

Abstract
Fatty acid synthase (FAS) activity is a potential therapeutic target to treat cancer and obesity. Here, we have identified a molecular link between FAS and HER2 (erbB-2) oncogene, a marker for poor prognosis that is overexpressed in 30% of breast and ovarian cancers. Pharmacological FAS inhibitors cerulenin and C75 were found to suppress p185HER2 oncoprotein expression and tyrosine-kinase activity in breast and ovarian HER2 overexpressors. Similarly, p185HER2 expression was dramatically down-regulated when FAS gene expression was silenced by using the highly sequence-specific mechanism of RNA interference (RNAi). Pharmacological and RNAi-mediated silencing of FAS specifically down-regulated HER2 mRNA and, concomitantly, caused a prominent up-regulation of PEA3, a transcriptional repressor of HER2. A cytoplasmic redistribution of p185HER2 was associated with marked morphological changes of FAS RNAi-transfected cells, whereas chemical inhibitors of FAS promoted a striking nuclear accumulation of p185HER2. The simultaneous targeting of FAS and HER2 by chemical FAS inhibitors and the humanized antibody directed against p185HER2 trastuzumab, respectively, was synergistically cytotoxic toward HER2 overexpressors. Similarly, concurrent RNAi-mediated silencing of FAS and HER2 genes synergistically stimulated apoptotic cell death in HER2 overexpressors. p185HER2 was synergistically down-regulated after simultaneous inhibition of FAS and HER2 by either pharmacological inhibitors or small interfering RNA. These findings provide evidence of an active role of FAS in cancer evolution by specifically regulating oncogenic proteins closely related to malignant transformation, strongly suggesting that HER2 oncogene may act as the key molecular sensor of energy imbalance after the perturbation of tumor-associated FAS hyperactivity in cancer cells.
Keywords: trastuzumab, small interfereing RNA, chemotherapy, lipogenesis, Herceptin
The biosynthetic enzyme fatty acid synthase (FAS) is the major enzyme required for the anabolic conversion of dietary carbohydrates to fatty acids, and it functions normally in cells with high lipid metabolism. Under normal physiological conditions, any FAS increase is tightly regulated by a number of environmental, hormonal, and nutritional signals (1, 2). However, human tissue studies have demonstrated that infiltrating carcinomas of the breast constitutively express high levels of FAS compared to nontransformed human epithelial tissue (3–8). Furthermore, increased levels of FAS accompany the development of in situ carcinoma of the breast, suggesting a potential link between increased expression and increased risk of breast cancer development (9). Remarkably, overexpression and hyperactivity of FAS is associated with more aggressive breast and ovarian cancers (3–8, 10). The early and nearly universal up-regulation of FAS in many human cancers and its association with poor clinical outcome both strengthen the hypothesis that FAS is involved in the development, maintenance, and enhancement of the malignant phenotype (11). However, FAS overexpression in tumor cells appears to be part of a more general change in the genetic program controlling lipogenesis, as evidenced by the concomitant increase of other enzymes of the same lipogenic pathway (9, 12, 13). Thus, it remains to be addressed whether tumor-associated FAS is a mere manifestation of early and common cancer-associated epigenetic changes or actively contributes to the cancer phenotype.
This study was undertaken to test the hypothesis that increased FAS activity plays an active role in cancer evolution by regulating oncogenic proteins closely related to malignant transformation. We show that FAS-dependent signaling regulates the expression, activity, and cellular localization of HER2 (c-erbB-2) oncogene in breast and ovarian cancer cells. We demonstrate that pharmacological and small interference RNA (siRNA)-mediated inhibition of FAS negatively regulates the expression of HER2 at the transcriptional level, and identify the ets transcription factor PEA3 as a molecular mechanism through which FAS blockade transcriptionally represses HER2 gene expression (14, 15). We further prove that simultaneous targeting of FAS and HER2 synergistically down-regulates p185HER2 and inhibits tumor cell proliferation by promoting apoptosis. We suggest that HER2 plays a previously uncharacterized role as a cellular energy sensor in the response of tumor cells to a nongenotoxic metabolic stress, such as the perturbation of FAS-dependent endogenous fatty acid biosynthesis, thus offering a rationale for a therapeutic targeting of FAS in HER2-overexpressing carcinomas.
Materials and Methods
Materials. Cerulenin and C75 were obtained from Sigma and Alexis Biochemicals (San Diego), respectively. Trastuzumab (Herceptin) was provided by the Evanston Northwestern Healthcare Hospital Pharmacy (Evanston, IL).
FAS Ab (M3 clone) was generously provided by E. Pizer (Johns Hopkins University, Baltimore). Anti-phosphotyrosine Ab (4G10 clone) was from Upstate Biotechnology (Lake Placid, NY). Abs against p185HER2 (Ab-3 and Ab-5 clones) were from Oncogene. Anti-cyclin D1, anti-β-actin, and anti-PEA3 Abs were from Santa Cruz Biotechnology.
Cell Culture. Cell lines were obtained from the American Type Culture Collection, and they were routinely grown in improved MEM (Biosource International, Camarillo, CA) containing 5% FBS and 2 mM l-glutamine as described (16, 17). MDA-MB-231/HER2 cells were constructed by transfection of MDA-MB-231 cells with pRC/CMV containing the full-length HER2 cDNA, and then selected by the addition of 200 μg/ml G418 (Sigma) into the medium.
FAS Activity. FAS activity was assayed by recording, spectrophotometrically at 25°C, the decrease of A340 because of oxidation of NADPH essentially as described by Dils and Carey (18).
Cell Viability. Concentration–effect curves were generated as a plot of the fraction of surviving cells versus drug concentration by using a colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) reduction assay (16, 17).
Synergy Analysis. The cytotoxic interaction between cerulenin and trastuzumab was evaluated by the isobologram technique (19).
Immunoblotting. Electrophoresis and Western blotting analyses were performed as described (17).
HER2-Specific ELISA. For quantitative determination of p185HER2, the Human neu Quantitative ELISA System (Oncogene) was applied according to the manufacturer's protocols.
Further Details. For details concerning RT-PCR, RNA interference (RNAi)-mediated silencing of FAS and HER2 genes, immunofluorescent staining, flow cytometry, apoptosis, and statistical analysis, see Supporting Text, which is published as supporting information on the PNAS web site.
Results
Pharmacological Blockade of FAS Activity Down-Regulates p185HER2 Expression and Activity in HER2-Overexpressing Breast and Ovarian Cancer Cells. We first examined how blockade of FAS activity affected p185HER2 expression in HER2- and FAS-overexpressing SK-Br3 cells. We used two well characterized chemical inhibitors of FAS activity: the natural mycotoxin cerulenin (20, 21), or a more stable synthetic FAS inhibitor, the α-methylene-γ-butyrolactone C75 (22). A dose-dependent decrease in the expression and tyrosine phosphorylation of p185HER2 was observed in SK-Br3 cells after 48 h of treatment with cerulenin as assessed by Western blotting. In fact, SK-Br3 cells did not express a detectable level of p185HER2 in the presence of 10 μg/ml cerulenin, whereas the levels of β-actin remained unchanged (Fig. 1A). We next performed a quantitative measurement of p185HER2 in cerulenin-treated SK-Br3 cells by using a HER2-specific ELISA. p185HER2 expression was dose-dependently reduced by cerulenin and the magnitude of this decrease ranged from 14% at 1.25 μg/ml cerulenin to 78% at 10 μg/ml cerulenin, relative to untreated cells (Fig. 1B).
Fig. 1.

Pharmacological blockade of FAS activity down-regulates expression and activity of p185HER2 oncoprotein. (A) SK-Br3 cells were cultured in the presence of cerulenin for 48 h. Twenty micrograms of protein was subjected to Western blot analyses with the p185HER2 Ab-3. Activation status of HER2 was analyzed by assessing tyrosine phosphorylation of p185HER2 using the anti-phosphotyrosine Ab 4G10. (B) HER2 concentration in cell lysates from cerulenin-treated SK-Br3 cells was quantified by using the Human neu Quantitative ELISA System per the manufacturer's instructions. Data are presented are means ± SD (bars) (n = 3, in duplicate). (C) SK-Ov3 cells was cultured in the presence of C75 for 48 h. Twenty micrograms of protein was subjected to Western blot analyses for HER2 expression as described in A. (D) BT-474, MDA-MB-453, and T47-D breast cancer cells were cultured in the presence of 10 μg/ml cerulenin for 48 h. Twenty micrograms of protein was subjected to Western blot analyses for HER2 expression as described in A.
We next tested the ability of chemical FAS inhibitors to modulate p185HER2 expression in SK-Ov3 cells, an ovarian cancer model for HER2 overexpression. Incubation of SK-Ov3 cells with C75 resulted in a dose-related down-regulation of the p185HER2, also reducing its tyrosine phosphorylation (Fig. 1C). In fact, a complete suppression of p185HER2 expression and activity was seen when high concentrations of C75 were used.
A significant decrease in the levels of p185HER2 upon treatment with chemical FAS inhibitors was also found in HER2 overexpressors BT-474 and MDA-MB-453, and in moderately HER2-expressing T47-D cells (Fig. 1D). These data demonstrate that the suppression of p185HER2 expression and activity is a previously uncharacterized cellular response that follows pharmacological inhibition of FAS activity in breast and ovarian cancer cells expressing high levels of HER2.
Pharmacological Inhibition of FAS Activity Up-Regulates the Expression of PEA3, a Transcriptional Repressor of HER2, and Down-Regulates HER2 mRNA Expression. To gain additional insight into the molecular mechanisms underlying the down-regulation of HER2 induced by cerulenin and C75, we examined the expression of HER2 regulators PEA3 and Cyclin D1 after pharmacological blockade of FAS activity in SK-Br3 cells. PEA3 is an ets DNA-binding protein that has been shown to inhibit HER2 promoted tumorigenesis by down-regulating HER2 promoter activity (14, 15). Cyclin D1 is required for HER2 induced transformation and could reciprocally up-regulate HER2 expression (23, 24). Cerulenin exposure up-regulated PEA3 expression in a dose- and time-dependent manner as assessed by Western blotting (Fig. 2A). Additionally, comparison of the intensity and appearance of PEA3 revealed an obvious difference as assessed by immunofluorescence staining and confocal analysis. Cell nuclei of SK-Br3 cells exposed to low doses of cerulenin yielded considerably brighter punctuate patterns of PEA3 than cell nuclei of untreated cells (Fig. 2A i–iv). Cyclin D1 expression was slightly decreased when high concentrations of cerulenin were used (Fig. 2A).
Fig. 2.

Accumulation of PEA3 and inhibition of HER2 gene transcription after pharmacological blockade of FAS activity. (A)(Upper) SK-Br3 cells were cultured either with graded concentrations of cerulenin for 48 h (Left) or exposed to 5 μg/ml cerulenin for 96 h (Right). Fifty micrograms of protein was subjected to Western blot analyses with anti-PEA3 and Cyclin D1 Abs. (Lower) SK-Br3 cells were treated with 2.5 μg/ml cerulenin for 48 h, and subcellular localization of immunofluorescent PEA3 was assessed with a Leica DMIRE2 confocal microscope. PEA3-stained cell nuclei are presented at two magnifications (control, i and ii; and cerulenin-treated cells, iii and iv). (B) Total RNA from cerulenin- and C75-treated SK-Br3 cells was isolated and RT-PCR analyses for HER2 and GADPH transcripts and expression were performed as described in Materials and Methods.
We next tested whether cerulenin- and C75-induced accumulation of PEA3 correlated with a transcriptional response of HER2 gene. HER2 mRNA expression was down-regulated when SK-Br3 cells were treated with cerulenin, as demonstrated by RT-PCR analyses. Similarly, no HER2 mRNA signal could be detected upon treatment with C75 (Fig. 2B). Taken together, these findings establish that PEA3 accumulation is a strong and early molecular change triggered by chemical FAS inhibitors. Moreover, the accumulation of PEA3 that follows FAS inhibition was concomitant with a down-regulation of HER2 expression, whereas the amplitude of the cerulenin and C75 effects on HER2 mRNA levels closely paralleled those observed for p185HER2 protein expression. Therefore, PEA3 up-regulation after FAS blockage may dictate the extent of HER2 down-regulation in cancer cells.
Silencing Expression of FAS Gene by siRNA Suppresses HER2 Oncogene Overexpression. To unquestionably establish a role for FAS in the regulation of HER2, we used an entirely different approach to interfere with FAS activity. This approach was based on selective FAS gene silencing, using the potent and highly sequence-specific mechanism of RNAi. RNAi is a cellular process resulting in enzymatic cleavage and breakdown of mRNA, guided by sequence-specific double-stranded RNA oligonucleotides (siRNA; see refs. 25 and 26). To specifically silence the FAS gene, SK-Br3 cells were transfected with siRNA targeting FAS mRNA or with a nonspecific control pool of RNAis. Fig. 3A shows a Western blot analysis for one representative experiment (n = 4) in which three different concentrations were used of the siRNA targeted to FAS mRNA, as well as a nonspecific targeted siRNA. FAS RNAi severely suppressed expression of FAS (up to ≈80% reduction) when compared with control cells transfected with nonspecific siRNA. Importantly, transfection of SK-Br3 cells with increasing concentrations of siRNA targeted to FAS gene resulted in an important decrease of p185HER2 expression (up to ≈60% reduction when compared with p185HER2 levels in cells transfected with a nonspecific pool of RNAi; Fig. 3A). No effect of FAS-targeted siRNA transfection was observed on β-actin expression, which was used as an internal control for FAS RNAi specificity and protein loading.
Fig. 3.

(A–C) RNAi-mediated silencing of the FAS gene suppresses HER2 expression. (A) SK-Br3 cells were transfected with siRNA targeting FAS gene or with a nonspecific control siRNA pool for 72 h. Twenty micrograms of protein was subjected to Western blot analyses with specific Abs against FAS, p185HER2, or β-actin. (B) The amount of p185HER2 in FAS RNAi-transfected cells was quantified by flow cytometry using the p185HER2 Ab-5. The mean fluorescence signal ± SD (n = 3) was quantified by using the Geo Mean fluorescence parameter provided with cellquest software. (C)(Upper) Fifty micrograms of protein from FAS RNAi-transfected SK-Br3 cells was subjected to Western blot analyses for PEA3. (Lower) Total RNA from FAS RNAi-transfected SK-Br3 cells was isolated and RT-PCR analyses for HER2 and β-actin transcripts and expression were performed as described in Materials and Methods. (D) Impact of FAS RNAi and pharmacological blockade of FAS activity on p185HER2 cellular localization. Nonspecific RNAi- and FAS RNAi-transfected cells were fixed and labeled with the p185HER2 Ab-5 (a–d) or the p185HER2 Ab-3 (a′–d′). SK-Br3 cells were treated for 72 h with DMSO vol/vol (a″), 2.5 μg/ml cerulenin (b″), or 2.5 μg/ml C75 (c″), and then fixed and labeled with the p185HER2 Ab-3. Cellular localization of p185HER2 was detected by indirect immunofluorescence by incubating with TRITC (a–d)- or FITC (a′–d′)-conjugated anti-mouse IgG. After counterstaining with 4′,6-diamidino-2-phenylindole, cells were examined and photographed by using a Zeiss fluorescent microscope. A representative immunostaining analysis (n = 3) is shown.
Flow cytometric analysis of p185HER2 demonstrated a very significant 53% reduction of p185HER2 in FAS siRNA-transfected SK-Br3 breast cancer cells (Fig. 3B Left). HER2-overexpressing SK-Ov3 ovarian cancer cells were also very sensitive to the down-regulatory effects on p185HER2 expression after RNAi-mediated FAS silencing (63% reduction; Fig. 3B Center). In MCF-7 cells, which express low levels of HER2, the differences in p185HER2 expression between FAS siRNA-transfected cells and cells transfected with a nonspecific siRNA pool were not detectable (Fig. 3B Right). These findings clearly demonstrate that FAS inhibition selectively suppresses HER2 overexpression in breast and ovarian cancer cells, and further support the contention that this effect occurs solely in HER2 overexpressors, but not in cells expressing physiological amounts of HER2.
We further assessed whether RNAi-mediated FAS silencing regulated PEA3 and HER2 mRNA. PEA3 expression was significantly up-regulated in a dose-dependent manner in SK-Br3 cells transfected with increasing concentrations of FAS siRNA (Fig. 3C). Moreover, FAS siRNA caused a significant transcriptional repression of HER2 expression as demonstrated by RT-PCR analyses of the HER2 mRNA in FAS siRNA-transfected SK-Br3 cells, whereas no effect was observed on the levels of β-actin mRNA (Fig. 3C). In agreement with the results employing chemical FAS inhibitors, the up-regulation of PEA3 that follows RNAi-mediated silencing of the FAS gene was concomitant with a down-regulation of HER2 expression at the transcriptional level, strongly suggesting that PEA3 plays a key role determining the extent of p185HER2 down-regulation in response to FAS blockade.
Pharmacological and siRNA-Mediated Inhibition of FAS Differently Modulates p185HER2 Cellular Localization and HER2 Overexpressor Cell Morphology. Although p185HER2 is a transmembrane oncoprotein, its cytoplasmic domain can also function as a nuclear transcriptional activator (27). Therefore, we analyzed the impact of RNAi-mediated gene silencing of FAS in the subcellular localization of p185HER2. To address this question, FAS siRNA-transfected SK-Br3 cells were permeabilized with Triton X-100 for the intracellular delivery of two monoclonal Abs directed against the extracellular domain and the carboxyl terminal 14 amino acids of p185HER2 (Ab-5 and Ab-3, respectively; Fig. 3D Upper). Untreated (data not shown) and siRNA negative control-transfected cells (Fig. 3Da–a′) showed a prominent cell-surface staining of p185HER2. Upon siRNA FAS transfection, p185HER2 membrane staining was markedly reduced as assessed by immunofluorescent staining by using either Ab-5 (Fig. 3D b–d) or Ab-3 (Fig. 3D b′–d′), with p185HER2 showing some distribution throughout the cytoplasm. In the presence of low concentrations of chemical FAS inhibitors, the membrane staining of p185HER2 was clearly diminished when the subcellular localization of p185HER2 was assessed with Ab-5 (not shown). Interestingly, when p185HER2 staining was analyzed by using Ab-3, we found that p185HER2 was distributed throughout the cytoplasm and, in some cases, concentrated in the peri- and nuclear areas (Fig. 3Db″). A more prominent nuclear accumulation of p185HER2 was observed in C75-treated SK-Br3 cells (Fig. 3Dc″).
It is noteworthy that both the down-regulation and the cellular redistribution of p185HER2 were associated with marked morphological changes in siRNA FAS-transfected SK-Br3 cells, including an astrocyte-like phenotype with the occurrence of numerous extrusions, a significant loss of cell–cell contacts, and a less packed cell growth. Interestingly, De Schrijver et al. (28) recently reported similar morphological changes in prostate cancer cells. Cerulenin- and C75-induced down-regulation and cellular redistribution of p185HER2 was also related with marked morphological changes in SK-Br3 cells, including an increase in cell volume (giant cells), and a significant loss of cell–cell contacts. These observations indicate that FAS signaling modulates not only the expression of p185HER2 but also its cellular localization.
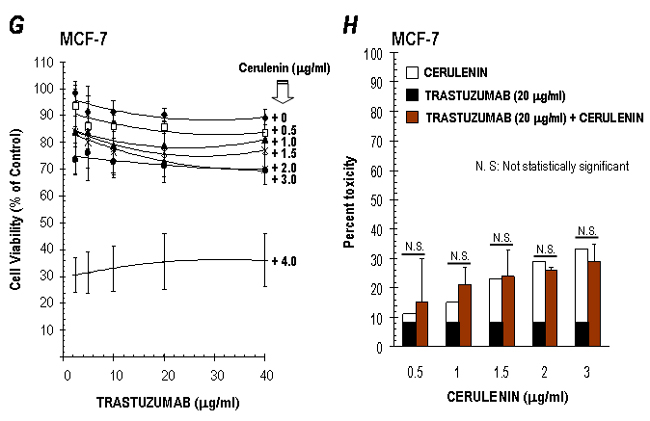
Combination of FAS Inhibitor Cerulenin and Anti-p185HER2 Ab Trastuzumab Synergistically Decreases Cell Viability. Trastuzumab (Herceptin) is a recombinant humanized anti-HER2 monoclonal Ab directed at the p185HER2 ectodomain that is active against HER2-overexpressing cancer cells (29–31). However, not all HER2-overexpressing tumors respond to treatment with trastuzumab, and its clinical benefit is limited by the occurrence of resistance (32–34). It has been suggested that tumor cell response to trastuzumab may correlate with HER2 expression profiles and/or the presence of compensatory prosurvival signaling pathways (32). To evaluate whether FAS signaling actively modulates the efficacy of trastuzumab, we analyzed the cytotoxic interactions between cerulenin and trastuzumab in cancer cell lines expressing high, moderate, and low levels of HER2. The IC30 values of trastuzumab were measured after a 72-h treatment in the presence or absence of a given concentration of cerulenin, and the degree of potentiation of trastuzumab efficacy was expressed as a sensitization factor by dividing the IC30 values in the absence of cerulenin by those in the presence of cerulenin. Cerulenin coexposure enhanced, in a dose-dependent manner, the efficacy of trastuzumab toward HER2-overexpressors (see Table 1, which is published as supporting information on the PNAS web site). The most significant changes were seen in SK-Br3 cells, in which cerulenin sensitized cells to trastuzumab by 200-fold. For BT-474 cells, cerulenin coexposure increased trastuzumab efficacy up to 52-fold. Similarly, the degree of potentiation of cerulenin efficacy by trastuzumab coexposure was evaluated by dividing the IC30 values of cerulenin in the absence of trastuzumab by those in the presence of trastuzumab (see Table 2, which is published as supporting information on the PNAS web site). A 72-h coexposure to trastuzumab sensitized SK-Br3 and BT-474 cells to cerulenin by 8- and 38-fold, respectively. When the precise nature of the interaction between cerulenin and trastuzumab was investigated by using the isobologram analysis (19) and the experimental isoeffect points (the concentrations of cerulenin and trastuzumab, which combined produced 30% reduction in cell survival) were plotted and compared to the additive line, the data points fell on the left side of the line, thus demonstrating a synergistic interaction of the two agents (see Fig. 5 A–D, which is published as supporting information on the PNAS web site). The cytotoxic interaction between cerulenin and trastuzumab was found to be synergistic in moderately HER2-expressing T47D cells, whereas an additive effect was observed in low-HER2-expressing MCF-7 cells (see Fig. 5 E–H).
Combination of FAS Inhibitor Cerulenin and Anti-HER2 Ab Trastuzumab Synergistically Down-Regulates p185HER2 Expression. To examine whether the synergistic inhibition of breast cancer cell viability that follows the concomitant targeting of FAS and p185HER was accompanied by changes in HER2 expression, p185HER2 levels were measured by ELISA after treatment with suboptimal doses of cerulenin and/or trastuzumab. In HER2 overexpressors, cerulenin and trastuzumab coexposure reduced p185HER2 levels more than either drug administered alone (see Fig. 6 A and B, which is published as supporting information on the PNAS web site). Cerulenin (1 μg/ml) and trastuzumab (10 μg/ml) showed a 48% decrease on p185HER2 expression when combined in SK-Br3 cells, whereas when used alone, they caused a 15% and 27% down-regulation of p185HER2, respectively. In BT-474 cells, combined treatment with cerulenin resulted in a synergistic increase of the trastuzumab-induced down-regulation of p185HER2 from 30% to 60%, whereas only a 15% reduction was observed in the presence of cerulenin. Moderately HER2-expressing T47-D cells cotreated with cerulenin and trastuzumab also displayed a synergistic down-regulation of p185HER2 (see Fig. 6C). These results illustrate that a synergistic augmentation of trastuzumab-induced down-regulation of p185HER2 occurs after pharmacological FAS blockade, and further show that HER2 down-regulation is a molecular mechanism of action for chemical FAS inhibitors.
Pharmacological Inhibition of FAS Activity Synergistically Enhances Trastuzumab-Induced Apoptosis. To determine whether the synergy between cerulenin and trastuzumab was accompanied with an increase in the extent of apoptosis, a combination of both agents was used to treat SK-Br3 cells, and cell death was measured by the Cell Death Detection ELISA, which allows the specific determination of mono- and oligo-nucleosomes released into the cytoplasm of cells dying from apoptosis. The combination of cerulenin and trastuzumab synergistically enhanced the basal level of cell death in untreated SK-Br3 cells. Thus, cerulenin (2 μg/ml) and trastuzumab (10 μg/ml) combined caused six times more cell death than trastuzumab alone, and twice the cell death than cerulenin alone (Figs. 4 A and 6A).
Fig. 4.

Concurrent inhibition of FAS and HER2 signalings synergistically enhances apoptotic cell death. (A) SK-Br3 cells were treated with 10 μg/ml trastuzumab, 2 μg/ml cerulenin, or a combination of trastuzumab and cerulenin. Seventy-two hours later, quantification of apoptotic cell death was determined by Cell Death ELISA (Roche Molecular Biochemicals) per the manufacturer's instructions. Data are expressed as absorbance by using the formula [A405 - A490]TREATED/[A405 - A490]UNTREATED. (B) SK-Br3 cells were grown in eight-well chamber slides and treated with 5 μg/ml trastuzumab, 1 μg/ml cerulenin, or a combination of 5 μg/ml trastuzumab plus 1 μg/ml cerulenin. Seventy-two hours later, TUNEL analyses were performed by using the DeadEnd Fluorometric TUNEL System per the manufacturer's protocol. The immunofluorescence photomicrographs (i–iii) of cells undergoing apoptosis (green staining) and the corresponding 4′,6-diamidino-2-phenylindole-counterstained cells are shown. The number in the lower right of each panel represents the percentage of TUNEL-positive cells. (C) (Upper) SK-Br3 cells were grown in eight-well chambers and transfected with 50 nM FAS siRNA, 50 nM HER2 siRNA, or a combination of 50 nM FAS siRNA plus 50 nM HER2 siRNA. Seventy-two hours later, TUNEL analyses were performed by using the DeadEnd Fluorometric TUNEL System. The number in the upper right of each panel represents the percentage of TUNEL-positive cells. (Lower) The amount of p185HER2 in FAS RNAi- and/or HER2 RNAi-transfected SK-Br3 cells was quantified by flow cytometry with the anti-p185HER2 Ab-5. The mean fluorescence signal ± SD (n = 3) was quantified by using the Geo Mean fluorescence parameter provided with cellquest software.
To assess whether the cell death observed above represented apoptosis, and also to assess the minimal concentration of cerulenin that, when combined with trastuzumab, caused synergistic cell death, we assessed apoptosis as measured by the enzymatic in situ labeling of DNA double-strand breaks [terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL)]. When cells were simultaneously treated with both drugs, there was a significant increase in apoptosis as measured by the number of TUNEL-positive cells (28%; Fig. 4Biii), whereas, individually, cerulenin (1 μg/ml) and trastuzumab (5 μg/ml) caused slight increases in the number of apoptotic cells (13% and 2% of TUNEL-positive cells, respectively; Fig. 4B i and ii). These results establish that the combined treatment with a chemical FAS inhibitor and trastuzumab synergistically induces apoptosis in HER2 overexpressors.
Simultaneous siRNA-Mediated Inhibition of FAS and HER2 Genes Synergistically Stimulates Apoptosis in HER2-Overexpressing Cancer Cells. To definitely establish that the synergistic apoptosis observed after cerulenin and trastuzumab coexposure specifically occurred through the inhibition of FAS and HER2, we examined the percentage of TUNEL-positive cells when the expression of FAS and HER2 genes was knocked down by RNAi-mediated silencing. Because of the exquisite sensitivity of SK-Br3 cells to either RNAi-mediated gene silencing, we were unable to demonstrate a synergistic effect when optimal concentrations of siRNA targeting FAS and HER2 (200 nM) were used (data not shown). Remarkably, concurrent RNAi-induced attenuation of FAS and HER2 expression with suboptimal concentrations (50 nM) of RNAi targeting FAS or HER2 was found to synergistically induce apoptosis in SK-Br3 cells (up to 32% of TUNEL-positive cells; Fig. 4Civ), whereas the number of apoptotic cells slightly increased after single transfections (Fig. 4Ci–iii). These results strongly suggest that FAS and HER2 intimately interact to maintain cell survival in HER2 overexpressors.
We next measured the levels of p185HER2 by flow cytometry after treatment with suboptimal doses of siRNAs directed against FAS and HER2. Double attenuation of FAS and HER2 expression dramatically decreased p185HER2 expression (>70% reduction when compared with cells transfected with nonspecific siRNA; Fig. 4Civ′), whereas transfection with suboptimal concentrations of siRNAs targeting either FAS or HER2 caused ≈30% down-regulation of p185HER2 expression, respectively (Fig. 4C i′–iii′). These findings demonstrating a nearly synergistic down-regulation of p185HER2 by RNAi-induced enzymatic cleavage and breakdown of FAS and HER2 mRNAs further support the hypothesis that FAS signaling plays a key role regulating the expression of HER2 oncogene in cancer cells.
Discussion
The present study demonstrates that chemical FAS inhibitors dramatically reduce the expression and activity of p185HER2 oncoprotein in HER2-overexpressing breast and ovarian cancer cells. Moreover, the specific suppression of FAS expression by RNAi-mediated silencing of the FAS gene also results in the suppression of HER2 overexpression. Because no effect was observed on cancer cells expressing physiological levels of HER2, these results reveal that tumor-associated FAS hyperactivity specifically regulates p185HER2 oncoprotein in HER2 overexpressors. We previously reported a positive correlation between high levels of FAS expression and the amplification and/or overexpression of HER2 (17), whereas HER2 overexpression was recently found to up-regulate FAS in breast cancer cells (35). Therefore, it is reasonable to propose that a bidirectional cross-talk between FAS and HER2 is taking place in cancer cells.
We have identified that pharmacological and RNAi-mediated inhibition of FAS represses HER2 expression at the transcriptional level, concomitantly up-regulating the expression of PEA3, an Ets factor that specifically reverses the in vitro transformed phenotype of HER2-overexpressing cancer cells through the attenuation of the HER2 oncogene promoter activity (14, 15). Therefore, FAS blockade indirectly suppresses HER2 overexpression through the ability of PEA3 to repress HER2 promoter activity.
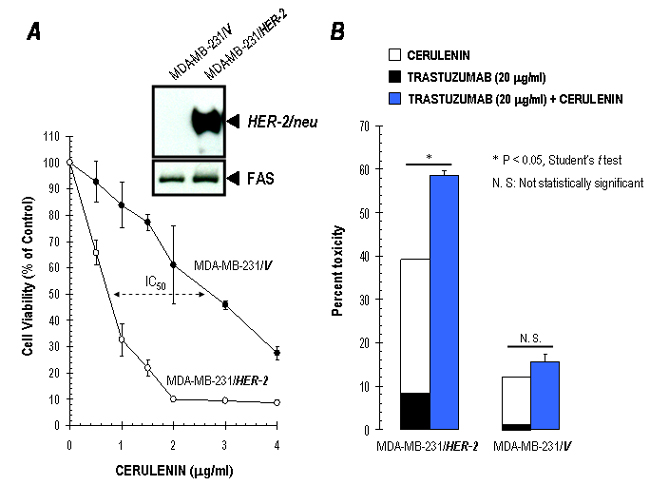
Interestingly, pharmacological inhibition of FAS provokes the redistribution of p185HER2 from membrane to cytoplasmic, perinuclear, and nuclear compartments, whereas FAS RNAi-induced silencing of FAS expression cannot induce a relocalization of p185HER2. Given that the specificity of pharmacological inhibitors in general, cannot be conclusively determined, it is possible that chemical FAS blockers modulate HER2 localization through a FAS-independent mechanism. However, the fact that an analogous nuclear accumulation of p185HER2 takes place after treatment with two different FAS blockers operating through different mechanisms of action (20–22) argues against a nonspecific pleiotropic effect. Therefore, it is tempting to suggest a previously undescribed working model connecting FAS activity and functioning of HER2 oncogene in tumor cells. Thus, a nuclear relocalization and accumulation of p185HER2 transmembrane tyrosine kinase receptor may represent a cellular response to an acute energy imbalance induced by pharmacological FAS inhibition, compared with a chronic metabolic effect derived from siRNA-mediated inhibition of FAS expression. Because the bulk of endogenously synthesized fatty acids are incorporated into membrane lipids by proliferating tumor cells (8, 36), pharmacological FAS blockade might result in rapid changes in the lipid composition of tumor cell membrane that could impair a correct cellular localization of p185HER2. Interestingly, the cytoplasmic domain of p185HER2 functions as a nuclear transcriptional activator, and nuclear HER2 exhibits a much higher autophosphorylation than its nonnuclear counterpart (27). Cerulenin and C75 may promote HER2 to enter the nucleus and to induce transcriptional activation of HER2-targeted genes. Indeed, we have shown that cerulenin- and C75-induced FAS inhibition is accompanied by the accumulation, activation, and/or cellular relocalization of multiple pro- and antiapoptotic proteins (17). Moreover HER2-negative MDA-MB-231 cells engineered to overexpress HER2 became hypersensitive to chemical FAS inhibitors in the absence of FAS protein levels comparable to those found in SK-Br3 cells (see Fig. 7 A and B, which is published as supporting information on the PNAS web site). Because these findings could not be explained on the basis of a transcriptional repression of HER2 promoter, HER2 overexpression on its own probably determines a greater sensitivity to chemical FAS inhibitors, suggesting a previously undescribed role of HER2 as a molecular sensor of the energy imbalance that follows FAS blockade.
Another implication of our findings is for the proposed use of FAS and HER2 for selective anticancer therapies. We present in vitro evidence of a clinically relevant synergistic interaction between chemical FAS inhibitors and the humanized anti-HER2 Ab trastuzumab, which, in addition, synergistically suppresses HER2 overexpression. These results suggest a promising therapeutic approach against HER2-overexpressing carcinomas using combinations of chemical FAS inhibitors, which could repress HER2 expression at the transcriptional level by up-regulating PEA3, and monoclonal Abs to HER2, which target the ectodomain of p185HER2 and promote HER2 protein degradation (37). We also report that the concurrent combination of chemical FAS inhibitors and trastuzumab is more effective than the single treatments in inducing apoptosis toward HER2-overexpressing cancer cells. Similarly, a concomitant attenuation of FAS and HER2 genes by siRNA synergistically induces both the down-regulation of p185HER2 expression and apoptotic cell death. Furthermore, we recently found that FAS blockade synergistically enhances the efficacy of taxanes against HER2 overexpressors (38). Altogether, these data are compatible with the hypothesis that FAS activity is necessary to integrate a number of signaling pathways that regulate metabolism, proliferation, and survival in HER2-overexpressing cancer cells.
The results presented here demonstrate that tumor cells' response to metabolic stress after perturbation of FAS activity is accompanied by the specific suppression of HER2 oncogene. Although these findings may solely delineate a previously undescribed mechanism of action for chemical FAS inhibitors in HER2 overexpressors, the specific down-regulation of HER2 that follows RNAi-mediated silencing of the FAS gene further reveal that HER2 could act as a molecular sensor of energy imbalance that participates actively in the maintenance of an abnormally elevated endogenous fatty acid metabolism in cancer cells. In terms of a clinical perspective, our current results provide a molecular rationale to the design of novel therapies directed against FAS in HER2-overexpressing carcinomas.
Supplementary Material
Abbreviations: FAS, fatty acid synthase; siRNA, small interfering RNA; RNAi, RNA interference; TUNEL, terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling.
References
- 1.Weiss, L., Hoffmann, G. E., Schreiber, R., Andres, E., Fuchs, E., Korber, E. & Kolb, H. J. (1986) Biol. Chem. Hoppe Seyler 367, 905-912. [DOI] [PubMed] [Google Scholar]
- 2.Sul, H. S. & Wang, D. (1998) Annu. Rev. Nutr. 18, 331-351. [DOI] [PubMed] [Google Scholar]
- 3.Kuhajda, F. P., Katumuluwa, A. I. & Pasternack, G. R. (1989) Proc. Natl. Acad. Sci. USA 86, 1188-1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuhajda, F. P. Piantadosi, S. & Pasternack, G. R. (1989) N. Engl. J. Med. 321, 636-641. [DOI] [PubMed] [Google Scholar]
- 5.Jensen, V., Holm-Nielsen, P. & Melsen, F. (1993) Breast Cancer Res. Treatment 27, 1-2. [Google Scholar]
- 6.Alo, P. L., Visca, P., Marci, A., Mangoni, A., Botti, C. & Di Tondo, U. (1996) Cancer 77, 474-482. [DOI] [PubMed] [Google Scholar]
- 7.Alo, P. L., Visca, P., Trombetta, G., Mangoni, A., Lenti, L., Monaco, S., Botti, C., Serpieri, D. E. & Di Tondo, U. (1999) Tumori 85, 35-40. [DOI] [PubMed] [Google Scholar]
- 8.Kuhajda, F. P., Jenner, K., Wood, F. D., Hennigar, R. A., Jacobs, L. B., Dock, J. D. & Pasternack, G. R. (1994) Proc. Natl. Acad. Sci. USA 91, 6379-6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milgraum, L. Z., Witters, L. A., Pasternack., G. R. & Kuhajda, F. P. (1997) Clin. Cancer Res. 3, 2115-2120. [PubMed] [Google Scholar]
- 10.Gansler, T. S., Hardman, W., III, Hunt, D. A., Schaffel, S. & Hennigar, R. A. (1997) Hum. Pathol. 28, 686-692. [DOI] [PubMed] [Google Scholar]
- 11.Kuhajda, F. P. (2000) Nutrition 16, 202-208. [DOI] [PubMed] [Google Scholar]
- 12.Swinnen, J. V., Ulrix, W., Heyns, W. & Verhoeven, G. (1997) Proc. Natl. Acad. Sci. USA 94, 12975-12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swinnen, J. V., Vanderhoydonc, F., Elgamal, A. A., Eelen, M., Vercaeren, I., Joniau, S., Van Poppel, H., Baert, L., Goossens, K., Heyns, W. & Verhoeven, G. (2000) Int. J. Cancer 88, 176-179. [DOI] [PubMed] [Google Scholar]
- 14.Xing, X., Wang, S. C., Xia, W., Zou, Y., Shao, R., Kwong, K. Y., Yu, Z., Zhang, S., Miller, S., Huang, L. & Hung, M. C. (2000) Nat. Med. 6, 189-195. [DOI] [PubMed] [Google Scholar]
- 15.Wang, S. C. & Hung, M. C. (2000) Drugs Today (Barcelona) 36, 835-843. [PubMed] [Google Scholar]
- 16.Tsai, M. S., Shamon-Taylor, L. A., Mehmi, I., Tang, C. K. & Lupu, R. (2003) Oncogene 22, 761-768. [DOI] [PubMed] [Google Scholar]
- 17.Menendez, J. A., Mehmi, I., Atlas, E., Colomer, R. & Lupu, R. (2004) Int. J. Oncol. 24, 591-608. [PubMed] [Google Scholar]
- 18.Dils, R. & Carey, E. M. (1975) Methods Enzymol. 35, 74-83. [DOI] [PubMed] [Google Scholar]
- 19.Berenbaum, M. C. (1989) Pharmacol. Rev. 41, 93-141. [PubMed] [Google Scholar]
- 20.Vance, D., Goldberg, I., Mitsuhashi, O. & Bloch, K. (1972) Biochem. Biophys. Res. Commun. 48, 549-565. [DOI] [PubMed] [Google Scholar]
- 21.Omura, S. (1976) Bacteriol. Rev. 40, 681-697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuhajda, F. P., Pizer, E. S., Li, J. N., Mani, N. S., Frehywot, G. L. & Townsend, C. A. (2000) Proc. Natl. Acad. Sci. USA 97, 3450-3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu, Q., Gen, Y. & Sicinski, P. (2001) Nature 411, 1017-1021. [DOI] [PubMed] [Google Scholar]
- 24.Nahta, R., Iglehart, D., Kempkes, B. & Schmidt, E. V. (2002) Cancer Res. 62, 2267-2271. [PubMed] [Google Scholar]
- 25.Zamore, P. D., Tuschl, T., Sharp, P. A. & Bartel, D. P. (2000) Cell 101, 25-33. [DOI] [PubMed] [Google Scholar]
- 26.Elbashir, S. M., Harborth, J., Lendeckel, W., Yalcin, A., Weber, K. & Tuschl, T. (2001) Nature 411, 494-498. [DOI] [PubMed] [Google Scholar]
- 27.Xie, Y. & Hung, M. C. (1994) Biochem. Biophys. Res. Commun. 203, 1589-1598. [DOI] [PubMed] [Google Scholar]
- 28.De Schrijver, E., Brusselmans, K., Heyns, W., Verhoeven, G. & Swinnen, J. V. (2003) Cancer Res. 63, 3799-3804. [PubMed] [Google Scholar]
- 29.Carter, P., Presta, L., Gorman, C. M., Ridgway, J. B., Henner, D., Wong, W. L., Rowland, A. M., Kotts, C., Carver, M. E. & Shepard, H. M. (1992) Proc. Natl. Acad. Sci. USA 89, 4285-4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vogel, C. L., Cobleigh, M. A., Tripathy, D., Gutheil, J. C., Harris, L. N., Fehrenbacher, L., Slamon, D. J., Murphy, M., Novotny, W. F., Burchmore, M., et al. (2002) J. Clin. Oncol. 20, 719-726. [DOI] [PubMed] [Google Scholar]
- 31.Baselga, J., Norton, L., Albanell, J., Kim, Y. M. & Mendelshon, J. (1998) Cancer Res. 58, 2825-2831. [PubMed] [Google Scholar]
- 32.Lane, H. A., Beuvink, I., Motoyama, A. B., Daly, J. M., Neve, R. M. & Hynes, N. E. (2000) Mol. Cell. Biol. 20, 3210-3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baselga, J. (2001) Eur. J. Cancer 37, Suppl. 1, 18-24. [PubMed] [Google Scholar]
- 34.Slamon, D. J., Leyland-Jones, B., Shak, S., Fuchs, H., Paton, V., Bajamonde, A., Fleming, T., Eiermann, W., Wolter, J., Pegram, M., et al. (2001) N. Engl. J. Med. 344, 783-779. [DOI] [PubMed] [Google Scholar]
- 35.Kumar-Sinha, C., Ignatoski, K. W., Lippman, M. E., Ethier, S. P. & Chinnaiyan, A. M. (2003) Cancer Res. 63, 132-139. [PubMed] [Google Scholar]
- 36.Pizer, E. S., Wood, F. D., Pasternack, G. R. & Kuhajda, F. P. (1996) Cancer Res. 56, 745-751. [PubMed] [Google Scholar]
- 37.Baselga, J. & Albanell, J. (2001) Ann. Oncol. 12, Suppl. 1, S35-S41. [DOI] [PubMed] [Google Scholar]
- 38.Menendez, J. A., Lupu, R. & Colomer, R. (2004) Breast Cancer Res. Treatment 84, 183-195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}