

Abstract
For many important pathogens, mechanisms promoting antigenic variation, such as mutation and recombination, facilitate immune evasion and promote strain diversity. However, mathematical models have shown that host immune responses to polymorphic antigens can structure pathogen populations into discrete strains with nonoverlapping antigenic repertoires, despite recombination. Until now, models of strain evolution incorporating host immunity have assumed a randomly mixed host population. Here, we illustrate the effects of different host contact networks on strain diversity and dynamics by using a stochastic, spatially heterogeneous analogue of this model. For randomly mixed populations, our model confirms that cross-immunity to strains sharing alleles at antigenic loci may structure the pathogen population into discrete, nonoverlapping strains. However, this structure breaks down once the assumption of random mixing is relaxed, and an increasingly diverse pathogen population emerges as contacts between hosts become more localized. These results imply that host contact network structure plays a significant role in mediating the emergence of pathogen strain structure and dynamics.
Many important pathogens, such as Neisseria meningitidis and Plasmodium falciparum, display structured strain diversity: highly diverse genotypes are organized into distinct, persisting strains, which can be detected as linkage disequilibrium between particular genes (for example, see refs. 1 and 2). Strains can often show cyclical temporal dynamics, with successive types dominating in prevalence within the host population. Understanding the maintenance of diversity within pathogen populations, and the dynamics of multiple strains, has been a focus for many theoretical studies. Previous studies have shown that interference between strains, either through the prevention of superinfection (3) or from cross-immunity gained by exposure to `similar' strains (4, 5), can allow for the stable coexistence of different strains, as well as sustained oscillations, under certain conditions. The latter studies emphasized the importance of cross-immunity as a mechanism for structuring pathogen populations, but assumed that the “similarity” between strains was based on a single genetic locus.
For pathogens that undergo antigenic variation, such as malaria, trypanosomes, and meningitis, multiple genetic loci are often important in generating host immune responses. Gupta et al. (6, 7) explicitly accounted for multiple, polymorphic immunogenic loci, by using the overlap between allelic profiles of different strains to determine the extent of host cross-immunity. They showed that even high levels of cross-immunity can result in stable, diverse pathogen populations. For very low levels, no strain structure is observed. As it increases, unstable structure can emerge, displaying cyclic or chaotic patterns of strain dominance. At sufficiently high levels of cross-immunity, selection by the immune system will result in the dominance of a set of strains with nonoverlapping antigenic repertoires (which will not be competing for susceptible hosts). This structure will persist despite recombination events that generate different variants, because immune selection against strains that share alleles at antigenic loci will suppress their prevalence. Gomes et al. (8) defined antigenic distance between strains in continuous strain space, showing analogous dynamical results for varying levels of cross-immunity, stable homogeneous and heterogeneous pathogen populations at low and high levels of cross-immunity, respectively, and traveling wave patterns through strain space at intermediate levels. A different approach has been taken to keep track of multiple strains (9), where the immune status of the hosts at any point in time is taken into account rather than the history of infection for each individual. Although sustained oscillations were not observed, the structuring of the pathogen population was still dependent on mechanisms of host immunity.
Despite differences in the formulation of these deterministic models, they produce similar outcomes in terms of the polarization of strains in strain space for higher levels of cross-immunity. However, they all assume that host populations are well mixed, and do not take stochastic effects and spatial heterogeneities into account. Studies have shown that network structure can significantly affect the processes occurring social networks, including the dynamics and evolution of infectious diseases (10–13). For example, some have investigated the effect of network structure on the evolution of disease traits such as infectious period and transmission rates (10), as well invasion thresholds for epidemics (11). Others have explored the role of spatial contact structure in the evolution of virulence (12). To date, there have been no studies explicitly investigating the effects of host contact networks on the interaction multiple strains incorporating host cross-immunity, however. Many important multistrain pathogens exist in diverse geographical environments and in different types of host populations. therefore follows that, for directly transmitted diseases, the social network structure of the host population may impact the pathogen population by affecting the extent of strain mixing, and therefore the level of competition and recombination between different strains. In communities where local contacts are the primary means of transmission, the population genetics of the pathogen may be very different from in large cities where individuals mix with large numbers of random contacts.
Here we use a stochastic individual-based model (IBM), based on the framework of Gupta et al. (6) described above, investigate the effects of social network structure on the evolution of pathogen diversity and strain structure. We first restrict our analyses to regular and random host contact networks, caricatures of two extreme social network scenarios, and compare these networks to each other as well as to stochastic mean-field approximations of the IBM to analyze the effect structured host contact networks on the dynamics of the strains. We then further analyze several small-world host contact networks and argue that the extent of host clustering is the primary network characteristic affecting pathogen strain structure and diversity. The results highlight the importance of considering social network structure in the analysis of pathogen population structuring and dynamics.
The Model
Hosts. The individual-based model simulates each potential host as a separate entity including its contacts, the strains it is infected with, and its immune response (memory of infection). Each individual has a position in a ring lattice. A host contact network is created at the beginning of a simulation, with every individual in contact with a fixed number of other individuals. This contact network remains constant throughout the simulation for all host contact networks modeled, except the mean-field approximation host network (described below). The structure of the contact network, ranging from regular through small-world to random, is determined by the ρ parameter, as in Watts and Strogatz (14). ρ is the probability that an individual will come into contact with a randomly chosen individual rather than a local neighbor in the ring lattice. Hence, a ρ of 0 means that an individual will only interact with its immediate neighbors, whereas a ρ of 1 means that the host contact network is a random network, wherein every fixed interaction is with a randomly chosen individual. To approximate the mean-field ordinary differential equation (ODE) model, the stochastic IBM model uses a host contact network that is random (i.e., ρ = 1) and changes at every time step (henceforth referred to as the “mean-field approximation” host contact network). Contact between hosts occurs once in each time step and changes in host infection and immunity status are updated synchronously at the end of each time step.
One important difference between the stochastic model and the mean-field ODE is the possibility of the stochastic loss of an allele in the IBM. In the deterministic ODE simulations, mutation was unnecessary because alleles could not be lost. However, there was a need for mutation to reintroduce alleles in the stochastic IBM. This was especially evident in small populations where demographic stochasticity frequently resulted in allelic extinction (data not shown).
Pathogens. Pathogens were represented as bit-strings, with each bit being one immunodominant locus coding for an antigen on the surface of the pathogen. We limited each locus to two alleles, designated as a “1” or a “0”. There are therefore 2n different configurations (“genotypes”) that a pathogen can have. A strain is defined as a pathogen subpopulation with one of these distinct configurations. To measure the genetic variability within a heterogeneous pathogen population, we introduce two metrics: diversity (D) and discordance (H).
Diversity measures the evenness with which a pathogen population is partitioned into all of its possible different strains. We calculate diversity by dividing the entropy of the pathogen population (also known as the Shannon–Weaver diversity index, ref. 15) by the maximum possible entropy of the population:
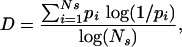 |
[1] |
where pi is the frequency of strain i in the population, and Ns, the number of strains, = 2n. Therefore, for a pathogen population, D ranges between 0 and 1, with D = 1 indicating that all of the possible strain types in the population are equally represented.
In addition to diversity, a metric that describes the average allelic difference between any two pathogens picked at random from a heterogeneous pathogen population is necessary to measure antigenic discordance between strains. We use a taxonomic distinctness measure, previously used in calculating the average phylogenetic distance between species within a community (16). Here, instead of using weights to quantify phylogenetic distances between species, we use weights to quantify allelic differences between strains. The weights can therefore simply be the Hamming distances between strains, where the Hamming distance between two strains is the number of bits by which they differ. Because the maximum Hamming distance possible in a pathogen population is known, we adjust the taxonomic distinctness measure by dividing by the maximum Hamming distance (the number of loci) to get a discordance (H) measure between 0 and 1:
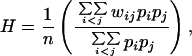 |
[2] |
where wij is the number of loci with different alleles for strains i and j. pi and pj are the frequencies of strain i and j in the pathogen population, respectively, and n is the number of loci. Fig. 1 illustrates the differences between diversity and discordance.
Fig. 1.

Strain histograms illustrating diversity and discordance metrics. In this example, with a three-allele pathogen, eight possible strains can exist in a population at any point in time (each consisting of a unique combination of three immunodominant loci). Populations in A and B both have the same discordance value (H = 0.5), but population in A has a more diverse distribution of strains present (D = 1.0) than population in B (D = 0.33). Populations in C and D, although having identical diversity levels (D = 0.79), differ in the extent of the allelic similarities of the strains present, with population in D having a higher discordance level (H = 0.68) than population in C (H = 0.51).
Dynamics. Pathogens are assumed to only exist within the modeled hosts. A host infected with a pathogen contacting an individual with no immunity to that pathogen will infect that individual with probability β. Although a host may be infected by several strains at once, it may only infect one individual with a single strain in any one time step. Upon infection, individuals remain infected by that pathogen for a period, such that the average duration of infection with a pathogen is 1/μ, where μ is the probability that the host rids itself of the pathogen in a time step (Table 1). After infection, individuals remain immune to that pathogen for a period, such that the average duration of immunity to that pathogen is 1/σ, where σ is the probability of the host losing its immunity to a pathogen in a time step (Table 1). The duration of infection and of immunity therefore exhibit exponential decay. When an infection event occurs, there is also the chance that the strain will undergo mutation or recombine with another strain in the same host. Both of these events occur with defined probabilities (τ and r, respectively, Table 1).
Table 1. Key model parameters, including symbol, description, and sample ranges used in the LHS sensitivity analysis.
| Parameter | Description | LHS range, min/max |
|---|---|---|
| C | Mean number of contacts per host | 4:12 |
| ρ | Degree of randomness in the host contact network | NA |
| 1/μ | Average duration of infection | 3:10 |
| 1/σ | Average duration of immunity | 10:30 |
| β | Probability of transmission (to a completely susceptible host) | 0.2:0.8 |
| R | Probability of recombination per allele | 0.01:0.1 |
| τ | Probability of allelic mutation per allele | 0.001:0.005 |
| γ | Degree of cross-immunity | 0.02:4 |
| N | Number of immunodominant loci | 2:4 |
| P | Host population size | 100:500 |
For the LHS analysis, we used a uniform probability distribution function for each range of parameter values. NA, not applicable; min/max, ratio of minimum to maximum values.
The strains that a host is immune to influences the host's probability of infection, given contact with an infected neighbor has occurred, depending on the similarity of the strains. We model this cross-immunity by assuming that a host's vulnerability to infection by a strain depends on the similarity between that strain and the strains in the host's immune memory, an assumption also made in Gupta et al. (6). Given this (reasonable) assumption, the fraction of identical bits between the host's immune memory and the infecting strain can therefore be converted into a vulnerability of infection (v), between 0 and 1, by using
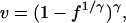 |
[3] |
where f is the fraction of identical bits and γ is a positive number scaling the level of cross-immunity (Table 1).
The measures of genetic variability used to quantify a pathogen population at one point in time, outlined above, can also be used to interpret the dynamics of a pathogen population on a host network and for comparisons between different networks. Pathogen populations that have only one discordant pathogen set present have a low mean diversity value [D ≈ log (2)/log(Ns)0] and a high mean degree of discordance (H ≈ 1). Pathogen populations with no strain structure have a high mean diversity value (D ≈ 1) and a low discordance value (H ≈ 0.5). Pathogen populations with stochastic cycling exhibit intermediate mean values of diversity and discordance.
Experimental Approach. Parameter space was explored by using the statistical technique of Latin hypercube sampling (LHS) (17), which selects combinations of parameter values without replacement, given parameter value ranges and probability distribution functions. The key model parameters that were sampled by using LHS can be found in Table 1.
We used 1,000 LHS to cover parameter space. For each of these, three simulations differing in host contact network structure were run for 3,000 time steps (sufficiently long to remove transient dynamics): one on a regular host network (ρ = 0), one on a random host network (ρ = 1), and one on a mean-field approximation network. In addition, small-world simulations were run for two sets of parameter values, with values of ρ between 0 and 1.
Results
Stochastic Extinctions. Of the 1,000 parameter combinations in the LHS sensitivity analysis, 255 resulted in the extinction of all pathogen strains on the host network in one or several of the three host network scenarios. Analysis of variance of the parameter combinations for these simulations, in contrast to those in which extinction did not occur, revealed that the main factors contributing to extinction were, in declining order of importance, a short infectious period (P < 0.001, F = 79.57), a high degree of cross-immunity (P < 0.001, F = 75.70), a small host population (P < 0.001, F = 69.45), and low numbers of contacts between individuals (P < 0.001, F = 42.65). The results described below are based on the 745 LHS sensitivity analysis parameter combinations in which stochastic extinction did not occur in any of the network scenarios.
Comparison with Mean-Field Models. The stochastic IBM reproduces many of the features present in the original mean-field ODE formulation (6, 7). The effect of varying cross-immunity is particularly clear, with pathogen populations having no strain structure at low cross immunity, displaying strain cycling or chaotic fluctuations at intermediate cross-immunity, and populations with one dominant, discordant set occurring at high levels of cross-immunity (Fig. 2). Fig. 2 shows that for both models strong host cross-immunity is sufficient to structure the pathogen population into discrete strains; in our model, this occurs regardless of the rate of recombination or mutation. As in the deterministic model, the changes in dynamics seen in Fig. 2 occur at critical values of γ, corresponding to the reduction in diversity and increase in discordance. In addition to the expected effect of γ on strain diversity and discordance, increasing the number of immunodominant loci (n) also affected these metrics by increasing diversity and decreasing discordance (Table 2).
Fig. 2.

An illustration of the changing dynamics for both the original deterministic model and our stochastic mean-field approximation model for pathogen populations with two loci (i.e., four strains). Two strains comprising one discordant set are plotted in black; the other discordant set is plotted in gray. Plotted in all simulations is the proportion of the host population immune to each of the four strains. Parameter values [corresponding to the parameter notation of Gupta et al. (6)] used for the ODE simulations (A–C) were μ = 0.02, σ = 10, R0 = 2, α = 1, with the only difference in parameter values being the degree of cross-immunity γ (0.3 in A, 0.7 in B, 0.9 in C). The mean-field approximation of the stochastic IBM (D–F) used parameter values corresponding to our model's parameter notation in Table 1: C = 12, r = 0.0953, τ = 0.0042, β = 0.2472, 1/μ = 7, 1/σ = 23, P = 223, n = 2. The degree of cross-immunity γ was 0.01 in D, 0.95 in E, and 2.00 in F. (Note that γ is defined slightly differently in our model compared with its definition in ref. 6). A and D, with the lowest values ofγ, both have no strain structure, with the mean diversity in D being 0.9882 and the mean discordance being 0.6723. B and E have intermediate values of γ, and both exhibit cyclical strain dynamics. Mean diversity in E is 0.8733, and mean discordance is 0.7496. C and F have high values of γ, and both exhibit strong strain structure, with one discordant set being dominant. Mean diversity in F is 0.5480, and mean discordance is 0.9720. Simulations were run for 2,000 time steps for the IBM and 500 time steps for the ODE.
Table 2. The effects of model parameters on diversity and discordance of the pathogen population.
| Regular
|
Random
|
Mean-field
|
Combined
|
|||||
|---|---|---|---|---|---|---|---|---|
| H | D | H | D | H | D | H | D | |
| C | 7.9 | 1.8 | 0.6 | 1.0 | 0.7 | |||
| r | ||||||||
| τ | 1.4 | 1.3 | 0.6 | |||||
| γ | 7.1 | 57.0 | 65.0 | 27.1 | 61.0 | 57.2 | 24.8 | 38.6 |
| β | ||||||||
| 1/μ | 2.1 | 4.2 | 1.0 | 1.1 | 1.2 | |||
| 1/σ | 0.7 | 0.7 | 0.9 | |||||
| P | 4.5 | 6.4 | 1.1 | |||||
| n | 15.6 | 2.3 | 1.5 | 13.5 | 10.7 | 12.1 | 3.3 | 10.0 |
| Network structure | NA | NA | NA | NA | NA | NA | 29.0 | 15.2 |
Results of linear regression analysis on the effects of variation in the model parameters on mean diversity (D) and mean discordance (H) for 3,000 iterations and for 745 simulations. Values are the percentage of the variance in D or H explained by the model parameter (determined by using Pearson's product moment correlation coefficient), with blank cells representing non-significant correlations (P < 0.05, two-tailed distribution). Data for the Combined column result from analyzing the data for Regular, Random, and Mean-field together, comparing the effect of network structure with that of the other model parameters.
Effect of Host Network Structure. Fig. 3 shows a comparison of mean diversity and mean discordance of simulations from the mean-field approximation versus the random fixed network and from the random fixed network versus the regular network. The results indicate that host contact network structure clearly affects pathogen strain structure and dynamics, with the discordant strain structure seen in the mean-field approximation breaking down in the more regular networks and strain diversity increasing. As the random mixing of the network decreases and contacts between hosts become more localized, the genetic structuring of the pathogen population decreases; the diversity of strains present increases and the dominance of sets of antigenically discordant strains declines. These results are robust for different parameter values (Table 2 and Fig. 3), and emphasize that the evolutionary dynamics of a pathogen may reflect the nature of the interactions between hosts rather than characteristics of the hosts or pathogen species themselves. Analysis into the relative effect of contact network structure in the LHS sensitivity analysis reveals that network structure describes a significant and comparatively large part of the variation in pathogen diversity and discordance (Table 2). Within a certain network type, however, the degree of cross-immunity (γ) and the number of loci (n) again account for most of the variance in discordance and diversity. The probability of recombination (r) and pathogen transmissibility (β) conspicuously do not significantly affect strain diversity or discordance in any of the three network types, a point to which we will return in the discussion.
Fig. 3.

A comparison of mean diversity and mean discordance for all 745 (of 1,000) simulations for which stochastic extinction did not occur within the first 3,000 time steps of either the mean-field approximation or the random or regular ring simulation. Mean diversity slightly increased from the mean-field approximation simulations to the random simulations (A), whereas mean discordance slightly decreased (B). A large increase in mean diversity is evident when regular ring network dynamics are compared with random host contact network dynamics (C), as is the large decrease in mean discordance levels (D).
We conjecture that the higher degree of host clustering in regular contact networks compared to random contact networks cause these patterns in mean diversity and mean discordance. To evaluate this hypothesis further, we simulated the strain dynamics with the IBM for two LHS samples, using 14 different values of ρ between 0 and 1. When ρ is between 0 and 1, the networks are considered to be “small-world” networks (14), and several quantities, such as characteristic path lengths and clustering coefficients, can be used as metrics to describe their structure (14). In Fig. 4, mean discordance values (Fig. 4A), mean diversity values (Fig. 4B), and the clustering coefficients characterizing the host networks (Fig. 4B) are plotted against ρ. Fig. 4 clearly illustrates that the systematic changes in mean diversity and mean discordance values as the host network goes from being regular to being random occur at the ρ values where the degree of clustering changes. Further analysis into the degree of strain clustering in the host contact network (a strain cluster is defined as a group of connected hosts who are currently either infected with, or immune to, a given strain) indicates that, as contacts between hosts become more localized (Fig. 4B), the average size of the largest strain cluster diminishes (Fig. 4C). As discordant sets occur together spatially, this trend indicates that discordant sets grow in cluster size as host contacts become more random.
Fig. 4.

The effects of transitioning from regular to random networks on strain diversity and discordance. (A) The effect of ρ (the degree of host mixing) on mean discordance (dashed line) and mean diversity (solid line) for two simulations. The simulation denoted with the open circle has parameter values C = 10, r = 0.0681, τ = 0.0049, γ = 2.8178, β = 0.4093, 1/μ = 8, 1/σ = 11, P = 481, n = 4. The simulation denoted with the open square has parameter values C = 8, r = 0.0554, τ = 0.0021, γ = 3.5578, β = 0.7262, 1/μ = 9, 1/σ = 15, P = 371, n = 3. (B) The degree of host clustering, measured by the clustering coefficient, as a function of ρ. The clustering coefficient is defined and computed as in Watts and Strogatz (14). (C) The average size of the largest strain cluster as a function of ρ. The decrease in discordance and the increase in diversity with more localized interactions (lower ρ) is strongly correlated to the degree of host clustering and the growth in the size of the largest strain cluster. Both simulations were run for 5,000 time steps, for each of the 14 ρ values, ranging from 0.0001 to 1. The first 2,000 time steps were discarded to remove the effect of transients. Note the logarithmic scale on the x axis.
Discussion
Our stochastic IBM model illustrates that network contact structure of the host population can play a major part in determining the strain structure and evolutionary dynamics of a pathogen population. For pathogens with polymorphic, immunodominant antigens, regular host networks with localized interactions may allow for a more diverse pathogen population to exist, whereas well mixed host populations promote genetic structuring by the host immune system.
Our mean-field approximation supports the findings of deterministic models, and reproduces the three types of dynamics found previously within this type of framework (5, 8, 9): no strain structure at low levels of cross-immunity, discrete, nonoverlapping strain structure at high levels, and cyclical dominance of nonoverlapping sets of strains at intermediate levels. The addition of a stochastic framework to these mean-field models has allowed for the inclusion of mutation events, a varied population size, and an increased number of strains, in addition to the exploration of different host networks. The fact that the effects of host cross-immunity are reproduced accurately even in relatively small populations, with large numbers of strains, and with high rates of mutation and recombination, provides strong support for the hypothesis that immunity of the host may dictate the structure and dynamics of the pathogen population when pathogens are antigenically variable.
The variance in strain discordance and diversity for all networks was primarily affected by the degree of cross-immunity (γ) and, to a lesser extent, the number of immunodominant loci (n). Across networks, the host contact structure also played a key role in determining these metrics (Table 2). Within a given host network type, as well as in the combined analysis, other factors, such as the average number of contacts per host (C), the average duration of infection and immunity (1/μ and 1/σ, respectively), and the host population size (P), only contributed slightly to explaining the variance in diversity and discordance. Interestingly, neither the probability of transmission (β) nor the probability of recombination (r) significantly explained any variance in these metrics. Although we would not expect the probability of transmission to necessarily affect these metrics, because all strains are equally fit, it is at first surprising that the probability of recombination does not contribute to explaining either of the metrics' variance. High rates of recombination, which should promote diversity and disrupt discordant strain structure, do not have this effect because a recombinant pathogen inherits immunodominant loci from its “parent” strains. Because discordant sets cluster together in the host networks, recombinants are generated in host environments in which the hosts are likely to have already experienced, and therefore be immune to, all of the immunodominant loci of the recombinant pathogen. Therefore, recombinant strains cannot establish themselves regardless of how often they are generated, because they are immediately suppressed by herd immunity to their parent strains. Therefore, higher recombination rates do not significantly affect strain diversity or discordance.
The fact that localized interactions may promote diversity in phenomena occurring on networks is well established (18–20). A number of loosely connected “islands of contacts” can result in the emergence of different dynamics occurring in different parts of the network, because local densities equilibrate more rapidly than global densities (13). As a result, models that have incorporated space have often produced differing results from their mean-field counterparts. This study is no exception. What makes our finding of particular importance, however, is the discovery of the primary importance of host contact network structure in controlling the dynamics of pathogen strain evolution and diversity. Unlike mean-field models, in which the selective force of the host immune system impacts the whole system equally, incorporating constraints on the spatial distribution of different strains allows for the build up of spatial clustering. Qualitative analyses suggest that discordant sets do arise locally, but that herd immunity is not established over the entire network when contacts between hosts are local. Moreover, the upward trend in the average size of the largest strain cluster associated with more random host networks highlights the importance of contact networks in controlling the establishment of widespread herd immunity. These observations argue for further investigation into the role that contact–network structure may play in generating these dynamics, in relation to these other key variables, especially considering that our general results appear over a large range of other parameter values.
Although mean-field models can provide valuable insight into the mechanisms driving pathogen evolution, we have shown that relaxing the assumption of random mixing within host populations may have profound effects on the interpretation of clinical data. Caution must be exercised when inferring mechanisms of selection from models that assume random host mixing, because the “environmental” contexts in which pathogen evolution occurs may be important in shaping their dynamics. Spatial patchiness, having been shown to be of great relevance in understanding ecological data, needs to be further addressed in the field of epidemiology, where the nonrandom connectivity of hosts provides the spatial backdrop for understanding and controlling disease dynamics.
Acknowledgments
This study was conceived and initiated as part of the Complex Systems Summer School 2003 at the Santa Fe Institute. We thank the Santa Fe Institute, Jonathan Shapiro, Tom Carter, and the participants of the summer school for advice and support during this study. We also thank two anonymous reviewers for suggestions.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: IBM, individual-based model; LHS, Latin hypercube sampling; ODE, ordinary differential equation.
References
- 1.Jolley, K. A., Kalmusova, J., Feil, E. J., Gupta, S., Musilek, M., Kriz, P. & Maiden, M. C. J. (2000) J. Clin. Microbiol. 38, 4492-4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gupta, S., Trenholme, K., Anderson, R. M. & Day, K. P. (1994) Science 263, 961-963. [DOI] [PubMed] [Google Scholar]
- 3.Dietz, K. (1979) J. Math. Biol. 8, 291-300. [DOI] [PubMed] [Google Scholar]
- 4.Castillo-Chavez, C., Hethcote, H. W., Andreasen, V., Levin, S. A. & Liu, W. M. (1989) J. Math. Biol. 27, 233-258. [DOI] [PubMed] [Google Scholar]
- 5.Andreasen, V., Lin, J. & Levin, S. A. (1997) J. Math. Biol. 35, 825-842. [DOI] [PubMed] [Google Scholar]
- 6.Gupta, S., Ferguson, N. & Anderson, R. M. (1998) Science 280, 912-915. [DOI] [PubMed] [Google Scholar]
- 7.Gupta, S., Maiden, M., Feavers, I. M., Nee, S., May, R. M. & Anderson, R. M. (1996) Nat. Med. 2, 437-442. [DOI] [PubMed] [Google Scholar]
- 8.Gomes, M. G., Medley, G. F. & Nokes, D. J. (2002) Proc. R. Soc. London Ser. B 269, 277-233. [Google Scholar]
- 9.Gog, J. R & Swinton, J. (2002) J. Math. Biol. 44, 169-184. [DOI] [PubMed] [Google Scholar]
- 10.Read, J. M. & Keeling, M. J. (2003) Proc. R. Soc. London Ser. B 270, 699-708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keeling, M. J. (1999) Proc. R. Soc. London Ser. B 266, 859-867. [Google Scholar]
- 12.O'Keefe, K. J. & Antonovics, J. (2002) Am. Nat. 159, 579-605. [DOI] [PubMed] [Google Scholar]
- 13.van Baalen, M. (2002) in The Adaptive Dynamics of Infectious Diseases: In Pursuit of Virulence Management, eds. Dieckman, U., Metz, J. A. J., Sabelis, M. W. & Sigmund, K. (Cambridge Univ. Press, Cambridge, U.K.), pp. 85-103.
- 14.Watts, D. J. & Strogatz, S. H. (1998) Nature 393, 440-442. [DOI] [PubMed] [Google Scholar]
- 15.Shannon, C. E. & Weaver, W. (1949) The Mathematical Theory of Communication (Univ. of Illinois Press, Urbana).
- 16.Warwick, R. M. & Clarke, K. R. (1995) Mar. Ecol. Prog. Ser. 129, 301-305. [Google Scholar]
- 17.Blower, S. M. & Dowlatabadi, H. (1994) Int. Stat. Rev. 2, 229-243. [Google Scholar]
- 18.Tilman, D. & Kareiva, P., eds. (1997) Spatial Ecology: The Role of Space in Population Dynamics and Interspecific Interactions (Princeton Univ. Press, Princeton)
- 19.Hassel, M. (2000) Spatial and Temporal Dynamics of Host–Parasitoid Interactions (Oxford Univ. Press, Oxford)
- 20.Newman, M. E. J. (2003) SIAM Rev. 45, 167-256. [Google Scholar]