

Abstract
The AHR is a ligand-activated transcription factor that mediates gene-environment interactions. Genome-wide expression profiling during differentiation of mouse ES cells into cardiomyocytes showed that AHR activation by 2,3,7,8-tetrachlorodibenzo-p-dioxin; Dioxin (TCDD), its prototypical ligand, disrupted the expression of multiple homeobox transcription factors and inhibited cardiomyocyte contractility. Here we treated ES cells with TCDD at daily differentiation intervals to investigate whether TCDD-induced loss of contractility had a developmental window of sensitivity. Surprisingly, contractility was an AHR-dependent TCDD target solely between differentiation days 0 and 3 during the period of panmesoderm development, when TCDD also disrupted expression of genes in the TGFβ/BMP2/4 and wingless-type MMTV integration site (WNT)signaling pathways, suppressed the secretion of bone morphogenetic protein (BMP4), WNT3a, and WNT5a and elevated the secretion of Activin A, as determined by ELISA of the secreted proteins in the culture medium. Supplementing the culture medium with BMP4, WNT3a, or WNT5a during the first 3 days of differentiation successfully countered TCDD-induced impairment of contractility, while anti-WNT3a, or anti-WNT5a antibodies or continuous Noggin (a BMP4 antagonist) or Activin A treatment inhibited the contractile phenotype. In Ahr+/+, but not in Ahr−/− ES cells, TCDD treatment significantly increased mitochondrial copy number, suggestive of mitochondrial stress and remodeling. Sustained AHR activation during ES cell differentiation appears to disrupt the expression of signals critical to the ontogeny of cardiac mesoderm and cause the loss of contractility in the resulting cardiomyocyte lineage.
Keywords: aryl hydrocarbon receptor, dioxin, embryonic stem cells, differentiation, cardiomyogenesis, mitochondrial dysfunction
The Barker theory often referred to as The Developmental Origins of Human Adult Disease, states that adverse influences early in development, and particularly during the embryonic stage, can lead to permanent changes in physiology and metabolism and result in increased disease risk in adulthood (Barker, 2007). In the context of environmental exposures, the prenatal and perinatal periods of development are particularly sensitive to toxicants, and both the nature and the severity of health outcomes may depend on the developmental time-period during which toxicant exposure occurs (Damstra, 2002). In the case of dioxin, a widespread toxicant, data on fish, birds, and mammals show that the developing tissues are more sensitive to 2,3,7,8-tetrachlorodibenzo-p-dioxin; Dioxin (TCDD)—the prototypical dioxin—than the adult ones (Kopf et al., 2009; Plavicki et al., 2013; Walker and Catron, 2000).
Epidemiological and biological studies support the concept that the developing cardiovascular system is a distinct target of TCDD (Puga, 2011). Work in multiple species including humans, rodents, fish, and avian embryos have shown that developmental exposure to TCDD results in cardiac defects, including reduced cardiomyocyte proliferation, altered fetal heart size, disruption of neovascularization, and ultimately cardiac hypertrophy (Ivnitski-Steele et al., 2005; Thackaberry et al., 2005). In utero exposure to TCDD also increases the susceptibility to cardiovascular dysfunction in adult life (Aragon et al., 2008). In humans, infants had born to mothers living near incinerators that emit complex mixtures of dioxins, furans, particulates, and heavy metals exhibit a higher incidence of lethal congenital heart diseases (Dummer et al., 2003). More recent epidemiological studies also showed an association between the incidence of hypoplastic left heart syndrome and maternal proximity during pregnancy to industrial release of halogenated hydrocarbons, dioxins, and polychlorinated biphenyls (Kuehl et al., 2006).
Most of the toxic endpoints observed in organisms exposed to TCDD are mediated by the Aryl Hydrocarbon Receptor (AHR), a ligand activated transcription factor that belongs to the bHLH/PAS superfamily of transcription factors (Gu et al., 2000). The ligand-activated AHR dimerizes in the nucleus with the Aryl Hydrocarbon Receptor Translocator (ARNT, also termed HIF1β) and the heterodimer binds to AHR/ARNT response elements located in the promoters of target genes, causing chromatin bending, recruitment of transcription coactivators, RNA polymerase II and associated chromatin remodeling factors, and initiation of gene transcription (Schnekenburger et al. 2007). In addition to the best known xenobiotic metabolism genes in the cytochrome P450 Cyp1 family, there are many other AHR/ARNT transcriptional targets, including genes involved in cell-cycle regulation and morphogenetic processes, suspected of playing key roles during cardiovascular development (Sartor et al., 2009).
Cardiomyocyte differentiation is a tightly orchestrated and dynamic process (Rajala et al., 2011). It requires sequential expression of multiple transcription factors and proper temporal and spatial integration of multiple signal transduction pathways, including those controlled by the wingless-type MMTV integration site (WNT), TGFβ, and FGF superfamilies (Liu and Foley, 2011; Rajala et al., 2011). These factors do not function alone; their cooperation and interdependent regulation are essential for cardiac development such that subtle disruption by haploinsufficient, hypomorphic, or dominant-negative mutations lead to malfunction of the heart at later times during the lifespan of the organism (Chien, 2000; Schott et al., 1998; Srivastava and Olson, 2000). Perturbation of the action of these regulatory factors by xenobiotic agents during cardiac development may account for the impairment of cardiogenesis caused by the compounds.
Genetic knockout of the Ahr gene in mice disrupts cardiovascular homeostasis, ultimately causing pathological cardiac hypertrophy (Lund et al., 2003). In wild-type mice, AHR expression is detectable in the heart of the post implantation mouse embryo as early as gestation days (GD) 7.5–9.5 (Abbott et al., 1995; Wang et al., 2013). Expression is evident in all 3 embryonic germ layers in vivo and on day 3 of the differentiation of mouse ES cells into embryoid bodies (EBs) (Wang et al., 2013). TCDD treatment during ES cell differentiation disrupts the concerted expression of genes involved in cardiac morphogenesis, including dozens of genes encoding homeobox transcription factors and polycomb and trithorax group genes (Wang et al., 2010, 2013). Interestingly, TCDD treatment at early differentiation time points disrupts WNT and bone morphogenetic protein (BMP) signaling pathways (Schneider et al., 2014; Wang et al. 2013). In addition, AHR activation, inhibition, or knockdown during mouse ES cell differentiation significantly inhibits cardiomyocyte contractility (Wang et al., 2013).
To investigate whether there was a critical time window of development for TCDD-induced impairment of cardiogenesis, we treated ES cells with TCDD at daily differentiation intervals and, as a measure of TCDD-induced cardiogenesis, assessed the loss of the ability of cardiomyocytes to contract spontaneously. Our results reveal that the early phase of EB formation, between days 0 and 3 of non-directed, ie, spontaneous, differentiation, is a critical developmental time window for TCDD impairment of cardiogenesis. In addition, loss of contractility due to AHR-disruption is accompanied by functional interference with genes in the TGFβ/BMP and WNT signaling pathways.
MATERIALS AND METHODS
Culture of embryonic stem cells, in vitro differentiation, and treatments.
AHR wild-type (Ahr+/+) ES cells were the C57BL/6N C-2 line previously described by others (Gertsenstein et al., 2010; Wang et al., 2013). The AHR-knockout (Ahr−/−) ES cells were established in our laboratory by standard procedures (Doetschman et al., 1987) from GD3.5 blastocysts of C57BL6/J Ahr−/− pregnant dams. Both undifferentiated ES cells were maintained in ES medium, consisting of high glucose Dulbecco’s minimal essential medium (DMEM; Gibco, Grand Island, New York) supplemented with 15% ES cell qualified fetal bovine serum (knockout serum replacement; Gibco), 2 mM glutamine, 1% nonessential amino acids, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.1 mM β-mercaptoethanol, and 1000 U/ml ESGRO mLIF (Millipore, Billerica, Massachusetts). Cells were seeded in 0.1% gelatin-coated plates at 37°C, 95% humidity with 5% CO2, and passaged every second or third day. Non-directed differentiation (hereinafter referred to simply as differentiation) was initiated on day 0 by transferring the cells to DMEM medium without LIF supplemented with 15% non-ES qualified fetal bovine serum and suspended at a concentration of 40 000–70 000 cells/ml to form EBs in hanging drops. Sixty 20 -μl aliquots were pipetted onto the inner surface of a bacterial Petri dish lid and the lid was inverted over the bottom plate containing 15 ml phosphate-buffered saline to provide humidity. Plates were incubated at 37°C for 3 days and thereafter the EBs were flushed with differentiation medium and incubated in 24-well or 10-cm plates for varying periods of time.
When needed, cultures were treated with 1 nM TCDD, the concentration commonly used for tissue culture work with the high-affinity AHR of C57BL/6 mice and established in our prior work to be effective in AHR activation while causing no overt cytotoxicity (Wang et al., 2010, 2013). TCDD was dissolved in DMSO and diluted in DMEM to reach the desired concentration when added to growth medium. DMSO in DMEM served as the vehicle control, the final concentration of DMSO was ≤ 0.05 % of the final volume. TCDD treatment was kept in the culture throughout the experiment, except when indicated otherwise. As needed, some cultures were also treated with 5 ng/ml Activin A (R&D Systems, Minneapolis, Minnesota), 5 ng/ml BMP4 (R&D Systems), 150 ng/ml Noggin (Stemgent, Cambridge, Massachusetts), 20 ng/ml recombinant mouse WNT3a (R&D Systems), 20 ng/ml WNT5a (R&D Systems), 1 μg/ml anti-WNT3a IgG (R&D Systems) or 1 μg/ml anti-WNT5a IgG (R&D Systems). These antibodies were certified by the manufacturer not to cross-react with other mouse WNTs. The exposure to ligands, antagonists or neutralizing antibodies was conducted either during differentiation days 2–3, 0–3, or throughout days 0–12. Media and treatments were replaced every 48 h, as needed.
Determination of cardiomyocyte contractility
To measure cardiomyocyte contractility, EBs were individually plated on wells of 24-well plates, allowed to differentiate for the indicated length of time in the presence of vehicle, TCDD, growth factors or antibodies, and visually scored daily for the presence of beating cell clusters by a researcher blinded to treatment. Beating became evident starting on days 6–7 with an experimental variation of ± 2 days, and was maximal by day 11–13. As previously determined (Wang et al., 2010, 2013), approximately 40% of the cells in the EBs were positive for cardiac troponin-T and were considered to be cardiomyocytes. If a well had more than 1 beating cluster, it was scored as a single beating EB.
Determination of activin A, BMP4, and WNT levels in EB culture medium
Ahr+/+ ES cells were allowed to differentiate by forming EBs in the presence of DMSO or TCDD. To determine extracellular Activin A, BMP4, and WNT levels, the culture medium of days 1–3 EBs was obtained from triplicate hanging drops of each treated culture. Culture levels of these signal proteins were determined using ELISA kits for Activin A (DAC00B, R&D Systems, MN), BMP4 (MBS703931, MyBioSource), WNT3a (DL-WNT3A-Mu, DongLin Sci&Tech, China), and WNT5a (MBS932852, MyBioSource). All determinations were conducted according to the manufactures’ protocols.
Transmission electron microscopy
Three-day old EBs treated with or without TCDD were collected and immediately fixed in phosphate-buffered 3% glutaraldehyde for 24 h and submitted to the Pathology Research Core at Cincinnati Children’s Hospital Medical Center (Cincinnati, Ohio) for sample processing and sectioning for electron microscopic examination. Samples were washed 3 times in 0.1 M cacodylate buffer and postfixed in 1% osmium tetroxide buffered with cacodylate, pH 7.2, at 4°C for 1 h. After dehydration in serial alcohol and propylene oxide solutions, samples were infiltrated with and embedded in LX112. Thin sections were stained with uranyl acetate and lead citrate. Imaging was performed on a transmission electron microscope (7600; Hitachi). Five non-overlapping ultraphotomicrographs per grid were taken at 15 000×, and 70 000× magnifications and evaluated using Image J 1.47 h software.
Mitochondria quantification
The relative amounts of mitochondria on days 2 and 4 EBs or day 11 beating cardiomyocytes were determined using real time-polymerase chain reaction (RT-PCR) to measure the ratio of mitochondrial DNA [mtDNA] to nuclear DNA [nDNA], as previously described (Stites et al., 2006). Day 11 beating cardiomyocytes were manually dissected from beating areas of differentiated EBs under a dissecting microscope. Total DNA was extracted with the DNeasy Blood & Tissue Kit (Qiagen, Valencia, California) following the manufacturer’s specification. The PCR reactions were conducted in triplicate in a total volume of 25 μl containing SYBR Green PCR Master Mix (Applied Biosystems, Grand Island, New York). Amplification was performed in a Stratagene M×3000P RT-PCR system (Agilent Technologies, Santa Clara, California); the reaction was heated to 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 15 s and annealing elongation at 60°C for 60 s. Detection of the fluorescent product was carried out during the 72°C extension period, and emission data were quantified using threshold cycle (Ct) values. The targets were the genes coding for the nuclear cytochrome P450 Cyp1a1 and the mitochondrial nicotinamide adenine dinucleotide dehydrogenase-5 (Nd5). Primers for the Cyp1a1 promoter region were: forward: 5′-AGGCTCTTCTCACGCAACTC-3′; reverse: 5′-TAAGCCTGCTCCATCCTCTG-3′. Primers for Nd5 were: forward 5′-TGGATGATGGTACGGACGAA-3′; reverse 5′-TGCGGT TATAGAGGATTGCTTGT-3′.
Total RNA isolation, reverse transcription, and RT reverse transcription PCR
Total RNA was extracted with the RNeasy Mini Kit (Qiagen) according to the manufacturer’s specifications. First-strand complementary DNAs (cDNAs) were synthesized from 0.5 μg of total RNA using the RT2 First Strand Kit (Qiagen) according to the manufacturer’s instruction. The resulting cDNA products were dissolved in a final volume of 111 μl, and 102 μl was subjected to RT-PCR detecting genes involved in TGFβ/BMP or WNT signaling pathways. The RT2 profiler PCR Arrays focusing on mouse TGFβ/BMP or mouse WNT signaling pathways were purchased from Qiagen (Qiagen). The PCR reactions were conducted according to the manufacturer’s protocols in a total volume of 25 μl containing RT2 SYBR Green ROX qPCR Master Mix (Qiagen). Amplification was performed in a Stratagene Mx3000P RT-PCR system (Agilent Technologies); the reaction was heated to 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 15 s and annealing elongation at 60°C for 60 s. Detection of the fluorescent product was carried out during the 72°C extension period, and emission data were quantified using threshold cycle (Ct) values. All Ct were then normalized to values for Gapdh mRNA.
RNA.seq data analysis
All steps of library construction, cluster generation, and HiSeq (Illumina, San Diego, California) sequencing were performed with biological duplicate samples by the Genomics, Epigenomics and Sequencing Core of the Department of Environmental Health, University of Cincinnati. One microgram of total RNA with RNA integrity number ≥ 7.0 (Agilent 2100 Bioanalyzer; Agilent Technologies, Santa Clara, California) was used as input. Apollo 324 NGS Library Prep System (Wafergen, Fremont, California) was used for automatic polyA RNA purification, library construction and PCR indexed library clean up. In brief, the isolated polyA-RNA was RNase III fragmented, adaptor-ligated and Superscript III reverse transcriptase (Lifetech, Grand Island, New York) converted into cDNA, followed by Apollo 324 automatic purification using Agencourt AMPure XP beads (Beckman Coulter, Indianapolis, Indiana). Next, using the cDNA as template and paired universal/index-specific primers for PCR under 12 cycles, sample-specific index was added to each adaptor-ligated cDNA sample and the amplified library was enriched by using Apollo 324 automatic AMPure XP beads purification. To check the quality and yield of the purified library, a library aliquot was analyzed by Bioanalyzer (Agilent, Santa Clara, California) using DNA high sensitivity chip. To accurately quantify the library concentration for the clustering, the library was 1:104 diluted in dilution buffer (10 mM Tris-HCl, pH 8.0 with 0.05% Tween 20), and qPCR measured by Kapa Library Quantification kit (Kapabiosystem, Woburn, Massachusetts) using ABI’s 9700HT RT-PCR system (Lifetech). Individually indexed libraries were proportionally pooled for clustering in cBot system (Illumina, San Diego, California). Libraries at the final concentration of 12 pM was clustered onto a flow cell using Illumina’s TruSeq SR Cluster kit v3, and sequenced for 50 cycles using TruSeq SBS kit on Illumina HiSeq system.
Sequence reads were demultiplexed and exported to fastq files using CASAVA 1.8 software (Illumina). The reads were then aligned to the reference genome (mm 10) using TopHat aligner (Trapnell et al., 2009). The counts of reads aligning to each gene’s coding region were summarized using ShortRead (Morgan et al., 2009) and associated Bioconductor packages for manipulating and analysis of next-generation sequencing data and custom-written R programs. Differential gene expression analysis was performed separately at each of the 3 different time points (days 1–3). We performed statistical analysis to identify differentially expressed genes for each comparison using the negative-binomial model of read counts as implemented in the Bioconductor DESeq package (Anders and Domenech, 2010).
To analyze gene expression changes across time, we compared days 1 and 3 EBs treated with 1 nM TCDD with days 1 and 3 vehicle-treated EBs, respectively. Genes in the TGFβ/BMP and WNT signaling pathways were hierarchically clustered based on the log2 fold change with positive values denoting up-regulation and negative values denoting down-regulation. Clustering was performed using custom-written R programs.
Statistical analysis
Data are presented as mean ± SEM from at least 3 independent experiments. Significant differences between groups were calculated using 2-way repeated measures ANOVA. P-values are indicated in the corresponding figure legends.
RESULTS
Early Differentiation Period Between Days 0 and 3 Is a Critical Time Window for TCDD-Induced Impairment of Cardiogenesis
Pluripotent ES cells can develop into most embryonic lineages in culture (Keller, 1995; Smith, 2001), including cardiomyocytes (Yamashita et al., 2005). Differentiation of ES cells into cardiomyocytes in vitro is readily identifiable microscopically by visual examination of differentiating EBs that spontaneously develops a contractile phenotype. Those ES cell-derived beating cardiomyocytes show all the properties characteristic of cardiac cells, such as electrophysiologic phenotypes and formation of stable intracardiac grafts when injected into mice (Klug et al., 1996). In our previous studies, continuous treatment of differentiating ES cells with TCDD disrupted the cardiomyocyte beating phenotype in a dose-dependent manner that was dependent on AHR activation (Wang et al., 2013). Here, we extended this observation by further characterizing whether there was a critical developmental time window when differentiating ES cells were most sensitive to TCDD exposure. Wild-type Ahr+/+ differentiating ES cells exposed to TCDD during the period ranging from day minus-2 or days 0–11, showed significantly lower numbers of beating EB-derived cultures that never reached >25% of control values (Figure 1A). In contrast, when TCDD treatment started at the end of days 3, 5, or 7, there was no significant decrease in the percent of beating EBs in TCDD-treated groups compared with controls (Figure 1A). Unlike wild type ES cells, differentiating Ahr−/− ES cells were resistant to TCDD-induced loss of contractility, although they showed a reproducible 1-day delay in establishing the phenotype, possibly an effect of TCDD’s hydrophobicity on cellular processes (Figure 1B). These results indicate that there is a critical time window when the differentiating ES cells are most vulnerable to AHR-dependent TCDD-induced impaired cardiogenesis and that this window is between days 0 and 3, the period when the ES cells in the hanging drop aggregate to form EBs. Beyond differentiation day 3, the cells in the EBs appear to be no longer sensitive to TCDD-induced loss of beating phenotype.
FIG. 1.
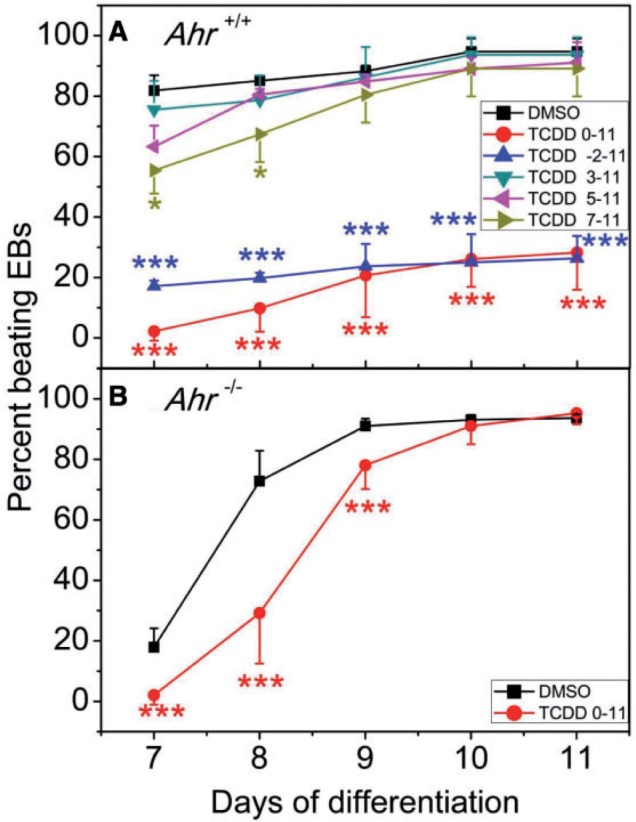
Determination of a window of susceptibility to TCDD-mediated loss of cardiomyocyte contractility. Ahr+/+ and Ahr−/− ES cells were incubated at 37°C for 3 days in hanging drops. On the third day the EBs were flushed with differentiation medium and individually incubated in 24-well plates for the duration of the experiment. TCDD treatment was (A) for the periods of time indicated for the Ahr+/+ cells, or (B) during the whole term of differentiation from days 0 to 11 for the Ahr−/− cells. EBs were examined daily under the microscope for the presence of a rhythmic beating phenotype. Data represent the mean ± SD of 3 independent experiments. The repeated measures 2-way ANOVA P-values for panel (A) were 1.15 × 10−12 for ‘Day of Differentiation’ ;1.86 × 101.86e-05 for ‘Treatment’ and 2.83 × 10−2 for ‘Day of Differentiation’ and ‘Treatment’ combined. The P-values for panel (B) were 2 × 10−16 for ‘Day of Differentiation’; 6.5 × 10−4 for ‘Treatment’ and 3.8 × 10−13 for ‘Day of Differentiation’ and ‘Treatment’ combined. The asterisks denote significant differences at the time points indicated relative to control.
Early TCDD Treatment Disrupts the Expression of Genes Involved in TGFβ/BMP and WNT Signaling Pathways
Our earlier studies on the effect of TCDD on ES cell gene expression were focused on the regulatory consequences of TCDD treatment on the late differentiation days comprised between 5 and 14 days post removal of LIF. At the earliest time point on day 5, analysis of gene expression profiling using the Ingenuity Knowledge Base (Ingenuity System-IPA) revealed that the most significant changes caused by TCDD were to the canonical WNT3A/β-Catenin signaling pathway and to the non-canonical WNT1. Several other signaling pathways were also significantly affected by TCDD treatment, including the BMP2/4 and TGFβ pathways (Wang et al., 2013). The signaling pathways controlled by TGFβ/BMP/Activin and WNT are essential for panmesoderm induction and resulting cardiomyocyte differentiation (Fukuda and Yuasa, 2006; Laflamme and Murry 2011); to test the hypothesis that these pathways cross-talked with the AHR/TCDD elicited signals, we used global gene expression profiling during the 3 days of EB differentiation to characterize the effect of TCDD on the expression of genes regulated by TGFβ/BMP and WNT. Ahr+/+ ES cells were allowed to differentiate in hanging-drops and samples were collected on days 1 and 3 for RNA.seq analysis. A significant number of genes in the TGFβ/BMP pathway, including those coding for Nodal, anti-Mullerian hormone receptor type 2, Activin receptor-1, Smad3/4/5 and various TGFβ superfamily inhibitors were down-regulated by TCDD at both day 1 and 3 of differentiation; others, like tgfbr1 and Bmp2 were upregulated at day 1 and down-regulated at day 3 (Figure 2A). Interestingly, Inhba, encoding the Inhibin βA subunit of both Inhibin and Activin A, was down-regulated on day 1 and upregulated on day 3. A homodimer of 2 Inhibin βA subunits forms Activin A, while a heterodimer of one Inhibin βA and one Inhibin α subunit forms Inhibin. In contrast to Inhibin βA, Inhibin α expression was depressed by 50% on day 3, suggesting that Activin A, not Inhibin βA, was the one upregulated by TCDD on day 3, while Inhibin A was not significantly changed.
FIG. 2.

Gene expression clustering of RNA.seq data in (A) TGFβ/BMP4 and (B) WNT signaling pathways. The heatmaps show the hierarchical clustering of RNA.seq data for days 1 and 3 of Ahr+/+ differentiating cells treated with 1 nM TCDD relative to DMSO vehicle. C, RNA.seq expression changes were verified by qRT-PCR for 30% of the genes shown in (A) and (B).
TCDD exposure also deregulated the expression of genes in the WNT pathway. A group of transcription factors including Ahr, Sox4, Nanog, and Tcf4, the latter 2 controlling maintenance of cell proliferation, were up-regulated on both days 1 and 3. On the other hand, Wnt3a, Wnt9a, and genes involved in WNT signal transduction, such as Wsp2 and Btrc were downregulated at both time points (Figure 2B). Gene expression changes determined by RNA.seq were confirmed by qRT-PCR for 30% of the TGFβ- and WNT-regulated genes. Changes determined by RNA.seq were confirmed with 3 exceptions, Gdf7, Fos and Gsc, in which the direction of change was different for RNA.seq and RT-PCR (Figure 2C). These results indicate that early developmental signaling mediated by the TGFβ/BMP and canonical and non-canonical WNT pathways are deregulated by TCDD treatment during the critical time window when TCDD disrupts cardiomyocyte beating.
TCDD-Dependent Increase in Activin A Secretion Suppresses Contractility
Both TGFβ and WNT are highly conserved secreted factors with roles in development and normal homeostasis. Knockout of the AHR elevates the secretion of active TGFβ in primary hepatocytes (Gonzalez and Fernandez-Salguero, 1998; Zaher et al., 1998), suggesting that AHR activation by TCDD may repress the secretion of other members of the TGFβ superfamily. Cross-talk between AHR and WNT pathways has been well established (Schneider et al., 2014) and the temporal modulation of canonical WNT signaling in human pluripotent stem cells has been shown to result in robust cardiomyocyte differentiation (Lian et al., 2013). Hence, it is reasonable to hypothesize that the expression changes in genes of the TGFβ/BMP and WNT pathways could be the consequence of the suppressed expression of TGFβ/BMP and WNT proteins, which would cause the dysfunction of the pertinent signal transduction pathways by depressing the secretion of the early signaling factors, including Activin A, BMP4, and WNT. To test this hypothesis and determine the role that TCDD treatment might have on Activin A levels and function, we measured Activin A in the medium of differentiating Ahr+/+ EBs by ELISA.
The level of secreted Activin A in the differentiation culture medium was unchanged by TCDD treatment during differentiation days 1 and 2, but was significantly increased in TCDD treated cells on day 3 (Figure 3A). To determine whether elevated Activin A levels had an effect on contractility, we supplemented the medium with recombinant Activin A during the whole term of differentiation, between days 0 and 12. This addition significantly decreased the number of beating EB-derived cardiomyocyte cultures in vehicle control treated groups but not so much in TCDD treated ones (Figure 3B), suggesting that TCDD and Activin A might have common targets and that elevation of Activin A levels in the medium by TCDD treatment was at least partly responsible for suppression of cardiomyocyte contractility.
FIG. 3.
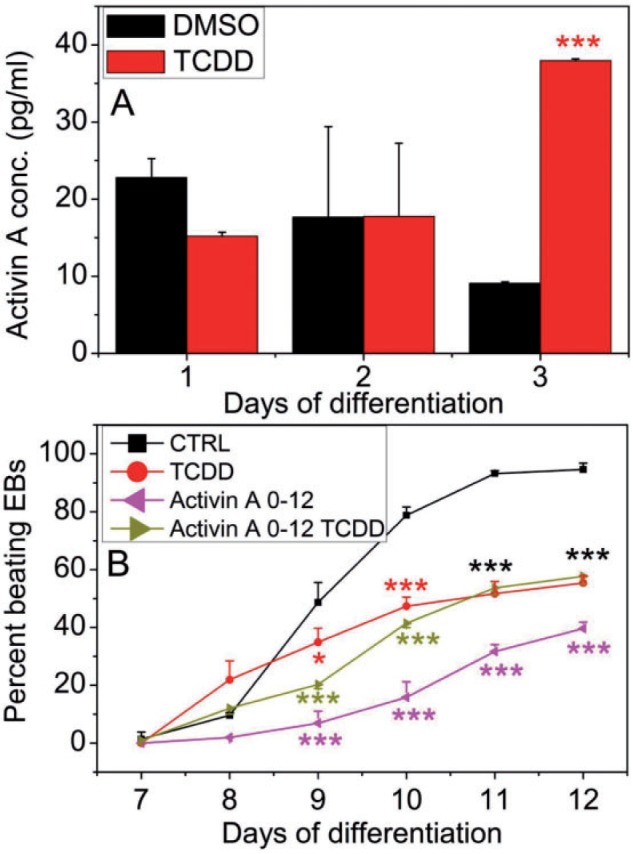
Activin A effect on Ahr+/+ cardiomyocyte contractility. A, Secreted Activin A levels in Ahr+/+ EB culture medium determined by ELISA. B, Effect of supplementing the differentiation medium with Activin A on the contractility of Ahr+/+ EB-derived cultures treated with or without TCDD. The number of EBs that developed a rhythmic beat was scored daily under the microscopy. TCDD and Activin A treatment were maintained from days 0–12. Data represent the mean ± SD of independent experiments. The repeated measures 2-way ANOVA P-values were 2 × 10−16 for ‘Day of Differentiation’; 1.32 × 10−5 for ‘Treatment’ and 2 × 10−16 for ‘Day of Differentiation’ and ‘Treatment’ combined. The asterisks denote significant differences at the time points indicated relative to control.
TCDD-Mediated Suppression of BMP4 Secretion Also Suppresses Cardiomyocyte Contractility
BMP4 plays a central role in many organ developmental processes, including cardiogenesis (Hogan 1996a,b). Studies to characterize the role of BMP signaling in cardiomyocyte differentiation have shown that administration of soluble BMP2 or BMP4 to explant chick embryo cultures induces cardiac differentiation (Schultheiss et al., 1997). We also measured BMP4 levels in the differentiation medium to determine whether TCDD treatment altered BMP4 secretion during the first 3 days of differentiation. We found that BMP4 secretion was not changed relative to control on differentiation day 1 but was significantly suppressed by TCDD on differentiation days 2 and 3 (Figure 4A). If TCDD induced impaired cardiogenesis were mediated by suppression of BMP4 secretion, we might expect that supplementing the culture medium with added ligand during the first 3 days of differentiation might be sufficient to rescue the cells from TCDD-induced impaired cardiogenesis. To test this hypothesis, ES cells were allowed to differentiate in the presence or absence of TCDD supplemented with soluble BMP4 or with its repressor, Noggin. Administration of BMP4 on differentiation days 2 and 3 significantly increased the number of beating EB-derived cardiomyocyte cultures exposed to TCDD, bringing the number of contractile cultures up to the level of the control group (Figure 4B). In contrast, repression of BMP4 by added Noggin significantly decreased the number of beating cultures (Figure 4C). These results indicate that suppression of BMP4 secretion by TCDD exposure during the first 3 days of differentiation may be at least in part responsible for the loss of cardiomyocyte contractility, which can be reversed by BMP4 supplementation.
FIG. 4.
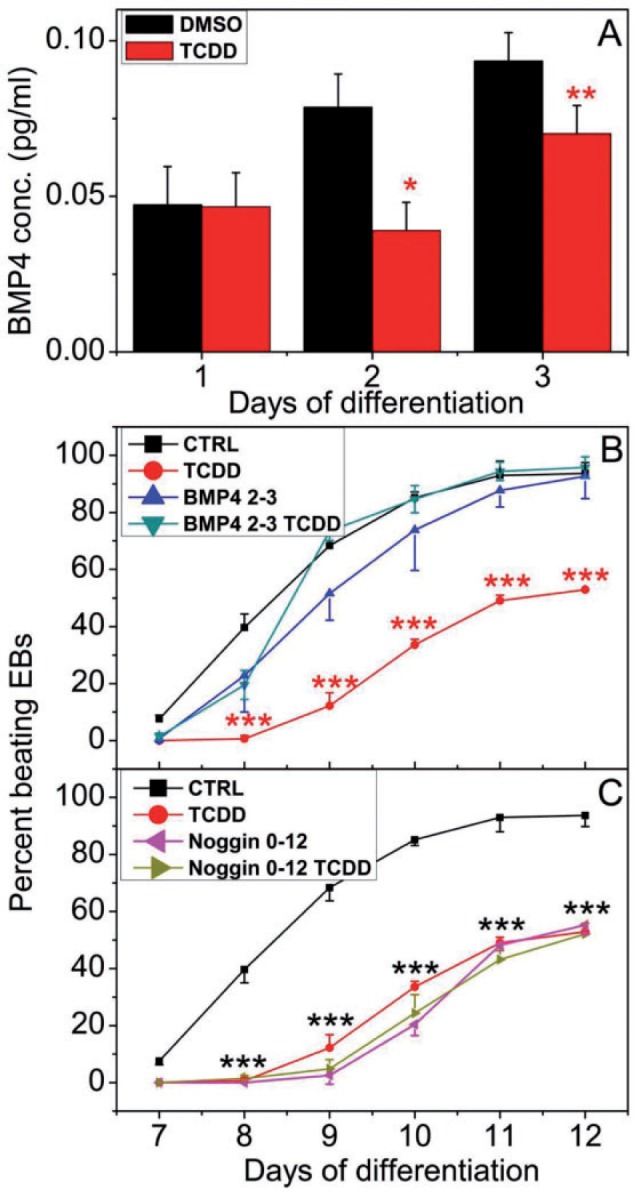
BMP4 effect on Ahr+/+ cardiomyocyte contractility. A, Secreted BMP4 level in Ahr+/+ EB culture medium determined by ELISA. B, Effect of supplementing differentiation medium with BMP4 or (C) BMP4 antagonist Noggin, on the contractility of Ahr+/+ EB-derived cultures treated with or without TCDD. Experimental details as in Figure 3. Data represent the mean ± SD of independent experiments. The repeated measures 2-way ANOVA P-values for panel (B) were 2 × 10−16 for ‘Day of Differentiation’; 1.43 × 10−4 for ‘Treatment’ and 3.01 × 10−9 for ‘Day of Differentiation’ and ‘Treatment’ combined. The P-values for panel (C) were 2 × 10−16 for Day of Differentiation’; 5.7 × 10−7 for ‘Treatment’ and 1.12 × 10−10 for ‘Day of Differentiation’ and ‘Treatment’ combined. The asterisks denote significant differences at the time points indicated relative to control.
TCDD-Mediated Suppression of WNT Secretion Also Suppresses Cardiomyocyte Contractility
WNT proteins, a large family of secreted signaling molecules, are one of the key regulators of cardiac specification, progenitor expansion and myocardial growth both in vivo and in vitro (Cohen et al., 2008; Gessert and Kuhl, 2010). To test whether disruption of contractility by TCDD was due to altered secretion of WNT proteins, we measured secreted canonical WNT3a and non-canonical WNT5a levels by ELISA in the culture medium of EBs during the first 3 days of differentiation. TCDD treatment significantly suppressed WNT3a secretion on days 2 and 3, while WNT5a secretion was also significantly decreased on day 3 (Figs. 5A and B).
FIG. 5.

Effect of WNT3a and WNT5a on Ahr+/+ cardiomyocyte contractility. Secreted levels of (A) WNT3a and (B) WNT5a in Ahr+/+ EB culture medium were determined by ELISA. Effect of supplementing the Ahr+/+ EB medium for the first 3 days of differentiation with (C) WNT3a or (D) WNT5a; or with (E) anti-WNT3a IgG or (F) anti-WNT5a IgG on the contractility of EB-derived cultures treated with or without TCDD. Experimental details as in Figure 3. Data represent the mean ± SD of independent experiments. The repeated measures 2-way ANOVA p-values for panel (C) were 2 × 10−16 for ‘Day of Differentiation’; 1.5 × 10−6 for ‘Treatment’ and 3.5 × 10−6 for ‘Day of Differentiation’ and ‘Treatment’ combined. The P-values for panel (D) were 2 × 10-16 for ‘Day of Differentiation’; 8.6 × 10-6 for ‘Treatment’ and 7.2 × 10−6 for ‘Day of Differentiation’ and ‘Treatment’ combined. The P-values for panel (E) were 2 × 10 − 16 for ‘Day of Differentiation’; 1.6 × 10−8 for ‘Treatment’ and 2 × 10−16 for’Day of Differentiation’ and ‘Treatment’ combined. The P-values for panel (F) were 2 × 10−16 for ‘Day of Differentiation’; 1.5 × 10−8 for ‘Treatment’ and 2 × 10−16 for ‘Day of Differentiation’ and ‘Treatment’ combined. The asterisks denote significant differences at the time points indicated relative to control.
If suppressed secretion of WNT proteins during the first 3 days of differentiation resulted in inhibited contractility, we would expect that addition of WNT back to the culture medium would counter the TCDD-mediated impaired cardiogenesis. To test this hypothesis, we supplemented the culture medium with either WNT3a or WNT5a for the first 3 days of differentiation. In agreement with our hypothesis, supplementation with either soluble WNT3a or WNT5a increased the percent of beating TCDD-treated EBs, completely countering the effect of TCDD on contractility (Figs. 5C and D). In confirmation of these results, addition of neutralizing WNT3a or WNT5a antibodies during the same period of time inhibited contractility almost completely in both control and TCDD-treated EBs (Figs. 5E and F). These data indicate that WNT is also a TCDD target during cardiomyocyte differentiation, and illustrate the signaling complexity responsible for the cardiac contractility.
Developmental TCDD Treatment Disrupts Mitochondrial Structure and Abundance
Our Ingenuity Knowledge Base (Ingenuity System-IPA) analysis of gene expression pathways also showed a significant disruption of mitochondrial function as a consequence of TCDD treatment. To determine whether there were AHR-dependent structural consequences of mitochondrial dysfunction, we examined Ahr+/+ and Ahr−/− EBs for the abundance and quality of mitochondria. The relative copy number of mitochondrial mtDNA to nuclear nDNA was measured by comparing the qRT-PCR ratio of the mitochondrial Nd1 gene to the nuclear Cyp1a1 gene. Two-day old wild-type EBs showed no difference in mitochondrial abundance between TCDD treated and control groups, but by day 4 TCDD-treated EBs showed a significant 30% increase in the mtDNA/nDNA ratio compared with vehicle control (Figure 6A). In contrast, on both days 2 and 4, Ahr−/− EBs had a significantly lower mitochondria copy number, approximately 50% of the wild-type, a number that was refractory to TCDD-induced copy number increase (Figure 6A).
FIG. 6.
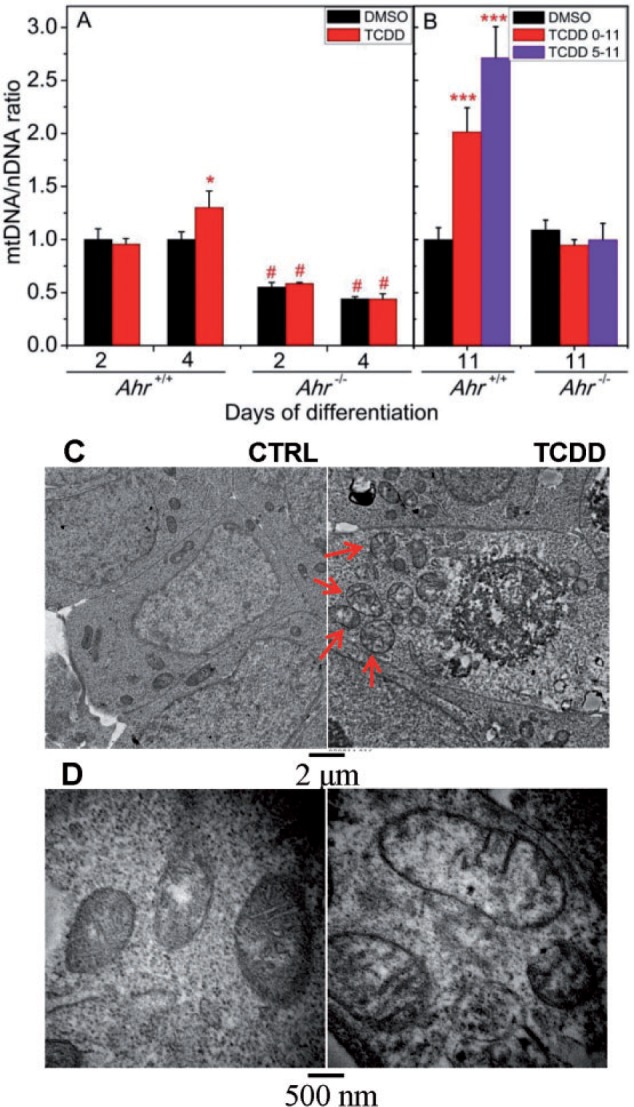
TCDD treatment disrupts mitochondrial structure and abundance. A, MtDNA/nDNA ratio on days 2 and 4 of differentiation for both Ahr+/+ and Ahr−/− EBs treated with TCDD or DMSO control. Data represent the mean ± SD of 3 independent experiments. TCDD treated was significantly different from vehicle control at *P < .05 on day 4. Ahr+/+ and Ahr−/− were significantly different (#P < .001) for the same treatment and time point. B, mtDNA/nDNA ratio on day 11 beating cardiomyocytes from either Ahr+/+ or Ahr−/− ES cells treated by TCDD or vehicle control from days 0 to 5 of differentiation. C, Ultra-thin sections of 3-day old EBs from Ahr+/+. ES cells treated with DMSO vehicle or TCDD were used for ultrastructural evaluation by transmission electron microscopy. Illustrative ultraphotomicrographs of higher density of mitochondria (arrows) within EBs treated with TCDD. Individual or clusters of mitochondria showed ultrastructural features of stress and degeneration (arrows), as evidenced by focal to global swelling, loss of matrix density, cristae unpacking, disorganization, and cristolysis.
The observed effect of TCDD on contractility of Ahr+/+ cardiomyocytes was never absolute; even when treatment was done during the critical window of days 0–3, roughly 25% of EBs retained contractility (Figure 1A). Furthermore, when EB treatment was begun beyond the critical window, on days 3, 5, or 7, 100% of the EBs were contractile. To examine whether beating cardiomyocytes that were treated with TCDD also suffered mitochondrial dysfunction, we micro-dissected day 11 beating cardiomyocytes from both wild-type and knockout cells that had been untreated or treated with TCDD from day 0 or day 5 to day 11. In the day 11 differentiated Ahr+/+ cardiomyocytes, the ratio of mtDNA to nDNA was also affected by the earlier TCDD treatment, which increased the mtDNA/nDNA ratio 2- to 2.5-fold over control (Figure 6B). In contrast, there was no significant change of mtDNA copy number in TCDD-treated Ahr−/− beating cardiomyocyte compared with control (Figure 6B), suggesting that TCDD-induced mitochondrial dysfunction is also AHR-dependent. Ultrastructurally, the increase of mitochondria DNA copies correlated with their higher density in the cytoplasm. Furthermore, mitochondria, individually or in clusters, showed ultrastructural features of stress and degeneration, as evidenced by focal to global swelling, loss of matrix density, cristae unpacking, disorganization, and cristolysis which affected higher numbers of mitochondria in TCDD-treated Ahr+/+ cells (Figs. 6C and D).
DISCUSSION
The results presented here show that AHR activation by TCDD during the early differentiation period comprised between days 0 and 3 of embryoid body formation after removal of LIF, significantly suppressed the development of the contractile cardiomyocyte phenotype, defining a developmental window of impaired cardiogenesis. At this time, we can only speculate as to the mechanisms responsible for the narrow window of sensitivity, which await direct hypothesis-driven analyses. This early period is the time when EBs are experiencing self-organization and axis formation to progress, in the context of cardiogenesis, from pluripotent ES cells to panmesoderm and cardiac mesoderm, a critical time window that requires not only the activation of multiple signaling pathways, including WNT, TGFβ/BMP, and Activin among others, but also the combinatorial integration of developmental signals elicited by the cross-talk among them (Ten Berge et al., 2008). Consistent with the participation of these pathways, inhibition of the beating phenotype is accompanied by parallel disruption of the TGFβ/BMP and WNT signaling pathways. Furthermore, TCDD treatment led to significant changes in the levels of these agents in the culture medium, causing an increase of Activin A and a decrease of BMP4 and WNTs secretion. Efficient cardiac differentiation requires the optimal combination of Activin A and BMP4 signaling (Kattman et al., 2011), hence it is plausible that inhibition of the beating phenotype may result from the imbalance of Activin A, BMP4, and WNT signals caused by TCDD treatment. In support of this concept, administration of BMP4, WNT3a, or WNT5a on the first 3 days of differentiation is sufficient to counter the effects of TCDD-induced impaired cardiogenesis and blocking BMP4 or WNT by an antagonist or by specific antibodies suppresses the beating phenotype even more than TCDD. These results strongly suggest a causal connection between early TCDD treatment, interference with early BMP, Activin A, and WNT signaling, and later disruption of cardiomyocyte function. Thus, it is not unexpected that single supplementation with either BMP4 or WNT is sufficient to counter TCDD toxicity, further documenting the intertwined cross-talk of those signaling pathways. Conversely, disruption of just one of the factors—Activin A, in this case—may cause the imbalance of the whole regulatory network. Surprisingly, increases of Activin A levels due to TCDD treatment are not detected until day 3 post-differentiation, a time when the effect of TCDD on contractility is no longer evident. One possible explanation for this apparent discrepancy is that inhibition of contractility might be the cumulative effect of subtle accompanying increases during days 1 and 2 of other members of the TGFβ family, such as Nodal or Integrin, or possibly, TGFβ itself.
Increased WNT signaling is a potent activator of mitochondrial biogenesis and reactive oxygen species generation (Wang et al., 2005). In hematopoietic progenitor cells, conditional expression of an active form of β-catenin resulted in a loss of mitochondrial membrane potential and induction of the intrinsic mitochondrial apoptotic pathway. Furthermore, non-canonical WNT5a regulates mitochondrial dynamics in neurons (Godoy et al., 2014; Serrat et al., 2013). In this study, we find that beating cardiomyocytes that were treated with TCDD as panmesoderm cells or cardiomyocyte precursors show changes in mtDNA copy number. These cells however, retain expression of cardiomyocyte markers, including Nkx2-5, Myh6, Myh7, Mlc2v, and Cx40 (Wang et al., 2013). MtDNA copy number is cell type specific and tightly controlled and regulated during development, with changes constituting a biological response to damage and dysfunction (Spikings et al., 2007; Thundathil et al., 2005). In this study, TCDD treatment caused an increase in mtDNA copy number as early as differentiation day 4 and the effect appeared to be cumulative, increasing to more that 2.5-fold over control by day 11. TCDD treatment has long been shown to cause mitochondrial dysfunction, being considered as a biomarker of exposure to polycyclic aromatic hydrocarbons (Kim et al., 2014). Effects have been reported in multiple organs and cell types due to multiple mechanisms, including inhibited mitochondrial transcription, disrupted mitochondrial transmembrane potential, inhibition of the mitochondrial electron chain, increased generation of reactive oxygen species, decreased ATP synthesis, and altered mitochondrial copy number (Aly and Domenech, 2009; Biswas et al., 2008; Chen et al., 2010; Kennedy et al., 2013; Park et al., 2013; Pavanello et al., 2013; Shertzer et al., 2006). In the context of cardiac consequences, zebrafish embryos exposed to TCDD show increased expression levels of mitochondrial energy transfer genes and subtle induction of several proteins coded by mitochondrial genes, which may be related to cardiomyopathy (Handley-Goldstone et al., 2005). Acute cardiac mitochondrial oxidative damage has also been reported in Wistar rats after TCDD exposure (Pereira et al., 2013).
Interplay between the TGFβ/BMP and WNT pathways has been known for quite a long time and is important for cell fate determination; the 2 pathways are intertwined at multiple levels, reciprocally regulating each other’s ligand production, critical for establishing extracellular gradients of these morphogens during embryonic development, sharing common target genes in a synergistic manner (Guo and Wang, 2009). During mesoderm differentiation, cross-talk between these 2 pathways control differentiation into panmesoderm, cardiac progenitor cells, and cardiomyocytes (Figure 7). AHR cross-talk with the WNT pathway has long been recognized (Schneider et al., 2014). By interference with each developmental stage, TCDD not only derails the regulatory pathways, but it also alters the expression of signature genes in each developmental stage, from Brachyury in the panmesoderm to Nkx2.5, Gata4, and various other genes in the cardiomyocyte progenitor cells (Figure 7). The factors that regulate development are not fixed in a static signaling state, but are in a dynamic state that receives and sends signals to one another giving rise to the changing phenotype of the developing embryo. Hence, the regulatory elements of relevant genes is in a constant change of functional states and it may be expected that interference with their dynamic progress by an environmental agent such as TCDD might alter the concerted pace and outcome of development. AHR might be a master upstream regulator that controls the expression of genes in signaling pathways crucial for embryonic development and adult tissue homeostasis.
FIG. 7.

ES cell differentiation trajectory into cardiomyocytes and impact of TCDD. Modified from (Laflamme and Murry, 2011; Rajala et al., 2011). The specification of the cardiomyocyte lineage involves a transition through a sequence of increasingly restricted progenitor cells, proceeding from a pluripotent state to mesoderm and to cells committed to cardiovascular fates. Growth factors that regulate fate choices are listed at branch points in blue. Antagonists for each growth factor are listed in black. Key transcription factors and surface markers for each cell state are listed under the corresponding cell types in purple. Red stands for upregulation while green stands for down-regulation by TCDD treatment at the specific time point. The critical time window when the cells are most vulnerable to TCDD-induced impaired cardiogenesis is the early time period between days 0 and 3 (shown by a double-lined red box), a time when pluripotent ES cells differentiate into panmesoderm. Within this time window, TCDD treatment increases Activin A and suppresses BMP4 and WNT secretion. Administration of BMP4, WNT3a or WNT5a counters the TCDD effects. Activin A increase on days 3–4 leads to the inhibition of the transition from cardiac mesoderm to cardiac progenitor cells. The AHR/TCDD axis also inhibits the WNT signals that promote the cardiac progenitor lineage. TCDD-induced mitochondrial dysfunction is a late event, independent from this critical time window (shown by a single-lined red box). Full color version available online.
CONCLUSIONS
Results from this study add to the growing body of evidence that developing organisms are more sensitive to toxicant exposure than their more developed counterparts. TCDD exposure during the initial differentiation stages in mouse ES cells disrupts TGFβ/BMP and WNT signaling pathways essential for cardiac development. Many studies in fish, birds, and mammals have shown that the embryo and the fetus are more sensitive to TCDD and TCDD-like chemicals than the adult (Lanham et al., 2012; Peterson et al., 1993; Walker and Catron, 2000). Identification of the critical window of developmental exposure to an environmental toxicant is of extreme importance to our understanding of the health outcome to that exposure (Selevan et al., 2000). Awareness of a developmental period of susceptibility is highly informative, as this knowledge makes it possible to avoid exposure during that critical time. Untimely activation of the Ah receptor may result in the imbalance of the complex regulatory network responsible for attainment and maintenance of cardiovascular homeostasis. The significant role that the AHR plays in cardiovascular development makes the heart a very sensitive target of fetal environmental injury.
FUNDING
This work was supported by Nation Institute of Environmental Health Sciences grants (R01 ES06273, R01 ES10807, R01 ES024744), and the NIEHS Center for Environmental Genetics grant (P30 ES06096).
ACKNOWLEDGMENTS
We thank Dr Ying Xia, Matthew de Gannes, and Andrew Vonhandorf for a critical reading of the article.
REFERENCES
- Abbott B. D., Birnbaum L. S., Perdew G. H. (1995). Developmental expression of two members of a new class of transcription factors: I. Expression of aryl hydrocarbon receptor in the C57BL/6N mouse embryo. Dev. Dyn. 204, 133–143. [DOI] [PubMed] [Google Scholar]
- Aly H. A., Domenech O. (2009). Cytotoxicity and mitochondrial dysfunction of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in isolated rat hepatocytes. Toxicol. Lett. 191, 79–87. [DOI] [PubMed] [Google Scholar]
- Anders S., Huber W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragon A. C., Kopf P. G., Campen M. J., Huwe J. K., Walker M. K. (2008). In utero and lactational 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure: effects on fetal and adult cardiac gene expression and adult cardiac and renal morphology. Toxicol. Sci. 101, 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker D. J. (2007). The origins of the developmental origins theory. J. Intern. Med. 261, 412–417. [DOI] [PubMed] [Google Scholar]
- Biswas G., Srinivasan S., Anandatheerthavarada H. K., Avadhani N. G. (2008). Dioxin-mediated tumor progression through activation of mitochondria-to-nucleus stress signaling. Proc. Natl. Acad. Sci. U. S. A. 105, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S. C., Liao T. L., Wei Y. H., Tzeng C. R., Kao S. H. (2010). Endocrine disruptor, dioxin (TCDD)-induced mitochondrial dysfunction and apoptosis in human trophoblast-like JAR cells. Mol. Hum. Reprod. 16, 361–372. [DOI] [PubMed] [Google Scholar]
- Chien K. R. (2000). Genomic circuits and the integrative biology of cardiac diseases. Nature 407, 227–232. [DOI] [PubMed] [Google Scholar]
- Cohen E. D., Tian Y., Morrisey E. E. (2008). Wnt signaling: an essential regulator of cardiovascular differentiation, morphogenesis and progenitor self-renewal. Development 135, 789–798. [DOI] [PubMed] [Google Scholar]
- Damstra T. (2002). Potential effects of certain persistent organic pollutants and endocrine disrupting chemicals on the health of children. J. Toxicol. Clin. Toxicol. 40, 457–465. [DOI] [PubMed] [Google Scholar]
- Doetschman T., Gregg R. G., Maeda N., Hooper M. L., Melton D. W., Thompson S., Smithies O. (1987). Targetted correction of a mutant HPRT gene in mouse embryonic stem cells. Nature 330, 576–578. [DOI] [PubMed] [Google Scholar]
- Dummer T. J., Dickinson H. O., Parker L. (2003). Adverse pregnancy outcomes around incinerators and crematoriums in Cumbria, north west England, 1956-93. J. Epidemiol. Commun. Health 57, 456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda K., Yuasa S. (2006). Stem cells as a source of regenerative cardiomyocytes. Circ. Res. 98, 1002–1013. [DOI] [PubMed] [Google Scholar]
- Gessert S., Kuhl M. (2010). The multiple phases and faces of wnt signaling during cardiac differentiation and development. Circ. Res. 107, 186–199. [DOI] [PubMed] [Google Scholar]
- Godoy J. A., Arrazola M. S., Ordenes D., Silva-Alvarez C., Braidy N., Inestrosa N. C. (2014). Wnt-5a ligand modulates mitochondrial fission-fusion in rat hippocampal neurons. J. Biol. Chem. 289, 36179–36193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez F. J., Fernandez-Salguero P. (1998). The aryl hydrocarbon receptor: studies using the AHR-null mice. Drug Metab. Dispos. 26, 1194–1198. [PubMed] [Google Scholar]
- Gu Y. Z., Hogenesch J. B., Bradfield C. A. (2000). The PAS superfamily: Sensors of environmental and developmental signals. Annu. Rev. Pharmacol. Toxicol. 40, 519–561. [DOI] [PubMed] [Google Scholar]
- Guo X., Wang X. F. (2009). Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 19, 71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handley-Goldstone H. M., Grow M. W., Stegeman J. J. (2005). Cardiovascular gene expression profiles of dioxin exposure in zebrafish embryos. Toxicol. Sci. 85, 683–693. [DOI] [PubMed] [Google Scholar]
- Hogan B. L. (1996). Bone morphogenetic proteins in development. Curr. Opin. Genet. Dev. 6, 432–438. [DOI] [PubMed] [Google Scholar]
- Hogan B. L. (1996). Bone morphogenetic proteins: multifunctional regulators of vertebrate development. Genes Dev. 10, 1580–1594. [DOI] [PubMed] [Google Scholar]
- Ivnitski-Steele I. D., Friggens M., Chavez M., Walker M. K. (2005). 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) inhibition of coronary vasculogenesis is mediated, in part, by reduced responsiveness to endogenous angiogenic stimuli, including vascular endothelial growth factor A (VEGF-A). Birth. Defects. Res. A Clin. Mol. Teratol. 73, 440–446. [DOI] [PubMed] [Google Scholar]
- Kattman S. J., Witty A. D., Gagliardi M., Dubois N. C., Niapour M., Hotta A., Ellis J., Keller G. (2011). Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell 8, 228–240. [DOI] [PubMed] [Google Scholar]
- Keller G. M. (1995). In vitro differentiation of embryonic stem cells. Curr. Opin. Cell. Biol. 7, 862–869. [DOI] [PubMed] [Google Scholar]
- Kennedy L. H., Sutter C. H., Leon Carrion S., Tran Q. T., Bodreddigari S., Kensicki E., Mohney R. P., Sutter T. R. (2013). 2,3,7,8-Tetrachlorodibenzo-p-dioxin-mediated production of reactive oxygen species is an essential step in the mechanism of action to accelerate human keratinocyte differentiation. Toxicol. Sci. 132, 235–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. Y., Kim H. R., Kang M. G., Trang N. T., Baek H. J., Moon J. D., Shin J. H., Suh S. P., Ryang D. W., Kook H., et al. (2014). Profiling of biomarkers for the exposure of polycyclic aromatic hydrocarbons: lamin-A/C isoform 3, poly[ADP-ribose] polymerase 1, and mitochondria copy number are identified as universal biomarkers. Biomed. Res. Int. 2014, 605135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klug M. G., Soonpaa M. H., Koh G. Y., Field L. J. (1996). Genetically selected cardiomyocytes from differentiating embronic stem cells form stable intracardiac grafts. J. Clin. Invest. 98, 216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopf P. G., Walker M. K. (2009). Overview of developmental heart defects by dioxins, PCBs, and pesticides. J. Environ. Sci. Health. C Environ. Carcinog. Ecotoxicol. Rev. 27, 276–285. [DOI] [PubMed] [Google Scholar]
- Kuehl K. S., Loffredo C. A. (2006). A cluster of hypoplastic left heart malformation in Baltimore, Maryland. Pediatr. Cardiol. 27, 25–31. [DOI] [PubMed] [Google Scholar]
- Laflamme M. A., Murry C. E. (2011). Heart regeneration. Nature 473, 326–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanham K. A., Peterson R. E., Heideman W. (2012). Sensitivity to dioxin decreases as zebrafish mature. Toxicol. Sci. 127, 360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian X., Zhang J., Azarin S. M., Zhu K., Hazeltine L. B., Bao X., Hsiao C., Kamp T. J., Palecek S. P. (2013). Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta-catenin signaling under fully defined conditions. Nat. Protoc. 8, 162–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Foley A. C. (2011). Signaling pathways in early cardiac development. Wiley Interdiscip Rev. Syst. Biol. Med. 3, 191–205. [DOI] [PubMed] [Google Scholar]
- Lund A. K., Goens M. B., Kanagy N. L., Walker M. K. (2003). Cardiac hypertrophy in aryl hydrocarbon receptor null mice is correlated with elevated angiotensin II, endothelin-1, and mean arterial blood pressure. Toxicol. Appl. Pharmacol. 193, 177–187. [DOI] [PubMed] [Google Scholar]
- Morgan M., Anders S., Lawrence M., Aboyoun P., Pages H., Gentleman R. (2009). ShortRead: a bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics 25, 2607–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park W. H., Jun D. W., Kim J. T., Jeong J. H., Park H., Chang Y. S., Park K. S., Lee H. K., Pak Y. K. (2013). Novel cell-based assay reveals associations of circulating serum AhR-ligands with metabolic syndrome and mitochondrial dysfunction. Biofactors 39, 494–504. [DOI] [PubMed] [Google Scholar]
- Pavanello S., Dioni L., Hoxha M., Fedeli U., Mielzynska-Svach D., Baccarelli A. A. (2013). Mitochondrial DNA copy number and exposure to polycyclic aromatic hydrocarbons. Cancer Epidemiol. Biomarkers. Prev. 22, 1722–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira S. P., Pereira G. C., Pereira C. V., Carvalho F. S., Cordeiro M. H., Mota P. C., Ramalho-Santos J., Moreno A. J., Oliveira P. J. (2013). Dioxin-induced acute cardiac mitochondrial oxidative damage and increased activity of ATP-sensitive potassium channels in Wistar rats. Environ. Pollut. 180, 281–290. [DOI] [PubMed] [Google Scholar]
- Peterson R. E., Theobald H. M., Kimmel G. L. (1993). Developmental and reproductive toxicity of dioxins and related compounds: cross-species comparisons. Crit. Rev. Toxicol. 23, 283–335. [DOI] [PubMed] [Google Scholar]
- Plavicki J., Hofsteen P., Peterson R. E., Heideman W. (2013). Dioxin inhibits zebrafish epicardium and proepicardium development. Toxicol. Sci. 131, 558–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puga A. (2011). Perspectives on the potential involvement of the AH receptor-dioxin axis in cardiovascular disease. Toxicol. Sci. 120, 256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajala K., Pekkanen-Mattila M., Aalto-Setala K. (2011). Cardiac differentiation of pluripotent stem cells. Stem Cells Int. 2011, 383709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartor M. A., Schnekenburger M., Marlowe J. L., Reichard J. F., Wang Y., Fan Y., Ma C., Karyala S., Halbleib D., Liu X., et al. (2009). Genomewide analysis of aryl hydrocarbon receptor binding targets reveals an extensive array of gene clusters that control morphogenetic and developmental programs. Environ. Health. Perspect. 117, 1139–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A. J., Branam A. M., Peterson R. E. (2014). Intersection of AHR and Wnt signaling in development, health, and disease. Int. J. Mol. Sci. 15, 17852–17885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnekenburger M., Peng L., Puga A. (2007). HDAC1 bound to the Cyp1a1 promoter blocks histone acetylation associated with Ah receptor-mediated trans-activation. Biochim. Biophys. Acta 1769, 569–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott J. J., Benson D. W., Basson C. T., Pease W., Silberbach G. M., Moak J. P., Maron B. J., Seidman C. E., Seidman J. G. (1998). Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 281, 108–111. [DOI] [PubMed] [Google Scholar]
- Schultheiss T. M., Burch J. B., Lassar A. B. (1997). A role for bone morphogenetic proteins in the induction of cardiac myogenesis. Genes Dev. 11, 451–462. [DOI] [PubMed] [Google Scholar]
- Selevan S. G., Kimmel C. A., Mendola P. (2000). Identifying critical windows of exposure for children's health. Environ. Health. Perspect. 108(Suppl 3), 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrat R., Lopez-Domenech G., Mirra S., Quevedo M., Garcia-Fernandez J., Ulloa F., Burgaya F., Soriano E. (2013). The non-canonical Wnt/PKC pathway regulates mitochondrial dynamics through degradation of the arm-like domain-containing protein Alex3. PLoS One 8, e67773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shertzer H. G., Genter M. B., Shen D., Nebert D. W., Chen Y., Dalton T. P. (2006). TCDD decreases ATP levels and increases reactive oxygen production through changes in mitochondrial F(0)F(1)-ATP synthase and ubiquinone. Toxicol. Appl. Pharmacol. 217, 363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. G. (2001). Embryo-derived stem cells: of mice and men. Annu. Rev. Cell Dev. Biol. 17, 435–462. [DOI] [PubMed] [Google Scholar]
- Spikings E. C., Alderson J., St John J. C. (2007). Regulated mitochondrial DNA replication during oocyte maturation is essential for successful porcine embryonic development. Biol. Reprod. 76, 327–335. [DOI] [PubMed] [Google Scholar]
- Srivastava D., Olson E. N. (2000). A genetic blueprint for cardiac development. Nature 407, 221–226. [DOI] [PubMed] [Google Scholar]
- Stites T., Storms D., Bauerly K., Mah J., Harris C., Fascetti A., Rogers Q., Tchaparian E., Satre M., Rucker R. B. (2006). Pyrroloquinoline quinone modulates mitochondrial quantity and function in mice. J. Nutr. 136, 390–396. [DOI] [PubMed] [Google Scholar]
- Ten Berge D., Koole W., Fuerer C., Fish M., Eroglu E., Nusse R. (2008). Wnt signaling mediates self-organization and axis formation in embryoid bodies. Cell Stem Cell 3, 508–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thackaberry E. A., Nunez B. A., Ivnitski-Steele I. D., Friggins M., Walker M. K. (2005). Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on murine heart development: alteration in fetal and postnatal cardiac growth, and postnatal cardiac chronotropy. Toxicol. Sci. 88, 242–249. [DOI] [PubMed] [Google Scholar]
- Thundathil J., Filion F., Smith L. C. (2005). Molecular control of mitochondrial function in preimplantation mouse embryos. Mol. Reprod. Dev. 71, 405–413. [DOI] [PubMed] [Google Scholar]
- Trapnell C., Pachter L., Salzberg S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker M. K., Catron T. F. (2000). Characterization of cardiotoxicity induced by 2,3,7, 8-tetrachlorodibenzo-p-dioxin and related chemicals during early chick embryo development. Toxicol. Appl. Pharmacol. 167, 210–221. [DOI] [PubMed] [Google Scholar]
- Wang Q., Chen J., Ko C. I., Fan Y., Carreira V., Chen Y., Xia Y., Medvedovic M., Puga A. (2013). Disruption of aryl hydrocarbon receptor homeostatic levels during embryonic stem cell differentiation alters expression of homeobox transcription factors that control cardiomyogenesis. Environ. Health. Perspect. 121, 1334–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Fan Y., Puga A. (2010). Dioxin exposure disrupts the differentiation of mouse embryonic stem cells into cardiomyocytes. Toxicol. Sci. 115, 225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Shu W., Lu M. M., Morrisey E. E. (2005). Wnt7b activates canonical signaling in epithelial and vascular smooth muscle cells through interactions with Fzd1, Fzd10, and LRP5. Mol. Cell Biol. 25, 5022–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita J. K., Takano M., Hiraoka-Kanie M., Shimazu C., Peishi Y., Yanagi K., Nakano A., Inoue E., Kita F., Nishikawa S. (2005). Prospective identification of cardiac progenitors by a novel single cell-based cardiomyocyte induction. FASEB J. 19, 1534–1536. [DOI] [PubMed] [Google Scholar]
- Zaher H., Fernandez-Salguero P. M., Letterio J., Sheikh M. S., Fornace A. J., Jr, Roberts A. B., Gonzalez F. J. (1998). The involvement of aryl hydrocarbon receptor in the activation of transforming growth factor-beta and apoptosis. Mol. Pharmacol. 54, 313–321. [DOI] [PubMed] [Google Scholar]