

Abstract
The concept of using crack propagation in polymeric materials to control drug release and its first demonstration are reported. The composite drug delivery system consists of highly-textured superhydrophobic electrosprayed microparticle coatings, composed of biodegradable and biocompatible polymers poly(caprolactone) and poly(glycerol monostearate carbonate-co-caprolactone), and a cellulose/polyester core. The release of entrapped agents is controlled by the magnitude of applied strain, resulting in a graded response from water infiltration through the propagating patterned cracks in the coating. Strain-dependent delivery of the anticancer agents cisplatin and 7-ethyl-10-hydroxycamptothecin to esophageal cancer cells (OE33) in vitro is observed. Finally the device is integrated with an esophageal stent to demonstrate delivery of fluorescein diacetate, using applied tension, to an ex vivo esophagus.
Keywords: drug delivery, stimuli-responsive, superhydrophobic, composites, polymers
Graphical Abstact
Drug release is controlled by the magnitude of applied tensile strain through superhydrophobic composites. Strain-dependent in vitro delivery of anticancer agents (cisplatin and 7-ethyl-10-hydroxycamptothecin) to OE33 esophageal cancer cells and ex vivo delivery of fluorescein diacetate with esophageal stent integrated device are demonstrated. This system provides mechanoresponsive delivery for both hydrophilic and lipophilic compounds.
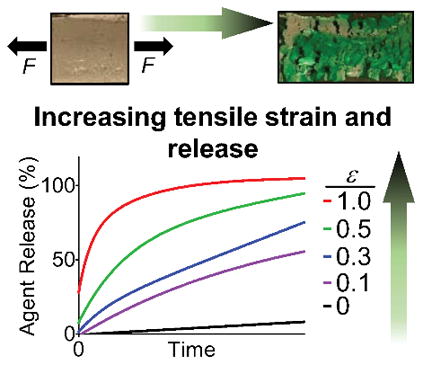
Mechanoresponsive polymeric materials are of significant interest as key functional elements in self-healing assemblies,[1] sensors and electronics,[2] and biology/medicine.[3] Consequently, mechanoresponsive materials are actively being developed that respond to mechanical stimuli such as compression,[4] tension,[5] shear,[6] or ultrasound.[7] Implanted medical devices also experience many of these forces, and even exert their own mechanical forces during use (e.g., stents).[8] Our approach to designing functional mechanoresponsive materials for drug delivery uses crack propagation failure modes of composite materials to control drug release. We hypothesized that crack formation could be initiated and propagated through superhydrophobic coatings on a multilayered drug delivery system by applying tension, with consequent device wetting and drug release. Given our interests in triggered drug release from polymeric[9] and superhydrophobic[10] materials, we realized an opportunity to design and evaluate such a new drug delivery system for an esophageal stent. Herein, we report: (1) the fabrication of a multilayered electrosprayed polymeric device; (2) the entrapment and subsequent controlled release of both hydrophilic and lipophilic agents under various applied strains; (3) analysis of the crack propagation mechanism with determination of the fracture toughness and critical strain energy release rate; (4) the demonstration of in vitro tension-mediated delivery of cisplatin and 7-ethyl-10-hydroxycamptothecin (SN-38) to cancer cells; and (5) the integration of the device with an esophageal stent to demonstrate fluorescein diacetate delivery, using applied tension, to an ex vivo esophagus.
By design, the device consists of a hydrophilic mesh core (containing release agent) encased by two superhydrophobic coatings that resist overall wetting. The mismatch in mechanical properties, resulting from a strong core and weaker coating, ensures mechanical failure of the coatings in the presence of applied tension, with crack propagation leading to water infiltration and release of the agent. Specifically, the absorbent cellulose/polyester core is rendered water-impermeable by electrospraying its entire surface with a low surface energy polymer blend. We selected electrospraying (Figure 1a) to produce superhydrophobic coatings, as opposed to other processing techniques[11], due to its ability to generate coatings of interconnected, hydrophobic micro- and nanoparticles on otherwise hydrophilic bulk materials, and because it is an industrial scalable technique.[12] The combination of low surface energy from the blend of two biodegradable, biocompatible polymers (polycaprolactone (PCL, Mw = 45 kg·mol−1) and poly(glycerol monostearate carbonate-co-caprolactone) (PGC-C18, Mw = 30 kg·mol−1)[13], (Figure 1a)), and high surface roughness from electrospraying, are requisites for eliciting superhydrophobicity (advancing water contact angles > 150°).[14] Hydrophilic meshes (250 μm thickness) homogenously loaded with dye or anticancer agents are electrosprayed with a 100 μm-thick coating (Figure 1b). Cross-sectional SEM image (Figure 1c) shows the three layers of the device, with coatings composed of interconnected particles of 2–7 μm diameter (Figure S1); these superhydrophobic surfaces exhibit advancing water contact angles approaching 170° as opposed to hydrophobic PCL electrosprayed surfaces with advancing contact angle of 119° (Figure 1d). Without these superhydrophobic coatings, the hydrophilic core rapidly absorbs water and organic solvents. This indiscriminate absorbency also permits a variety of molecular agents to be loaded and studied.
Figure 1.

Fabrication schematic and concept of tension-responsive system. (a) The PGC-C18 and PCL electrosprayed barrier coating (yellow) prevents release from the drug-loaded core (green). (b) Photographs of the native mesh with no dye, after dye-loading, and after subsequent coating. Tension is applied to the spray-coated system (bottom) longitudinally. (c) SEM image of device cross-section shows consistent morphology of microparticle coatings surrounding mesh core. (d) Advancing water contact angles for electrosprayed hydrophobic (top, PCL) and superhydrophobic (bottom, PCL:PGC-C18 1:1) coatings.
The tension-mediated release of agents from these multilayered devices is readily visualized using a hydrophilic green dye and quantified by UV-Visible spectrophotometry. These devices do not wet when submerged in simulated biological fluid (PBS with 10% FBS) for prolonged durations (> 24 hours). When subjected to tension, however, disruption of this otherwise superhydrophobic barrier occurs via coating fracture and causes subsequent dye efflux. Importantly, dye release rates are controlled by the magnitude of applied strain input (Figure 2a). All release aliquot concentrations after application of strain are significantly higher than those measured in the absence of strain (ε = 0), and aliquot concentrations of dye for ε = 0.1 and ε = 0.3 are significantly different from ε = 1.0 after 5 minutes and 10 minutes, respectively (ANOVA one-way, p < 0.05). Electrosprayed coatings that are merely hydrophobic (PCL, θadv = 119°, Figure 1d) rapidly wet even in the absence of tension, confirming the necessity of superhydrophobic coatings to prevent release. Water infiltration upon coating fracture is observed also using contrast-enhanced microcomputed tomography (μCT) by subjecting the superhydrophobic devices to tension (ε = 1.0) while submerged in a solution of PBS with 10% FBS and iodixanol. As shown in Figure 2b, μCT attenuation between un-stretched (left) and stretched (right) devices noticeably differ due to the compromised superhydrophobic coating and consequent core hydration after applying tension. Because bulk wetting of the coating does not occur, as visualized by the minimal attenuation at the outer layers of the device, the mechanism of release is attributed to core hydration by water infiltrating through the fractured superhydrophobic coatings.
Figure 2.

Strain-dependent release of model hydrophilic dye from a tension-responsive, superhydrophobic drug delivery system via crack propagation. (a) Cumulative dye release with electrosprayed coating of PCL:PGC-C18 1:1, or PCL (control, orange hexagon) as a function of tensile strain. Error bars denote + SD, n = 3 for each time point. (b) Contrast-enhanced microcomputed tomography (μCT) images before and after applied tensile strain (left and right, respectively), indicating water absorption (blue) after coating fracture. Arrowheads define the boundary of the device. (c) Image sequences from video as device is stretched along the x-axis showing crack development in the superhydrophobic barrier coating. (d) Coating fracture analysis in terms of number of cracks and total crack area as a function of increasing strain, n = 1. (e) Average crack area as a function of increasing strain, n = 1.
Next, we investigated the mechanism of release using fracture image analysis and tensile testing. The image sequence in Figure 2c shows the effect of increasing tensile strain (ε = 0 to 1.0) to initiate release. Mode I macroscopic crack initiation occurs at strain magnitudes of ~0.3, followed by propagation and additional crack formation as strain increases. As shown in Figure 2d and 2e, the number of cracks increases at first and then begins to decrease as the cracks merge with one another, resulting in greater mean crack area with increasing strain. As a result, more of the core surface area is exposed, leading to faster release rates. These fracture patterns are reminiscent of those found in thin films adhered to rigid, deformable substrates,[15] which consist of periodic parallel cracks formed perpendicular to the direction of applied strain. Mechanical analysis in accordance with ASTM Standard D 5049–99, using a polydimethylsiloxane (PDMS) substrate analog of our cellulose/polyester mesh system (see Supporting Information), estimates plane-strain fracture toughness (KIc) and critical strain energy release rate (GIc) at 0.0103 ± 0.00165 MPa·m1/2 and 1.253 ± 0.595 N·mm−1, respectively. These KIc and GIc values represent a lower limit of fracture resistance in terms of applied stress and strain of the coating, respectively. Using the KIc and GIc values, and accounting for the volume fraction of the two materials, the Young’s modulus (E) of the coating is estimated through the relation
| (Equation 1) |
resulting in a value of 0.0303 ± 0.014 MPa—two orders of magnitude lower than the core substrate (3.2 MPa). Together with KIc and GIc, E of the coating and substrate describe the favorable conditions for forming cracks,[16] and will aid in the design considerations for future tension responsive systems.
Mechanoresponsive drug delivery systems can be integrated with and controlled by current medical devices. Specifically, radially expanded esophageal stents are used for the palliative treatment of esophageal cancer. Esophageal cancer is the sixth deadliest cancer worldwide with a 5-year survival rate of 17.5%.[17] Patients typically have difficulty swallowing solid and liquid food due to tumor ingrowth. In order to mitigate symptoms, esophageal stents are often used to keep the esophagus open to improve intake of nutrients, increasing patient comfort, and quality of life. Our composite system can be an outer polymeric sheath that is stretched with stent expansion, enabling mechano-triggered control over drug delivery while minimizing side effects, eliminating drug loss during stent deployment, enabling increased drug dose, and providing easier patient nutrient intake. As the first steps towards this demonstration, we studied the release of two chemotherapeutic agents (cisplatin or SN-38) from the device, and integrated the device with an esophageal stent to deliver localized fluorescein diacetate ex vivo using applied tension.
In vitro efficacy of cisplatin or SN-38 delivery (2.4 wt% and 0.1 wt% loading capacity, 97.8+/−8.8% and 97.1+/−4.8% encapsulation efficiency, respectively) from the multilayer tension responsive device is evaluated against the OE33 human esophageal cancer cell line. As shown in Figure 3a, the release profile for cisplatin is similar to the hydrophilic dye. Statistically significant differences in release from the control (ε = 0) are achieved starting at 2 minutes at ε = 1.0 and after 40 minutes for both ε = 0.1 and 0.3. Likewise, strain-dependent release rates are observed with SN-38-loaded devices, but over a longer duration because SN-38 is lipophilic (Figure 3b). Statistically significant increases of SN-38 release occur after 0.5 hour and 2.5 hours between the control and ε = 1.0 or ε = 0.5, respectively. Release aliquots from drug-loaded devices elicit efficacy (cytotoxicity) against OE33 cells only after applying strain (Figure 3c and 3d). In vitro dose response is reflected in the significantly different cell viabilities achieved by varying the applied strain magnitude of cisplatin-loaded devices from 0 (control), 0.3, and 1.0 after 30 minutes. Similar strain-dependent dose-response behavior is observed using SN-38-loaded devices subjected to strain magnitudes of 0, 0.5, and 1.0 after 15 minutes. All tests were conducted with ANOVA one-way (p < 0.05). The difference in release kinetics of these two chemotherapeutic agents are attributed to inherent differences in aqueous solubilities, as cisplatin is hydrophilic and SN-38 is lipophilic.
Figure 3.

Tension-responsive release of chemotherapeutic agents from the superhydrophobic multilayered device. Cumulative release of (a) cisplatin, and (b) SN-38. Corresponding strain-dependent dose response of in vitro (c) cisplatin and (d) SN-38 delivery to esophageal cancer cells (OE33) after 30 minutes and 15 minutes, respectively. Error bars denote + SD with * = p < 0.05, n = 3 for each time point.
Localized ex vivo delivery is accomplished by integrating the tension-responsive device with a metal esophageal stent. Fluorescein diacetate was chosen as the release agent due to its strong fluorescent signal after hydrolysis into fluorescein (λex = 490 nm, λem = 525 nm) to enable visualization of dye delivery. The fluorescein diacetate loaded devices were sutured around a self-expanding Ni-Ti alloy esophageal stent to form an enveloping sheath, capable of undergoing radial expansion with the stent. Next, the stent was inserted into excised bovine esophagi, and expanded to ε = 1.0. Current esophageal stents on the market can be expanded to a broad range of strains, from ε = 0.14 to 2.83, via selection based on various diameters of stents and introducers.[18] After 8 hours, the esophagi were dissected longitudinally to reveal a fluorescent band corresponding to the tension-responsive device under UV light (Figure 4a and 4b). The fluorescence from the expanded systems (ε = 1.0) is greater than the unexpanded controls (ε = 0) in subsequent esophageal cross-sections (100 μm thickness) with better lumenal localization to the esophageal epithelial mucosa layer (Figure 4c and 4d).
Figure 4.

Ex vivo delivery of fluorescein diacetate from superhydrophobic devices. Photographs of tension-responsive device integrated with esophageal stent (ε = 1.0) and fluorescein diacetate delivery (yellow, green) to bovine esophagus under (a) white and (b) UV light. Scale bar (white) = 20 mm. (c) Representative bright field (left) and fluorescent (right) microscopy images of esophagus cross-section after delivery. T denotes esophageal tissue and L denotes lumen space. Scale bar (red) = 2.5 mm; yellow arrow represents analysis profile. (d) Fluorescence intensity profile across esophagus cross-section. Blue dotted line represents mucosal-device boundary; error bars denote ± SD for n = 4.
While selective wetting of superhydrophobic materials and stimuli-responsive drug delivery are active areas of functional materials research,[7a, 19] the tension-induced wetting of a superhydrophobic material via crack propagation departs from previously reported approaches. For example Zhang et al.[20] used a triangular polyamide mesh to reversibly transition from superhydrophobic to superhydrophilic (i.e., wetting) states using equibiaxial strains greater than 120%. This wetting transition was due to an increase in the average side length of the triangular net-like pores, which reduced surface roughness, thus overcoming the droplet surface tension and causing its collapse. Recently, Huang et al.[21] demonstrated the ability to reversibly switch between superhydrophobic to superhydrophilic states and achieved wetting triggered by increases in strain, pH, or temperature due to expansion of various hydrogels coated with silanized glass particles. Specifically, the amount of strain applied controlled hydrophilic dye penetration into the alginate-acrylamide hydrogel. Choi and coworkers[22] demonstrated a similar approach using absorbent fabrics that were rendered omniphobic by dip-coating in a solution of fluorodecyl polyhedral oligomeric silsesquioxanes. Wetting of these omniphobic surfaces by a variety of polar and non-polar solvents was achieved using equibiaxial strains greater than ~20%, depending on droplet surface tension. Alternatively, Di et al.[23] reported a mechanism for controlling release by increasing the available surface area for drug diffusion. The multicomponent system consists of drug-loaded poly(lactic-co-glycolic acid) nanoparticles incorporated in alginate microdepots, which are elongated as the underlying elastomeric substrate is stretched, to demonstrate cycle-dependent release. While these approaches provide a basis of controlling release through changes in microscale surface features to alter wettability or diffusivity, our study employs a more macroscopic approach—specifically, by introducing fractures within composite materials.
In conclusion, we describe the fabrication, characterization, and evaluation of a tension-responsive drug delivery system. The superhydrophobic microparticle coatings are applied to core substrates using a facile and scalable electrospraying process, which provides flexibility in substrate choice and the potential to incorporate these mechanoresponsive systems onto existing medical devices to enhance their current functionality. The key design feature responsible for the tension-triggered release of entrapped agents is facilitating crack initiation and propagation within these coatings due to their mismatch in mechanical properties with the core material. The device is amenable to delivery of both lipophilic and hydrophilic agents, suggesting widespread utility for a number of drug delivery applications where mechanical force, such as tensile strain or device expansion, are experienced.
Experimental Section
Device Fabrication
Solutions of dye, cisplatin, SN-38, or fluorescein diacetate were adsorbed onto cellulose/polyester meshes and electrosprayed with a solution of 1:1 PGC-C18 (Mw = 30 kg·mol−1, PDI = 1.4) and PCL (Mw = 45 kg·mol−1) (flow rate = 5 mL hr−1, voltage = 20 kV, tip-to-collector distance = 10 cm).
Tension-Mediated Release Studies
Devices were stretched at 7% strain·s−1 in a bath PBS or RPMI, with 10% v/v FBS.
Cell Culture Assays
OE33 esophageal cancer cells were incubated with release aliquots for 96 or 72 hours (SN-38 or cisplatin, respectively). Cell viability was measured via MTS assay.
Ex vivo delivery via esophageal stent
Esophageal stents integrated with fluorescein diacetate loaded devices were inserted into excised bovine esophagi and allowed to expand to either strains of 1.0 or left unexpanded (ε = 0). After dissection, esophagi lumen were imaged under white and UV light. 100 μm cross sections were imaged on an inverted fluorescence microscope (Olympus IX 81; λex = 490 nm, λex = 525 nm).
See Supporting Information for more details.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institutes of Health T32 EB006359 (MWG, JW) and a National Science Foundation Graduate Research Fellowship GRF DGE-1247312 (JW). The authors thank Dr. Jonathan D. Freedman for assistance in μCT acquisition and analysis for determining water infiltration.
Footnotes
Supporting information for this article is given via a link at the end of the document
References
- 1.a) Caruso MM, Davis DA, Shen Q, Odom SA, Sottos NR, White SR, Moore JS. Chem Rev. 2009;109:5755–5798. doi: 10.1021/cr9001353. [DOI] [PubMed] [Google Scholar]; b) Ghosh B, Urban MW. Science. 2009;323:1458–1460. doi: 10.1126/science.1167391. [DOI] [PubMed] [Google Scholar]; c) White SR, Sottos NR, Geubelle PH, Moore JS, Kessler MR, Sriram SR, Brown EN, Viswanathan S. Nature. 2001;409:794–797. doi: 10.1038/35057232. [DOI] [PubMed] [Google Scholar]; d) Yan X, Wang F, Zheng B, Huang F. Chem Soc Rev. 2012;41:6042–6065. doi: 10.1039/c2cs35091b. [DOI] [PubMed] [Google Scholar]; e) Burnworth M, Tang L, Kumpfer JR, Duncan AJ, Beyer FL, Fiore GL, Rowan SJ, Weder C. Nature. 2011;472:334–337. doi: 10.1038/nature09963. [DOI] [PubMed] [Google Scholar]; f) Sijbesma RP, Beijer FH, Brunsveld L, Folmer BJ, Hirschberg JK, Lange RF, Lowe JK, Meijer E. Science. 1997;278:1601–1604. doi: 10.1126/science.278.5343.1601. [DOI] [PubMed] [Google Scholar]; g) Wathier M, Grinstaff MW. J Am Chem Soc. 2008;130:9648–9649. doi: 10.1021/ja803248q. [DOI] [PubMed] [Google Scholar]; h) Lin X, Grinstaff MW. Isr J Chem. 2013;53:498–510. [Google Scholar]
- 2.a) Schäfer CG, Gallei M, Zahn JT, Engelhardt J, Hellmann GP, Rehahn M. Chem Mater. 2013;25:2309–2318. [Google Scholar]; b) Feng H, Lu J, Li J, Tsow F, Forzani E, Tao N. Adv Mater. 2013;25:1729–1733. doi: 10.1002/adma.201204105. [DOI] [PubMed] [Google Scholar]; c) Roberts DRT, Holder SJ. J Mater Chem. 2011;21:8256–8268. [Google Scholar]; d) Davis DA, Hamilton A, Yang J, Cremar LD, Van Gough D, Potisek SL, Ong MT, Braun PV, Martinez TJ, White SR, Moore JS, Sottos NR. Nature. 2009;459:68–72. doi: 10.1038/nature07970. [DOI] [PubMed] [Google Scholar]; e) Wang Q, Gossweiler GR, Craig SL, Zhao X. Nat Commun. 2014;5 doi: 10.1038/ncomms5899. [DOI] [PubMed] [Google Scholar]
- 3.a) Chan A, Orme RP, Fricker RA, Roach P. Adv Drug Del Rev. 2013;65:497–514. doi: 10.1016/j.addr.2012.07.007. [DOI] [PubMed] [Google Scholar]; b) Rapoport N. Prog Polym Sci. 2007;32:962–990. [Google Scholar]; c) Riehl BD, Park JH, Kwon IK, Lim JY. Tissue Eng, Part B. 2012;18:288–300. doi: 10.1089/ten.teb.2011.0465. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yang Y, Magnay JL, Cooling L, El Haj AJ. Biomaterials. 2002;23:2119–2126. doi: 10.1016/s0142-9612(01)00342-8. [DOI] [PubMed] [Google Scholar]; e) Zhu X, Mills KL, Peters PR, Bahng JH, Liu EH, Shim J, Naruse K, Csete ME, Thouless MD, Takayama S. Nat Mater. 2005;4:403–406. doi: 10.1038/nmat1365. [DOI] [PubMed] [Google Scholar]
- 4.a) Lee KY, Peters MC, Anderson KW, Mooney DJ. Nature. 2000;408:998–1000. doi: 10.1038/35050141. [DOI] [PubMed] [Google Scholar]; b) Izawa H, Kawakami K, Sumita M, Tateyama Y, Hill JP, Ariga K. J Mater Chem B. 2013;1:2155–2161. doi: 10.1039/c3tb00503h. [DOI] [PubMed] [Google Scholar]; c) Xiao L, Tong Z, Chen Y, Pochan DJ, Sabanayagam CR, Jia X. Biomacromolecules. 2013;14:3808–3819. doi: 10.1021/bm4011276. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yang Y, Tang G, Zhang H, Zhao Y, Yuan X, Fan Y, Wang M. Mater Sci Eng C Mat Biol Appl. 2011;31:350–356. [Google Scholar]
- 5.a) Barthes J, Mertz D, Bach C, Metz-Boutigue MH, Senger B, Voegel JC, Schaaf P, Lavalle P. Langmuir. 2012;28:13550–13554. doi: 10.1021/la302550q. [DOI] [PubMed] [Google Scholar]; b) Zhang Y, Chen Q, Ge J, Liu Z. Chem Commun. 2013;49:9815–9817. doi: 10.1039/c3cc45837g. [DOI] [PubMed] [Google Scholar]; c) Vogt C, Mertz D, Benmlih K, Hemmerlé J, Voegel JC, Schaaf P, Lavalle P. ACS Macro Lett. 2012;1:797–801. doi: 10.1021/mz3000896. [DOI] [PubMed] [Google Scholar]
- 6.a) Korin N, Kanapathipillai M, Matthews BD, Crescente M, Brill A, Mammoto T, Ghosh K, Jurek S, Bencherif SA, Bhatta D, Coskun AU, Feldman CL, Wagner DD, Ingber DE. Science. 2012;337:738–742. doi: 10.1126/science.1217815. [DOI] [PubMed] [Google Scholar]; b) Holme MN, Fedotenko IA, Abegg D, Althaus J, Babel L, Favarger F, Reiter R, Tanasescu R, Zaffalon PL, Ziegler A. Nat Nanotechnol. 2012;7:536–543. doi: 10.1038/nnano.2012.84. [DOI] [PubMed] [Google Scholar]; c) Ramirez ALB, Kean ZS, Orlicki JA, Champhekar M, Elsakr SM, Krause WE, Craig SL. Nat Chem. 2013;5:757–761. doi: 10.1038/nchem.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Yohe ST, Kopechek JA, Porter TM, Colson YL, Grinstaff MW. Adv Healthc Mater. 2013;2:1204–1208. doi: 10.1002/adhm.201200381. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang CY, Yang CH, Lin YS, Chen CH, Huang KS. Biomaterials. 2012;33:1547–1553. doi: 10.1016/j.biomaterials.2011.10.047. [DOI] [PubMed] [Google Scholar]; c) Husseini GA, Diaz de la Rosa MA, Richardson ES, Christensen DA, Pitt WG. J Control Release. 2005;107:253–261. doi: 10.1016/j.jconrel.2005.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Adler DG. Video J Encycl GI Endosc. 2013;1:66–68. [Google Scholar]; b) Harewood FJ, McHugh PE. Ann Biomed Eng. 2007;35:1539–1553. doi: 10.1007/s10439-007-9326-6. [DOI] [PubMed] [Google Scholar]
- 9.a) Griset AP, Walpole J, Liu R, Gaffey A, Colson YL, Grinstaff MW. J Am Chem Soc. 2009;131:2469–2471. doi: 10.1021/ja807416t. [DOI] [PubMed] [Google Scholar]; b) Colson YL, Grinstaff MW. Adv Mater. 2012;24:3878–3886. doi: 10.1002/adma.201200420. [DOI] [PubMed] [Google Scholar]
- 10.a) Yohe ST, Herrera VLM, Colson YL, Grinstaff MW. J Control Release. 2012;162:92–101. doi: 10.1016/j.jconrel.2012.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yohe ST, Colson YL, Grinstaff MW. J Am Chem Soc. 2012;134:2016–2019. doi: 10.1021/ja211148a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Nayak BK, Caffrey PO, Speck CR, Gupta MC. Appl Surf Sci. 2013;266:27–32. [Google Scholar]; b) Cui Y, Paxson AT, Smyth KM, Varanasi KK. Colloids Surf, A. 2012;394:8–13. [Google Scholar]; c) Manna U, Kratochvil MJ, Lynn DM. Adv Mater. 2013;25:6405–6409. doi: 10.1002/adma.201302561. [DOI] [PubMed] [Google Scholar]; d) Li XM, Reinhoudt D, Crego-Calama M. Chem Soc Rev. 2007;36:1350–1368. doi: 10.1039/b602486f. [DOI] [PubMed] [Google Scholar]; e) Sas I, Gorga RE, Joines JA, Thoney KA. J Polym Sci, Part B: Polym Phys. 2012;50:824–845. [Google Scholar]; f) Yohe ST, Freedman JD, Falde EJ, Colson YL, Grinstaff MW. Adv Funct Mater. 2013;23:3628–3637. doi: 10.1002/adfm.201203111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yohe ST, Grinstaff MW. Chem Commun. 2013;49:804–806. doi: 10.1039/c2cc38012a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolinsky JB, Yohe ST, Colson YL, Grinstaff MW. Biomacromolecules. 2012;13:406–411. doi: 10.1021/bm201443m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Crick CR, Parkin IP. Chem Eur J. 2010;16:3568–3588. doi: 10.1002/chem.200903335. [DOI] [PubMed] [Google Scholar]; b) Zhang X, Shi F, Niu J, Jiang Y, Wang Z. J Mater Chem. 2008;18:621–633. [Google Scholar]
- 15.Thouless MD, Li Z, Douville NJ, Takayama S. J Mech Phys Solids. 2011;59:1927–1937. doi: 10.1016/j.jmps.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Beuth JL, Klingbeil NW. J Mech Phys Solids. 1996;44:1411–1428. [Google Scholar]; b) Kim BC, Moraes C, Huang J, Thouless MD, Takayama S. Biomater Sci. 2014;2:288–296. doi: 10.1039/C3BM60276A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]; b) Jemal A, Siegel R, Xu J, Ward E. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 18.Hindy P, Hong J, Lam-Tsai Y, Gress F. Gastroenterol Hepatol. 2012;8:526. [PMC free article] [PubMed] [Google Scholar]
- 19.a) Chang JH, Hunter IW. Macromol Rapid Commun. 2011;32:718–723. doi: 10.1002/marc.201000716. [DOI] [PubMed] [Google Scholar]; b) Hersey JS, Freedman JD, Grinstaff MW. J Mater Chem B. 2014;2:2974–2977. doi: 10.1039/C4TB00143E. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Verplanck N, Coffinier Y, Thomy V, Boukherroub R. Nanoscale Res Lett. 2007;2:577–596. [Google Scholar]
- 20.Zhang J, Lu X, Huang W, Han Y. Macromol Rapid Commun. 2005;26:477–480. [Google Scholar]
- 21.Huang X, Sun Y, Soh S. Adv Mater. 2015;27:4062–4068. doi: 10.1002/adma.201501578. [DOI] [PubMed] [Google Scholar]
- 22.Choi W, Tuteja A, Chhatre S, Mabry JM, Cohen RE, McKinley GH. Adv Mater. 2009;21:2190–2195. [Google Scholar]
- 23.Di J, Yao S, Ye Y, Cui Z, Yu J, Ghosh TK, Zhu Y, Gu Z. ACS Nano. 2015;9:9407–9415. doi: 10.1021/acsnano.5b03975. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.