

Abstract
Many hereditary and sporadic neurodegenerative disorders are characterized by the accumulation of aberrant proteins. In sporadic Parkinson's disease, representing the most prevalent movement disorder, oxidative and nitrosative stress are believed to contribute to disease pathogenesis, but the exact molecular basis for protein aggregation remains unclear. In the case of autosomal recessive-juvenile Parkinsonism, mutation in the E3 ubiquitin ligase protein parkin is linked to death of dopaminergic neurons. Here we show both in vitro and in vivo that nitrosative stress leads to S-nitrosylation of wild-type parkin and, initially, to a dramatic increase followed by a decrease in the E3 ligase-ubiquitin-proteasome degradative pathway. The initial increase in parkin's E3 ubiquitin ligase activity leads to autoubiquitination of parkin and subsequent inhibition of its activity, which would impair ubiquitination and clearance of parkin substrates. These findings may thus provide a molecular link between free radical toxicity and protein accumulation in sporadic Parkinson's disease.
Accumulation of misfolded proteins is thought to be a predominant factor in many neurodegenerative disorders (1–3). Rare hereditary mutations can simulate the commonplace sporadic forms of the disease, resulting in fibrillization or frank aggregation of aberrant proteins in disorders as diverse as Parkinson's disease (PD), Alzheimer's disease, amyotrophic lateral sclerosis, polyglutamine triple repeat disorders, and tauopathies such as frontotemporal dementia (4, 5). In the case of PD, mutations in genes including parkin or α-synuclein can simulate the sporadic phenotype, which is believed to be induced by insults to mitochondrial complex I resulting in oxidative/nitrosative stress to the endoplasmic reticulum and ubiquitin–proteasome systems (UPS), and consequent aberrant protein accumulation; dopaminergic neurons appear to be particularly vulnerable to such insults, possibly explaining the selective cell destruction in the substantia nigra that occurs in PD (6–9). Mutation in parkin, an E3 ubiquitin ligase that normally participates in ubiquitination and possibly chaperone activity, results in autosomal recessive-juvenile Parkinsonism (10). We noted that such E3 ligases contain cysteine residues in their RING domains, raising the possibility that cysteine thiol could react with nitric oxide (NO) species to form an S-nitrosylated derivative and thus influence biological activity (11–13). In this manner, S-nitrosylation of wild-type parkin would affect the ubiquitin–proteasome system degradative pathway and contribute to protein aggregation and thus the parkinsonian phenotype (2, 6, 8).
Materials and Methods
Fluorometric Detection of S-Nitrosothiols. The concentration of S-nitrosothiol formation was detected by conversion of the fluorescent compound 2,3-naphthyltriazole (NAT) from 2,3-diaminonaphthalene (DAN) at an emission wavelength of 360 nm and an excitation wavelength of 260 nm by using a Fluoro-Max-2 spectrofluorometer and datamax software (Instruments SA, Edison, NJ). Conversion to S-nitrosothiol is linear over the range 0.05–50 μM, as described (14). NAT was prepared by dissolving 500 mg of DAN in 20 ml of glacial acetic acid, diluting the solution to 100 ml with distilled water on ice, then rapidly reacting it with 2.5 ml of 1.2 M NaNO2. The precipitated NAT was recrystallized three times from boiling water containing decolorizing carbon. The white needle-like crystal NAT had a melting point of 194°C. The fluorescence intensity curve of serial NAT dilutions was used to construct a standard curve.
Biotin-Switch Assay for Detection of S-Nitrosylated Proteins. Cell lysates and brain tissue extracts were prepared in HENC or HENT buffers (250 mM Hepes, pH 7.5/1 mM EDTA/0.1 mM neocuproine/0.4% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate or 1% Triton X-100). Various amounts of protein were used for the assay, typically 500 μg for cell lysates and up to 7.5 mg for tissues. Blocking buffer [2.5% SDS/20 mM methyl methane thiosulfonate (MMTS) in HEN buffer] was mixed with the samples and incubated for 30 min at 50°C to block free thiol groups. After removing excess MMTS by acetone precipitation, nitrosothiols were then reduced to thiols with 1 mM ascorbate. The newly formed thiols were then linked with sulhydryl-specific biotinylating reagent N-[6-biotinamido)-hexyl]-1′-(2′pyridyldithio) propionamide. We then pulled down the biotinylated proteins with Streptavidin-agarose beads and performed Western blot analysis to detect the amount of parkin remaining in the samples (15).
Ubiquitination Assays. In vitro ubiquitination reactions were carried out by mixing immunoprecipitated parkin with 100 ng of E1 ubiquitin-activating enzyme (Calbiochem)/250 ng of E2 ubiquitin-conjugating enzyme (UbcH7, Boston Biochem, Cambridge, MA)/10 μg of ubiquitin (Sigma) in ubiquitination buffer (2 mM ATP/50 mM Tris/2.5 mM MgCl2 at pH 7.5) for 2 h at room temperature. Autoubiquitination of parkin was then detected by antiubiquitin antibody (Zymed). For the ex vivo ubiquitination assays, parkin-overexpressing SH-SY5Y neuroblastoma cells were transfected with the hemagglutinin (HA)-tagged ubiquitin and then exposed to S-nitrosocysteine (SNOC) or rotenone. The proteasome inhibitor MG132 (10 μM) was added to block the rapid degradation of polyubiquitinated proteins during the assay. Cell lysates were subjected to immunoprecipitation with anti-myc antibody, followed by Western blot analysis with anti-HA antibody. For assay of synphilin ubiquitination by parkin, we used HA-tagged synphilin-1 and myc-tagged ubiquitin.
Rotenone and 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Administration. All experiments were performed according to guidelines set by The Burnham Institute Animal Care Committee. Sprague–Dawley rats, weighing 200–250 g, were injected i.p. with freshly prepared rotenone (1 mg/kg per day in 50% DMSO and 50% polyethylene glycol) for 5 days. One group of animals received 3-bromo-7-nitroindazole (30 mg/kg per day) 30 min before rotenone injection. For the mouse experiments, neuronal NO synthase knockout and wild-type littermates, weighing 15–30 g, were injected with rotenone as above. Another group of C57BL/6 mice were injected with MPTP (30 mg/kg) i.p. for 4 days. Control animals were injected with vehicle only (5–10 animals per group).
Mass Spectrometry (MS). In-gel digestions were performed as described (14). The peptides were extracted, dried under vacuum, and redissolved in 20 μl of 2% acetonitrile/0.1% formic acid in water. Nano-liquid chromatography (LC) MS analyses were carried out by using an LC Packings nano-LC system (Amsterdam) and a Micromass quadrupole time-of-flight (Q-TOF) API-US mass spectrometer (Manchester, U.K.). The analytical column was a PepMap C18 (LC Packings) with dimensions of 75 μm i.d. × 15 cm. Mobile phase A was 2% acetonitrile/0.1% formic acid, whereas mobile phase B was 80% acetonitrile/0.1% formic acid. The gradient was 2%∼ 40% mobile phase B in 75 min, followed by 100% mobile phase B for 5 min. The flow rate was ≈200 nl/min. The mass spectra of parkin and SNOC-exposed parkin were analyzed for cysteine alkylation, S-nitrosylation, and sulfination/sulfonation modifications, as well as for methionine oxidation by Mascot (Matrix Science, London).
Molecular Modeling. We modeled the parkin RING I domain structure with the jackal molecular modeling package (16), using the related human c-Cbl crystal structure (PDB ID code 1FBV) as a template. The alignment of c-Cbl and parkin RING I protein sequences was generated by segment alignment algorithm (sea) software (17–19).
Results
S-Nitrosylation of Parkin in Vitro. Initially, we gathered three lines of chemical, biochemical, and immunochemical evidence to show that parkin was S-nitrosylated by NO-related species in vitro. First, in a specific fluorescence assay for S-nitrosylated proteins, we demonstrated the reaction of recombinant parkin with the physiological NO donor, SNOC. This assay detects the formation of S-nitrosothiol by measurement of the fluorescent compound NAT (14, 20). NAT is stoichiometrically converted from 2,3-diaminonaphthalene by NO released from S-nitrosylated proteins and thus provided a quantitative measure of S-nitrosothiol formation on parkin. SNOC-treated recombinant GST-parkin resulted in significant S-nitrosothiol formation, whereas GST alone did not under these conditions (Fig. 1A). To ensure that the S-nitrosothiol generated under these conditions represented S-nitroso-parkin (SNO-PARK) rather than residual SNOC, the stability of S-nitrosothiols was examined at different incubation times. SNO-PARK was much more stable than SNOC alone; within 15 min of incubation, >95% of the SNOC had decayed, whereas SNO-PARK remained (14). This temporal separation allowed us to distinguish SNOC from SNO-PARK in the fluorescent S-nitrosothiol assay. Second, we showed that parkin was S-nitrosylated by SNOC by using the biotin-switch biochemical method (Fig. 1B). In this assay, nitrosylated proteins are identified by Western blot after substituting SNO for a more stable biotin group via chemical reduction with ascorbate, an assay pioneered by Snyder and colleagues (15). Third, we obtained immunoblot evidence for SNO-PARK formation by colabeling of the same band with an antibody specific for parkin and an antibody specific for S-nitrosylated protein (Fig. 1C) (21).
Fig. 1.

S-nitrosylation of parkin in vitro. (A) Recombinant protein GST-parkin (0.2μM) was incubated with SNOC (200μM) for 30 min at room temperature (RT). The SNO-PARK thus generated was assessed by release of NO, causing the conversion of 2,3-diaminonaphthalene to the fluorescent compound NAT. GST protein alone was used as negative control. BSA (5 μM) was used as positive control (25). SNOC itself quickly decayed and thus resulted in insignificant S-nitrosothiol readings in this assay. (B)(Upper) Cell lysates from human neuroblastoma SH-SY5Y cells overexpressing myc-tagged parkin were incubated with 200 μM SNOC at RT. Control samples were subjected to decayed SNOC under the same conditions. Thirty minutes after SNOC exposure, SNO-PARK was detected by the biotin-switch assay (15). w/o Biotin-HPDP represents the control without biotin linker [N-[6-biotinamido)hexyl]-1′-(2′pyridyldithio) propionamide] (Lower) Total parkin in cell lysates identified by immunoblot. (C) (Upper) Cell lysates were immunoprecipitated with anti-myc antibody. The immunoprecipitates were incubated with or without SNOC and subjected to SDS/PAGE Western analysis under nonreducing conditions. The membrane was blotted with anti-S-nitrosylated protein antibody to identify nitrosylated parkin and revealed an ≈2-fold increase after SNOC exposure by densitometry. (Lower) Same blot reprobed with anti-parkin antibody to verify equal loading.
Regulation of Parkin E3 Ubiquitin Ligase Activity by S-Nitrosylation. To determine whether S-nitrosylation of parkin affected its E3 ligase activity, we performed an in vitro E3 ligase assay comparing activity of wild-type parkin and SNO-PARK. We found that exposure of parkin to SNOC resulted in a dramatic increase in its E3 ligase activity, monitored by the autoubiquitination of parkin (Fig. 2A). To confirm that these in vitro findings were relevant to the cellular milieu, we incubated parkin-transfected, dopaminergic SH-SY5Y cells with SNOC or with the mitochondrial complex I inhibitor rotenone, which is known to induce the parkinsonian phenotype (22). We then monitored SNO-PARK formation and parkin E3 ligase activity. We found that either SNOC or rotenone led to the generation of SNO-PARK by the biotin-switch assay (Fig. 2B). In both cases, within a few hours of exposure to SNOC or rotenone, the resulting S-nitrosylation significantly increased the E3 ligase activity of parkin in SH-SY5Y cells lysates (Fig. 2C). Interestingly, if assayed 12–24 h after exposure to the physiological NO donors SNOC or S-nitrosoglutathione, or to rotenone, parkin E3 ligase activity began to decrease (Fig. 2D). Similarly, parkin E3 ligase activity increased 2 h after NO exposure but began to decrease by 24 h with synphilin-1 as the substrate (Fig. 2E). This biphasic action would be expected if S-nitrosylation initially activated E3 ligase activity, and the resulting autoubiquitination of parkin subsequently led to its inactivation. Moreover, marked increases in ubiquitinated proteins can potentially overwhelm the proteasome and result in aberrant protein accumulation, as has been observed with parkin in Lewy bodies of PD patients (6, 7, 23). As controls, Hoechst staining revealed that the cells were still alive at the time of the parkin E3 ligase activity assay and that there was no effect on the assay after incubation of purified parkin directly with rotenone (data not shown).
Fig. 2.

Regulation of parkin E3 ubiquitin ligase activity by S-nitrosylation. (A) In vitro S-nitrosylation of parkin up-regulates its E3 ligase activity. (Upper) Lysates from parkin-overexpressing SH-SY5Y cells were immunoprecipitated with anti-myc antibody. The immunoprecipitates were incubated with SNOC (200 μM) and then subjected to in vitro ubiquitination reaction. SNOC increased the autoubiquitination of parkin, detected by anti-ubiquitin antibody. (Lower) Parkin loading control. (B) S-nitrosylation of parkin after SNOC or rotenone exposure. (Upper) Cell lysates from SH-SY5Y cells that had been exposed to SNOC (SNOC Ex Vivo) or rotenone (1 μM for 6 h) were subjected to the biotin-switch assay to detect SNO-PARK. Also, cell lysates were exposed to SNOC directly (SNOC In Vitro). Rotenone exposure was supplemented with l-arginine (labeled l-Arg) as a NOS substrate. Both SNOC and rotenone increased SNO-PARK in these assays. Nω-nitro-l-arginine (NNA) was used as a NOS inhibitor and suppressed SNO-PARK formation. (Lower) Parkin loading control. (C) Ex vivo S-nitrosylation of parkin up-regulates its E3 ligase activity. SH-SY5Y cells were transfected with HA-tagged ubiquitin and exposed to SNOC or rotenone to nitrosylate parkin. Cell lysates were then subjected to immunoprecipitation with anti-myc followed by Western blot analysis with anti-HA to detect autoubiquitinated parkin. NNA prevented the rotenone-induced increase in E3 ligase activity. (D) Effect of NO donor SNOC and rotenone on parkin E3 activity at different times. SH-SY5Y cells were exposed to SNOC or rotenone. At early time points (2 h after SNOC or 6 h after rotenone), parkin autoubiquitination increased compared with control but decreased several hours later. (E) Effect of NO donor on ubiquitination of synphilin-1 (Syn-1) by parkin. SH-SY5Y cells were cotransfected with HA-synphilin-1 and myc-ubiquitin and exposed to SNOC. Synphilin-1 ubiquitination increased 2 h after SNOC exposure but decreased 24 h later. Old SNOC, decayed SNOC.
S-Nitrosylation of Parkin in PD Animal Models and in Human PD Brain. We next asked whether the SNO-PARK that we encountered in vitro and in cell-based assays after S-nitrosylation was also present in vivo in rodent parkinsonian models induced by the mitochondrial complex I inhibitors rotenone or MPTP, as well as in human brains with sporadic PD. We first examined the substantia nigra and striatum of rats that had been injected with rotenone and found SNO-PARK formation by the biotin-switch assay compared to vehicle-injected control rats (Fig. 3A). Additionally, SNO-PARK was observed in the brains of MPTP-injected mice (Fig. 3B). Finally, we examined human brains with sporadic PD with diffuse Lewy bodies (versus autopsy brains from patients who died of other causes) and found evidence for SNO-PARK formation by the biotin-switch method (Fig. 3C and Fig. 6, which is published as supporting information on the PNAS web site).
Fig. 3.

S-nitrosylation of parkin in rodent models and human PD brain. (A) Vehicle (control), rotenone, or rotenone plus the relatively specific neuronal NO synthase inhibitor 3-bromo-7-nitroindazole (3br7NI) was i.p. injected into Sprague–Dawley rats (26). (Upper) Brain extracts from substantia nigra and striatum were subjected to the biotin-switch assay to detect SNO-PARK. Rotenone resulted in increased SNO-PARK that was partially abrogated by 3br7NI. (Lower) Parkin loading control. (B) SNO-PARK was detected in MPTP-, but not control-, injected mice. (C) SNO-PARK was detected in representative temporal cortex of human brain extracts from two sporadic PD patients with diffuse Lewy bodies but not from control brain by using the biotin-switch assay. In total, we tested four brain samples from PD, two normal controls with no CNS disease, two controls with Alzheimer's disease, and two controls with Huntington's disease (see Fig. 6). Each of the PD brain samples, but none of the control brains, manifests evidence for SNO-PARK formation (P < 0.01 by χ2 test).
If S-nitrosylation of parkin and consequent increase in E3 ligase activity occur because of reaction with NO in neurons, then inhibition of NO synthase (NOS) activity should abrogate the effect. Therefore, we pretreated with a NOS inhibitor an aliquot of rotenone-exposed SH-SY5Y cells stably transfected with parkin to see whether we could block SNO-PARK formation and attenuate the increase in E3 ligase activity. Indeed, we found that NOS inhibition prevented these effects (Fig. 2 B and C). Additionally, pharmacological inhibition or genetic disruption of neuronal NO synthase prevented the formation of SNO-PARK observed in vivo in rotenone-exposed rodents (Fig. 3A and data not shown).
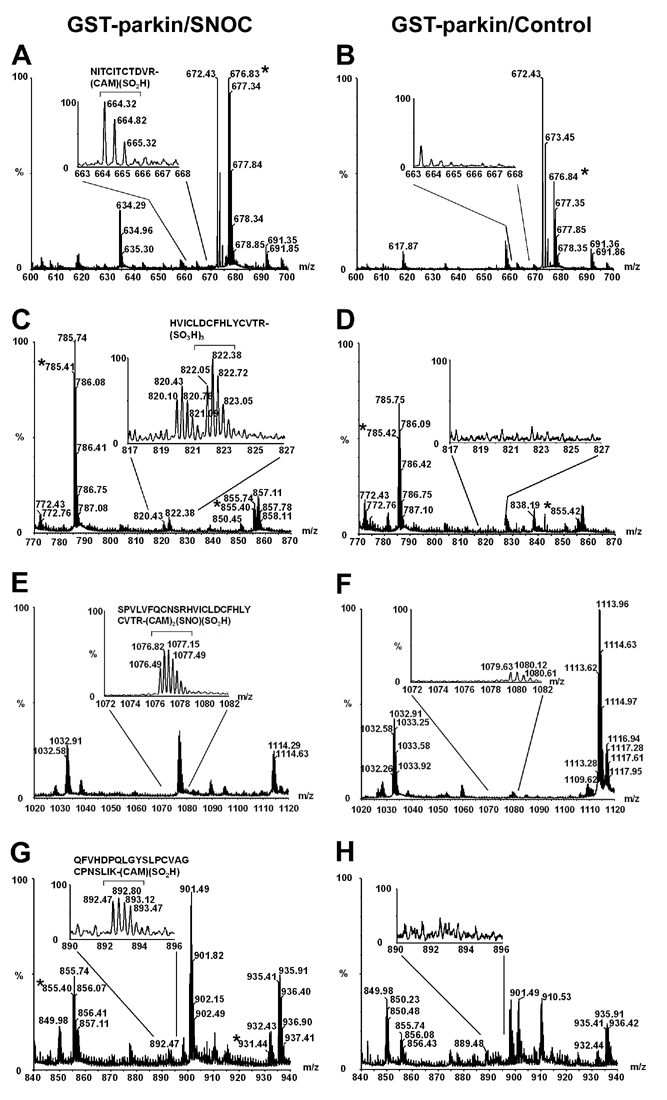
Peptide Mass Fingerprinting Analysis of Recombinant Human Parkin. It is most important to link structure with function, that is, to identify the critical cysteine residue(s) in parkin whose S-nitrosylation regulates the E3 ligase activity. Therefore, as a first step to further identify the chemical nature of the NO-triggered-modification of the parkin E3 ligase responsible for activation and assess the possibility of additional oxidative products, we conducted peptide mass fingerprinting. Mass spectra were obtained after in-gel digestion by trypsin of human recombinant parkin using quadrupole time-of-flight (Q-TOF) MS with methods that we have described (14). With MS, we found that five of seven cysteines in the RING I domain of parkin are candidates to be S-nitrosylated; however, it will be important in the future to delineate which of these nitrosylated cysteines are responsible for the altered E3 ligase activity. Moreover, as we have described for activation of matrix metalloproteinase enzymes (14), these cysteine residues can undergo further oxidation to sulfinic or sulfonic acid derivatives (Fig. 4 and Fig. 7, which is published as supporting information on the PNAS web site); whether these oxidized products are relevant to the functional activity of the endogenous E3 ligase, however, remains unknown. Additionally, we observed that five other cysteines in the in-between-RING and RING II domains were potentially S-nitrosylated and/or sulfonated (Table 1, which is published as supporting information on the PNAS web site). Analysis of crystal structure models of the parkin RING I domain revealed that two of the cysteines lie within the consensus motif for S-nitrosylation that we had identified, making them likely candidates for physiological nitrosylation (Fig. 5) (13).
Fig. 4.

Peptide mass fingerprinting analysis of the modified MS thiol group of cysteine residues within the highly conserved RING I domain of recombinant human parkin exposed to SNOC. (Upper) Quadrupole time-of-flight (Q-TOF) results for RING I domain of recombinant parkin exposed to SNOC. Sequences containing modified cysteines. Carbamidomethyl (CAM) signifies modification by iodoacetamide alkylation to protect free cysteines before trypsin digestion. Cys-SNO indicates S-nitrosothiol formation; Cys-SO2H or Cys-SO3H indicates sulfinic or sulfonic acid derivatization, respectively. Previously, we had shown that nitrosylation can precede further oxidation to these derivatives (14). Five of the seven cysteine residues in the RING I domain underwent modification. Single-letter abbreviations for the amino acid residues are as follows: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, Trp; and Y, Tyr. (Lower) Schematic representation of parkin and sample mass spectra of parkin modifications in RING I domain (bars indicate tryptic fragments containing modified cysteine residues). Ubl, ubiquitin-like domain; IBR, in-between RING domain. For additional mass spectra results on the RING II and IBR domains, see Fig. 7 and Table 1.
Fig. 5.

Model of S-nitrosylation of parkin. Ribbon structure of a partial sequence of human parkin showing RING I domain (residues 237Thr to 291Ala). α-Helix shown in red, β-sheet in cyan, cysteine residues in green, histidine in yellow, aspartate in red, and arginine in blue. 241Cys and 260Cys match the nitrosylation consensus motif (13).
Discussion
In summary, we found that S-nitrosylation of parkin can stimulate its E3 ligase activity, resulting in an initial increase in enzyme activity, autoubiquitination, and thereafter subsequent decrease in activity. We speculate that the initial increase in parkin's ubiquitin E3 ligase activity and its autoubiquitination may result in proteasome dysfunction and protein accumulation. Moreover, the subsequent decrease in parkin's ubiquitin E3 ligase activity may impair ubiquitination of parkin substrates and hence the neuroprotective effect that has been observed with parkin overexpression. This biphasic process may repeat itself throughout the course of the disease as additional molecules of parkin are nitrosylated and oxidized. We found evidence for SNO-PARK formation in both animal models of mitochondrial complex I deficiency and in human brains of patients with sporadic PD and diffuse Lewy body disease.
Dopaminergic neurons are particularly vulnerable to ubiquitin-proteasome system (UPS) dysfunction, and interference with this degradation system may lead to aberrant protein accumulation and cell death. Hence, nitrosative and oxidative stress resulting in parkin dysfunction may provide a mechanistic link between free radical production, UPS deficits, protein accumulation, and dopaminergic neuronal demise in sporadic PD. The elucidation of an NO-mediated pathway to neuronal apoptosis involving parkin dysfunction may contribute to the development of new therapeutic approaches for sporadic PD and other disorders associated with protein accumulation due to nitrosative and oxidative stress. Additionally, the activity of other RING-finger E3 ligases may also be affected by S-nitrosylation, providing a heuristic framework for the pathogenesis of other neurodegenerative and systemic disorders.
Note. After submission of this manuscript, another group reported that parkin was S-nitrosylated in sporadic PD (24). These authors also found that overexpression of parkin rescued neuronal cell lines subjected to proteasome inhibition and this protective effect was prevented by S-nitrosylation of parkin, supporting the pathophysiological relevance of SNO-PARK formation. However, that work reported that parkin E3 ligase activity was decreased after S-nitrosylation. We feel that the discrepancy with our observation of an initial increase in parkin E3 ligase activity followed by a decrease can be explained by our looking at earlier time points after S-nitrosylation and by other methodological differences in the E3 ligase assays, such as our inclusion of a proteasome inhibitor in the E3 ligase assays.
Supplementary Material
Acknowledgments
We thank Dr. Donger Zhang (The Scripps Research Institute, La Jolla, CA) for the constructs of ubiquitin and Dr. Yuzhen Ye for modeling and illustration of the parkin crystal structure. This work was supported in part by National Institutes of Health Grants P01 HD29587, R01 EY09024, R01 EY05477, and R01 NS41207 and the American Parkinson's Disease Association, San Diego Chapter.
Abbreviations: NAT, 2,3-naphthyltriazole; SNOC, S-nitrosocysteine; SNO-PARK, S-nitroso-parkin; HA, hemagglutinin; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; PD, Parkinson's disease; NOS, NO synthase.
References
- 1.Mayer, R. J., Lowe, J., Lennox, G., Doherty, F. & Landon, M. (1989) Prog. Clin. Biol. Res. 317, 809-818. [PubMed] [Google Scholar]
- 2.Bence, N. F., Sampat, R. M. & Kopito, R. R. (2001) Science 292, 1552-1555. [DOI] [PubMed] [Google Scholar]
- 3.Goldberg, A. L. (2003) Nature 426, 895-899. [DOI] [PubMed] [Google Scholar]
- 4.Perutz, M. F. & Windle, A. H. (2001) Nature 412, 143-144. [DOI] [PubMed] [Google Scholar]
- 5.Yong, V. W., Power, C., Forsyth, P. & Edwards, D. R. (2001) Nat. Rev. Neurosci. 2, 502-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dawson, T. M. & Dawson, V. L. (2003) Science 302, 819-822. [DOI] [PubMed] [Google Scholar]
- 7.Giasson, B. I. & Lee, V. M. (2003) Cell 114, 1-8. [DOI] [PubMed] [Google Scholar]
- 8.Masliah, E., Rockenstein, E., Veinbergs, I., Mallory, M., Hashimoto, M., Takeda, A., Sagara, Y., Sisk, A. & Mucke, L. (2000) Science 287, 1265-1269. [DOI] [PubMed] [Google Scholar]
- 9.Zhang, Y., Dawson, V. L. & Dawson, T. M. (2000) Neurobiol. Dis. 7, 240-250. [DOI] [PubMed] [Google Scholar]
- 10.Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., Yokochi, M., Mizuno, Y. & Shimizu, N. (1998) Nature 392, 605-608. [DOI] [PubMed] [Google Scholar]
- 11.Lipton, S. A., Choi, Y. B., Pan, Z. H., Lei, S. Z., Chen, H. S., Sucher, N. J., Loscalzo, J., Singel, D. J. & Stamler, J. S. (1993) Nature 364, 626-632. [DOI] [PubMed] [Google Scholar]
- 12.Stamler, J. S. (1994) Cell 78, 931-936. [DOI] [PubMed] [Google Scholar]
- 13.Stamler, J. S., Toone, E. J., Lipton, S. A. & Sucher, N. J. (1997) Neuron 18, 691-696. [DOI] [PubMed] [Google Scholar]
- 14.Gu, Z., Kaul, M., Yan, B., Kridel, S. J., Cui, J., Strongin, A., Smith, J. W., Liddington, R. C. & Lipton, S. A. (2002) Science 297, 1186-1190. [DOI] [PubMed] [Google Scholar]
- 15.Jaffrey, S. R., Erdjument-Bromage, H., Ferris, C. D., Tempst, P. & Snyder, S. H. (2001) Nat. Cell Biol. 3, 193-197. [DOI] [PubMed] [Google Scholar]
- 16.Xiang, Z. & Honig, B. (2001) J. Mol. Biol. 311, 421-430. [DOI] [PubMed] [Google Scholar]
- 17.Ye, Y., Jaroszewski, L., Li, W. & Godzik, A. (2003) Bioinformatics 19, 742-749. [DOI] [PubMed] [Google Scholar]
- 18.Zheng, N., Wang, P., Jeffrey, P. D. & Pavletich, N. P. (2000) Cell 102, 533-539. [DOI] [PubMed] [Google Scholar]
- 19.Zheng, N., Schulman, B. A., Song, L., Miller, J. J., Jeffrey, P. D., Wang, P., Chu, C., Koepp, D. M., Elledge, S. J., Pagano, M., et al. (2002) Nature 416, 703-709. [DOI] [PubMed] [Google Scholar]
- 20.Wink, D. A., Kim, S., Coffin, D., Cook, J. C., Vodovotz, Y., Chistodoulou, D., Jourd'heuil, D. & Grisham, M. B. (1999) Methods Enzymol. 301, 201-211. [DOI] [PubMed] [Google Scholar]
- 21.Gow, A. J., Chen, Q., Hess, D. T., Day, B. J., Ischiropoulos, H. & Stamler, J. S. (2002) J. Biol. Chem. 277, 9637-9640. [DOI] [PubMed] [Google Scholar]
- 22.Betarbet, R., Sherer, T. B., MacKenzie, G., Garcia-Osuna, M., Panov, A. V. & Greenamyre, J. T. (2000) Nat. Neurosci. 3, 1301-1306. [DOI] [PubMed] [Google Scholar]
- 23.Schlossmacher, M. G., Frosch, M. P., Gai, W. P., Medina, M., Sharma, N., Forno, L., Ochiishi, T., Shimura, H., Sharon, R., Hattori, N., et al. (2002) Am. J. Pathol. 160, 1655-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chung, K. K., Thomas, B., Li, X., Pletnikova, O., Troncoso, J. C., Marsh, L., Dawson, V. L. & Dawson, T. M. (2004) Science, 304, 1328-1331. [DOI] [PubMed] [Google Scholar]
- 25.Stamler, J. S., Simon, D. I., Osborne, J. A., Mullins, M. E., Jaraki, O., Michel, T., Singel, D. J. & Loscalzo, J. (1992) Proc. Natl. Acad. Sci. USA 89, 444-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He, Y., Imam, S. Z., Dong, Z., Jankovic, J., Ali, S. F., Appel, S. H. & Le, W. (2003) J. Neurochem. 86, 1338-1345. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}