

Abstract
Using the carcinogen 3-methylcholanthrene (MCA), we demonstrate with Fourier transform-infrared spectroscopy that a cancer DNA phenotype is produced well in advance of palpable tumors. We further demonstrate that the administration of cyclophosphamide markedly inhibits the development of the cancer phenotype and concomitantly delays tumor formation. MCA, injected into the hind legs of mice, produced a variety of significant structural changes in the nucleotide bases and phosphodiester-deoxyribose backbone, as reflected in a substantial (34%) difference between the mean DNA spectra of the control and the MCA-injected mice. Strikingly, 57 days before the mean appearance of tumors, we could not distinguish the DNA structure of the histologically normal tissues of the MCA-injected mice from the DNA structure of the tumor tissues. This finding indicates the early development of a cancer phenotype. Confirmatory evidence was obtained when tissues from a group of mice injected with both MCA and cyclophosphamide did not manifest the cancer phenotype, and their mean DNA structure closely resembled that of the control mice. Accordingly, we propose that the cancer DNA phenotype, as evinced by Fourier transform-infrared spectroscopy, is a promising early indicator of tumor formation, and we postulate that agents capable of inhibiting this phenotype may delay or prevent carcinogenesis.
Keywords: 3-methylcholanthrene, antineoplastic agents, cyclophosphamide, cancer inhibition, cancer prediction
The carcinogen 3-methylcholanthrene (MCA) readily produces primary sarcomas in mice. The biochemistry associated with this transformation is complex and diverse. Initially, MCA binds with the Ah (aromatic hydrocarbon) receptor and activates cytochrome P450 (1) and other xenobiotic-induced enzymes (2). Resulting metabolites react with the 2-amino group of guanine to produce bulky carcinogen-DNA adducts, which are associated with G·C → T·A transversions (3). MCA is also hydroxylated at the C1 position to form a major oxidation product, 1-hydroxy-3-methylchloranthrene (4). Esterification of this metabolite produces 1-sulfooxy-3-methylchloranthrene, which generates a highly reactive carbonium ion capable of damaging the nucleotide base structure (4), thus introducing mutagenic and carcinogenic properties into DNA (5).
The prodrug cyclophosphamide (CPA) is a commonly used antineoplastic agent (6) that can reduce the rate of MCA-induced tumor formation (7). CPA is metabolized to 4-hydroxy-cyclophosphamide, which breaks down into the reactive metabolites phosphoramide mustard and acrolein, both of which are known to damage DNA (8). Phosphoramide mustard is a DNA-reactive alkylating agent that binds to the N7 position of guanine (6, 9). Phosphoramide mustard may also react with the O6 position of guanine (6) and has been implicated in DNA cross-linking (10). Acrolein is believed to contribute to DNA damage primarily by creating abasic sites and single-strand breaks (11–14). The DNA damage described probably contributes to apoptosis, which reduces the population of MCA-damaged cells (15, 16). This finding may explain why administration of CPA to MCA-injected mice has been found to delay tumor formation (7).
Using the BALB/c mouse model and the same dose of MCA and CPA as reported in ref. 7, we investigated changes in DNA structure associated with MCA exposure. Fourier transform-infrared (FTIR) spectral models (17–21) were used to identify alterations in the nucleotide bases and phosphodiester-deoxyribose backbone during tumorigenesis. Moreover, we looked for the early development of a DNA structure (cancer DNA phenotype), indistinguishable from that of tumor DNA, which was found in the prostates of older, cancer-free men (18). We also investigated whether the delay in MCA-induced tumor formation after systemic treatment with CPA is reflected in a delayed appearance of the cancer phenotype. We have used FTIR spectral models for predicting primary tumor formation and metastasis in the human breast (22) and prostate (18) and for the early diagnosis of the clonal hematopoietic stem-cell disorder, myelodysplastic syndrome (17).
Materials and Methods
Tissue Acquisition and Histology. Six- to 8-week-old female BALB/c mice were obtained from Charles River Laboratories. The mice were kept on a 12-h light-and-dark cycle and were allowed free access to food and water. The control group (n = 10) received no treatment. On day 1, 40 mice were injected intramuscularly in the right hind thigh with 0.1 mg of MCA dissolved in trioctanoin (which we found to produce no effect on the spectra of isolated DNA). The MCA group (n = 10) was killed on day 70. The tumor group (n = 10) was killed when the mice had tumors ≤15 mm (13.7 ± 1.3 mm; 1.4 ± 0.6 g; average time of killing, 127 days). Two additional groups each received a 2-mg i.p. injection of CPA 2 weeks before the MCA injection. These CPA injections continued every 10 days until killing. The MCA + CPA group (n = 10) was killed on day 70 together with the MCA group. The CPA tumor group (n = 10) was killed when the mice had tumors ≤15 mm (13.2 ± 1.2 mm; 1.1 ± 0.5 g; average time of killing, 167 days). The dose and schedule of CPA administration were based on an earlier study showing delayed formation of primary MCA-induced sarcomas in BALB/c mice (7). The control, MCA, and MCA + CPA mice were 5–6 months of age at the time of killing. Halothane was used in a sealed chamber to kill the animals. The right hind-leg muscle or tumor was removed, and a small piece of tissue was fixed in neutral buffered formalin for histology. The remaining tissue was immediately frozen in liquid nitrogen and maintained at -80°C until DNA extraction. Formalin-fixed tissues were passed through a series of alcohol and xylene solutions, then placed into paraffin blocks. Sections of the tissues were stained with hematoxylin and eosin and examined microscopically. No histological evidence was found for cancer cells or no recognizable premalignant changes were found in any of the leg tissues before tumor formation (any small submicroscopic clusters of cancer cells would be insufficient to influence the spectral measurements obtained). Minor inflammatory or scarring changes affected <10% of the tissues sampled and would not be detected spectrally in the context of the >90% of tissues showing no inflammatory changes. The experimental protocols were approved by the Institutional Animal Care and Use Committee of the Pacific Northwest Research Institute.
DNA Extraction. DNA (≈50 μg) was extracted from the leg muscle or tumor tissue (≈100 mg) by using Qiagen 100/G Genomic-tips (Qiagen, Valencia, CA) and following a slightly modified Qiagen extraction protocol: after elution, the DNA solution (eluate) was passed through a 5.0-μm Cameo 25N syringe filter (GE Osmonics, Minnetonka, MN). The Qiagen protocol was then resumed, and the isolated DNA was washed three times with ice-cold 70% ethanol. The DNA was subsequently dissolved in 10–40 μl of Optima Grade water (Fisher Scientific) in preparation for FTIR spectral analysis. All samples were randomly selected for analysis to avoid batch effects.
FTIR Spectroscopy. DNA structural analyses were conducted by using FTIR microscope spectroscopy as described in refs. 18 and 23. Briefly, a 0.2-μl aliquot of DNA solution was spotted on a BaF2 plate and allowed to spread. As the spots dried, an outer ring containing the DNA was formed. Two separate spots were created for each DNA sample. Spotting was repeated until the ring was at least 100 μm wide, the width of the aperture of the microscope spectrometer (System 2000, PerkinElmer). Initially, a background energy reading (percent transmittance) was determined from a blank area of the BaF2 plate. Points for spectral determinations were selected by where the energy readings were 15–25% less than the background energy (optimally close to 15% less). Ten spectral determinations were made around the two rings for each sample, and the percent transmittance values were converted (Fourier-transformed) to absorbance values. To adjust for varying film thickness, each spectrum was baselined and normalized as described in ref. 18.
Statistical Analyses. All statistical procedures used in this study are described in refs. 18, 19, and 23. The representative FTIR spectrum of each sample was obtained by determining the mean of 20 spectral determinations obtained from the two spots on the BaF2 plate. It is often difficult to visually discern subtle differences between mean DNA spectra for different groups of samples. Therefore, a t test was performed to determine the statistical significance (P value) of differences at each wavenumber between mean absorbance values for each pair of groups (e.g., control vs. tumor DNA). Whereas the P values over ≈1,000 consecutive wavenumbers in the spectral range used were not statistically independent, regions of the spectrum with P ≤ 0.05 likely indicate real structural differences between the groups. P values ≤0.05 constituting ≤5% of the entire spectral range are expected by chance (20).
Principal components analysis (PCA) (19, 20, 23) integrates ≈1 million correlations of spectral properties (e.g., varying peak heights, peak locations, and various combinations thereof) and is thus a highly discerning tool for elucidating spectral differences between DNA groups. PCA was performed on the mean spectra, resulting in 10 principal component (PC) scores for each sample. Significant differences in the PC scores between tissue groups were determined by using t tests (18). Those PC scores with significant differences (P ≤ 0.05) were used to construct 2D scatter plots. The spatial location and distribution (e.g., clustering) of PC scores in scatter plots served to identify structural differences and/or similarities between the groups.
Results and Discussion
MCA-Induced DNA Damage Before Tumor Formation. We have demonstrated that the FTIR statistical models of DNA used in this study are capable of identifying a variety of subtle changes in base functional group and backbone structures as well as alterations in conformational properties (20) associated with carcinogenesis (19, 21, 23–25). Specifically, this technology has been used most recently to discriminate between DNA structures of normal prostate tissues, primary tumors, metastasizing primary tumors, and distant metastases of the prostate (18). In addition, the technology has been used to differentiate between the DNA of normal granulocytes and granulocytes from patients with myelodysplastic syndrome (17).
In the present study, we compared the mean FTIR spectra of DNA of the control group and the MCA group (harvested 10 weeks after MCA injection, at which time the leg tissues were histologically normal) (Fig. 1A). P values calculated for each wavenumber (Fig. 1B) revealed significant differences between the groups over ≈34% of the spectral range (1,750–700 cm-1). This percentage is far greater than the 5% expected by chance (20). To better visualize these differences, a portion of the spectral comparison (1,700–1,625 cm-1) has been magnified (Fig. 1C) along with the corresponding P values (Fig. 1D). The spectral differences (Fig. 1 A and B) were associated with the nucleotide bases (≈1,750–1550 cm-1) and the phosphodiester-deoxyribose backbone (≈1,275–750 cm-1) (26). Specifically, they were reflected in the shoulder at ≈1,681 cm-1 (P = 0.04), which is attributed to NH2 scissoring vibrations of the nucleotide bases; the strong peak at ≈1,653 cm-1 (P = 0.03), which is assigned to in-plane ring vibrations of the bases; and the peak at ≈1,228 cm-1 (P = 0.04), which represents antisymmetric stretching vibrations of the 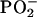 (26). Significant differences (P ≤ 0.05) between bands below ≈1,050 cm-1 are attributed to ribose-phosphate main-chain vibrations (26). The structural differences induced by MCA would be expected to be highly complex, reflecting perturbations in vertical base stacking and conformational properties (e.g., via changes in torsion angles) of the phosphodiester-deoxyribose moiety (20).
(26). Significant differences (P ≤ 0.05) between bands below ≈1,050 cm-1 are attributed to ribose-phosphate main-chain vibrations (26). The structural differences induced by MCA would be expected to be highly complex, reflecting perturbations in vertical base stacking and conformational properties (e.g., via changes in torsion angles) of the phosphodiester-deoxyribose moiety (20).
Fig. 1.

Significant spectral differences relating to base and backbone structures are shown between the DNA of the control and MCA groups. (A) Comparison of mean DNA spectra of the control (black, n = 10) and MCA (red, n = 10) groups. (B) Corresponding P values at each wavenumber. (C) Magnification of wavenumbers between 1,700 and 1,625 cm-1. (D) Corresponding P values for this spectral range.
MCA-Induced DNA Damage in Tumor Tissues. The mean DNA spectra from the control and tumor groups are compared in Fig. 2A. We found significant differences between the means across 34% of the spectral range (Fig. 2B), indicating that the tumor DNA was markedly different in structure from that of the control group. These differences involve both the nucleotide bases and the phosphodiester-deoxyribose backbone. A slight wavenumber shift in the peak for the 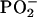 antisymmetric stretching vibrations between the two groups may explain the significant differences found for the left (1,266–1,233 cm-1) and right (1,201–1,158 cm-1) slopes of this band. There were also significant differences <≈1,050 cm-1 associated with ribose-phosphate main-chain vibrations (26). These differences demonstrate that substantial modifications in the base and backbone structures occur in the progression of normal tissues to MCA-induced sarcomas. Considering that the enzymatic machinery “reads” coded information at the level of hydrogen bonds, these changes in DNA structure would be expected to influence gene expression and the fidelity of transcription and replication (27), thus having a pivotal effect on the neoplastic transformation of normal tissues.
antisymmetric stretching vibrations between the two groups may explain the significant differences found for the left (1,266–1,233 cm-1) and right (1,201–1,158 cm-1) slopes of this band. There were also significant differences <≈1,050 cm-1 associated with ribose-phosphate main-chain vibrations (26). These differences demonstrate that substantial modifications in the base and backbone structures occur in the progression of normal tissues to MCA-induced sarcomas. Considering that the enzymatic machinery “reads” coded information at the level of hydrogen bonds, these changes in DNA structure would be expected to influence gene expression and the fidelity of transcription and replication (27), thus having a pivotal effect on the neoplastic transformation of normal tissues.
Fig. 2.

Significant spectral differences relating to base and backbone structures are shown between the DNA of the control and tumor groups and between the MCA and tumor groups. (A) Comparison of mean DNA spectra of the control (black, n = 10) and tumor (red, n = 10) groups. (B) Corresponding P values. (C) Comparison of mean DNA spectra of the MCA (black, n = 10; obtained 57 days prior to mean tumor formation) and tumor (red, n = 10) groups. (D) Corresponding P values.
Evidence for a Cancer DNA Phenotype. Strikingly, when we compared the mean DNA spectrum of the MCA-injected mice that had not yet developed tumors with the tumor DNA (Fig. 2C), there was no significant difference (4%) over the entire spectral range (Fig. 2D), indicating that the two DNA groups were statistically indistinguishable. That is, 57 days before the mean time of tumor harvest, the histologically normal tissues of the MCA group had DNA modifications similar to those of the tumor group. These findings suggest that MCA exposure resulted in the early development of a readily identifiable cancer phenotype in the DNA of histologically normal cells. The cancer phenotype would be expected to comprise a multiplicity of structural changes reflecting perturbations in the base and backbone structures. These structural changes would almost certainly incorporate a wide array of mutations, which may well be consistent with the concept of a mutator phenotype (28, 29).
We found confirmatory evidence for the cancer phenotype after performing the sensitive and highly discriminating statistical technique of PCA on the spectral data (20). We constructed a 2D scatter plot by using PC4 and PC6, both of which showed significant differences (P = 0.01 and P = 0.006, respectively) between the control and tumor groups (Fig. 3). The scatter plot shows that the DNA structures from the control group were substantially more diverse than those of the tightly clustered MCA and tumor groups. These results indicate that the MCA reduced DNA structural diversity, which was reflected in a subgroup of 6 of 10 mice with a DNA structure indistinguishable from that of the tumor group. The mean spectrum for these six mice (whose tissues were found to be histologically normal) was found to be identical to the mean spectrum for the tumor group (data not shown), thus providing the most precise depiction of the cancer phenotype obtained based on the FTIR spectral models. The DNA structures of the other four mice appear to be in transition between the structural diversity of the control group and the tight clustering of the cancer phenotype and tumor groups.
Fig. 3.

Plot of PC4 vs. PC6 from PCA of FTIR spectra from control (open diamonds, n = 10), MCA (filled triangles, n = 10), and tumor (filled circles, n = 10) groups (see text for details).
The cancer DNA phenotype identified by PCA and observed long before (57 days) there was histological evidence of tumors in the MCA-injected mouse leg, is comparable with the phenotype we identified in the histologically normal prostate tissues of ≈42% of cancer-free men (ages 55–80) (18). That is, in both the mouse and prostate studies, the structure of the cancer phenotype could not be distinguished from that of the tumor. The important distinction between these two studies is that the prostate study lacked direct evidence linking the cancer phenotype to subsequent tumor formation. However, in this mouse study, 100% of the MCA-injected mice followed for tumor formation developed tumors. The findings with the present mouse model and the human prostate (18) imply that identification of the cancer phenotype is a promising basis for the early prediction of tumor formation in cancer-prone tissues. By using the described statistical models (e.g., based on PCA), such predictions can be made by comparing the DNA of suspect tissues with the DNA profile established for tumors.
CPA Suppresses the Cancer DNA Phenotype. CPA has been shown to substantially reduce the rate of sarcoma formation in BALB/c mice receiving MCA in the hind leg (7). In our experiments, the mean time to tumor harvest after the MCA injection was 127 ± 32 days without CPA treatment vs. 167 ± 30 days with treatment (P < 0.001). That is, CPA-treated mice took ≈30% longer to develop tumors, consistent with previous findings (7). The mean spectra of the DNA from the control and the MCA + CPA groups are compared in Fig. 4A. Significant differences (P ≤ 0.05) between the two groups (Fig. 4B) were limited to 9% of the spectral range, compared with 34% for the comparison between the control and MCA groups. In addition, all of the peak and shoulder changes were eliminated in the MCA + CPA group, with the exception of several weak backbone-related bands appearing <≈1,050 cm-1.
Fig. 4.

Spectral differences are shown in the base and backbone structures between the DNA of the control and MCA + CPA groups (4-fold less than the differences obtained between the control and MCA groups). (A) Comparison of mean DNA spectra of the control (black, n = 10) and MCA + CPA (red, n = 10) groups. (B) Corresponding P values.
Importantly, CPA produced a marked reduction in the DNA structural damage found with the MCA group (Fig. 1 A and B); that is, the cancer phenotype was virtually eliminated. This result was likely a major factor in the substantial (≈40 days) delay in tumor formation, although additional effects of CPA cannot be excluded. However, the outcome fell short of complete inhibition. CPA requires activation by hepatic microsomal enzymes to form cytotoxic species, such as phosphoramide and ifosfamide mustards (30). These species are alkylating agents forming DNA cross-links that inhibit DNA synthesis, which contributes to apoptosis (7, 15, 16) through a complex cascade of events involving the activation of cysteine proteases (caspases) (31). Our findings are consistent with the ability of CPA to inhibit MCA-induced DNA damage and/or increase the mortality of DNA-damaged cells, as evidenced by the almost 4-fold reduction in spectral/structural differences in the CPA-treated mice (Figs. 1B and 4B).
Conclusions
Our findings may explain why a cancer DNA phenotype associated with prostate carcinomas was detected in ≈42% of older, cancer-free men (18) by showing that a similar cancer phenotype was found in histologically normal mouse tissues, all of which would later become cancerous. We propose that the cancer phenotype is an early, readily identifiable marker that signals a high risk for tumor development. In support of this concept, we showed that CPA concomitantly delays tumor formation and inhibits the formation of the cancer phenotype. On the basis of the evidence presented, we postulate that the FTIR technique can be used to identify agents that may well be more efficacious than CPA and may serve to delay or prevent development of some cancers in humans either by removing cells that have the cancer phenotype or by preventing their appearance. Additional studies are needed to further test the hypothesis that the cancer DNA phenotype is an early predictor of tumor formation, and that agents capable of delaying or preventing the appearance of the cancer phenotype will likewise delay or prevent tumorigenesis.
Acknowledgments
We thank Drs. Krystyna Frenkel, Lawrence A. Loeb, Evangelos Moudrianakis, Jose Russo, and George J. Todaro for helpful comments and Brenda Duer, Erica Strobl, and Nhan Vo for technical assistance. This work was supported by National Institutes of Health Grant CA79479.
Abbreviations: CPA, cyclophosphamide; FTIR, Fourier transform-infrared; MCA, 3-methylcholanthrene; PCA, principal components analysis; PC, principal component.
References
- 1.Quattrochi, L. C., Vu, T. & Tukey, R. H. (1994) J. Biol. Chem. 269, 6949-6954. [PubMed] [Google Scholar]
- 2.Atlas, S. A., Taylor, B. A., Diwan, B. A. & Nebert, D. W. (1976) Genetics 83, 537-550. [PMC free article] [PubMed] [Google Scholar]
- 3.Shimokado, K., Watanabe, H., Sumii, M., Miyagawa, K., Kamiya, K., Dohi, K. & Niwa, O. (1998) Jpn. J. Cancer Res. (GANN) 89, 269-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flesher, J. W., Horn, J. & Lehner, A. F. (1998) Biochem. Biophys. Res. Commun. 243, 30-35. [DOI] [PubMed] [Google Scholar]
- 5.King, H. W. S., Osborne, M. R. & Brookes, P. (1977) Int. J. Cancer 20, 564-571. [DOI] [PubMed] [Google Scholar]
- 6.Boddy, A. V. & Yule, S. M. (2000) Clin. Pharmacokinet. 38, 291-304. [DOI] [PubMed] [Google Scholar]
- 7.Hellström, I. & Hellström, K. E. (1978) Nature 275, 129-130. [DOI] [PubMed] [Google Scholar]
- 8.Baumann, F. & Preiss, R. (2001) J. Chromatogr. B Biomed. Sci. Appl. 764, 173-192. [DOI] [PubMed] [Google Scholar]
- 9.Kohn, K. W., Hartley, J. A. & Mattes, W. B. (1987) Nucleic Acids Res. 15, 10531-10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Springer, J. B., Colvin, M. E., Colvin, O. M. & Ludeman, S. M. (1998) J. Org. Chem. 63, 7218-7222. [DOI] [PubMed] [Google Scholar]
- 11.Chung, F. L., Young, R. & Hecht, S. S. (1984) Cancer Res. 44, 990-995. [PubMed] [Google Scholar]
- 12.Smith, R. A., Williamson, D. S. & Cohen, S. M. (1989) Chem. Res. Toxicol. 2, 267-271. [DOI] [PubMed] [Google Scholar]
- 13.Smith, R. A., Williamson, D. S., Cerny, R. L. & Cohen, S. M. (1990) Cancer Res. 50, 3005-3012. [PubMed] [Google Scholar]
- 14.Sodum, R. S. & Shapiro, R. (1988) Bioorg. Chem. 16, 272-282. [Google Scholar]
- 15.O'Connor, P. M., Wassermann, K., Sarang, M., Magrath, I., Bohr, V. A. & Kohn, K. W. (1991) Cancer Res. 51, 6550-6557. [PubMed] [Google Scholar]
- 16.Hickman, J. A. (1992) Cancer Metastasis Rev. 11, 121-139. [DOI] [PubMed] [Google Scholar]
- 17.Malins, D. C., Anderson, K. M., Polissar, N. L., Ostrander, G. K., Knobbe, E. T., Green, V. M., Gilman, N. K. & Spivak, J. L. (2004) Proc. Natl. Acad. Sci. USA 101, 5008-5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malins, D. C., Johnson, P. M., Barker, E. A., Polissar, N. L., Wheeler, T. M. & Anderson, K. M. (2003) Proc. Natl. Acad. Sci. USA 100, 5401-5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia-Closas, M., Hankinson, S. E., Ho, S., Malins, D. C., Polissar, N. L., Schaefer, S. N., Su, Y. & Vinson, M. A. (2000) J. Natl. Cancer Inst. 27, 147-156. [DOI] [PubMed] [Google Scholar]
- 20.Malins, D. C., Polissar, N. L., Ostrander, G. K. & Vinson, M. A. (2000) Proc. Natl. Acad. Sci. USA 97, 12442-12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malins, D. C., Polissar, N. L., Schaefer, S., Su, Y. & Vinson, M. (1998) Proc. Natl. Acad. Sci. USA 95, 7637-7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malins, D. C., Polissar, N. L., Nishikida, K., Holmes, E. H., Gardner, H. S. & Gunselman, S. J. (1995) Cancer (Philadelphia) 75, 503-517. [DOI] [PubMed] [Google Scholar]
- 23.Malins, D. C., Polissar, N. L., Su, Y., Gardner, H. S. & Gunselman, S. J. (1997) Nat. Med. 3, 927-930. [DOI] [PubMed] [Google Scholar]
- 24.Malins, D. C., Polissar, N. L. & Gunselman, S. J. (1997) Proc. Natl. Acad. Sci. USA 94, 259-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malins, D. C., Polissar, N. L. & Gunselman, S. J. (1996) Proc. Natl. Acad. Sci. USA 93, 14047-14052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsuboi, M. (1969) Appl. Spectrosc. Rev. 3, 45-90. [Google Scholar]
- 27.Turner, B. M. (2001) Chromatin and Gene Regulation: Mechanisms in Epigenetics (Blackwell, Oxford), pp. 25-41.
- 28.Loeb, L. A. (1991) Cancer Res. 51, 3075-3079. [PubMed] [Google Scholar]
- 29.Loeb, L. A. & Christians, F. C. (1996) Mutat. Res. 350, 279-286. [DOI] [PubMed] [Google Scholar]
- 30.Fleming, R. A. (1997) Pharmacotherapy 17, S146-S154. [Google Scholar]
- 31.Schwartz, P. S. & Waxman, D. J. (2001) Mol. Pharmacol. 60, 1268-1279. [DOI] [PubMed] [Google Scholar]