

Abstract
Tumor Necrosis Factor alpha (TNFα) is an inflammatory cytokine that plays a central role in the pathogenesis of chronic inflammatory disease. Here we describe the chemical synthesis of l-TNFα along with the mirror-image d-protein for use as a phage display target. The synthetic strategy utilized native chemical ligation and desulfurization to unite three peptide segments, followed by oxidative folding to assemble the 52 kDa homotrimeric protein. This synthesis represents the foundational step for discovering an inhibitory d-peptide with the potential to improve current anti-TNFα therapeutic strategies.
Graphical abstract
Introduction
Tumor Necrosis Factor alpha (TNFα) is an inflammatory cytokine released by immune cells in response to injury or infection.1 Following its release, TNFα serves as a master regulator of the inflammatory response, influencing the release of additional cytokines such as IL-6, IL-8, and GM-CSF.2-4 Improper regulation of TNFα production and release is a major contributing factor to chronic inflammatory disease.2, 5, 6 Sequestration of extracellular TNFα with antibodies (e.g., infliximab, adalimumab, golimumab, and certolizumab pegol) or the receptor fusion protein etanercept has revolutionized the treatment of these diseases. However, use of these injectable biologic drugs is not without risks and challenges. Of particular concern, they cause systemic immunosuppression, which leaves patients vulnerable to opportunistic infections.7 Current anti-TNFα biologics are also immunogenic,8 which can reduce efficacy and require transitioning to another treatment.
An alternative anti-TNFα agent that overcomes these issues has been a major goal of the drug discovery community for many years. The major challenge towards achieving this goal is that TNFα interacts with TNF receptors over a broad surface devoid of well-defined, targetable hydrophobic pockets. With a few exceptions,9-12 almost all known small molecule antagonists are indirect inhibitors, targeting TNFα production and/or release, or downstream signaling cascades.13, 14
Direct inhibitors of TNFα have been developed both by structurally mimicking a natural TNFα binding partner (TNFR1)15 or by peptide phage display.16, 17 Unfortunately, peptides comprised of natural l-amino acids are typically rapidly degraded and cleared from the body. These poor pharmacokinetic properties are a substantial barrier to clinical use.
On the other hand, d-peptides (comprised solely of d-amino acids and achiral glycine) are inherently resistant to proteolysis by natural enzymes.18 Additionally, d-peptides exhibit reduced immunogenicity as they are not processed for MHC antigen presentation.19 However, the discovery of inhibitory d-peptides represents a considerable challenge compared to their l-counterparts. Inhibitory d-peptides are discovered by utilizing the symmetry relationship between l- and d-peptides.20 In mirror-image phage display, randomized genetically encoded l-peptide libraries are screened against a mirror-image target protein (chemically synthesized from d-amino acids). The highest affinity phage clone sequences are then synthesized as d-peptides, and by the law of symmetry, these d-peptides will possess the same binding interaction with the natural l-target as the l-peptide with the mirror-image (d-) target. This technique has been successfully used to discover d-peptide inhibitors of HIV,21-23 the p53-MDM2 complex,24 and VEGF.25, 26 However, with the exception of VEGF (a covalent homodimer of 102 aa subunits27), the requirement for chemical synthesis of the d-target has limited the application of this technology to relatively small protein targets (<100 residues).28
The clinical importance of anti-TNFα therapies for treating chronic inflammation, combined with the potential to reduce drug-related side effects, makes TNFα an ideal target for mirror-image phage display. In this report, we first describe the chemical synthesis and biological evaluation of natural l-TNFα. Using the same strategy, we then assembled and validated the mirror-image d-protein for use as a mirror-image phage display target.
Results and discussion
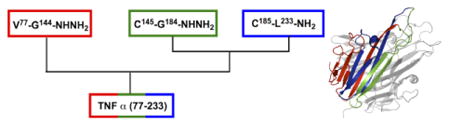
We initiated our work by identifying the shortest sequence of TNFα that was capable of proper folding and interaction with TNF receptors. Initially expressed as a membrane-bound homotrimer, TNFα is cleaved by TNFα converting enzyme, releasing a soluble 52 kDa homotrimer.3 Both soluble TNFα and membrane-anchored TNFα bind the two distinct TNF receptors (TNFR1 and TNFR2), and they are structurally equivalent with regard to their receptor-binding interface. Therefore, the 52 kDa soluble domain (77-233) was chosen as our mirror-image phage display target.
In order to access a mirror-image phage display target of this size (157 residues per monomer), we developed a synthetic strategy that utilized sequential native chemical ligation reactions to assemble the protein from three segments. Native chemical ligation (NCL)29 is a robust method for uniting peptide segments produced by either Fmoc or Boc solid-phase peptide synthesis (SPPS) and has enabled the chemical synthesis of numerous proteins,30-37 even as large as 312 residues.38 For this project, we used Fmoc SPPS to synthesize individual peptide segments on TentaGel® R RAM resin to generate C-terminal amides and 2-chlorotrityl hydrazine resin to generate C-terminal hydrazides. Peptide hydrazides were chosen because they provide a convenient, Fmoc-compatible method for generating a C-terminal thioester suitable for NCL (via the chemoselective oxidation of a C-terminal hydrazide followed by thiolysis with MPAA).39, 40
To divide soluble TNFα into manageable segments for SPPS, we first identified cysteine residues that could be used for NCL. TNFα contains two natural cysteines, located at residues 145 and 177 (Fig. 1A). Unfortunately, using Cys 177 would require ligation to a proline thioester, which is known to be a poor ligation junction because of slow ligation kinetics41, 42 and intramolecular cyclization to form a C-terminal diketopiperazine.43 In spite of these challenges, successful proline ligations have been demonstrated by employing bis(2-sulfanylethyl)amido (SEA) thioesters at elevated temperature.44 Therefore, we first attempted to utilize these two cysteines in our synthetic strategy. Unfortunately, the C-terminal segment required for the proline ligation strategy demonstrated poor solubility, further complicating the intermolecular reaction. Additionally, both segments 177-233 and the ligated product 145-233 demonstrated similar HPLC retention properties and considerable peak tailing, preventing efficient isolation of the ligated product from an excess of 177-233.
Figure 1.

Synthesis of TNFα by native chemical ligation. (A) Sequence of soluble ectodomain using full-length sequence numbering. N-terminal, middle, and C-terminal segments are red, green and blue, respectively. Native cysteine residues are depicted in bold, and ligation junctions are bold and underlined. X is hydrogen (unprotected N-terminus) in l-TNF or Biotin-PEG2 in d-TNF. (B) Assembly strategy for the three segments; NCL includes oxidation (hydrazide to azide), conversion to MPAA thioester, and ligation to an N-terminal Cys-containing peptide.
To circumvent these problems, we redesigned our retrosynthetic strategy, shifting the ligation junction to A185. Here, position 185 is originally introduced as Cys and then desulfurized45, 46 after ligation to produce the native sequence. Using Cys145 and Ala185 as ligation junctions allowed the protein to be divided into three segments of 68, 40, and 49 residues that could be assembled using highly reactive glycine thioesters (Fig. 1B).42 Acetamidomethyl (Acm) thioethers47 were used for orthogonal protection of the two native cysteine residues during the desulfurization step.48
Execution of this strategy began with the solid-phase synthesis and RP-HPLC purification of each segment. Using optimized synthesis conditions, the N-terminal and middle segments could be obtained in excellent purity following HPLC purification (see supplementary information). However, the C-terminal segment was highly prone to aspartimide formation (Fig. 2).49 Poor solubility in any solvents other than highly denaturing 6 M guanidine hydrochloride (GuHCl) buffers prevented adequate removal of the aspartimide impurity by RP-HPLC. We explored adding a C-terminal solubilizing poly-lysine tag,50 which improved solubility and facilitated HPLC purification. Unfortunately, removal of the auxiliary following assembly of the full-length sequence proved to be problematic, requiring exposure to strong base (1 M NaOH) for several hours. Strategies involving combining the C-terminal and middle segment during SPPS, or splitting the C-terminal segment were also explored with limited success. Ultimately, the most effective solution was to directly suppress aspartimide formation during SPPS. Addition of 0.1 M Oxyma Pure to the Fmoc deprotection solution51 effectively prevented aspartimide formation, providing a sufficiently pure C-terminal segment that did not require additional HPLC purification (Fig. 2).
Figure 2.

Deconvoluted mass spectrum of the crude TNFα C-terminal segment (185-233) synthesized with (blue) and without (red) 0.1 M Oxyma Pure added to the Fmoc deprotection solution (20% piperidine).
At this point, protein assembly (Fig. 1B) was conducted in the C to N direction by first ligating the crude C-terminal segment (185-233) to the middle segment (145-184). This ligated product demonstrated improved handling properties compared to the C-terminal segment alone, allowing for high resolution HPLC purification using a Phenomenex C4 Jupiter column (5 μm, 10 × 250 mm) at 50°C. Following desulfurization of the non-native cysteine residue at position 185 and HPLC purification, the Acm groups were cleaved to reveal the native cysteine residues at 145 and 177. Finally, ligation with the N-terminal segment (77-144) provided our target sequence in a total of four steps with an overall yield of ∼2% (see supplemental information for yields at every step).
For synthesis of the d-protein, a biotin-PEG2 tag was incorporated on the N-terminus to enable immobilization on streptavidin-coated beads (for future application in mirror-image phage display). N-terminal biotinylation was selected due to the distal location from the receptor-binding interface and because other N-terminal tags have been well tolerated without impacting TNFα trimerization or biological activity.52
After assembling the linear sequence, the next challenge was developing disulfide oxidation and folding conditions to produce the native trimer. The refolding of guanidine-denatured TNFα has previously been described,53 but, to the best of our knowledge, there are no reported protocols for refolding fully reduced and denatured TNFα. Our initial attempts to refold a reduced recombinant sample by dialysis from a 6 M GuHCl buffer to non-denaturing aqueous buffers failed to produce any properly folded material. Successful folding of reduced and denatured TNFα was ultimately accomplished by first forming the disulfide bond under denaturing conditions (6 M GuHCl, 50 mM phosphate pH 8, and 2% DMSO as an oxidant) at <100 μM protein concentrations. Disulfide bond formation was monitored by a shift in HPLC retention (Fig. 3A, folding of biotin- d-TNFα), as well as a shift in the relative population of MS ion charge states (Fig. 3B) and a 2 Da shift in the deconvoluted mass (Fig. 3B). The oxidation was complete within 4 h, at which time folding was initiated by stepwise dialysis into 2 M and 0.5 M GuHCl (in 50 mM phosphate, pH 8) and finally into 50 mM ammonium bicarbonate. After dialysis, the properly folded trimeric protein was isolated by size exclusion chromatography (SEC) in 50 mM ammonium bicarbonate, pH 8. After folding and final SEC purification, approximately 10% of the synthetic material could be recovered. This low recovery is largely due to the poor efficiency of the folding step, which results in ∼30% recovery (starting from reduced and denatured recombinant TNFα). Further optimization of this folding procedure is likely possible, but these yields are adequate for phage display, which requires <50 μg of target protein.
Figure 3.

HPLC retention and MS charge states of reduced and oxidized synthetic biotin- d-TNFα. (A) The oxidation reaction was monitored by a shift in RP-HPLC retention (2 h time point). (B) Formation of the disulfide bond results in a shift to lower charge states and a 2 Da shift in the deconvoluted mass.
To evaluate our synthetic proteins, we first compared the l-enantiomer to recombinantly produced TNFα. Both synthetic and recombinant l-TNFα have identical HPLC retention times (Fig. 4A), and the synthetic protein has the expected molecular weight (Fig. 4B and 4C). Both elute as ∼50 kDa trimers by SEC (Fig. 4D) and show similar circular dichroism (CD) spectra (Fig. 4E). As a final validation, we tested our synthetic product in an L929 cell viability assay. In the presence of actinomycin D, murine L929 fibroblast cells are highly susceptible to TNFα-induced cytotoxicity.54, 55 In this assay, the chemically synthesized TNFα meets the expected level of activity (ED50 < 0.1 ng/mL) and is as potent as recombinant TNF-α (Fig. 5). The mirror-image d-TNFα is not capable of interacting with natural l-TNFR, preventing its biological validation. However, the d-protein has the expected mass (Fig. 3B) and elutes as a trimer by SEC (Fig. 4D). Furthermore, the biotin- d-TNFα had a matching, but inverse, CD spectrum compared to the synthetic and recombinant l-proteins (Fig. 4E).
Figure 4.
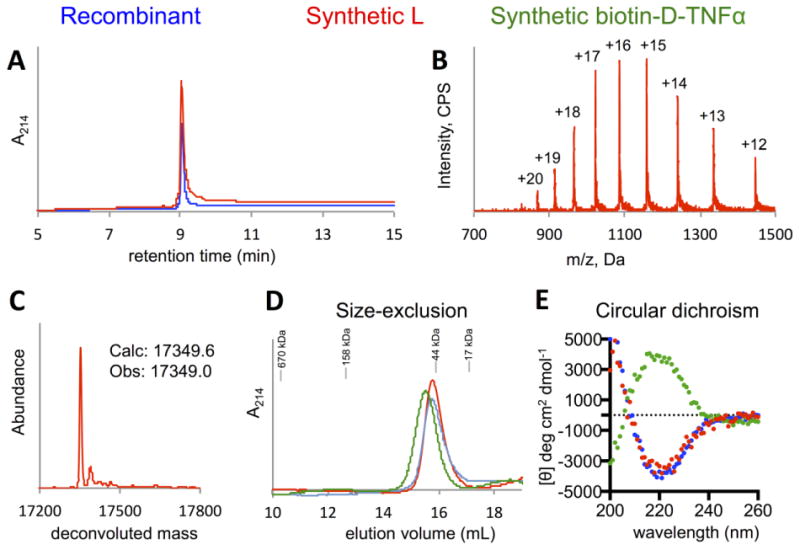
Biophysical characterization of folded synthetic l-TNFα (red) and biotin- d-TNFα (green). (A) RP-HPLC comparison of synthetic l-TNFα with recombinant TNFα (blue) shows that both proteins elute at ∼9.1 min (Phenomenex C4 Aeris column). (B) Mass spectrum of oxidized, folded, and SEC-purified synthetic l-TNFα. (C) Deconvoluted mass spectrum of synthetic l-TNFα matches the expected values. (D) Analytical SEC traces show that recombinant TNFα, synthetic l-TNFα, and synthetic biotin- d-TNFα show elution volumes on a Superdex 200 column consistent with trimeric protein. (E) Circular dichroism indicates a minimum at 218 nm and maximum around 200 nm consistent with the primarily β-sheet structure of TNFα (<3% α–helix). The mirror-image d-protein has an equivalent, but inverse, spectrum to the synthetic l- and recombinant proteins.
Figure 5.

L929 cells were treated for 20 h with a serial 3-fold dilution of either synthetic or recombinant l-TNFα, with a maximum dose of 10 ng/mL (measured by quantitative SDS-PAGE, see supplemental information). Cell viability was measured using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega) visualized at 490 nm, 4 h post treatment.
Conclusions
The overall goal of this project is to develop a d-peptide inhibitor of TNFα, an inflammatory cytokine involved in the pathogenesis of several chronic inflammatory diseases.2 Here we report the formative step towards achieving this goal, the chemical synthesis and biophysical validation of d-TNFα. A challenge encountered in this synthesis was the inability to use the native cysteine 177 in a ligation reaction with a proline thioester. Failure of this reaction required repositioning the ligation junction to alanine 185, which resulted in two additional handling steps, desulfurization and Acm cleavage. Through our optimized synthetic route, we produced a sufficient quantity (∼300 μg) of the natural l-enantiomer to confirm proper folding by SEC and CD and demonstrate biological activity in a cell viability assay. Finally, we synthesized a biotinylated version of d-TNFα, which was validated by HPLC, MS, SEC, and CD, and is suitable for use as a mirror-image phage display target.
The next phase of this project will be to employ the d-TNFα generated here to discover inhibitory d-peptides by mirror-image phage display. d-peptide TNFα inhibitors have the potential to overcome some of the challenges associated with existing anti-TNFα biologics and improve the quality of life for individuals with a chronic inflammatory disease. A particularly promising application of such inhibitors would be their use as an oral treatment for inflammatory bowel disease (IBD). In IBD, misrecognition of luminal material by the immune system leads to elevated levels of TNFα in the gut mucosa.56 Oral administration of a non-degradable, non-absorbable d-peptide could provide local suppression of TNFα at sites of gut inflammation and avoid the toxicities associated with systemic TNFα blockade. Such an approach has already been shown to be effective in a mouse IBD model using very high doses of a bovine anti-TNFα antibody.57 A d-peptide could provide a smaller and lower cost solution, with the potential for better gut tissue penetration and lower dosing. Also, the low immunogenicity of d-peptides could enable less toxic intermittent systemic dosing to treat acute flare-ups (not recommended with current agents due to increased risk of an anti-drug immune response),8 allowing the immune system to recover while the disease is in remission. In addition to current indications, a d-peptide could also open new possibilities for anti-TNF therapy, such as short-term or localized therapy (e.g., to treat acute inflammation resulting from traumatic brain or lung injury). Broad application of the techniques described here will also facilitate the use of mirror-image drug discovery for larger protein targets.
Supplementary Material
Acknowledgments
We would like to thank the Mathew Mulvey lab for providing the L929 cell line, Amanda Smith for assistance with tissue culture, and Debra Eckert for assistance with recombinant protein expression and critical review of the manuscript. Funding for this research was provided by the NIH Cardiovascular Training Grant T32 HL007576 (MEP), F31 CA171677 (MTJ), and R01 AI76168 (MSK).
Footnotes
Electronic supplementary information (ESI) available: Detailed description of the materials, experimental procedures, and analytical data. See DOI:
References
- 1.Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. Proc Natl Acad Sci U S A. 1975;72:3666–3670. doi: 10.1073/pnas.72.9.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feldmann M, Maini RN. Nat Med. 2003;9:1245–1250. doi: 10.1038/nm939. [DOI] [PubMed] [Google Scholar]
- 3.Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Pharm Therapeut. 2008;117:244–279. doi: 10.1016/j.pharmthera.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Wajant H, Pfizenmaier K, Scheurich P. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 5.Neurath MF. Nat Rev Immunol. 2014;14:329–342. doi: 10.1038/nri3661. [DOI] [PubMed] [Google Scholar]
- 6.Bradley JR. J Pathol. 2008;214:149–160. doi: 10.1002/path.2287. [DOI] [PubMed] [Google Scholar]
- 7.Doran MF, Crowson CS, Pond GR, O'Fallon WM, Gabriel SE. Arthritis & Rheumatism. 2002;46:2287–2293. doi: 10.1002/art.10524. [DOI] [PubMed] [Google Scholar]
- 8.van Schouwenburg PA, Rispens T, Wolbink GJ. Nat Rev Rheumatol. 2013;9:164–172. doi: 10.1038/nrrheum.2013.4. [DOI] [PubMed] [Google Scholar]
- 9.Alexiou P, Papakyriakou A, Ntougkos E, Papaneophytou CP, Liepouri F, Mettou A, Katsoulis I, Maranti A, Tsiliouka K, Strongilos A, Chaitidou S, Douni E, Kontopidis G, Kollias G, Couladouros E, Eliopoulos E. Archiv der Pharmazie. 2014;347:798–805. doi: 10.1002/ardp.201400198. [DOI] [PubMed] [Google Scholar]
- 10.He MM, Smith AS, Oslob JD, Flanagan WM, Braisted AC, Whitty A, Cancilla MT, Wang J, Lugovskoy AA, Yoburn JC, Fung AD, Farrington G, Eldredge JK, Day ES, Cruz LA, Cachero TG, Miller SK, Friedman JE, Choong IC, Cunningham BC. Science. 2005;310:1022–1025. doi: 10.1126/science.1116304. [DOI] [PubMed] [Google Scholar]
- 11.Ma L, Gong H, Zhu H, Ji Q, Su P, Liu P, Cao S, Yao J, Jiang L, Han M, Ma X, Xiong D, Luo HR, Wang F, Zhou J, Xu Y. J Biol Chem. 2014;289:12457–12466. doi: 10.1074/jbc.M113.521708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun H, Yost GS. Chemical Research in Toxicology. 2008;21:374–385. doi: 10.1021/tx700294g. [DOI] [PubMed] [Google Scholar]
- 13.Palladino MA, Bahjat FR, Theodorakis EA, Moldawer LL. Nat Rev Drug Discov. 2003;2:736–746. doi: 10.1038/nrd1175. [DOI] [PubMed] [Google Scholar]
- 14.Paul AT, Gohil VM, Bhutani KK. Drug Discovery Today. 2006;11:725–732. doi: 10.1016/j.drudis.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 15.Takasaki W, Kajino Y, Kajino K, Murali R, Greene MI. Nat Biotech. 1997;15:1266–1270. doi: 10.1038/nbt1197-1266. [DOI] [PubMed] [Google Scholar]
- 16.Jonsson A, Wallberg H, Herne N, Stahl S, Frejd FY. Biotechnol Appl Biochem. 2009;54:93–103. doi: 10.1042/BA20090085. [DOI] [PubMed] [Google Scholar]
- 17.Luzi S, Kondo Y, Bernard E, Stadler LKJ, Vaysburd M, Winter G, Holliger P. Prot Eng Des Sel. 2015;28:45–52. doi: 10.1093/protein/gzu055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Milton RCd, Milton SCF, Kent SBH. Science. 1992;256:1445–1448. doi: 10.1126/science.1604320. [DOI] [PubMed] [Google Scholar]
- 19.Dintzis HM, Symer DE, Dintzis RZ, Zawadzke LE, Berg JM. Proteins. 1993;16:306–308. doi: 10.1002/prot.340160309. [DOI] [PubMed] [Google Scholar]
- 20.Schumacher TNM, Mayr LM, M DL, Jr, Milhollen MA, Burgess MW, Kim PS. Science. 1996;271:1854–1857. doi: 10.1126/science.271.5257.1854. [DOI] [PubMed] [Google Scholar]
- 21.Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS. Cell. 1999;99:103–115. doi: 10.1016/s0092-8674(00)80066-5. [DOI] [PubMed] [Google Scholar]
- 22.Welch BD, Francis JN, Redman JS, Paul S, Weinstock MT, Reeves JD, Lie YS, Whitby FG, Eckert DM, Hill CP, Root MJ, Kay MS. J Virol. 2010;84:11235–11244. doi: 10.1128/JVI.01339-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. Proc Natl Acac sci USA. 2007;104:16828–16833. doi: 10.1073/pnas.0708109104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu M, Li C, Pazgier M, Li C, Mao Y, Lv Y, Gu B, Wei G, Yuan W, Zhan C, Lu WY, Lu W. Proc Natl Acad Sci. 2010;107:14321–14326. doi: 10.1073/pnas.1008930107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mandal K, Uppalapati M, Ault-Riché D, Kenney J, Lowitz J, Sidhu SS, Kent SBH. Proc Natl Acad Sci USA. 2012;109:14779–14784. doi: 10.1073/pnas.1210483109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uppalapati M, Lee DJ, Mandal K, Li H, Miranda LP, Lowitz J, Kenney J, Adams JJ, Ault-Riché D, Kent SBH, Sidhu SS. ACS Chem Biol. 2016;11:1058–1065. doi: 10.1021/acschembio.5b01006. [DOI] [PubMed] [Google Scholar]
- 27.Mandal K, Kent SBH. Angew Chem Int Ed. 2011;50:8029–8033. doi: 10.1002/anie.201103237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao L, Lu W. Isr J Chem. 2011;51:868–875. [Google Scholar]
- 29.Dawson PE, Muir TW, Clark-Lewis l, SBH Kent. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 30.Kent SB. Chem Soc Rev. 2009;38:338–351. doi: 10.1039/b700141j. [DOI] [PubMed] [Google Scholar]
- 31.Pentelute BL, Gates ZP, Dashnau JL, Vanderkooi JM, Kent SBH. J Am Chem Soc. 2008;130:9702–9707. doi: 10.1021/ja801352j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang P, Dong S, Shieh JH, Peguero E, Hendrickson R, Moore MA, Danishefsky SJ. Science. 2013;342:1357–1360. doi: 10.1126/science.1245095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Torbeev VY, Kent SB. Angew Chem Int Ed Engl. 2007;46:1667–1670. doi: 10.1002/anie.200604087. [DOI] [PubMed] [Google Scholar]
- 34.Haj-Yahya M, Fauvet B, Herman-Bachinsky Y, Hejjaoui M, Bavikar SN, Karthikeyan SV, Ciechanover A, Lashuel HA, Brik A. Proc Natl Acad Sci USA. 2013;110:17726–17731. doi: 10.1073/pnas.1315654110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bondalapati S, Jbara M, Brik A. Nat Chem. 2016;8:407–418. doi: 10.1038/nchem.2476. [DOI] [PubMed] [Google Scholar]
- 36.Dawson PE, Kent SBH. Annu Rev Biochem. 2000;69:923–960. doi: 10.1146/annurev.biochem.69.1.923. [DOI] [PubMed] [Google Scholar]
- 37.Malins LR, Payne RJ. Curr Opin Chem Biol. 2014;22:70–78. doi: 10.1016/j.cbpa.2014.09.021. [DOI] [PubMed] [Google Scholar]
- 38.Weinstock MT, Jacobsen MT, Kay MS. Proc Natl Acac sci USA. 2014;111:11679–11684. doi: 10.1073/pnas.1410900111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fang GM, Li YM, Shen F, Huang YC, Li JB, Lin Y, Cui HK, Liu L. Angew Chem Int Ed. 2011;50:7645–7649. doi: 10.1002/anie.201100996. [DOI] [PubMed] [Google Scholar]
- 40.Fang GM, Wang JX, Liu L. Angew Chem Int Ed. 2012;51:10347–10350. doi: 10.1002/anie.201203843. [DOI] [PubMed] [Google Scholar]
- 41.Pollock SB, Kent SBH. Chemical Communications. 2011;47:2342–2344. doi: 10.1039/c0cc04120c. [DOI] [PubMed] [Google Scholar]
- 42.Hackeng TM, Griffin JH, Dawson PE. Proc Natl Acad Sci USA. 1999;96:10068–10073. doi: 10.1073/pnas.96.18.10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakamura T, Shigenaga A, Sato K, Tsuda Y, Sakamoto K, Otaka A. Chem Commun. 2014;50:58–60. doi: 10.1039/c3cc47228k. [DOI] [PubMed] [Google Scholar]
- 44.Raibaut L, Seeberger P, Melnyk O. Org Lett. 2013;15:5516–5519. doi: 10.1021/ol402678a. [DOI] [PubMed] [Google Scholar]
- 45.Yan LZ, Dawson PE. J Am Chem Soc. 2001;123:526–533. doi: 10.1021/ja003265m. [DOI] [PubMed] [Google Scholar]
- 46.Wan Q, Danishefsky SJ. Angew Chem Int Ed. 2007;46:9248–9252. doi: 10.1002/anie.200704195. [DOI] [PubMed] [Google Scholar]
- 47.Veber D, Milkowski J, Varga S, Denkewalter R, Hirschmann R. J Am Chem Soc. 1972;94:5456–5461. doi: 10.1021/ja00770a600. [DOI] [PubMed] [Google Scholar]
- 48.Pentelute BL, Kent SBH. Org Lett. 2007;9:687–690. doi: 10.1021/ol0630144. [DOI] [PubMed] [Google Scholar]
- 49.Mergler M, Dick F, Sax B, Stahelin C, Vorherr T. J Pept Sci. 2003;9:518–526. doi: 10.1002/psc.473. [DOI] [PubMed] [Google Scholar]
- 50.Hossain MA, Belgi A, Lin F, Zhang S, Shabanpoor F, Chan L, Belyea C, Truong HT, Blair AR, Andrikopoulos S, Tregear GW, Wade JD. Bioconjugate Chem. 2009;20:1390–1396. doi: 10.1021/bc900181a. [DOI] [PubMed] [Google Scholar]
- 51.Subirós-Funosas R, El-Faham A, Albericio F. J Pept Sci. 2012;98:89–97. doi: 10.1002/bip.21713. [DOI] [PubMed] [Google Scholar]
- 52.Curnis F, Sacchi A, Borgna L, Magni F, Gasparri A, Corti A. Nat Biotech. 2000;18:1185–1190. doi: 10.1038/81183. [DOI] [PubMed] [Google Scholar]
- 53.Hlodan R, Pain RH. Eur J Biochem. 1995;231:381–387. doi: 10.1111/j.1432-1033.1995.tb20710.x. [DOI] [PubMed] [Google Scholar]
- 54.Promega Corporation. Tumor Necrosis Factor-α, Human, Recombinant, Part #9PIG524. http://www.promega.com/∼/media/files/resources/protocols/productinformation sheets/g/tumor necrosisfactoralphahumanrecombinantprotocol.pdf)
- 55.Shultz S, Niles A, Cheng J, Allard STM. Promega Notes Number 100. 2008 [Google Scholar]
- 56.Hanauer SB. Inflamm Bowel Dis. 2006;12 Suppl 1:S3–9. doi: 10.1097/01.mib.0000195385.19268.68. [DOI] [PubMed] [Google Scholar]
- 57.Bhol KC, Tracey DE, Lemos BR, Lyng GD, Erlich EC, Keane DM, Quesenberry MS, Holdorf AD, Schlehuber LD, Clark SA, Fox BS. Inflamm Bowel Dis. 2013;19:2273–2281. doi: 10.1097/MIB.0b013e3182a11958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.