

Abstract
Tumor suppressor genes (TSGs) are important gatekeepers that protect against somatic evolution of cancer. Losing both alleles of a TSG in a single cell represents a step toward cancer. We study how the kinetics of TSG inactivation depends on the population size of cells and the mutation rates for the first and second hit. We calculate the probability as function of time that at least one cell has been generated with two inactivated alleles of a TSG. We find three different kinetic laws: in small, intermediate, and large populations, it takes, respectively, two, one, and zero rate-limiting steps to inactivate a TSG. We also study the effect of chromosomal and other genetic instabilities. Small lesions without genetic instability can take a very long time to inactivate the next TSG, whereas the same lesions with genetic instability pose a much greater risk for cancer progression.
In 1971, Knudson (1) performed a statistical analysis of the incidence of retinoblastoma in young children. This analysis and subsequent work eventually led to the model invoking two hits of the retinoblastoma gene as rate-limiting steps in tumorigenesis (2–6). In the inherited form, the first mutation is already present in the germ line, whereas the second mutation emerges during somatic cell divisions. These observations led to the concept of a tumor suppressor gene (TSG). In the meantime, a large number of TSGs have been discovered that are involved in various human cancers (7–10). Here, we will calculate the dynamics of inactivating TSGs in populations of dividing cells with or without genetic instability (11–15).
A normal cell has two alleles of a TSG. Inactivating the first allele is considered to be a neutral (or almost neutral) mutation. Inactivating the second allele provides the cell with an increased net reproductive rate. Point mutations, small insertions, deletions, structural changes of the chromosome, or chromosomal loss can constitute the first hit, whereas all of these events plus mitotic recombination can occur as the second hit. Usually large deletions or chromosome loss do not account both for the first and second step in one cell, because large homozygous deletions are often lethal for a cell. Denote by u1 and u2 the mutation rates for the first and second hit (including all possible mechanisms). It is natural to assume that u1 is less than u2, because there are more possibilities for the second hit.
We will now ask the most basic question regarding the somatic evolutionary dynamics of TSGs: how long does it take for a population of N cells to generate a single cell with two inactivated alleles of a TSG?
For small populations, 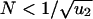 , the first hit takes over the population before the second hit occurs (Fig. 1). There is an intuitive explanation for this threshold: a cell with one hit is a neutral mutant that takes on average N generations to reach fixation, whereas the waiting time for the second hit is 1/(Nu2). From N < 1/(Nu2) we obtain
, the first hit takes over the population before the second hit occurs (Fig. 1). There is an intuitive explanation for this threshold: a cell with one hit is a neutral mutant that takes on average N generations to reach fixation, whereas the waiting time for the second hit is 1/(Nu2). From N < 1/(Nu2) we obtain 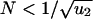 . In this case, the probability that a single cell with two hits emerges before time t is given by
. In this case, the probability that a single cell with two hits emerges before time t is given by
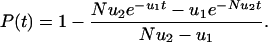 |
[1] |
Time, t, is measured in units of cell generations. For very short times, t < 1/Nu2, we have P(t) ≈ Nu1u2t2/2. The probability accumulates as second order of time. The 2 in the exponent is the same as in Knudson's two-hit hypothesis: it takes two rate-limiting hits to inactivate a TSG in this case.
Fig. 1.

Inactivating a TSG requires two, one, or zero rate-limiting steps depending on the population size. (a) Cells of type 0, 1, and 2 have, respectively, 0, 1, and 2 inactivated alleles of the TSG. The mutation rate for inactivating the first and second allele are given by u1 and u2. Initially all cells are of type 0. (b) In small populations, 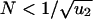 , type 1 cells will reach fixation before a cell of type 2 has been generated. The resulting kinetics have two rate-limiting steps (Eq. 1). (c) In intermediate populations, 1/u1 <
, type 1 cells will reach fixation before a cell of type 2 has been generated. The resulting kinetics have two rate-limiting steps (Eq. 1). (c) In intermediate populations, 1/u1 < 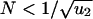 , a lineage of type 1 cells generates a type 2 cell before reaching fixation. The resulting kinetics have one rate-limiting step (Eq. 2). (d) In large populations, 1/u1 < N, type 1 cells are generated immediately and accumulate as a linear function of time. After some (short) time a type 2 cell will be generated. The kinetics involve two steps, none of which is rate-limiting for overall cancer progression (Eq. 3). This classification allows us to analyze the effect of cell number and mutation rate on the kinetics of TSG inactivation.
, a lineage of type 1 cells generates a type 2 cell before reaching fixation. The resulting kinetics have one rate-limiting step (Eq. 2). (d) In large populations, 1/u1 < N, type 1 cells are generated immediately and accumulate as a linear function of time. After some (short) time a type 2 cell will be generated. The kinetics involve two steps, none of which is rate-limiting for overall cancer progression (Eq. 3). This classification allows us to analyze the effect of cell number and mutation rate on the kinetics of TSG inactivation.
For larger populations, 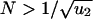 , the second hit occurs in a cell before the first hit has taken over the whole population. We have to subdivide into two cases. If
, the second hit occurs in a cell before the first hit has taken over the whole population. We have to subdivide into two cases. If 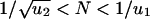 , then the kinetics are dominated by waiting for a single event, namely producing a cell with one hit that gives rise to a lineage that generates a cell with two hits (Fig. 1). We obtain
, then the kinetics are dominated by waiting for a single event, namely producing a cell with one hit that gives rise to a lineage that generates a cell with two hits (Fig. 1). We obtain
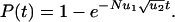 |
[2] |
For short times, the probability increases as a linear function of time, which means that it takes only one rate-limiting hit to inactivate a TSG in this parameter regime. The occurrence of 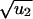 in the exponent is surprising, but a similar law has been found in a different context (16, 17).
in the exponent is surprising, but a similar law has been found in a different context (16, 17).
If, on the other hand, N > 1/u1, then the first hit will occur immediately and the waiting time is dominated by how long it takes for a cell with two hits to emerge (Fig. 1). This process is described by the two-hit kinetics
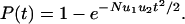 |
[3] |
Fig. 2 shows the perfect fit between our analytic calculations and numerical simulations of the underlying stochastic process. Thus, it takes two hits to inactivate a TSG in small and large populations, but only one hit in populations of intermediate size. Eliminating a TSG in a very large population of rapidly dividing cells is, however, not rate-limiting for the overall process of tumorigenesis. Therefore, in small, intermediate, and large populations of cells it takes two, one, and zero rate-limiting hits to inactivate a TSG.
Fig. 2.

Perfect agreement between exact numerical simulations and the analytic approximations given by Eqs. 1–3. Population sizes are (a) N = 2, (b) N = 100, and (c) N = 107. (d) T1/2 denotes the half-time of TSG inactivation, defined as the time until the probability of generating at least one cell of type 2 has become 1/2. Shown is logT1/2 vs. logN. The numerical data (dots) can be approximated by three straight lines derived from Eqs. 1–3. For small N, we have T1/2 = log2/u1. For intermediate N, we have 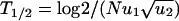 ). For large N, we have
). For large N, we have 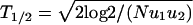 . For reasonably fast computing time, we used the mutation rates u1 = 10–5 and u2 = 0.002 and performed 104 independent runs for every N.
. For reasonably fast computing time, we used the mutation rates u1 = 10–5 and u2 = 0.002 and performed 104 independent runs for every N.
In Appendix we describe the derivation of our results and include the possibility that inactivation of the first allele is not neutral but leads to a selective advantage r > 1. This assumption accelerates the overall kinetics.
Let us consider some specific examples. The mutation rate of inactivating a TSG depends on its size and the type of possible mutations (5, 18, 19). The typical mutation rate per gene per cell division is u1 = 10–7. In a normal cell, the second allele of a TSG is thought to be inactivated at a similar or somewhat faster rate. In Table 1, we consider the two cases u2 = 10–6 and u2 = 10–5. In cells with chromosomal instability (CIN), the second step can be caused by loss of a whole chromosome or chromosome arm, a translocation, inversion, or deletion at a rate of u2 = 10–2 per cell division (12). In cells with microsatellite instability (MIN), the point mutation rate is increased 50- to 1,000-fold, suggesting values of u1 = u2 = 5 × 10–6 to 10–4 per gene per cell division (10, 14, 20). In Table 1, we use u1 = u2 = 10–5 as an example for MIN.
Table 1. Time until inactivation of a TSG in genetically stable and unstable neoplasias of various sizes ranging from N = 103 to N = 109 cells.
| Normal
|
Normal
|
CIN
|
MIN
|
|||||
|---|---|---|---|---|---|---|---|---|
| u1 = 10-7, | u2 = 10-6 | u1 = 10-7, | u2 = 10-5 | u1 = 10-7, | u2 = 10-2 | u1 = 10-5, | u2 = 10-5 | |
| N | r = 1 | r = 1.1 | r = 1 | r = 1.1 | r = 1 | r = 1.1 | r = 1 | r = 1.1 |
| 103 | >1,000 y | >100 y | >1,000 y | >100 y | >100 y | >100 y | 61.3 y | 2.3 y |
| 104 | >1,000 y | 21.3 y | >100 y | 21.3 y | 21.3 y | 13.4 y | 6.7 y | 145 d |
| 105 | >100 y | 2.4 y | 61.3 y | 2.3 y | 2.1 y | 1.4 y | 1.2 y | 69 d |
| 106 | 21.3 y | 170 d | 6.7 y | 145 d | 83 d | 56 d | 120 d | 43 d |
| 107 | 3.6 y | 93 d | 1.2 y | 69 d | 14 d | 11 d | 38 d | 23 d |
| 108 | 380 d | 66 d | 120 d | 43 d | 4 d | 3 d | 12 d | 10 d |
| 109 | 120 d | 43 d | 38 d | 23 d | 1 d | 1 d | 4 d | 3 d |
We have used Eq. 6 to calculate the time T1/2 (in units of cell generations, here assumed to be 1 day) until there is a 50% chance of having produced at least one cell with two inactivated alleles. For example, a lesion containing N = 106 cells without genetic instability, assuming u1 = 10-7 and u2 = 10-6, takes T1/2 = 21 years if the first hit is neutral (r = 1) and T1/2 = 170 days if the first hit has a 10% fitness advantage (r = 1.1). The same number of cells with CIN, assuming u1 = 10-7 and u2 = 10-2, needs only T1/2 = 83 days (with r = 1). For N = 106 cells with MIN (u1 = 10-5 and u2 = 10-5) we need T1/2 = 120 days (with r = 1). d, day; y, year.
Table 1 shows the half-life of TSG inactivation, defined as the time T until the probability of having produced at least one cell with two hits is 1/2. Consider a lesion of N = 106 cells dividing once per day and assume that the first hit is neutral, r = 1. For normal cells with u2 = 10–6 it takes 21 years. If u2 = 10–5 it takes 6.7 years. For CIN cells with u2 = 10–2 it takes 83 days. If the first hit is not neutral but confers a 10% selective advantage, r = 1.1, then the respective half-lives are reduced to 170, 145, and 56 days. In this case, the difference between normal cells and CIN cells is greatly reduced. A lesion consisting of n = 109 cells dividing once per day needs ≈120 days without genetic instability and of the order of 1 day with either CIN or MIN. These calculations do not include a cost of genetic instability. If every other CIN or MIN cell dies from receiving lethal mutations, then these lesions would need twice as long.
We can also calculate the probability that a lesion of a certain size has inactivated a TSG gene within a given time. Suppose N = 105 cells divide once per day. If these cells are genetically stable (assuming u2 = 10–6 and r = 1), then the probability of inactivating a TSG within 5 years is ≈1%; if these cells have CIN, then the probability is ≈80%. Therefore small lesions with CIN pose a much greater risk for cancer progression if the next step requires inactivation of a TSG (especially with a neutral first hit).
Two more extensions of the theory are discussed in Appendix. First, the population size, N, can change over time. Second, if the mutation rates u1 and u2 are very different, as is the case for CIN, then including the possibility of a fast first hit followed by a slow second hit increases the rate of TSG inactivation.
Genetic instability is one of the most active research areas of cancer biology (12, 13, 21–26). Loeb and colleagues (14, 27) were the first to suggest that somatic evolution of cancer might be driven by cells with increased mutation rates, so-called mutator phenotypes. CIN and MIN represent particular examples of such mutator phenotypes. Because genetic instability is a feature of almost all late-stage cancer cells, the main question is whether genetic instability arises early or late during tumorigenesis. The problem is unresolved both from an experimental (28–31) and theoretical perspective (15, 32–35). The present article contributes to the ongoing discussion by providing a quantitative framework for the kinetics of TSG inactivation. Such a frame-work is necessary to understand the consequences of genetic instability. In the present article we have not addressed the question of whether an additional CIN (or MIN) mutation might precede TSG inactivation. This has been discussed elsewhere (15, 32, 36, 37).
In summary, we have outlined a quantitative theory for the evolutionary dynamics of inactivating TSGs with and without genetic instability. In a healthy tissue consisting of small compartments that are renewed by tissue-specific stem cells, two rate-limiting hits are needed to eliminate a TSG: the overall rate is proportional to the second order of time. In small neoplasias, only one hit is required to inactivate both alleles of a TSG: the overall rate is proportional to the first order of time. In this case, the rate constant includes the square root of the mutation rate, u2. In large populations of cancer cells, it takes again two hits, but neither of them is rate-limiting for tumor progression. Therefore, as the population size increases, TSGs are inactivated in two, one, and zero rate-limiting steps.
Our results should stimulate experimental investigations in cell culture studying how the kinetics of TSG inactivation scales with mutation rates and population size. In particular, it should be possible to quantify the rates of the various mutational mechanisms that contribute to inactivating the first and second allele of a TSG. Furthermore, measuring the inactivation kinetics of TSGs in animal models and comparing the data with our equations will reveal the relevant tissue architecture for the process of cancer initiation in various organs (38). In addition, our findings have implications for linking cancer incidence curves to the molecular events of cancer progression (35).
Appendix
Types 0, 1, and 2 denote cells with 0, 1, and 2 inactivated alleles of a TSG. Type 1 cells have relative fitness r. In the main text we present results for r = 1. Here, we discuss the general case. The relative fitness of type 2 cells (which exceeds 1) does not enter into our calculations, because we are only interested in the time until at least one type 2 cell has been produced. Of course, the probability that a type 2 cell spreads in the population and the time for reaching a certain abundance does depend on the fitness of the cell (see below).
Small Populations. For 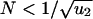 , the dynamics can be described as transition among homogeneous states. Denote by X0, X1, and X2, respectively, the probabilities that the population contains only unmutated cells, only cells where one allele has been inactivated, and at least one cell where both alleles have been inactivated. The time derivatives of these probabilities are given by
, the dynamics can be described as transition among homogeneous states. Denote by X0, X1, and X2, respectively, the probabilities that the population contains only unmutated cells, only cells where one allele has been inactivated, and at least one cell where both alleles have been inactivated. The time derivatives of these probabilities are given by
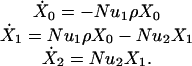 |
[4] |
Here ρ = (1–1/r)/(1–1/rN) is the probability that a single cell of type 1 will reach fixation in a population of type 0 cells. For r = 1 we have ρ = 1/N. The probability that at least one cell of type 2 has been produced before time t is given by
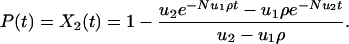 |
[5] |
For ρ = 1/N we obtain Eq. 1. For t > 1/(Nu2) we have P(t) ≈ 1–exp(–u1t). On this time scale, the second hit is fast and can be neglected. Only the first hit is rate-limiting.
Large Populations. For 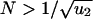 , a type 2 cell will be produced before the type 1 cells have reached fixation. We can use a branching process to describe the evolutionary dynamics. The population is dominated by type 0 cells. For each newly generated type 1 cell, we calculate the probability that the resulting lineage of type 1 cells will generate a type 2 cell. This probability can be calculated with a recursive formula assuming that lineages starting from different type 1 cells behave independently of each other, which is a good assumption if the abundance of type 1 cells remains much smaller than N. The probability that at least one cell of type 2 has been produced before time t is given by
, a type 2 cell will be produced before the type 1 cells have reached fixation. We can use a branching process to describe the evolutionary dynamics. The population is dominated by type 0 cells. For each newly generated type 1 cell, we calculate the probability that the resulting lineage of type 1 cells will generate a type 2 cell. This probability can be calculated with a recursive formula assuming that lineages starting from different type 1 cells behave independently of each other, which is a good assumption if the abundance of type 1 cells remains much smaller than N. The probability that at least one cell of type 2 has been produced before time t is given by
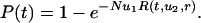 |
[6] |
Here
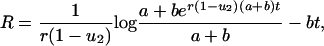 |
where a < b are the roots of the quadratic equation
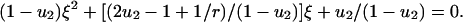 |
There are useful approximations in various limits. If N ≪ 1/u1 then (i) R = r/(1–r) for 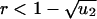 ; (ii)
; (ii) 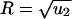 for
for 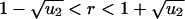 ; and (iii) R = 1–1/r for
; and (iii) R = 1–1/r for 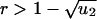 . If N ≫ 1 then R = ru2t. For r = 1 we obtain Eqs. 2 and 3. If r–1 ≫ u2 and 1/u1 < N < 1/(u1u2) we have R = u2[(e(r–1)t–1)/(r–1)–t]. All of these approximations are in excellent agreement with numerical simulations of the corresponding parameter regimes.
. If N ≫ 1 then R = ru2t. For r = 1 we obtain Eqs. 2 and 3. If r–1 ≫ u2 and 1/u1 < N < 1/(u1u2) we have R = u2[(e(r–1)t–1)/(r–1)–t]. All of these approximations are in excellent agreement with numerical simulations of the corresponding parameter regimes.
Changing Population Size. Suppose the population grows deterministically according to N(t) = N0eγt starting from N0 unmutated cells at time t = 0. For N(t) < 1/u1 we find 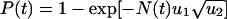 . For N(t) > 1/u1 we find P(t) = 1–exp[–u1u2N(t) logN(t)]. If the population grows stochastically, with an exponentially distributed waiting time with mean 1/[N(t)] between cell divisions, then P(t) = 1–1/[1–N(t)u1u2logu2] starting from one unmutated cell.
. For N(t) > 1/u1 we find P(t) = 1–exp[–u1u2N(t) logN(t)]. If the population grows stochastically, with an exponentially distributed waiting time with mean 1/[N(t)] between cell divisions, then P(t) = 1–1/[1–N(t)u1u2logu2] starting from one unmutated cell.
Very Different Mutation Rates. In CIN cells, the mutation rate u1 is determined by subtle nucleotide sequence changes, whereas the much faster mutation rate u2 is determined by loss of (arms of) chromosomes. It might be possible in some cases that a u2 step could precede a u1 step. Assuming r = 1, we have for small populations P(t) = 1–exp(–Nu1t) and for large populations P(t) = 1–exp(–Nu1u2t2). This effect further accelerates the rate of TSG inactivation in CIN cells.
Take Over Time. In large tumors, the waiting times until producing a cell with two hits becomes so short that we have to add the time it takes for an advantageous mutant cell to grow to a sizeable abundance. This time is approximately given by t = (log N)/(a–1), where a > 1 is the relative fitness of the cell with the inactivated TSG. The probability that the cell spreads in a large population is given by 1–1/a.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: TSG, tumor suppressor gene; CIN, chromosomal instability; MIN, microsatellite instability.
References
- 1.Knudson, A. G. (1971) Proc. Natl. Acad. Sci. USA 68, 820–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knudson, A. G. (2001) Nat. Rev. Cancer 1, 157–162. [DOI] [PubMed] [Google Scholar]
- 3.Cavenee, W. K., Dryja, T. P., Phillips, R. A., Benedict, W. F., Godbout, R., Gallie, B. L., Murphree, A. L., Strong, L. C. & White, R. L. (1983) Nature 305, 779–784. [DOI] [PubMed] [Google Scholar]
- 4.Friend, S. H., Bernards, R., Rogelj, S., Weinberg, R. A., Rapaport, J. M., Albert, D. M. & Dryja, T. P. (1986) Nature 323, 643–646. [DOI] [PubMed] [Google Scholar]
- 5.Vogelstein, B. & Kinzler, K. W. (1998) The Genetic Basis of Human Cancer (McGraw–Hill, Toronto).
- 6.Moolgavkar, S. H. & Knudson, A. G. (1981) J. Natl. Cancer Inst. 66, 1037–1052. [DOI] [PubMed] [Google Scholar]
- 7.Kinzler, K. W., Nilbert, M. C., Vogelstein, B., Bryan, T. M., Levy, D. B., Smith, K. J., Preisinger, A. C., Hamilton, S. R., Hedge, P., Markham, A., et al. (1991) Science 251, 1366–1370. [DOI] [PubMed] [Google Scholar]
- 8.Weinberg, R. A. (1991) Science 254, 1138–1146. [DOI] [PubMed] [Google Scholar]
- 9.Knudson, A. G. (1993) Proc. Natl. Acad. Sci. USA 90, 10914–10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levine, A. J. (1993) Annu. Rev. Biochem. 62, 623–651. [DOI] [PubMed] [Google Scholar]
- 11.Boyer, J. C., Umar, A., Risinger, J. I., Lipford, J. R., Kane, M., Yin, S., Barrett, J. C., Kolodner, R. D. & Kunkel, T. A. (1995) Cancer Res. 55, 6063–6070. [PubMed] [Google Scholar]
- 12.Lengauer, C., Kinzler, K. W. & Vogelstein, B. (1997) Nature 386, 623–627. [DOI] [PubMed] [Google Scholar]
- 13.Lengauer, C., Kinzler, K. W. & Vogelstein, B. (1998) Nature 396, 623–649. [DOI] [PubMed] [Google Scholar]
- 14.Loeb, L. A. (2001) Cancer Res. 61, 3230–3239. [PubMed] [Google Scholar]
- 15.Nowak, M. A., Komarova, N. L., Sengupta, A., Jallepalli, P. V., Shih, I.-M., Vogelstein, B. & Lengauer, C. (2002) Proc. Natl. Acad. Sci. USA 99, 16226–16231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karlin, S. & Tavare, S. (1983) SIAM J. Appl. Math. 43, 31–41. [Google Scholar]
- 17.Robertson, A. (1978) Genet. Res. 31, 255–264. [Google Scholar]
- 18.Lamlum, H., Ilyas, M., Rowan, A., Clark, S., Johnson, V., Bell, J., Frayling, I., Efstathiou, J., Pack, K., Payne, S., et al. (1999) Nat. Med. 5, 1071–1075. [DOI] [PubMed] [Google Scholar]
- 19.Nagase, H. & Hakamura, Y. (1993) Hum. Mut. 2, 425–434. [DOI] [PubMed] [Google Scholar]
- 20.Wheeler, J. M., Beck, N. E., Kim, H. C., Tomlinson, I. P., Mortensen, N. J. & Bodmer, W. F. (1999) Proc. Natl. Acad. Sci. USA 96, 10296–10301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kolodner, R. D., Putnam, C. D. & Myung, K. (2002) Science 297, 552–557. [DOI] [PubMed] [Google Scholar]
- 22.Nasmyth, K. (2002) Science 297, 559–565. [DOI] [PubMed] [Google Scholar]
- 23.Maser, R. S. & DePinho, R. A. (2002) Science 297, 565–569. [DOI] [PubMed] [Google Scholar]
- 24.Haigis, K. M., Caya, J. G., Reichelderfer, M. & Dove, W. F. (2002) Proc. Natl. Acad. Sci. USA 99, 8927–8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pihan, G. A., Wallace, J., Zhou, Y. & Doxsey, S. J. (2003) Cancer Res. 63, 1398–1404. [PubMed] [Google Scholar]
- 26.Hermsen, M., Postma, C., Baak, J., Weiss, M., Rapallo, A., Sciutto, A., Roemen, G., Arends, J. W., Williams, R., Giaretti, W., et al. (2002) Gastroenterology 123, 1109–1119. [DOI] [PubMed] [Google Scholar]
- 27.Loeb, L. A., Springgate, C. F. & Battula, N. (1974) Cancer Res. 34, 2311–2321. [PubMed] [Google Scholar]
- 28.Shih, I. M., Zhou, W., Goodman, S. N., Lengauer, C., Kinzler, K. W. & Vogelstein, B. (2001) Cancer Res. 61, 818–822. [PubMed] [Google Scholar]
- 29.Rajagopalan, H., Nowak, M. A., Vogelstein, B. & Lengauer, C. (2003) Nat. Rev. Cancer 3, 695–700. [DOI] [PubMed] [Google Scholar]
- 30.Sieber, O. M., Heinimann, K., Gorman, P., Lamlum, H., Crabtree, M., Simpson, C. A., Davies, D., Neale, K., Hodgson, S. V., Roylance, R. R., et al. (2002) Proc. Natl. Acad. Sci. USA 99, 16910–16905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sieber, O. M., Heinimann, K. & Tomlinson, I. P. M. (2003) Nat. Rev. Cancer 3, 701–708. [DOI] [PubMed] [Google Scholar]
- 32.Komarova, N. L., Lengauer, C., Vogelstein, B. & Nowak, M. A. (2002) Cancer Biol. Ther. 1, 685–692. [DOI] [PubMed] [Google Scholar]
- 33.Michor, F., Iwasa, Y., Komarova, N. L. & Nowak, M. A. (2003) Curr. Biol. 13, 581–584. [DOI] [PubMed] [Google Scholar]
- 34.Tomlinson, I. P., Novelli, M. R. & Bodmer, W. F. (1996) Proc. Natl. Acad. Sci. USA 93, 14800–14803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luebeck, E. G. & Moolgavkar, S. H. (2002) Proc. Natl. Acad. Sci. USA 99, 15095–15100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Michor, F., Iwasa, Y. & Nowak, M. A. (2004) Nat. Rev. Cancer 4, 197–206. [DOI] [PubMed] [Google Scholar]
- 37.Komarova, N. L., Sengupta, A. & Nowak, M. A. (2003) J. Theor. Biol. 223, 433–450. [DOI] [PubMed] [Google Scholar]
- 38.Nowak, M. A., Michor, F. & Iwasa, Y. (2003) Proc. Natl. Acad. Sci. USA 100, 14966–14969. [DOI] [PMC free article] [PubMed] [Google Scholar]