

Abstract
A novel, mixed-ligand chiral rhodium(II) catalyst, Rh2(S-NTTL)3(dCPA), has enabled the first enantioselective total synthesis of the natural product piperarborenine B. A crystal structure of Rh2(S-NTTL)3(dCPA) reveals a “chiral crown” conformation with a bulky dicyclohexylphenyl acetate ligand and three N-naphthalimido groups oriented on the same face of the catalyst. The natural product was prepared on large scale using rhodium-catalyzed bicyclobutanation/copper-catalyzed homoconjugate addition chemistry in the key step. The route proceeds in ten steps with an 8% overall yield and 92% ee.
Keywords: chiral rhodium catalyst, bicyclobutane, strained molecule, homoconjugate addition, piperarborenine B
Graphical abstract
Mixing Ligands: A new mixed-ligand chiral rhodium(II) catalyst, Rh2(S-NTTL)3(dCPA), has enabled the first enantioselective total synthesis of the natural product piperarborenine B. Key to the synthesis is a rhodium-catalyzed bicyclobutanation/copper-catalyzed homoconjugate addition for complex cyclobutane synthesis. The natural product was prepared on 400 mg scale and 8% overall yield.
Truxillate and truxinate natural products exhibit diverse biological activities. These molecules and their derivatives display potential as therapeutic leads in areas including oncology, neurology, infectious diseases, and respiratory diseases.[1] Despite the significant number of natural lead compounds, the underrepresentation[2] of cyclobutane-containing drugs is a reflection of an area in organic chemistry where concise, general and stereoselective methods for cyclobutane synthesis are still needed.
Piperarborenine B, a cyclobutane-containing natural product isolated from Piper arborescens, has exhibited activity against P-388, A-549, and HT-29 cancer cell lines (IC50 = 0.13, 1.4, and 2.4 μM, respectively).[3] Other cyclobutane-containing natural products such as incarvillateine,[4][5] pauferrol A[1d] and the synthetic SB-FI-26[6] have also exhibited exciting biological activities, with the cyclobutane core proving to be an essential feature.
There are several ways to access truxillate and truxinate natural products, but most are focused on the preparation of compounds with symmetrical cyclobutane cores. The photochemical [2+2] reaction has factored significantly in the total synthesis of symmetrical truxillic acid and related derivatives,[7] including elegant solutions from crystal engineering[8] and advances from redox photocatalysis.[9] Alternatively, truxinic acid derivatives have been formed in modest diastereoselectivity by [2+2] photocycloaddition in flow.[10] However, direct photodimerization is extremely limited for the construction of cyclobutane natural products where the core is chiral and the alkene precursors are non-equivalent such as the compounds displayed in Figure 1.[8b]
Figure 1.

Cyclobutane-containing natural products and drug leads.
In recent years, catalytic C–H activation methods have factored prominently in the elaboration of cyclobutane natural products.[11] Recently, Baran has shown that racemic tri- and tetrasubstituted cyclobutanes can be prepared from disubstituted cyclobutanes through sequential arylation and vinylation reactions,[12] using directing groups and catalytic conditions originally developed by Daugulis[11b, 13] and also popularized by Chen.[14] Baran’s synthesis of piperarborenine B showcased sequential cyclobutane C(sp3)–H activation to construct the unsymmetrical truxillate core. This approach provided racemic piperarborenine B in 7% overall yield in 7 steps.
Several enantioselective approaches to unsymmetrical cyclobutane cores have been described in recent years.[15–20] Tang has reported the synthesis of the initially assigned structures of pipercyclobutanamide A and piperchabamide G via Rh(I)-catalyzed conjugate addition of arylboronic acid to a cyclobutenoate derived from enantiospecific cyclopropane ring expansion.[15] Rh2(5S-MEPY)4-catalyzed cyclopropanation was used to set the stereochemistry of the core in 80% ee. This study revised the assignments to the six-membered ring isomers chabamide and nigramide F, respectively.
Recently, our group has introduced a modular new approach to chiral cyclobutane synthesis involving tandem Rh(II)-catalyzed enantioselective ‘bicyclobutanation’[21]/Cu(I)-catalyzed homoconjugate addition of α-cinnamyl-α-diazoesters (Scheme 1).[22, 23] Inspired by the unique bioactivities of a number of cyclobutane-containing natural products, we sought to demonstrate the power of our strategy for enantioselective synthesis of unsymmetrical cyclobutanes. Additionally, we realized this would potentially enable rapid diversification of complex cyclobutanes for SAR studies. Herein, we report the first enantioselective total synthesis of piperarborenine B (1). Critical to the synthesis was the development of a new, chiral Rh(II)-carboxylate complex that catalyzes the key bicyclobutanation step with high enantioselectivity and turnover number. Additional key aspects of the synthesis include improved ‘on water’ conditions for cyclobutane C–H activation and a streamlined route that was used to produce the natural product on 400 mg scale in only one week from veratraldehyde.
Scheme 1.

Bicyclobutanation/homoconjugate addition strategy for enantioselective cyclobutane synthesis.
Retrosynthetically, we envisioned the α-truxillate core of 7 arising from a Baran late stage C–H activation using a properly installed directing group. The stereotriad 5 would be constructed using our previously developed enantioselective bicyclobutanation/homoconjugate addition chemistry. The required diazoester 3 would arise from commercially available veratraldehyde using a Tsuji-Trost allylation in a key step.
The synthesis commenced by developing a synthesis of the diazoester (3). Several efficient routes to the compound were developed (see supporting information). In the most time and scale efficient route (Scheme 3), veratraldehyde was treated with vinylMgBr to provide a vinyl alcohol that was shown to be an effective substrate for an ‘on water’ Tsuji-Trost allylation of tert-butyl acetoacetate using Pd(OAc)2 (2 mol %), PPh3 (10 mol %), and adamantoic acid (10 mol %).[24] Treatment of ketoester 2 with p-ABSA, and NaOH in a mixture of acetonitrile/H2O provided the desired diazoester 3 in 47% yield over three steps. This three step process for diazoester synthesis was accomplished in one day and represents an efficient new strategy for α-allyl-α-diazoester synthesis.
Scheme 3.

An efficient route to the substituted diazoester, 3.
Recently, our group[22] and that of Davies[21] have described complementary catalyst systems for enantioselective bicyclobutanation. Our system provides access to tert-butyl bicyclobutane carboxylates that can be combined in situ with Grignard reagents to produce functionalized, enantiomerically enriched cyclobutanes.[22] In our previous work, we described Rh2(S-NTTL)4 as an effective catalyst for bicyclobutanation in terms of enantioselectivity and yield. However, our efforts to apply this catalyst produced the bicyclobutane intermediate 4 in only 84% ee (Scheme 4). Screening efforts with other symmetrical catalysts were unsuccessful (see SI for full optimization table).
Scheme 4.

Rh(II)-catalyst screening and enantioselective synthesis of vinylcyclobutane, 5.
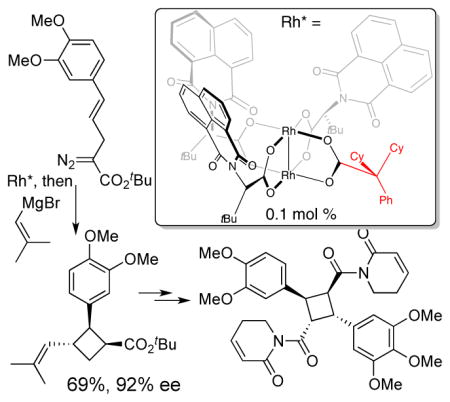
Recently, our group described the mixed-ligand Rh(II)-catalyst, Rh2(S-PTTL)3(TPA), a dirhodium paddlewheel complex that is substituted by three chiral ligands and one achiral ligand.[25] This catalyst displayed superior results in certain enantioselective cyclopropanation, cyclopropenation and indole functionalization reactions. Our computational and crystallographic studies showed that it adopts a ‘chiral crown’ conformation, where the N-phthalimido groups are all presented on the same face. In the bicyclobutanation of 3, Rh2(S-PTTL)3(TPA) gave the bicyclobutane 4 in a promising 60% ee. We speculated that replacing TPA with a ligand that makes non-covalent contacts between the chiral ligand and substrate may be beneficial to asymmetric induction. We further speculated that a large achiral ligand may rigidify the chiral crown arrangement of the chiral ligands. Accordingly we designed a new mixed-ligand Rh(II)-catalyst, Rh2(S-NTTL)3(dCPA), which contains a bulky dicyclohexylphenylacetate group (dCPA) as the achiral ligand. A crystal structure of Rh2(S-NTTL)3(dCPA) revealed that the chiral crown conformation was conserved with the bulky dCPA ligand. As shown in Figure 2, the N-naphthalimido groups are oriented on the same face of the catalyst with the sterically demanding tert-butyl groups on the opposite face blocking reactivity on the bottom face of the catalyst. Indeed, this new mixed-ligand complex gave 92% ee and 79% yield of the bicyclobutane. With this optimal catalyst in hand, we used it in the key step in a one-pot bicyclobutanation/homoconjugate addition sequence to assemble the core of piperarborenine B. Hence, treatment of diazoester 3 with 0.1 mol % Rh2(S-NTTL)3(dCPA) at −78 °C in toluene followed by cuprate addition using CuBr•SMe2, PPh3, and 2-methyl-1-propenylmagnesium bromide in THF provided the desired trisubstituted cyclobutane 5 in 69% yield, 92% ee and 4:1 dr after kinetic protonation with BHT.
Figure 2.

X-ray crystal structure of Rh2(S-NTTL)3(dCPA).
In order to set the desired stereochemical relationship of the cyclobutane ring, kinetic protonation with a bulky proton source was required. BHT was an effective proton source providing the desired diastereomer in 4:1 dr.[22] An even higher 6:1 dr was obtained with the sterically demanding 2,6-di(adamantan-1-yl)-4-(tert-butyl)phenol, but unfortunately this high selectivity was poorly reproducible upon scale up. For this reason, BHT was chosen for large scale synthesis of the enantiomerically enriched cyclobutane 5, which was prepared in gram quantities.
We next sought to install the amide directing group to facilitate subsequent Pd-catalyzed C–H activation. The carboxylate functionality was unveiled by an ozonolysis/Pinnick oxidation sequence. First, vinylcyclobutane 5 was treated with O3 in the presence of 1,3,5-trimethoxybenzene, an additive found to suppress over-oxidation of the electron-rich 1,2-dimethoxyphenyl group. After reductive quench with PPh3, the aldehyde intermediate was oxidized to the carboxylic acid by Pinnick oxidation. The crude carboxylic acid was used in a HATU-enabled coupling with 2-(methylthio)aniline to install the desired directing group providing cyclobutane 6 in 66% yield, along with an additional 14% yield of a separable diastereomer derived from the minor diastereomer of 5.
Initial attempts at C–H activation using known conditions[12a] (Ar-I, Pd(OAc)2, Ag2CO3, tert-BuOH, 75 °C) provided the arylated product in 45% yield and incomplete conversion of starting material. Gratifyingly, we discovered that the yield was significantly improved by running the reaction ‘on water.’[26] Thus, the combination of 3,4,5-trimethoxyiodobenzene, Pd(OAc)2, PivOH, and K2CO3 on water gave the desired cyclobutane 7 in 69% yield. These conditions avoid the use of Ag2CO3 and also proceed considerably faster with full consumption of starting material after 12 h (as opposed to 45% yield and 80% conversion after 36 h in tert-BuOH at 75 °C). Running the reaction neat led to a slightly diminished 56% yield. Interestingly, microwave heating at 100 °C resulted in 55% yield with incomplete conversion after only 20 minutes but was not further investigated due to scalability limitations of our microwave reactor.
The directing group was removed by converting to the Bocamide followed by hydrolysis with lithium hydroperoxide. [12a],[27] The remaining tert-butylester monoacid was treated with TFA to afford the diacid 8. The synthesis of piperarborenine B was completed by dihydropyridinone addition to in situ generated diacyl chloride in 75% yield (Scheme 6).
Scheme 6.

Completion of piperarborenine B.
In conclusion, the highly efficient, enantioselective total synthesis (8% overall yield) can be completed in only one week to produce 400 mg of the natural product, piperarborenine B. Key features include a new protocol for α-allyl-α-diazoester synthesis that can be completed in one day while avoiding unstable intermediates. A new mixed-ligand Rh(II)-catalyst was designed, synthesized and demonstrated to be effective for the key bicyclobutanation step in 92% ee with only 0.1 mol % of the catalyst. This catalytic enantioselective reaction can be employed in a one-pot bicyclobutanation/homoconjugate addition cascade to establish the cyclobutane core. ‘On water’ conditions for cyclobutane C–H activation were found to be particularly effective in forming the truxillate core. The use of aqueous media factored prominently in this total synthesis with two steps conducted ‘on water’ (Tsuji-Trost allylation and C–H activation) and three other steps conducted in aqueous/organic mixtures. The reported route is especially modular featuring several points of derivatization that can enable straightforward synthesis of enantiomerically enriched analogues.
Supplementary Material
Scheme 2.

Retrosynthetic strategy for the enantioselective synthesis of piperarborenine B.
Scheme 5.

Directing group installation, Pd-catalyzed C–H activation and hydrolysis via lithium hydroperoxide.
Acknowledgments
For funding we thank NSF CHE 1300329. RAP thanks NIH T32GM008550 for a training grant. For instrumentation we thank NIH P20GM104316, P30GM110758, S10RR026962-01, S10OD016267-01 and NSF grants: CHE 0840401, CHE-1229234 and CHE-1048367. We are grateful to Glenn Yap for the determining the crystal structure of Rh2(S-NTTL)3(dCPA).
Footnotes
Supporting information for this article is given via a link at the end of the document. CCDC 1449073 contains the supplementary crystallographic data for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.
References
- 1.a) Dembitsky VM. J Nat Med. 2008;62:1–33. doi: 10.1007/s11418-007-0166-3. [DOI] [PubMed] [Google Scholar]; b) Sergeiko A, Poroikov VV, Hanuš LO, Dembitsky VM. Open Med Chem J. 2008;2:26–37. doi: 10.2174/1874104500802010026. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tan JJ, Tan CH, Wang YQ, Jiang SH, Zhu DY. Helv Chim Acta. 2006;89:117–121. [Google Scholar]; d) Nozaki H, Hayashi KI, Kido M, Kakumoto K, Ikeda S, Matsuura N, Tani H, Takaoka D, Iinuma M, Akao Y. Tetrahedron Lett. 2007;48:8290–8292. [Google Scholar]
- 2.McGrath NA, Brichacek M, Njardarson JT. J Chem Ed. 2010;87:1348–1349. [Google Scholar]
- 3.a) Lee FP, Chen YC, Chen JJ, Tsai IL, Chen IS. Helv Chim Acta. 2004;87:463–468. [Google Scholar]; b) Tsai IL, Lee FP, Wu CC, Duh CY, Ishikawa T, Chen JJ, Chen YC, Seki H, Chen IS. Planta Med. 2005;71:535–542. doi: 10.1055/s-2005-864155. [DOI] [PubMed] [Google Scholar]
- 4.a) Nakamura M, Chi YM, Yan WM, Nakasugi Y, Yoshizawa T, Irino N, Hashimoto F, Kinjo J, Nohara T, Sakurada S. J Nat Prod. 1999;62:1293–1294. doi: 10.1021/np990041c. [DOI] [PubMed] [Google Scholar]; b) Nakamura M, Chi YM, Kinjo J, Yan WM, Nohara T. Phytochemistry. 1999;51:595–597. [Google Scholar]
- 5.a) Chi YM, Nakamura M, Yoshizawa T, Zhao XY, Yan WM, Hashimoto F, Kinjo J, Nohara T, Sakurada S. Biol Pharm Bull. 2005;28:1989–1991. doi: 10.1248/bpb.28.1989. [DOI] [PubMed] [Google Scholar]; b) Nakamura M, Chi YM, Yan WM, Yonezawa A, Nakasugi Y, Yoshizawa T, Hashimoto F, Kinjo J, Nohara T, Sakurada S. Planta Med. 2001;67:114–117. doi: 10.1055/s-2001-11512. [DOI] [PubMed] [Google Scholar]
- 6.Berger WT, Ralph BP, Kaczocha M, Sun J, Balius TE, Rizzo RC, Haj-Dahmane S, Ojima I, Deutsch DG. PLoS ONE. 2012;7:e50968. doi: 10.1371/journal.pone.0050968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Iriondo-Alberdi J, Greaney MF. Eur J Org Chem. 2007;2007:4801–4815. [Google Scholar]; b) Bach T, Hehn JP. Angew Chem Int Ed. 2011;50:1000–1045. doi: 10.1002/anie.201002845. [DOI] [PubMed] [Google Scholar]; c) Crimmins MT. Chem Rev. 1988;88:1453–1473. [Google Scholar]
- 8.a) Ichikawa M, Takahashi M, Aoyagi S, Kibayashi C. J Am Chem Soc. 2004;126:16553–16558. doi: 10.1021/ja0401702. [DOI] [PubMed] [Google Scholar]; b) Skiredj A, Beniddir MA, Joseph D, Leblanc K, Bernadat G, Evanno L, Poupon E. Angew Chem Int Ed. 2014;53:6419–6424. doi: 10.1002/anie.201403454. [DOI] [PubMed] [Google Scholar]; c) Biradha K, Santra R. Chem Soc Rev. 2013;42:950–967. doi: 10.1039/c2cs35343a. [DOI] [PubMed] [Google Scholar]
- 9.a) Ischay MA, Anzovino ME, Du J, Yoon TP. J Am Chem Soc. 2008;130:12886–12887. doi: 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]; b) Du J, Yoon TP. J Am Chem Soc. 2009;131:14604–14605. doi: 10.1021/ja903732v. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ischay MA, Ament MS, Yoon TP. Chem Sci. 2012;3:2807–2811. doi: 10.1039/C2SC20658G. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Riener M, Nicewicz DA. Chem Sci. 2013;4:2625–2629. doi: 10.1039/C3SC50643F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Telmesani R, Park SH, Lynch-Colameta T, Beeler AB. Angew Chem Int Ed. 2015;54:11521–11525. doi: 10.1002/anie.201504454. [DOI] [PubMed] [Google Scholar]
- 11.a) Engle KM, Mei TS, Wasa M, Yu JQ. Acc Chem Res. 2012;45:788–802. doi: 10.1021/ar200185g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nadres ET, Santos GI, Shabashov D, Daugulis O. J Org Chem. 2013;78:9689–9714. doi: 10.1021/jo4013628. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Neufeldt SR, Sanford MS. Acc Chem Res. 2012;45:936–946. doi: 10.1021/ar300014f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Gutekunst WR, Baran PS. J Am Chem Soc. 2011;133:19076–19079. doi: 10.1021/ja209205x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gutekunst WR, Baran PS. J Org Chem. 2014;79:2430–2452. doi: 10.1021/jo4027148. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gutekunst WR, Gianatassio R, Baran PS. Angew Chem Int Ed. 2012;51:7507–7510. doi: 10.1002/anie.201203897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Shabashov D, Daugulis O. J Am Chem Soc. 2010;132:3965–3972. doi: 10.1021/ja910900p. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zaitsev VG, Shabashov D, Daugulis O. J Am Chem Soc. 2005;127:13154–13155. doi: 10.1021/ja054549f. [DOI] [PubMed] [Google Scholar]
- 14.a) He G, Chen G. Angew Chem Int Ed. 2011;50:5192–5196. doi: 10.1002/anie.201100984. [DOI] [PubMed] [Google Scholar]; b) Feng Y, Chen G. Angew Chem Int Ed. 2010;49:958–961. doi: 10.1002/anie.200905134. [DOI] [PubMed] [Google Scholar]
- 15.a) Liu R, Zhang M, Wyche TP, Winston-McPherson GN, Bugni TS, Tang W. Angew Chem Int Ed. 2012;51:7503–7506. doi: 10.1002/anie.201203379. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xu H, Zhang W, Shu D, Werness JB, Tang W. Angew Chem Int Ed Engl. 2008;47:8933–8936. doi: 10.1002/anie.200803910. [DOI] [PubMed] [Google Scholar]
- 16.a) Albrecht L, Dickmeiss G, Cruz Acosta F, Rodriguez-Escrich C, Davis RL, Jørgensen KA. J Am Chem Soc. 2012;134:2543–2546. doi: 10.1021/ja211878x. [DOI] [PubMed] [Google Scholar]; b) Halskov KS, Kniep F, Lauridsen VH, Iversen EH, Donslund BS, Jørgensen KA. J Am Chem Soc. 2015;137:1685–1691. doi: 10.1021/ja512573q. [DOI] [PubMed] [Google Scholar]
- 17.Du J, Skubi KL, Schultz DM, Yoon T. Science. 2014;344:392–396. doi: 10.1126/science.1251511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson T, Choo K-L, Lautens M. Chem Eur J. 2014;20:14194–14197. doi: 10.1002/chem.201404896. [DOI] [PubMed] [Google Scholar]
- 19.a) Chen G, Shigenari T, Jain P, Zhang Z, Jin Z, He J, Li S, Mapelli C, Miller MM, Poss MA, Scola PM, Yeung KS, Yu JQ. J Am Chem Soc. 2015;137:3338–3351. doi: 10.1021/ja512690x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xiao KJ, Lin DW, Miura M, Zhu RY, Gong W, Wasa M, Yu JQ. J Am Chem Soc. 2014;136:8138–8142. doi: 10.1021/ja504196j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Y-J, Hu T-J, Feng C-G, Lin G-Q. Chem Commun. 2015;51:8773–8776. doi: 10.1039/c5cc02023a. [DOI] [PubMed] [Google Scholar]
- 21.Concomitant with our previous work Davies has independently described complementary bicyclobutanation conditions: Qin C, Davies HML. Org Lett. 2013;15:310–313. doi: 10.1021/ol303217s.
- 22.Panish R, Chintala SR, Boruta DT, Fang Y, Taylor MT, Fox JM. J Am Chem Soc. 2013;135:9283–9286. doi: 10.1021/ja403811t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Homoconjugate additions of arylsulfonyl-substituted bicyclobutanes: Gianatassio R, Lopchuck JM, Wang J, Pan C-M, Malins LR, Prieto L, Brandt TA, Collins MR, Gallego GM, Sach NW, Spangler JE, Zhu H, Zhu J, Baran PS. Science. 2016;351:241–246. doi: 10.1126/science.aad6252.
- 24.a) Manabe K, Kobayashi S. Org Lett. 2003;5:3241–3244. doi: 10.1021/ol035126q. [DOI] [PubMed] [Google Scholar]; b) Shibuya R, Lin L, Nakahara Y, Mashima K, Ohshima T. Angew Chem Int Ed. 2014;53:4377–4381. doi: 10.1002/anie.201311200. [DOI] [PubMed] [Google Scholar]
- 25.Boruta DT, Dmitrenko O, Yap GPA, Fox JM. Chem Sci. 2012;3:1589–1593. doi: 10.1039/C2SC01134D.For heteroleptic Rh(II) complexes composed solely of chiral ligands, see: Lindsay VNG, Charette AB. ACS Catal. 2012;2:1221–1225.
- 26.Li B, Dixneuf PH. Metal-Catalyzed Reactions in Water. Wiley-VCH Verlag GmbH & Co., KGaA; 2013. pp. 47–86. [Google Scholar]
- 27.Evans DA, Britton TC, Ellman JA. Tetrahedron Lett. 1987;28:6141–6144. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.