

Abstract
Defects in mitochondrial oxidative phosphorylation have frequently been associated with Alzheimer's disease (AD), and both inherited and somatic mtDNA mutations have been reported in certain AD cases. To determine whether mtDNA mutations contribute more generally to the etiology of AD, we have investigated the sequence of the mtDNA control region (CR) from AD brains for possible disease-causing mutations. Sixty-five percent of the AD brains harbored the T414G mutation, whereas this mutation was absent from all controls. Moreover, cloning and sequencing of the mtDNA CR from patient and control brains revealed that all AD brains had an average 63% increase in heteroplasmic mtDNA CR mutations and that AD brains from patients 80 years and older had a 130% increase in heteroplasmic CR mutations. In addition, these mutations preferentially altered known mtDNA regulatory elements. Certain AD brains harbored the disease-specific CR mutations T414C and T477C, and several AD brains between 74 and 83 years of age harbored the CR mutations T477C, T146C, and T195C, at levels up to 70–80% heteroplasmy. AD patient brains also had an average 50% reduction in the mtDNA L-strand ND6 transcript and in the mtDNA/nuclear DNA ratio. Because reduced ND6 mRNA and mtDNA copy numbers would reduce brain oxidative phosphorylation, these CR mutations could account for some of the mitochondrial defects observed in AD.
Alzheimer's disease (AD) is a progressive neurodegenerative disease resulting in dementia associated with the excessive deposition in the brain of Aβ amyloid peptide plaques and neurofibrillary tangles. Early-onset, familial AD has been associated with mutations in the Aβ precursor protein (APP) and the Presenilin-containing APP peptide processing complexes (1). However, mutations in these genes have not been identified in the majority of late-onset, sporadic AD patients, leaving the molecular basis of these cases undetermined (2).
One hypothesis for the etiology of late-onset, sporadic AD is that it is caused by defects in mitochondrial oxidative phosphorylation (OXPHOS). Structurally abnormal mitochondria have been observed in AD brains (3, 4), and deficiencies in mitochondrial OXPHOS enzymes, such as cytochrome c oxidase (COX or complex IV), have been repeatedly reported in the brains and other tissues of AD patients (5, 6). Moreover, mitochondrial OXPHOS defects have been recovered in cultured human cell cybrids by fusion of AD patient blood platelets to human cell lines that lack mtDNA (ρo cells) (5–13).
Defects in OXPHOS inhibit ATP production, but they also increase mitochondrial reactive oxygen species (ROS) production which, in turn, can activate the mitochondrial permeability transition pore (mtPTP) and destroy the surrounding cell by apoptosis. Electrons in the initial steps of the mitochondrial electron transport chain can be transferred directly to molecular oxygen (O2) to generate superoxide anion 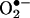 . This superoxide anion is converted to hydrogen peroxide (H2O2) by mitochondrial superoxide dismutase, and the hydrogen peroxide is converted to hydroxyl radical OH· by electron transfer from reduced transition metals. The resulting ROS (
. This superoxide anion is converted to hydrogen peroxide (H2O2) by mitochondrial superoxide dismutase, and the hydrogen peroxide is converted to hydroxyl radical OH· by electron transfer from reduced transition metals. The resulting ROS (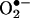 , H2O2, and OH·) damage mitochondrial proteins and membranes and mutagenize the mtDNA. This inhibits the electron transport chain, causing the electron carriers to become more reduced, further stimulating mitochondrial ROS production. The ultimate result is the activation of the mtPTP and destruction of the surrounding cell by apoptosis. Consistent with this scenario, the brains of late-onset, sporadic AD patients have been observed to have increased oxidative damage (14, 15), increased activated caspase activity (16), and increased numbers of terminal-deoxynucleotidyltransferase-mediated-dUTP-nick-end-labeling-positive cells (5).
, H2O2, and OH·) damage mitochondrial proteins and membranes and mutagenize the mtDNA. This inhibits the electron transport chain, causing the electron carriers to become more reduced, further stimulating mitochondrial ROS production. The ultimate result is the activation of the mtPTP and destruction of the surrounding cell by apoptosis. Consistent with this scenario, the brains of late-onset, sporadic AD patients have been observed to have increased oxidative damage (14, 15), increased activated caspase activity (16), and increased numbers of terminal-deoxynucleotidyltransferase-mediated-dUTP-nick-end-labeling-positive cells (5).
Inhibition of mitochondrial OXPHOS leading to AD could result from environmental intoxicants or from acquired or inherited mtDNA mutations. The mtDNA codes for 37 genes, 13 essential OXPHOS polypeptides, 22 tRNA genes, and a 12S and 16S rRNA gene.
Somatic mtDNA rearrangement mutations have been observed to be increased in AD brains. This includes the common 5-kb mtDNA rearrangement, which has been observed to be elevated about 15-fold in AD patient brains up to age 75 years (17). Certain germ-line mtDNA mutations have also been associated with late-onset AD. For example, the nucleotide pair (np) 4336 mutation in the tRNAGln gene has been observed in about 5% of late-onset AD patients (18, 19), and this association has been supported in three of four independent European studies (20–23).
AD has been further linked to the mitochondria through reports that the European mtDNA lineages (haplogroups) J and Uk are protective of AD and Parkinson's disease (24, 25) and are also associated with increased longevity (16). This protection has been proposed to result from ancient mtDNA variants that partially uncouple OXPHOS, increasing mitochondrial heat production and permitting adaptation to colder northern latitudes (6, 7). Both European subhaplogroup J1 and haplogroup Uk harbor the same cytochrome b mutation at np 14798, whereas subhaplogroup J2 harbors a different cytochrome b mutation at np 15257. Both of these cytochrome b mutations alter conserved amino acids in the two coenzyme Q10 binding sites, and thus could affect the efficiency of proton pumping by the Q cycle of complex III. Because uncoupling mutations that partially depolarize the mitochondrial inner membrane proton gradient would keep the electron-transport-chain carriers oxidized, these mutants would also reduce mitochondrial ROS production. This would reduce brain oxidative damage and neuronal somatic mtDNA mutations, explaining the protective effect of these mtDNA lineages for AD and Parkinson's disease (16, 26).
In addition to its mRNA, rRNA, and tRNA genes, the mtDNA encompasses a 1,112-np control region (CR). This CR includes the L- and H-strand promoters (PL and PH); their mitochondrial transcription factor A (mtTFA) binding sites; the downstream conserved sequence blocks (CSB) I, II, and III; and the origins of H-strand replication (OH1 and OH2) (27) (Fig. 1a). Recently, somatic mutations in this region of the mtDNA have been associated with aging. A T414G mutation in the mtTFA binding site of PL accumulates with age in cultured skin fibroblasts (28) and can be detected at low levels in skeletal muscle, but not in brain, by using our sensitive protein nucleic acid (PNA)-clamping (PCR) method (29). The A189G and T408A CR mutations accumulate with age in skeletal muscle (30), and a T150C mutation accumulates in white blood cells (31).
Fig. 1.

Representation of the somatic mtDNA CR mutation distribution in AD and control brains. (a) Schematic representation of nps 16000–570 of the mtDNA CR. The numbers below the line mark the mtDNA nps, and the boxes above the line represent the regulatory elements. The thick horizontal lines below the CR map represent the locations of the AD (red), control (blue), or common (gold) heteroplasmic mutations. (b) The number of heteroplasmic mutations in mtDNA CR regulatory elements in AD and control brains.
Although the risk of AD increases with age, to date no specific somatic mtDNA CR mutations have been reported for normal or AD brains (32–34). Consequently, we surveyed the mtDNA CR sequence of AD and control autopsy brains from subjects 59 to 94 years old. We now report that AD brains exhibit a striking increase in mtDNA CR mutations. Moreover, these mutations are located in elements known to be involved in mtDNA L-strand transcription and/or H-strand replication, and they are associated with reductions in the mtDNA L-strand ND6 mRNA and in the mtDNA copy number. Hence, somatic mtDNA CR mutations could account for the sporadic appearance and mitochondrial defects seen in late-onset AD and thus may contribute to the etiology of this disease.
Materials and Methods
Brain Samples from AD Patients and Controls. Frontal cortex brain samples from age-matched AD and control subjects were obtained from the Department of Neurology of Massachusetts General Hospital and Harvard Medical School (Boston) and from the Emory University Center for Neurodegenerative Disease Brain Bank (Atlanta). The clinical status, AD versus control, of all subject brain samples was confirmed by staining and counting senile plaques and neurofibrillary tangles. A total of 23 AD and 40 control brain samples, all pathologically confirmed, were used in this study. The mtDNA hypervariable region (np 16000–100) of each brain sample was sequenced, and those samples belonging to the common European mtDNA haplogroups H, U, J, and T were chosen for further cloning and sequencing studies (35). To eliminate the possibility that the observed CR variants were the product of the spurious amplification of nuclear-DNA-encoded, mtDNA pseudogenes, all PCR protocols were applied to cells lacking mtDNA (ρo cells) to ensure that no mtDNA-like sequences could be amplified.
Detection of the T414G Mutation in AD Brains. The T414G mutation was sought in the frontal cortex DNAs by the PNA-clamping PCR procedure (Fig. 2a). PNAs have a peptide bond backbone attached to nucleic acid bases. Because the protein backbone lacks negative charges, PNAs bind with high specificity and affinity to homologous DNA. In the PNA-clamping PCR procedure, a 15-base PNA that overlaps the target base (414) and was homologous to the common wild-type sequence (414T) was included in the PCR. This PNA was mixed with a pair of oligonucleotide primers, one of which binds just 3′ to the mutant base. During the PCR procedure, the PNA was first bound at high temperature, thus blocking all wild-type templates. The oligonucleotides were then annealed at a lower temperature and used to prime DNA synthesis, thus selectively amplifying the mutant sequence (414G). Through this procedure, we were able to detect one mutant mtDNA in 1,000 wild-type molecules (29). The presence of the T414G mutation in the resulting 334-np PCR product was confirmed through cleavage with FokI and by cloning and sequencing individual PCR molecules (Fig. 2 c and e).
Fig. 2.

PNA-clamping PCR assay for T414G mtDNA mutation in AD and control brains. (a and b) Agarose gel results of controls (a) or AD patients (b). The individual samples in a and b are identified by the age of the subject. Two PCRs are shown for each subject, one in the absence (-) and the other in the presence (+) of a PNA encompassing the 414 wild-type base, which suppresses amplification of the wild-type mtDNA. (c and d) FokI digestion of the PNA-clamping PCR products was used to confirm the presence of the T414G mutation from AD brains (c) and from AD and control brains run into same gel for comparison (d). Lanes in c are labeled with age of AD patients. -c, the FokI digestion result from the PCR product from wild-type plasmid; +c, the result from a T414G mutant plasmid. The arrow indicates the T414G FokI product. (e) Sequence analysis of CR fragments from a 74-year-old subject. The 414 region was PNA-clamping PCR-amplified, the resulting fragments were reamplified without PNA, and the final fragments were cloned and sequenced. The mutant nucleotide G (indicated with an arrow) is seen in three of five clones.
Identification of Novel mtDNA CR Mutations in AD Brains Through Cloning and Sequencing. Additional somatic mtDNA CR mutations were identified by PCR amplification of the mtDNA CR between nps 16527 and 636, cloning, and sequencing (Fig. 1). Frontal cortex genomic DNA was extracted by using the pure gene kit (Gentra Systems). The CR was amplified by using the primers np 16527–16546 (5′-CCT AAA TAG CCC ACA CGT TC-3′) and np 617–636 (5′-TGA TGT GAG CCC GTC TAA AC-3′), together with high fidelity Epicentre failsafe TaqDNA polymerase (Epicentre Technologies, Madison, WI). The desired PCR fragments were purified by using agarose gel electrophoresis, extracted by using the NucleoTrap gel kit (Clontech), and cloned by using the TOPO TA cloning protocol (Invitrogen), and the desired plasmids were purified by the minipreparation. Plasmid DNAs were cycle-sequenced by using BigDye dideoxy chain terminator chemistry (Applied Biosystems) on an ABI 3100 capillary sequencer, and the sequencing results were analyzed by using sequencer 4.0.5 (Gene Codes, Ann Arbor, MI).
Quantification of mtDNA Transcript Levels and Copy Number. To determine the ratio of mtDNA L-strand to H-strand transcripts, total RNA was extracted from the cortex tissue by using TRIzol (GIBCO/BRL) and the L-strand, ND6, mRNA and H-strand, ND2, mRNAs were reverse-transcribed and quantified by quantitative RT-PCR. ND6 was amplified by using forward primer np 14260–14279 (5′-ATC CTC CCG AAT GAA CCC TG-3′) and reverse primer np 14466–14485 (5′-GAT GGT TGT CTT TGG ATA TA-3′). ND2 mRNA was amplified by using the same primers that were used to determine the mtDNA/nuclear DNA ratio, which involved quantitative RT-PCR amplification of the ND2 mtDNA gene and the 18S rRNA nuclear DNA genes from genomic DNA (36).
Results
The T414G CR Mutation Is Found in AD Brains, but Not in Controls. When the frontal cortex genomic DNAs of 23 AD and 40 control subjects were tested for the presence of the T414G mutation by using PNA-clamping PCR, 65% of the AD brains were positive for the T414G mutation, but none of the controls was (Fig. 2 a and b). The presence of the T414G mutation was confirmed in the AD samples by FokI-restriction endonuclease digestion and through direct cloning and sequencing (Fig. 2 c–e).
Identification and Quantification of AD Brain CR Mutations. To determine the generality of the increased presence of somatic mtDNA CR mutations in AD brains, we PCR-amplified, cloned, and sequenced 10–20 CR clones from each of 16 AD and 17 control brain samples, giving a total of 250 AD and 235 control clones analyzed. This analysis revealed an overall 63% increase in the frequency of heteroplasmic mtDNA CR mutations in AD brains versus controls (P < 0.01) (Fig. 3a). Moreover, division of the AD cases into decade age groups revealed that the 59- to 69-year age group had a 79% increase, the 70- to 79-year age group had an 18% increase, and the 80-year-and-older age group had a 130% increase in mtDNA CR mutations relative to controls, with the difference between the 80-year-and-older AD patients and controls being highly significant (P < 0.001) (Fig. 3b).
Fig. 3.

Total number of heteroplasmic mtDNA CR mutations observed by cloning and sequencing CR clones from AD and control brain samples. (a) Number of mutants from all age groups (range 59–94); *, P < 0.01. (b) Number of mutants from three different age groups: 59–69, 70–79, and 80 & up; for 80 & up DNA mutation frequency, **, P < 0.001.
To determine the functional significance of these CR mutations, we correlated their position with that of the known functional elements of the mtDNA CR (Fig. 1). No clear difference in mutation distribution was seen between AD and control samples in the CR between nps 1 and 100, where few regulatory elements have been identified. By contrast, a striking increase in CR mutations was seen in the AD brain clones in the region between nps 101 and 570, which encompasses most of the known mtDNA regulatory elements (Fig. 1a).
Moreover, the AD mutants, but not the control mutants, were preferentially located in known functional transcription and replication elements. For example, seven heteroplasmic CR mutations were observed in AD brains in CSBI, but none was seen in controls (Fig. 1b). Likewise, 17 heteroplasmic mutations were found in the four mtTFA binding sites (two between PL and PH and two between CSBI and CSBII) in AD brains, whereas only five mutations were observed in the controls (P < 0.001) (Fig. 2b). Indeed, seven heteroplasmic mutations were present in the two mtTFA binding sites associated with PH and PL in AD brains, but none was found in these mtTFA sites in control brains. Therefore, mtDNA CR mutations are more common in AD patient brains, and they preferentially affect functionally important motifs.
High-Level mtDNA CR Mutant Heteroplasmy in AD Brains. Not only were CR mutations more prevalent in AD brains, they were also frequently present at exceptionally high proportion of the mtDNAs of the brain (Fig. 4). Although no marked differences were found between AD and control brains between nps 1 and 100 (Fig. 4 a versus b), multiple high-percentage heteroplasmic mutations were found in AD brains relative to controls in the region between np 100 and 570 (Fig. 4 c versus d).
Fig. 4.

Specific somatic mtDNA CR mutants and their percentage of heteroplasmy in AD and control brains. Subjects are listed by age. CR nps 1–100: (a) control brains; (b) AD brains. CR nps 101–570: (c) control brains; (d) AD patient brains. The specific mutations are listed below the abscissa lines and color-coded. The percentage of each mutation in each individual's brain is given by the height of the bar of that color. Homoplasmic germ-line mutations were also observed for these mutations. For the np 1–100 region, A73G was seen in six ADs and four controls. In the np 101–570 region, T146C was seen in three ADs and two controls; T152C was seen in three ADs and four controls; A189G was seen in no ADs and one control; and T195C was seen in two ADs and two controls. T414C and T477C were not found in the homoplasmic state in either AD or control samples.
Two of the identified higher percentage CR mutations proved to be specific for AD brains. One AD mutation, T414C, was found in 59-, 83-, and 84-year-old AD patients at about 10% mutant but was not present in any controls. The second AD-specific mutation, T477C, was found in the 76-, 78-, and 83-year-old AD patients at 70–80% mutant and in an 89-year-old patient at 20% mutant, but it was not found in controls (Fig. 4 c and d).
Four other high-percentage CR mutations were found predominantly in AD brains, but also in some controls, although at lower levels and later ages. A T146C mutation was found in 74- and 83-year-old AD patient brains at 70–80% mutant but also in one 94-year-old control at about 50% mutant. A T195C mutation was found in 74- and 83-year-old AD patients at 80% and 10% mutant, respectively, but also in one 77-year-old control at about 10% mutant. A T152C mutation was found in 67- and 76-year-old AD patient brains at 5–20% mutant and also in one 87-year-old control at 5% mutant. A A189G mutation was found in 62-, 67-, and 93-year-old AD brains at 5–20% mutant but also in 59- and 86-year-old control brains at less than 10% mutant (Fig. 4 d versus c).
These same CR mutants also co-occurred more often in AD brains than in controls. Four AD brains harbored more than one heteroplasmic mutation. The 67-year-old AD brain had both the T152C and A189G mutations, although at low percentages; the 74-year-old AD brain had the T146C and T195C mutations at very high levels; the 76-year-old AD brain harbored the T152C and T477C mutations at lower and higher percentages, respectively; and the 83-year-old AD brain harbored the T146C and T477C mutations at high percentages, as well as the T195C and T4141C mutations at low percentages. None of the control brains harbored more than one heteroplasmic CR mutation. Furthermore, six AD patients were homoplasmic for the T146C mutation, and four of these were also homoplasmic for the T195C mutation. By contrast, only two controls were homoplasmic for the T146C mutation, and none of these had the T195C mutation.
Finally, all of the AD patients that harbored individual mtDNA CR mutations with mutant levels of 70% or greater occurred in the age range of 74–83 years (four of seven cases, or 60%), whereas no patients were found with a very high percentage of mutant between ages 59 and 72 and between ages 84 and 93. Therefore, mtDNA CR mutations are more common, accumulate earlier, and can be present at higher percentages of the mtDNAs in AD patient brains than in control brains.
Reduced mtDNA L-Strand Transcripts and Copy Number in AD Brains. Most of the heteroplasmic mtDNA CR mutations observed in AD brains occurred in proximity to PL, from which L-strand transcription is initiated; in CSBI, after which the L-strand transcript is cleaved by the mitochondrial RNA-processing RNase to yield the 3′-OH replication primer; and around OH1 and OH2, where mtDNA polymerase γ (27) initiates H-strand replication. Therefore, we would expect that the CR mutations found in AD brains would reduce L-strand transcription and mtDNA copy number.
A reduction in AD brain L-strand transcription was confirmed by determining the ratio of the L-strand ND6 mRNA versus the H-strand ND2 mRNA by using quantitative RT-PCR. The ND6/ND2 mRNA ratio of 12 AD brains was 0.29 ± 0.18, but that of 11 controls was 0.67 ± 0.38, a twofold reduction in the ND6 mRNA level (P = 0.01). Similarly, analysis of the mtDNA/nuclear DNA ratio by quantitative RT-PCR of the ND2 and 18S rRNA gene copy numbers gave an average ratio of 12 ± 6.9 for 9 AD brains, but 22 ± 18 for 17 control brains, a 50% reduction in mtDNA copy number (P = 0.03).
Discussion
By analyzing the mtDNA CR sequence variation of the frontal cortex of pathologically confirmed AD and control brains, we have discovered that AD brains harbor a high frequency of heteroplasmic mtDNA CR mutations in key elements that regulate mtDNA L-strand transcription and H-strand replication. Consistent with the functional location of these mutations, AD brains have a marked reduction in the L-strand ND6 mRNA levels and in the cellular mtDNA copy number.
Mutations in PL could account for the reduction in ND6, because ND6 is the only L-strand mRNA. Additionally, mutations in PL and the downstream CSBI and OH1 and OH2 elements could account for the reduction in mtDNA copy number, because the L-strand transcript, processed at CSBI, has been proposed to provide the primer for initiating mtDNA H-strand synthesis at OH1 and OH2 (31).
A reduction in the ND6 mRNA would preferentially inhibit respiratory complex I, because ND6 is essential for complex I assembly (37, 38). Depletion of the mtDNA would diminish the activities of complexes I, III, IV, and V because the mtDNA encodes core subunits for each of these complexes (3, 39). Inhibition of OXPHOS would increase ROS production, sensitize the mtPTP, and enhance the apoptotic loss of synaptic connections between neurons (40). Thus, the accumulation of somatic mtDNA CR mutations provides an explanation for the neurological loss and resulting dementia associated with late-onset AD.
One unexpected finding about the CR mutations in AD brains was the apparent dichotomy between AD brains from subjects 74 to 83 years old and those from subjects over 83 years. Brains from 74- and 83-year-old subjects had fewer CR mutations (Fig. 3b), but with each present in a very high percentage of the brain mtDNAs (70–80%) (Fig. 4 c and d). AD brains from subjects over 83 years had many more CR mutations (Fig. 3b), but with each present at a lower percentage (<20%) of the brain mtDNAs (Fig. 4 c and d). This phenomenon might be explained by the time in brain development when the mtDNA CR mutation arose. In the 74- to 83-year-old cases, one or a few somatic mtDNA CR mutations could have arisen early in development and subsequently become widely distributed throughout the cells of the brain through cell replication. Because deleterious mtDNA mutations in postmitotic cells are selectively amplified (41–43), these early mtDNA mutants could come to predominate in each cell that contained mutant mtDNAs. Because virtually every cell in the brain would contain the same mutant mtDNA, the majority of the brain's mtDNAs would ultimately come to have that mutation.
In the case of the AD brains older than 83 years, somatic mtDNA mutations could have arisen later in brain development, with multiple different mtDNA CR mutations arising in different cells. Because each mutant would become dispersed into a smaller number of descendant cells and subsequently be amplified, these patients' brains would harbor several different mtDNA mutations, each in a lower percentage of the brain mtDNAs. Because these mutations arise later in development, they would be associated with later-onset symptoms. In the end, however, the severity of the dementia would be determined by the overall proportion of the patient's brain cells that had acquired a deleterious mtDNA mutation which subsequently became amplified. When the mutant mtDNA increased within a synapse sufficiently to cause an OXPHOS defect, then ROS production would increase, the mtPTP in the synaptic mitochondria would be activated, and the synaptic connections would be lost.
The question remains: is the increase in somatic mtDNA CR mutations seen in AD brains simply a reflection of accelerated aging, or is it a distinct pathological phenomenon? On the side of accelerated aging, we observed that some very old control brains also harbored some high-percentage mtDNA mutants. However, the fact that the T414G, T414C, and T477C mutations were only found in AD brains might suggest that AD is a distinct disease.
Still, the relationship of these three mutations to the AD brain pathology needs further clarification. The T414G mutation, although found in most AD brains, was present in only a small percentage of the brain mtDNAs. We consistently detected this mutation by using our highly sensitive PNA-clamping PCR technology. However, this mutation was not detected by others using the less sensitive primer extension (32) and CR cloning and sequencing strategies (34). Furthermore, while the T414C and T477C mutations were found in a much higher percentage of the mtDNAs of some AD brains, not all AD brains harbored high levels of these mutations. Therefore, the best interpretation is that the T414G mutation is a valuable marker for AD but that it is the total mtDNA CR mutation load that contributes to the brain pathology and that this may include mutations such as the T414C and T477C mutations.
Given this perspective, these two alternate views may not be very different. It seems likely that somatic mtDNA CR mutations accumulate with age in all individuals, but that the mutation rate of certain individuals is much higher. These later individuals, in turn, have a higher probability of acquiring one of the AD-specific mutations, thus enhancing the probability of dementia. Are, then, the mtDNA CR mutations a cause or an effect? We think that the accumulation of somatic mtDNA CR mutations is an important cause of neuronal mitochondrial energetic failure and synaptic loss. However, a variety of factors could modulate the mtDNA CR somatic mutation rate and thus increase the probability of dementia.
This mitochondrial hypothesis of AD would be consistent with the observation that individuals that harbor the ApoE ε4 allele have an increased risk of AD (44). The ε4 allele has been shown to be associated with increased oxidative stress (45–47). This would increase the mtDNA somatic mutation rate and exacerbate mitochondrial oxidative stress, mtPTP activation, and synaptic loss.
The association of Aβ amyloid plaques with AD could also be consistent with a mitochondrial etiology of late-onset AD. Aβ peptide has been proposed to be an antioxidant defense system to protect neuronal synapses from oxidative damage, but when the Aβ concentration becomes high, the peptide aggregates and becomes a toxic pro-oxidant (48). Moreover, recently, Aβ has been reported to enter the mitochondria, bind to mitochondrial alcohol dehydrogenase, and increase mitochondrial ROS production and propensity for cytochrome c release, presumably initiated by mtPTP activation (49). These observations suggest that in early-onset, familial AD, mutations in APP and the presenilin complex genes would increase Aβ peptide production, mitochondrial ROS production, mitochondrial damage, and apoptosis. By contrast, in late-onset, sporadic AD, somatic mtDNA CR mutations would arise by chance, and these would cause increased mitochondrial ROS production. The increased neuronal oxidative stress would then induce a compensatory production of Aβ peptide as an antioxidant defense. Ultimately, however, the increased ROS generated by the mutant mtDNAs and excessive Aβ would inhibit mitochondrial function, leading to mtPTP activation and synaptic loss.
In conclusion, our data suggest that a significant factor in the development of late-onset, sporadic AD could be the accumulation of somatic mtDNA CR mutations resulting from endogenous mitochondrial ROS damage. Once these mutations arise, they would become enriched in the postmitotic cells of the brain and ultimately result in mitochondrial OXPHOS deficiency, increased ROS production, activation of the mtPTP in the synaptic mitochondria, and loss of synaptic connections through apoptosis. Thus, the mitochondrial hypothesis could explain many of the unusual genetic and pathological features of late-onset, sporadic AD.
Acknowledgments
We thank Dr. Eduardo Ruiz-Pesini (University of California, Irvine) for his critical review of this manuscript and for the important scientific insights that he contributed and Dr. Marla Gearing (Emory University, Atlanta) for generously providing tissue samples for this study and for helpful consultation. This work was supported by National Institutes of Health Grants AG13154, AG20729, and NS21328, as well as an Ellison Medical Foundation Senior Investigator Award presented to D.C.W.
Abbreviations: AD, Alzheimer's disease; APP, Aβ precursor protein; CR, control region; CSB, conserved sequence block; mtPTP, mitochondrial permeability transition pore; mtTFA, mitochondrial transcription factor A; np, nucleotide pair; OXPHOS, oxidative phosphorylation; PL and PH, L- and H-strand promoters; PNA, protein nucleic acid; ROS, reactive oxygen species.
References
- 1.Tandon, A., Rogaeva, E., Mullan, M. & St. George-Hyslop, P. H. (2000) Curr. Opin. Neurol. 13, 377-384. [DOI] [PubMed] [Google Scholar]
- 2.Smith, M. A., Casadesus, G., Joseph, J. A. & Perry, G. (2002) Free Radical Biol. Med. 33, 1194-1199. [DOI] [PubMed] [Google Scholar]
- 3.Castellani, R., Hirai, K., Aliev, G., Drew, K. L., Nunomura, A., Takeda, A., Cash, A. D., Obrenovich, M. E., Perry, G. & Smith, M. A. (2002) J. Neurosci. Res. 70, 357-360. [DOI] [PubMed] [Google Scholar]
- 4.Harman, D. (2002) Ann. N.Y. Acad. Sci. 959, 384-395. [DOI] [PubMed] [Google Scholar]
- 5.Cottrell, D. A., Borthwick, G. M., Johnson, M. A., Ince, P. G. & Turnbull, D. M. (2002) Neuropathol. Appl. Neurobiol. 28, 390-396. [DOI] [PubMed] [Google Scholar]
- 6.Bosetti, F., Brizzi, F., Barogi, S., Mancuso, M., Siciliano, G., Tendi, E. A., Murri, L., Rapoport, S. I. & Solaini, G. (2002) Neurobiol. Aging 23, 371-376. [DOI] [PubMed] [Google Scholar]
- 7.Mutisya, E. M., Bowling, A. C. & Beal, M. F. (1994) J. Neurochem. 63, 2179-2184. [DOI] [PubMed] [Google Scholar]
- 8.Cardoso, S. M., Proenca, M. T., Santos, S., Santana, I. & Oliveira, C. R. (2004) Neurobiol. Aging 25, 105-110. [DOI] [PubMed] [Google Scholar]
- 9.Trimmer, P. A., Keeney, P. M., Borland, M. K., Simon, F. A., Almeida, J., Swerdlow, R. H., Parks, J. P., Parker, W. D., Jr., & Bennett, J. P., Jr. (2004) Neurobiol. Dis. 15, 29-39. [DOI] [PubMed] [Google Scholar]
- 10.Kish, S. J., Mastrogiacomo, F., Guttman, M., Furukawa, Y., Taanman, J. W., Dozic, S., Pandolfo, M., Lamarche, J., DiStefano, L. & Chang, L. J. (1999) J. Neurochem. 72, 700-707. [DOI] [PubMed] [Google Scholar]
- 11.Verwer, R. W., Jansen, K. A., Sluiter, A. A., Pool, C. W., Kamphorst, W. & Swaab, D. F. (2000) Exp. Neurol. 163, 440-451. [DOI] [PubMed] [Google Scholar]
- 12.Nagy, Z., Esiri, M. M., LeGris, M. & Matthews, P. M. (1999) Acta Neuropathol. 97, 346-354. [DOI] [PubMed] [Google Scholar]
- 13.Valla, J., Berndt, J. D. & Gonzalez-Lima, F. (2001) J. Neurosci. 21, 4923-4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pratico, D. (2002) Biochem. Pharmacol. 63, 563-567. [DOI] [PubMed] [Google Scholar]
- 15.Gibson, G. E. & Huang, H. M. (2002) Front. Biosci. 7, d1007-d1015. [DOI] [PubMed] [Google Scholar]
- 16.Coskun, P. E., Ruiz-Pesini, E. E. & Wallace, D. C. (2003) Proc. Natl. Acad. Sci. USA 100, 2174-2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Corral-Debrinski, M., Horton, T., Lott, M. T., Shoffner, J. M., McKee, A. C., Beal, M. F., Graham, B. H. & Wallace, D. C. (1994) Genomics 23, 471-476. [DOI] [PubMed] [Google Scholar]
- 18.Shoffner, J. M., Brown, M. D., Torroni, A., Lott, M. T., Cabell, M. R., Mirra, S. S., Beal, M. F., Yang, C., Gearing, M., Salvo, R., et al. (1993) Genomics 17, 171-184. [DOI] [PubMed] [Google Scholar]
- 19.Brown, M. D., Shoffner, J. M., Kim, Y. L., Jun, A. S., Graham, B. H., Cabell, M. F., Gurley, D. S. & Wallace, D. C. (1996) Am. J. Hum. Genet. 61, 283-289. [DOI] [PubMed] [Google Scholar]
- 20.Hutchin, T. & Cortopassi, G. (1995) Proc. Natl. Acad. Sci. USA 92, 6892-6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tysoe, C., Robinson, D., Brayne, C., Dening, T., Paykel, E. S., Huppert, F. A. & Rubinsztein, D. C. (1996) J. Med. Genet. 33, 1002-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Egensperger, R., Kosel, S., Schnopp, N. M., Mehraein, P. & Graeber, M. B. (1997) Neuropathol. Appl. Neurobiol. 23, 315-321. [PubMed] [Google Scholar]
- 23.Wragg, M. A., Talbot, C. J., Morris, J. C., Lendon, C. L. & Goate, A. M. (1995) Neurosci. Lett. 201, 107-110. [DOI] [PubMed] [Google Scholar]
- 24.Chagnon, P., Gee, M., Filion, M., Robitaille, Y., Belouchi, M. & Gauvreau, D. (1999) Am. J. Med. Genet. 85, 20-30. [DOI] [PubMed] [Google Scholar]
- 25.van der Walt, J. M., Nicodemus, K. K., Martin, E. R., Scott, W. K., Nance, M. A., Watts, R. L., Hubble, J. P., Haines, J. L., Koller, W. C., Lyons, K., et al. (2003) Am. J. Hum. Genet. 72, 804-811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruiz-Pesini, E., Mishmar, D., Brandon, M., Procaccio, V. & Wallace, D. C. (2004) Science 303, 223-226. [DOI] [PubMed] [Google Scholar]
- 27.Shadel, G. S. & Clayton, D. A. (1997) Annu. Rev. Biochem. 66, 409-435. [DOI] [PubMed] [Google Scholar]
- 28.Michikawa, Y., Mazzucchelli, F., Bresolin, N., Scarlato, G. & Attardi, G. (1999) Science 286, 774-779. [DOI] [PubMed] [Google Scholar]
- 29.Murdock, D. G., Christacos, N. C. & Wallace, D. C. (2000) Nucleic Acids Res. 28, 4350-4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang, Y., Michikawa, Y., Mallidis, C., Bai, Y., Woodhouse, L., Yarasheski, K. E., Miller, C. A., Askanas, V., Engel, W. K., Bhasin, S. & Attardi, G. (2001) Proc. Natl. Acad. Sci. USA 98, 4022-4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang, J., Asin-Cayuela, J., Fish, J., Michikawa, Y., Bonafe, M., Olivieri, F., Passarino, G., De Benedictis, G., Franceschi, C. & Attardi, G. (2003) Proc. Natl. Acad. Sci. USA 100, 1116-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chinnery, P. F., Taylor, G. A., Howell, N., Brown, D. T., Parsons, T. J. & Turnbull, D. M. (2001) Am. J. Hum. Genet. 68, 529-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin, M. T., Simon, D. K., Ahn, C. H., Kim, L. M. & Beal, M. F. (2002) Hum. Mol. Genet. 11, 133-145. [DOI] [PubMed] [Google Scholar]
- 34.Simon, D. K., Lin, M. T., Ahn, C. H., Liu, G. J., Gibson, G. E., Beal, M. F. & Johns, D. R. (2001) Genomics 73, 113-116. [DOI] [PubMed] [Google Scholar]
- 35.Richards, M., Macaulay, V., Hickey, E., Vega, E., Sykes, B., Guida, V., Rengo, C., Sellitto, D., Cruciani, F., Kivisild, T., et al. (2000) Am. J. Hum. Genet. 67, 1251-1276. [PMC free article] [PubMed] [Google Scholar]
- 36.Rodriguez-Santiago, B., Casademont, J. & Nunes, V. (2001) Eur. J. Hum. Genet. 9, 279-285. [DOI] [PubMed] [Google Scholar]
- 37.Bai, Y. & Attardi, G. (1998) EMBO J. 17, 4848-4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ugalde, C., Janssen, R. J., van den Heuvel, L. P., Smeitink, J. A. & Nijtmans, L. G. (2004) Hum. Mol. Genet. 13, 659-667. [DOI] [PubMed] [Google Scholar]
- 39.Chandrasekaran, K., Hatanpaa, K., Rapoport, S. I. & Brady, D. R. (1997) Mol. Brain Res. 44, 99-104. [DOI] [PubMed] [Google Scholar]
- 40.Rapoport, S. I. (2003) Neurotoxicol. Res. 5, 385-398. [Google Scholar]
- 41.Khrapko, K., Bodyak, N., Thilly, W. G., van Orsouw, N. J., Zhang, X., Coller, H. A., Perls, T. T., Upton, M., Vijg, J. & Wei, J. Y. (1999) Nucleic Acids Res. 27, 2434-2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coller, H. A., Bodyak, N. D. & Khrapko, K. (2002) Ann. N.Y. Acad. Sci. 959, 434-447. [DOI] [PubMed] [Google Scholar]
- 43.Trounce, I., Schmiedel, J., Yen, H. C., Hosseini, S., Brown, M. D., Olson, J. J. & Wallace, D. C. (2000) Nucleic Acids Res. 28, 2164-2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roses, A. D., Einstein, G., Gilbert, J., Goedert, M., Han, S. H., Huang, D., Hulette, C., Masliah, E., Pericak-Vance, M. A., Saunders, A. M., et al. (1996) Ann. N.Y. Acad. Sci. 777, 146-157. [DOI] [PubMed] [Google Scholar]
- 45.Ramassamy, C., Krzywkowski, P., Averill, D., Lussier-Cacan, S., Theroux, L., Christen, Y., Davignon, J. & Poirier, J. (2001) Brain Res. Mol. Brain Res. 86, 76-83. [DOI] [PubMed] [Google Scholar]
- 46.Ramassamy, C., Averill, D., Beffert, U., Theroux, L., Lussier-Cacan, S., Cohn, J. S., Christen, Y., Schoofs, A., Davignon, J. & Poirier, J. (2000) Neurobiol. Dis. 7, 23-37. [DOI] [PubMed] [Google Scholar]
- 47.Miyata, M. & Smith, J. D. (1996) Nat. Genet. 14, 55-61. [DOI] [PubMed] [Google Scholar]
- 48.Huang, X., Atwood, C. S., Hartshorn, M. A., Multhaup, G., Goldstein, L. E., Scarpa, R. C., Cuajungco, M. P., Gray, D. N., Lim, J., Moir, et al. (1999) Biochemistry 38, 7609-7616. [DOI] [PubMed] [Google Scholar]
- 49.Lustbader, J. W., Cirilli, M., Lin, C., Xu, H. W., Takuma, K., Wang, N., Caspersen, C., Chen, X., Pollak, S., Chaney, M., et al. (2004) Science 304, 448-452. [DOI] [PubMed] [Google Scholar]