

Abstract
Carbaboranes are increasingly studied as pharmacophores, particularly as replacements for aromatic systems. However, especially ortho-carbaborane is prone to degradation of the cluster, which hampers biological application. This study demonstrates that deboronation of the cluster may not only lead to a more active analogue but can also improve the solubility and stability of a carbaborane-containing inhibitor. Notably, introduction of a nido-dicarbaborate cluster into the cyclooxygenase (COX) inhibitor indomethacin results in a remarkably increased inhibitory potency and selectivity for COX-2 relative to the respective phenyl analogue. The first crystal structure of a carbaborane-containing inhibitor bound to COX-2 further reveals a novel binding mode for the inhibitor that is strikingly different from that of indomethacin. These results indicate that nido-dicarbaborate is a promising pharmacophore exhibiting properties which are also highly beneficial for its introduction into other inhibitor classes.
Keywords: carboranes, cyclooxygenases, drug design, enzyme inhibitor selectivity, inhibitors
Graphical abstract

Potency Boost: Replacement of a phenyl moiety in indomethacin with a nido-dicarbaborate results in a novel binding mode with markedly increased inhibitory activity and selectivity for COX-2, with a concomitant increase in stability and water solubility of the inhibitor while still ensuring strong hydrophobic interactions in the binding site of the enzyme. This shows nido-dicarbaborate to be a promising pharmacophore for a variety of inhibitors.
Carbaboranes are icosahedral boron clusters with extraordinary properties that have attracted increasing interest among medicinal chemists. Besides their proposed applications as boron delivery agents in boron neutron capture therapy (BNCT),[1] they have been the focus of pronounced attention as pharmacophores in recent years.[1c,d,2] The three isomers, 1,2-, 1,7-, and 1,12-dicarba-closo-dodecaborane(12) (i.e., ortho-, meta-, and para-carbaborane), serve as three-dimensional analogues of aromatic hydrocarbons, as their cluster electrons are delocalized, but they occupy a slightly larger volume than a rotating phenyl ring. Furthermore, compared to other pharmacophores, they offer several advantages, including a remarkably high hydrophobicity, nontoxicity, high metabolic stability, and a predominantly hydridic periphery, which enables unconventional proton–hydride interactions with biomolecules (dihydrogen bonds).[1c,2,3] Notably, the decapped anionic nido-dicarbaundecaborates have not aroused as much attention as their closo counterparts, from which they are formed by deboronation (cluster decapping). Thus far, they have been primarily applied in potential BNCT agents to achieve water solubility.[1b–d] The closely related metallacarbaboranes have been successfully used for radiolabeling and for the preparation of potent HIV protease inhibitors.[4] However, the potential of nido-dicarbaborates as individual pharmacophores has only been studied recently. Although their introduction into inhibitors has yielded promising bio-active compounds, these nido-carbaboranyl derivatives have not yet proved to be superior to the respective phenyl or adamantyl analogues.[5]
We recently reported unique structure–activity relationships when carbaboranes were used to replace phenyl rings in established cyclooxygenase (COX) inhibitors.[6] The pronounced isomer-dependent COX inhibitory potency of carbaborane-containing, indole-based inhibitors has not been described for any other class of inhibitors to date. Whereas the introduction of ortho-carbaborane yields active inhibitors, the use of meta- or para-carbaborane results in suppression of the COX inhibitory potency. In general, meta- and para-carbaborane are more stable towards decapping than the ortho isomer. The aim of this study was therefore to investigate the impact of cluster deboronation on the potency of carbaborane-containing COX inhibitors.
COX, an enzyme that catalyzes the key steps in the biosynthesis of prostanoids, exists in two isoforms.[7] Whereas COX-1 is constitutively expressed and primarily involved in normal physiological processes, inducible COX-2 is mainly associated with pathological functions such as inflammation and tumorigenesis. Both isozymes are homodimeric, integral membrane proteins sharing a similar structure and high sequence identity.[8] COX inhibitors are among the most used medications, primarily for the treatment of inflammatory diseases. The well-known nonsteroidal anti-inflammatory drugs (NSAIDs; e.g., aspirin, ibuprofen) are generally inhibitors of both COX isozymes. However, the inhibition of COX-1 leads to side effects, particularly in the gastrointestinal tract (bleeding, ulcers). This motivated the development of COX-2-selective inhibitors (COXIBs; e.g., celecoxib, rofecoxib) which exhibit a good anti-inflammatory potency with reduced gastrointestinal toxicity. However, cardiovascular toxicity resulting from long-term use of these drugs led to withdrawal of some of them from the market. Despite these concerns, COXIBs are promising antitumor medications and probes for in vivo imaging technologies, as COX-2 is overexpressed in various tumors.[7] Thus, the development of COXIBs with fewer side effects is still of significant interest. Although diaryl heterocycles have been studied intensively for COX-2 inhibition, only a few of the established NSAIDs, such as indomethacin, have been used as templates for the design of COXIBs.
Indomethacin (Scheme 1) is a potent, COX-1-selective inhibitor, mainly used for relief of pain associated with arthritis, but causes severe side effects. Directed derivatization of the molecule by conversion of the aryl acetic acid into neutral ester or amide analogues confers COX-2 selectivity.[9] Similarly, a slight enlargement of the 4-chlorobenzoyl ring by replacement with a 2,4,6-trichlorobenzoyl or 4-bromobenzyl group produces potent COX-2-selective inhibitors.[10] The selectivity shifts arise from substitutions in the amino acid sequences of the COX isoforms resulting in a larger and more flexible binding site in the case of COX-2.[8,9a] However, modifications at the benzoyl ring of indomethacin only allow a two-dimensional expansion of the moiety. In contrast, carbaboranes provide the possibility of a three-dimensional extension.
Scheme 1.

We have already shown that the replacement of the 4-chlorophenyl ring in indomethacin by an ortho-carbaborane generates highly potent and selective COX-2 inhibitor 1 (Scheme 1).[6c] However, the amide bond in indomethacin is rather labile and becomes more prone to hydrolysis on introduction of the carbaborane in 1, due to the high electron deficiency of the cluster. In turn, the electron-withdrawing effect of the carbonyl group adjacent to the carbaborane also increases the lability of the cluster,[11] which results in rapid deboronation of 1 in polar solvents such as methanol (see Supporting Information). Furthermore, 1 exhibits very low water solubility due to the high hydrophobicity of the carbaborane cluster, another property which may hamper medical application. Thus, the anionic 7,8-nido-dicarbaborate was introduced into indomethacin in place of the 4-chlorophenyl ring. As esterification of indomethacin was reported to confer COX-2 selectivity,[9] the respective methyl ester was used, with the expectectation of a synergistic effect of both moieties in 2 on the selectivity for COX-2 (Scheme 1). The anionic character of nido-dicarbaborate and the concomitant higher electron density of the cluster should furthermore stabilize the derivative against hydrolysis of the amide bond and degradation of the cluster. 2 was synthesized by controlled deboronation of the closo derivative 1. Due to the generally high reactivity of nido-dicarbaboranes, the hydronium ion of the respective conjugated acid was exchanged by a sodium ion. As expected, in contrast to 1, compound 2 was stable towards degradation of the cluster, and the amide bond proved to be even more stable towards hydrolysis than that of indomethacin (see Supporting Information). Besides stabilizing the compound, the nido cluster also led to high water solubility. Complete stabilization of the linkage between the indole and dicarbaborate can be achieved by replacing the amide bond with a methylene bridge. Therefore, congener 3 (Scheme 1) was synthesized to study the importance of the carbonyl group for binding to COX.[12] Both nido-dicarbaborato derivatives were tested for inhibition of COX-1 and COX-2 (Table 1).
Table 1.
IC50 values [μM] of different indomethacin derivatives for the inhibition of COX-1 and COX-2.
| Compound | COX-1[a] | COX-2[b] |
|---|---|---|
| indomethacin | 0.05[c] | 0.75[c] |
| indomethacin methyl ester | 33[c] | 0.25[c] |
| 1[d] | >4 | 0.072 |
| 2[e] | >4 | 0.051 |
| 3[e] | >4 | >4 |
| SC-299 | >66[c] | 0.060[c] |
Ovine COX-1.
Murine COX-2.
IC50 values as reported previously.[9b]
A stock solution of 1 was prepared in ethanol to prevent deboronation during the inhibition assay, whereas all other compounds were dissolved in methanol (see Supporting Information).
The nido-dicarbaborato derivatives were applied as racemates. The values are the mean of duplicates with standard deviation less than 10%.
Compound 2 is a highly potent inhibitor with a clearly increased selectivity for COX-2 compared to indomethacin. Although the selectivity is similar to that of the methyl ester of indomethacin, the potency is significantly enhanced by introduction of the nido-dicarbaborate. Its COX-2 inhibitory activity in the nanomolar range is comparable to that of SC-299, a well-characterized COXIB based on a diaryl oxazole (Scheme 1, Table 1).[13] To the best of our knowledge, this is the first nido-dicarbaborate-containing inhibitor which shows such a significantly increased potency relative to its phenyl analogue.
In contrast to 2, the methylene-bridged congener 3 did not show inhibition of COX (Table 1). This demonstrates the importance of the carbonyl group for binding to the enzyme. A corresponding interaction was confirmed by a crystal structure of COX-2 complexed with 2 (Figure 1). 2 was found to bind in the hydrophobic channel of the COX binding site in a completely different orientation compared to indomethacin.[14] The indole moiety of 2 resides in a similar position in the COX-2 channel to that of indomethacin, but in a flipped orientation. Whereas the carboxyl group of indomethacin forms a salt bridge with an arginine residue (Arg-120) at the entrance of the binding pocket, the ester group of 2 interacts with a serine residue (Ser-530) in the upper part of the channel. In the case of indomethacin, Ser-530 is involved in a hydrogen-bonding interaction with the carbonyl group linking the indole and 4-chlorophenyl ring. The respective carbonyl group in 2 instead forms polar interactions with Arg-120, which seem to be critical for the activity of the inhibitor (cf. 3). Furthermore, whereas the 4-chlorophenyl ring of indomethacin is bound in the apex of the binding channel (near the catalytic Tyr-385), the corresponding nido-dicarbaborate moiety of 2 protrudes into a hydrophobic subpocket, which is specifically opened up in this structure by rotation of a leucine residue (Leu-531). As the resolution of the structure (2.29 Å) does not provide the detailed location of the carbon and boron atoms within the cluster, an occupancy of 0.5 for each enantiomer was modeled into the final structure. However, docking studies predict a higher binding probability for the S enantiomer and suggest that the planar chirality of the nido-dicarbaborate may result in different binding affinities for the enantiomers (see Supporting Information).
Figure 1.
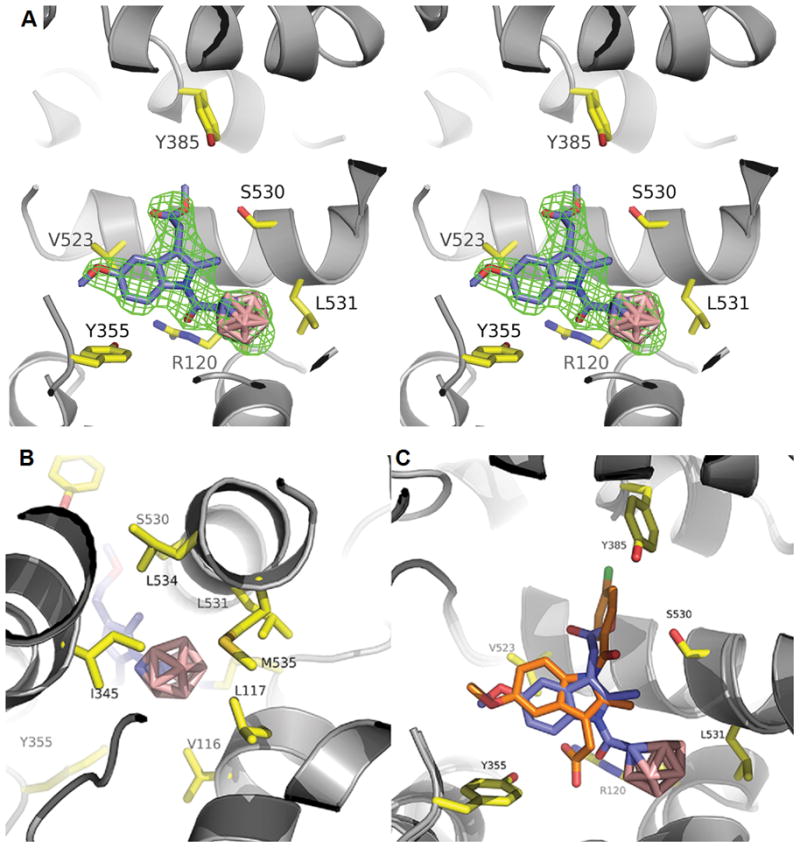
Binding modes of 2 and indomethacin in the COX-2 active site. A) COX-2 complexed with 2 (PDB ID: 4Z0L); stereo diagram; superposition of the R and S enantiomers; simulated annealing omit map [Fo–Fc] countered at 3σ in green mesh; key residues shown in yellow-stick representation. B) COX-2 complexed with 2; binding of the nido-dicarbaborate cluster in the hydrophobic subpocket; only the S enantiomer is shown. C) Superposition of the structures of 2 (blue; S enantiomer) and indomethacin (orange; PDB ID: 4COX; key residues not illustrated) in COX-2.
The binding mode of COX-2-selective ester and amide derivatives of indomethacin was shown to be analogous to that of indomethacin, and projection of the ester or amide group through the constriction at the entrance of the binding site was proposed to account for their COX-2 selectivity.[9,15] Thus, the novel binding mode of 2 suggests that both inhibitory potency and even COX-2 selectivity may arise from the nido-dicarbaborate moiety.
Compared to closo derivative 1, compound 2 exhibits a slightly increased COX inhibitory activity (Table 1). However, the rapid decapping of 1 hampers medical application and impedes biological evaluation. Therefore, the inhibition of COX by 1 was correlated with the deboronation of the compound (see Supporting Information). Whereas decapping of the cluster led to an increase in inhibitory activity, further degradation involving cleavage of the amide bond resulted in reduction of the potency. This trend confirmed 2 as the most active species of the carbaboranyl derivatives.
In conclusion, esterification and introduction of nido-dicarbaborate converts indomethacin to a highly potent and selective inhibitor of COX-2. The nido-dicarbaborate cluster leads to a remarkable increase in the inhibitory potency relative to the phenyl analogue. It also results in a novel binding mode of the inhibitor in the COX-2 active site with concomitant opening of a hydrophobic subpocket.
In contrast to its ortho-carbaboranyl congener, the nido-dicarbaborato indole exhibits high water solubility and stability towards degradation. The studies also indicate that deboronation of carbaborane-containing inhibitors may result in the formation of more potent species. As hydrophobicity of inhibitors is often important for affine binding but frequently accompanied by low solubility, the nido-dicarbaborate offers a promising pharmacophore for a variety of inhibitor classes.
Supplementary Material
Footnotes
This work was supported by the Fonds der Chemischen Industrie (doctoral grant for W.N.), the Graduate School “Building with Molecules and Nano-objects (BuildMoNa)” funded by the Deutsche Forschungsgemeinschaft, the US National Institutes of Health (CA89450), the German Academic Exchange Service (PPP USA), the European Union, and the Free State of Saxony (ESF-NFG 100148835). Part of the research was conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamline, which is supported by a grant from the National Institute of General Medical Sciences (P41 GM103403) of the National Institutes of Health. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. The authors thank Prof. Dr. Terry P. Lybrand for valuable discussions, Markus Hiller and Carlotta Taddei for experimental support, and Dr. Carol Rouzer for editorial assistance.
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Dr. Wilma Neumann, Institut für Anorganische Chemie, Universität Leipzig, Johannisallee 29, 04103 Leipzig (Germany)
Dr. Shu Xu, Department of Biochemistry, Vanderbilt University, School of Medicine, 23rdAvenue South & Pierce, Nashville, TN 37232 (USA)
Dr. Menyhárt B. Sárosi, Institut für Anorganische Chemie, Universität Leipzig, Johannisallee 29, 04103 Leipzig (Germany)
Dr. Matthias S. Scholz, Institut für Anorganische Chemie, Universität Leipzig, Johannisallee 29, 04103 Leipzig (Germany). (new address) Pharmazeutisches Institut, Rheinische Friedrich-Wilhelms-Universität Bonn, An der Immenburg 4, 53121 Bonn (Germany)
Brenda C. Crews, Department of Biochemistry, Vanderbilt University, School of Medicine, 23rdAvenue South & Pierce, Nashville, TN 37232 (USA)
Kebreab Ghebreselasie, Department of Biochemistry, Vanderbilt University, School of Medicine, 23rdAvenue South & Pierce, Nashville, TN 37232 (USA).
Dr. Surajit Banerjee, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14853 (USA). Northeastern Collaborative Access Team, Argonne National Laboratory, Argonne, IL 60439 (USA)
Prof. Dr. Lawrence J. Marnett, Department of Biochemistry, Vanderbilt University, School of Medicine, 23rdAvenue South & Pierce, Nashville, TN 37232 (USA)
Prof. Dr. Dr. h.c. Evamarie Hey-Hawkins, Institut für Anorganische Chemie, Universität Leipzig, Johannisallee 29, 04103 Leipzig (Germany)
References
- 1.a) Hawthorne MF. Angew Chem Int Ed. 1993;32:950. [Google Scholar]; Angew Chem. 1993;105:997. [Google Scholar]; b) Soloway AH, Tjarks W, Barnum BA, Rong FG, Barth RF, Codogni IM, Wilson JG. Chem Rev. 1998;98:1515. doi: 10.1021/cr980493e. [DOI] [PubMed] [Google Scholar]; c) Valliant JF, Guenther KJ, King AS, Morel P, Schaffer P, Sogbein OO, Stephenson KA. Coord Chem Rev. 2002;232:173. [Google Scholar]; d) Hosmane NS, editor. Boron Science: New Technologies and Applications. CRC Press; Boca Raton: 2011. [Google Scholar]
- 2.a) Issa F, Kassiou M, Rendina LM. Chem Rev. 2011;111:5701. doi: 10.1021/cr2000866. [DOI] [PubMed] [Google Scholar]; b) Scholz M, Hey-Hawkins E. Chem Rev. 2011;111:7035. doi: 10.1021/cr200038x. [DOI] [PubMed] [Google Scholar]
- 3.Fanfrlík J, Lepšík M, Horinek D, Havlas Z, Hobza P. ChemPhysChem. 2006;7:1100. doi: 10.1002/cphc.200500648. [DOI] [PubMed] [Google Scholar]
- 4.Řezáčoá P, Cígler P, Matĕjíček P, Lepšík M, Pokorná J, Grüner B, Konvalinka J. In: Boron Science: New Technologies and Applications. Hosmane NS, editor. CRC Press; Boca Raton: 2011. p. 41. [Google Scholar]
- 5.a) Reynolds RC, Campbell SR, Fairchild RG, Kisliuk RL, Micca PL, Queener SF, Riordan JM, Sedwick WD, Waud WR, Leung AKW, Dixon RW, Suling WJ, Borhani DW. J Med Chem. 2007;50:3283. doi: 10.1021/jm0701977. [DOI] [PubMed] [Google Scholar]; b) Brynda J, Mader P, Šícha V, Fábry M, Poncová K, Bakardiev M, Grüner B, Cígler P, Řezáčová P. Angew Chem Int Ed. 2013;52:13760. doi: 10.1002/anie.201307583. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2013;125:14005. [Google Scholar]; c) Wilkinson SM, Gunosewoyo H, Barron ML, Boucher A, McDonnell M, Turner P, Morrison DE, Bennett MR, McGregor IS, Rendina LM, Kassiou M. ACS Chem Neurosci. 2014;5:335. doi: 10.1021/cn500054n. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) El-Zaria ME, Genady AR, Janzen N, Petlura CI, Beckford Vera DR, Valliant JF. Dalton Trans. 2014;43:4950. doi: 10.1039/c3dt53189a. [DOI] [PubMed] [Google Scholar]
- 6.a) Scholz M, Bensdorf K, Gust R, Hey-Hawkins E. ChemMedChem. 2009;4:746. doi: 10.1002/cmdc.200900072. [DOI] [PubMed] [Google Scholar]; b) Scholz M, Blobaum AL, Marnett LJ, Hey-Hawkins E. Bioorg Med Chem. 2011;19:3242. doi: 10.1016/j.bmc.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Scholz M, Blobaum AL, Marnett LJ, Hey-Hawkins E. Bioorg Med Chem. 2012;20:4830. doi: 10.1016/j.bmc.2012.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Laube M, Neumann W, Scholz M, Lönnecke P, Crews B, Marnett LJ, Pietzsch J, Kniess T, Hey-Hawkins E. ChemMedChem. 2013;8:329. doi: 10.1002/cmdc.201200455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simmons DL, Botting RM, Hla T. Pharmacol Rev. 2005;56:387. doi: 10.1124/pr.56.3.3. [DOI] [PubMed] [Google Scholar]
- 8.Michaux C, Charlier C. Mini-Rev Med Chem. 2004;4:603. doi: 10.2174/1389557043403756. [DOI] [PubMed] [Google Scholar]
- 9.a) Kalgutkar AS, Crews BC, Rowlinson SW, Marnett AB, Kozak KR, Remmel RP, Marnett LJ. Proc Natl Acad Sci USA. 2000;97:925. doi: 10.1073/pnas.97.2.925. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kalgutkar AS, Marnett AB, Crews BC, Remmel RP, Marnett LJ. J Med Chem. 2000;43:2860. doi: 10.1021/jm000004e. [DOI] [PubMed] [Google Scholar]
- 10.Black WC, Bayly C, Belley M, Chan CC, Charleson S, Denis D, Gauthier JY, Gordon R, Guay D, Kargman S, Lau CK, Leblanc Y, Mancini J, Ouellet M, Percival D, Roy P, Skorey K, Tagari P, Vickers P, Wong E, Xu L, Prasit P. Bioorg Med Chem Lett. 1996;6:725. [Google Scholar]
- 11.Schaeck JJ, Kahl SB. Inorg Chem. 1999;38:204. [Google Scholar]
- 12.Neumann W, Frank R, Hey-Hawkins E. Dalton Trans. 2015;44:1748. doi: 10.1039/c4dt03218g. [DOI] [PubMed] [Google Scholar]
- 13.Talley JJ, Bertenshaw SR, Brown DL, Carter JS, Graneto MJ, Koboldt CM, Masferrer JL, Norman BH, Rogier DJ, Jr, Zweifel BS, Seibert K. Med Res Rev. 1999;19:199. doi: 10.1002/(sici)1098-1128(199905)19:3<199::aid-med1>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 14.Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, Gildehaus D, Miyashiro JM, Penning TD, Seibert K, Isakson PC, Stallings CW. Nature. 1996;384:644. doi: 10.1038/384644a0. [DOI] [PubMed] [Google Scholar]
- 15.Harman CA, Turman MV, Kozak KR, Marnett LJ, Smith WL, Garavito RM. J Biol Chem. 2007;282:28096. doi: 10.1074/jbc.M701335200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.