys filter, reabsorb, catabolize and excrete ApoA-I and other components of HDL, it is possible that low levels of circulating ApoA-I/HDL reflect prevailing kidney dysfunction perhaps even in advance of other established markers such as reduced rate of glomerular filtration or increased proteinuria. As noted above, low ApoA-I/HDL may be a marker of early/subtle disruption in the glomerular filtration barrier and/or tubular reabsorption capacity. In addition, because HDL and its components are metabolized in the kidney, HDL could potentially directly modulate renal resident cells, which express its transporters and receptors. Indeed, lipid-laden foam cells are seen in focal and segmental glomerulosclerosis (FSGS) and also in the interstitium in Alport syndrome. Many animal models of kidney disease including metabolic syndrome, diabetic nephropathy, aging nephrosclerosis and acute and chronic progressive kidney injury show lipid accumulation in kidney tissue [33, 34]. Currently, the origin of the foam cells and effects of lipids on specific cell types remains to be clarified.
In this connection, cholesterol loading and downregulation of ABCA1 has been observed in podocytes exposed to sera of albuminuric diabetic patients compared to sera of normalbuminuric diabetic patients, although both groups had similar lipid profiles and duration of diabetes [35]. Expansion in the podocyte lipid pool was not due to increased cholesterol uptake or synthesis and instead, was linked to impairment in cholesterol efflux, which was related to downregulation in ABCA1. Notably, induction of cholesterol efflux with cyclodextrin in cultured cells and in diabetic mice preserved podocyte functions and lessened albuminuria. Interestingly, renal expression of ABCA1 and ABCG1 correlated with progression of human diabetic nephropathy and deterioration of eGFR [36]. These observations suggest that altered kidney handling of HDL components may affect its structure and function and impact pathways involved in progressive kidney damage.
In addition to levels and functionality, abnormalities in particular components of HDL can cause kidney damage. Two haplotypes of APOL1, a lipoprotein found in the HDL3b and HDL3c subfractions, harbor three coding-sequence mutations as risk variants associated with non-diabetic CKD in African Americans. APOL1 renal risk variants confer higher risk to development of FSGS (OR=17), HIV-associated nephropathy (OR=29), and end-stage renal disease attributed to hypertension (OR=7.3) [37]. Several mechanisms have been proposed for how APOL1 might contribute to glomerulopathies, including lysosomal membrane permeabilization, autophagic cell death, apoptosis, and necrosis, all of which are related to cell death [38].
Conclusion
The kidneys have a significant role in metabolism of individual components of HDL, which affect the levels, composition and functionality of HDL particles. Alterations in kidney metabolism of HDL that follows glomerular or tubular injury can lead to reduced plasma concentration and/or increased urinary excretion of HDL particles. Thus, observed change in renal handling of HDL components, e.g., ApoA-I, may be a particularly sensitive marker of subtle kidney damage. Renal handling of HDL also suggests that intrarenal modifications of HDL can directly situate dysfunctional HDL in inflammatory, oxidative or proliferative pathways involved in progressive kidney damage or bring dysfunctional HDL into the circulation to promote adverse consequences in extra-renal tissues.
Key Points.
HDL is not a single molecule. Kidneys participate in homeostasis of HDL components, especially apolipoproteins and enzymes, which determine the level as well as composition of HDL particles.
HDL components including apolipoproteins, e.g., ApoA-I, and enzymes e.g., LCAT, cross the glomerular filtration barrier.
Apolipoproteins are taken up by proximal tubules and catabolized, modified and/or returned to the circulation or lost in the urine.
Urinary HDL components including apolipoproteins, e.g., ApoA-I, and enzymes, e.g., PLTP, may be markers of kidney damage.
Acknowledgments
Financial support and sponsorship: This research was supported in part by NIHP01HL116263.
Footnotes
Disclosures: None.
Conflicts of interest: There are no conflicts.
References
- 1.Woollett LA, Spady DK. Kinetic parameters for high density lipoprotein apoprotein AI and cholesteryl ester transport in the hamster. J Clin Invest. 1997;99:1704–1713. doi: 10.1172/JCI119334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhong J, Yang H, Tsuchida Y, et al. Podocyte and tubule injury have different effects on renal handling of apolipoprotein A-I (ApoA-I) and its receptors. ASN Kidney Week 2015; San Diego, CA, USA. 2015. [Google Scholar]
- 3.Hammad SM, Barth JL, Knaak C, et al. Megalin acts in concert with cubilin to mediate endocytosis of high density lipoproteins. J Biol Chem. 2000;275:12003–12008. doi: 10.1074/jbc.275.16.12003. [DOI] [PubMed] [Google Scholar]
- 4.Graversen JH, Castro G, Kandoussi A, et al. A pivotal role of the human kidney in catabolism of HDL protein components apolipoprotein A-I and A-IV but not of A-II. Lipids. 2008;43:467–470. doi: 10.1007/s11745-008-3169-2. [DOI] [PubMed] [Google Scholar]
- 5.Aseem O, Smith BT, Cooley MA, et al. Cubilin maintains blood levels of HDL and albumin. J Am Soc Nephrol. 2014;25:1028–1036. doi: 10.1681/ASN.2013060671. *The study shows that cubilin deficiency in the proximal tubule reduces uptake of ApoA-I and albumin which is tied to decreased levels of plasma albumin, ApoA-I, and HDL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee JY, Timmins JM, Mulya A, et al. HDLs in apoA-I transgenic Abca1 knockout mice are remodeled normally in plasma but are hypercatabolized by the kidney. J Lipid Res. 2005;46:2233–2245. doi: 10.1194/jlr.M500179-JLR200. [DOI] [PubMed] [Google Scholar]
- 7.Vaziri ND, Liang K, Parks JS. Down-regulation of hepatic lecithin: cholesterol acyltransferase gene expression in chronic renal failure. Kidney Int. 2001;59:2192–2196. doi: 10.1046/j.1523-1755.2001.00734.x. [DOI] [PubMed] [Google Scholar]
- 8.Calabresi L, Simonelli S, Conca P, et al. Acquired lecithin: cholesterol acyltransferase deficiency as a major factor in lowering plasma HDL levels in chronic kidney disease. J Intern Med. 2014 doi: 10.1111/joim.12290. [DOI] [PubMed] [Google Scholar]
- 9.Schaefer EJ, Blum CB, Levy RI, et al. Metabolism of high-density lipoprotein apolipoproteins in Tangier disease. N Engl J Med. 1978;299:905–910. doi: 10.1056/NEJM197810262991701. [DOI] [PubMed] [Google Scholar]
- 10.Craciun FL, Bijol V, Ajay AK, et al. RNA Sequencing Identifies Novel Translational Biomarkers of Kidney Fibrosis. J Am Soc Nephrol. 2015 doi: 10.1681/ASN.2015020225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Batista MC, Welty FK, Diffenderfer MR, et al. Apolipoprotein A-I, B-100, and B-48 metabolism in subjects with chronic kidney disease, obesity, and the metabolic syndrome. Metabolism. 2004;53:1255–1261. doi: 10.1016/j.metabol.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 12.Shearer GC, Newman JW, Hammock BD, et al. Graded effects of proteinuria on HDL structure in nephrotic rats. J Am Soc Nephrol. 2005;16:1309–1319. doi: 10.1681/ASN.2004080644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bulum T, Kolaric B, Duvnjak L. Lower levels of total HDL and HDL3 cholesterol are associated with albuminuria in normoalbuminuric Type 1 diabetic patients. J Endocrinol Invest. 2013;36:574–578. doi: 10.3275/8850. [DOI] [PubMed] [Google Scholar]
- 14.Soto-Miranda E, Carreon-Torres E, Lorenzo K, et al. Shift of high-density lipoprotein size distribution toward large particles in patients with proteinuria. Clin Chim Acta. 2012;414:241–245. doi: 10.1016/j.cca.2012.09.028. [DOI] [PubMed] [Google Scholar]
- 15.Newman JW, Kaysen GA, Hammock BD, et al. Proteinuria increases oxylipid concentrations in VLDL and HDL but not LDL particles in the rat. J Lipid Res. 2007;48:1792–1800. doi: 10.1194/jlr.M700146-JLR200. [DOI] [PubMed] [Google Scholar]
- 16.Godfrey L, Yamada-Fowler N, Smith J, et al. Arginine-directed glycation and decreased HDL plasma concentration and functionality. Nutr Diabetes. 2014;4:e134. doi: 10.1038/nutd.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chronic Kidney Disease Prognosis C. Matsushita K, van der Velde M, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet. 2010;375:2073–2081. doi: 10.1016/S0140-6736(10)60674-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarnak MJ, Astor BC. Implications of proteinuria: CKD progression and cardiovascular outcomes. Adv Chronic Kidney Dis. 2011;18:258–266. doi: 10.1053/j.ackd.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 19.Kopecky C, Haidinger M, Birner-Grunberger R, et al. Restoration of renal function does not correct impairment of uremic HDL properties. J Am Soc Nephrol. 2015;26:565–575. doi: 10.1681/ASN.2013111219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shroff R, Speer T, Colin S, et al. HDL in children with CKD promotes endothelial dysfunction and an abnormal vascular phenotype. J Am Soc Nephrol. 2014;25:2658–2668. doi: 10.1681/ASN.2013111212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holzer M, Schilcher G, Curcic S, et al. Dialysis Modalities and HDL Composition and Function. J Am Soc Nephrol. 2015;26:2267–2276. doi: 10.1681/ASN.2014030309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamamoto S, Yancey PG, Ikizler TA, et al. Dysfunctional high-density lipoprotein in patients on chronic hemodialysis. J Am Coll Cardiol. 2012;60:2372–2379. doi: 10.1016/j.jacc.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaseda R, Jabs K, Hunley TE, et al. Dysfunctional high-density lipoproteins in children with chronic kidney disease. Metabolism. 2015;64:263–273. doi: 10.1016/j.metabol.2014.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Speer T, Rohrer L, Blyszczuk P, et al. Abnormal high-density lipoprotein induces endothelial dysfunction via activation of Toll-like receptor-2. Immunity. 2013;38:754–768. doi: 10.1016/j.immuni.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 25.Annema W, von Eckardstein A. High-density lipoproteins. Multifunctional but vulnerable protections from atherosclerosis. Circ J. 2013;77:2432–2448. doi: 10.1253/circj.cj-13-1025. [DOI] [PubMed] [Google Scholar]
- 26.Khera AV, Cuchel M, de la Llera-Moya M, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rohatgi A, Khera A, Berry JD, et al. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014;371:2383–2393. doi: 10.1056/NEJMoa1409065. *The first demonstration that impaired cholesterol efflux from macrophages predicts incident CVD events in a large multiethnic clinical population. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saleheen D, Scott R, Javad S, et al. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: a prospective case-control study. Lancet Diabetes Endocrinol. 2015;3:507–513. doi: 10.1016/S2213-8587(15)00126-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Annema W, DA Freark, de Boer J, Dullaart RPF, Sanders JSF, Bakker JL, Tietge UJF. HDL Cholesterol Efflux Predicts Graft Failure in Renal Transplant Recipients. JASN. 2015 doi: 10.1681/ASN.2014090857. *The study demonstrates a strong inverse association between cholesterol efflux capacity and graft failure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Odden MC, Tager IB, Gansevoort RT, et al. Hypertension and low HDL cholesterol were associated with reduced kidney function across the age spectrum: a collaborative study. Ann Epidemiol. 2013;23:106–111. doi: 10.1016/j.annepidem.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rahman M, Yang W, Akkina S, et al. Relation of serum lipids and lipoproteins with progression of CKD: The CRIC study. Clin J Am Soc Nephrol. 2014;9:1190–1198. doi: 10.2215/CJN.09320913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baragetti A, Norata GD, Sarcina C, et al. High density lipoprotein cholesterol levels are an independent predictor of the progression of chronic kidney disease. J Intern Med. 2013;274:252–262. doi: 10.1111/joim.12081. [DOI] [PubMed] [Google Scholar]
- 33.Levi M, Wang X, Choudhury D. Nuclear hormone receptors as therapeutic targets. Contrib Nephrol. 2011;170:209–216. doi: 10.1159/000325668. [DOI] [PubMed] [Google Scholar]
- 34.Muroya Y, Ito O, Rong R, et al. Disorder of fatty acid metabolism in the kidney of PAN-induced nephrotic rats. Am J Physiol Renal Physiol. 2012;303:F1070–1079. doi: 10.1152/ajprenal.00365.2011. [DOI] [PubMed] [Google Scholar]
- 35.Merscher-Gomez S, Guzman J, Pedigo CE, et al. Cyclodextrin protects podocytes in diabetic kidney disease. Diabetes. 2013;62:3817–3827. doi: 10.2337/db13-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herman-Edelstein M, Scherzer P, Tobar A, et al. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J Lipid Res. 2014;55:561–572. doi: 10.1194/jlr.P040501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329:841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lan X, Jhaveri A, Cheng K, et al. APOL1 risk variants enhance podocyte necrosis through compromising lysosomal membrane permeability. Am J Physiol Renal Physiol. 2014;307:F326–336. doi: 10.1152/ajprenal.00647.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]