

Abstract
Introduction:
Nonsyndromic cleft palate is a common birth defect (1:700) with a complex etiology involving both genetic and environmental risk factors. Nicotine, a major teratogen present in tobacco products, was shown to cause alterations and delays in the developing fetus.
Methods:
To demonstrate the postpartum effects of nicotine on palatal development, we delivered three different doses of nicotine (1.5, 3.0, and 4.5mg/kg/d) and sterile saline (control) into pregnant BALB/c mice throughout their entire pregnancy using subcutaneous micro-osmotic pump. Dams were allowed to deliver (~day 21 of pregnancy) and neonatal assessments (weight, length, nicotine levels) were conducted, and palatal tissues were harvested for morphological and molecular analyses, as well as transcriptional profiling using microarrays.
Results:
Consistent administration of nicotine caused developmental retardation, still birth, low birth weight, and significant palatal size and shape abnormality and persistent midline epithelial seam in the pups. Through microarray analysis, we detected that 6232 genes were up-regulated and 6310 genes were down-regulated in nicotine-treated groups compared to the control. Moreover, 46% of the cleft palate-causing genes were found to be affected by nicotine exposure. Alterations of a subset of differentially expressed genes were illustrated with hierarchal clustering and a series of formal pathway analyses were performed using the bioinformatics tools.
Conclusions:
We concluded that nicotine exposure during pregnancy interferes with normal growth and development of the fetus, as well results in persistent midline epithelial seam with type B and C patterns of palatal fusion.
Implications:
Although there are several studies analyzing the genetic and environmental causes of palatal deformities, this study primarily shows the morphological and large-scale genomic outcomes of gestational nicotine exposure in neonatal mice palate.
Introduction
Cleft palate is the second most common birth defect in the world with a prevalence of approximately 1 in 700 depending on the population.1 Cleft palate is mostly caused by inadequate palatal shelf growth and subsequent failure of the palatal midline epithelial seam (MES) to disintegrate and become confluent.2 Both genetic and environmental components contribute to the complex etiology of cleft palate.3 Using knockout mice models, numerous genes have been identified associated with palatal cleft such as Tgf-β3, Tgf-α, Msx1, Irf6, p63, Tbx22, Shh, Snai1, Snai2, and Gabrb3 etc.4–7 However, none of these genes have been identified as single causal agent of cleft palate. Therefore an interaction between high-risk alleles and environmental risk factors during embryogenesis has been implied for the induction of cleft palate.8 Smoking, alcohol consumption, and lack of vitamins during pregnancy are among the mostly studied environmental risk factors to date.9–11
It is well documented that maternal smoking during pregnancy is associated with a wide variety of adverse reproductive outcomes, such as increased rates of spontaneous abortion, premature birth, smaller head size, and low birth weight (LBW) in newborns.12,13 Among smoke products, nicotine is considered to be the main teratogenic substance that alters and delays embryonic development.14,15 Animal studies confirmed nicotine’s teratogenic effects in disrupting development of several organs including brain and lungs, leading to adverse cognitive, emotional, and behavioral outcomes.12,16 Although smoking has been associated with the gene-environment interaction for orofacial clefs (OFC),17 nicotine’s capacity to alter the genes causing cleft palate remains as a gap in our knowledge. Considering various factors contributing to the palate development, we hypothesized that fetal nicotine exposure can induce differential expression of genes involved in development, particularly palatogenesis. Therefore, our major objective in this study was to determine the postpartum genetic and morphological effects of nicotine on palatogenesis in the BALB/c murine model system.
Materials and Methods
Surgical Procedure and Nicotine Administration
BALB/c mice (Charles River Laboratory, Cambridge, MA) were used in our studies due to their frequent use in teratogenic and genotoxic studies and their well-characterized genome and phenome.18 Following examination of female mice for the presence of a vaginal plug as a verification of pregnancy, animals were placed under general anesthesia at 1 days-post-coitum (dpc) using 2% isoflurane via inhalation. The surgical field (skin) over the implantation site was shaved and disinfected using 70% ethanol solution. A small incision of 0.5cm was then made in the skin posterior to the shoulder and a micro-osmotic pump (Model 1004—Alzet Corp, Cupertino, CA) containing either sterile physiological saline or nicotine (1.5mg/kg/d, 3.0mg/kg/d, or 4.5mg/kg/d) was implanted subcutaneously. The micro-osmotic pump delivered the solutions continuously as 0.11µL/h without the need for external connections or frequent handling of animals.19 The incision was closed with a wound clip (Stoelting, Wooddale, IL) and covered with an antibiotic. The clip was removed after 14 days and dams were allowed to give birth naturally at/around 21 days. Care and use of experimental animals described in this work comply with guidelines and policies of the Institutional Animal Care and Use Committee (IACUC # 0606404-FC) of the University of Nebraska Medical Center (UNMC).
The dose of nicotine was calculated in terms of free base, using nicotine tartrate (Sigma Chemical Co, St Louis, MO) dissolved in a sterile physiological saline solution. Pregnant mice were treated either with saline or one of the following nicotine concentrations: 1.5mg/kg/d, 3.0mg/kg/d, or 4.5mg/kg/d, which is equivalent to 1-, 2-, and 3-pack-a-day smoking in humans, respectively.20 Micro-osmotic pumps were implanted into eight pregnant mice from each group. Numbers of pups used to test nicotine level for the groups were as follows: 66, 58, 47, and 31 for saline control, 1.5, 3.0, and 4.5mg/kg/d nicotine, respectively. Success of nicotine administration throughout the pregnancy was confirmed by testing nicotine levels in tissue samples at birth as described in Supplementary Data (S1).
Sample Collection and Morphology Assessment
All pregnant females were allowed to deliver their litters by natural birth (full-term pregnancy: ~21 days), after which the total number of pups per litter and number of stillborn pups per litter were assessed. All newborn pups, including stillborns, were subjected to a number of evaluations, which included the determination of weight (mg) and length from tip of nose to the end of the tail (mm). The statistical analyses were performed as explained below. Newborn pups were also examined visually under the dissecting microscope for existence of any OFC and palatal fusion phenotypes were classified from type A to F as described in Supplementary Data (S2).37 Following the sacrifice of all newborn pups, palatal shelves were collected using a dissecting microscope, and analyzed in three sections. The middle one-third of the palate underwent fixation in 10% formalin and was prepared for hematoxyline and eosin staining using a Tissue Tek VIP processor (GMI Inc, Ramsey, MN). The dimensional analysis of hematoxyline and eosin–stained palatal sections were performed using the Photoshop software (Adobe Inc, San Jose, CA). The anterior one-third and posterior one-third of the palate were immediately snap-frozen in liquid nitrogen for nicotine concentration assay and RNA extraction, respectively.
Microarray Processing and Bioinformatics Analysis
RNA samples from each treatment group were extracted and quantitated as described in Supplementary Data (S3). RNA samples were profiled using Affymetrix GeneChip Mouse Genome (MG) 430 2.0 arrays (Platform: GPL11180, Affymetrix, Inc, Santa Clara, CA) according to the standard Affymetrix gene chip analysis protocol. Gene Chip Hybridization Oven 320 (Affymetrix) and Gene Chip Fluidics Station 400 (Affymetrix) at UNMC Microarray Core were used for hybridization and scanning. The GeneChip HT MG-430 PM Array Plate is comprised of 45 000 probe sets to analyze the expression level of more than 39 000 transcripts and variants from more than 34 000 well-characterized mouse genes. A total of eight microarray chips were studied (two per each treatment group: saline, 1.5, 3.0, and 4.5mg/kg/d nicotine). Scanned array images were analyzed by dChip as described in Supplementary Data (S4).
The effects of nicotine on fetal development were examined in the context of detailed molecular interaction networks using Ingenuity Pathway Analysis (IPA; Ingenuity Systems, Redwood City, CA), a web-delivered application used to discover, visualize, and explore relevant networks, as described in Supplementary Data (S5).
Statistical Analysis
Postnatal data were presented as mean ± standard error of the mean. The pups from each dam (litter) were considered to represent a single determination. The data for pup weight at birth, pup length at birth, palate width, and palate height were analyzed for statistical significance using random effects analysis of variance with interpretation using adjusted P values. The P values were adjusted by the Dunnett multiple comparison (differences in the least square means for the three nicotine groups compared with the control = “saline”). A P value less than .05 was considered statistically significant.
Results
Validation of Nicotine Delivery in the Dams and Pups
Following the sacrifice of newborn pups and dams, the level of nicotine in two of the sample was determined by gas chromatography. The average blood nicotine concentration of the control dams was 3.82 (±0.89ng/mL). In the 1.5, 3.0, and 4.5mg/kg/d nicotine-treated dams, average blood nicotine concentrations was 17.88ng/mL, 35.58ng/mL, and 48.98ng/mL, respectively (Figure 1A). In terms of the offspring, the pups born to the control group were found to have no nicotine concentration within their tissues. Similar to the pregnant dams, the pups born to nicotine-treated mothers were found to have significantly higher average tissue nicotine concentrations (22.4, 36.1, and 47.2ng/mL respectively, P < .01; Figure 1A).
Figure 1.
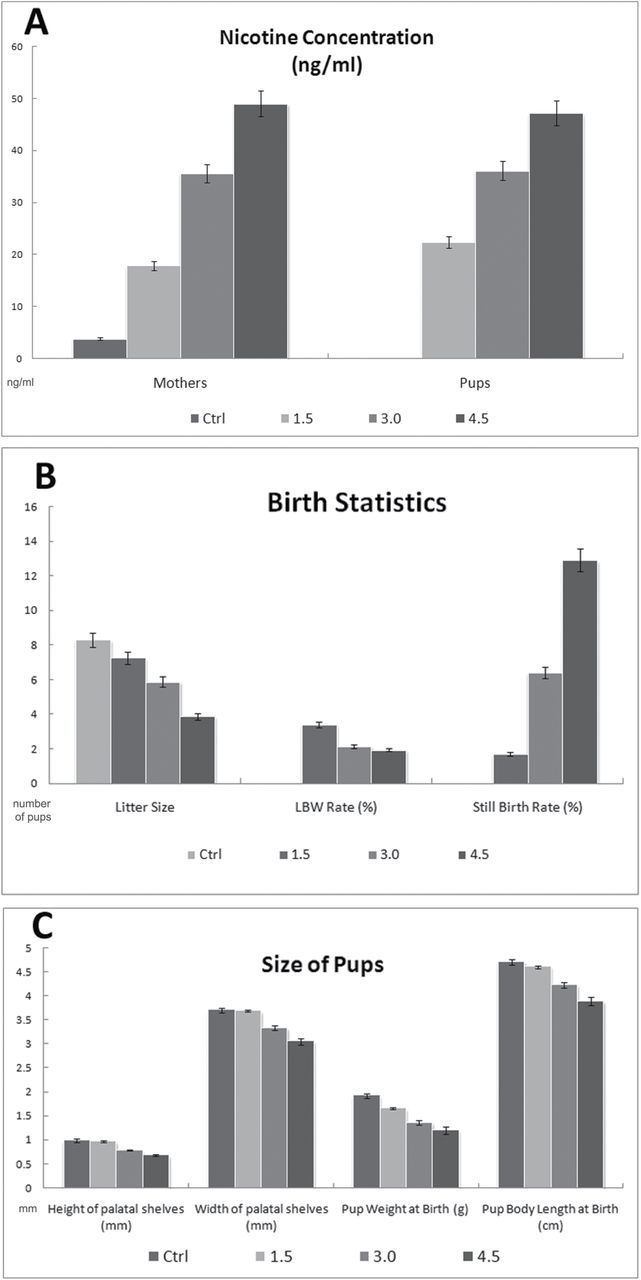
Nicotine concentration and morphological features of pups with nicotine exposure. (A) Concentrations of nicotine (ng/mL) in the dams and newborn pups of all treatment groups were determined by using either the blood or the homogenized palate tissue, respectively. (B) Number of pups at one delivery (litter size), low birth weight (LBW), and still birth rate among treatment groups are represented as an average of duplicates. (C) The width and height of the middle one third of all palates were measured using image software (Adobe PhotoShop 6.0). Birth weight and body size of pups within all treatment groups were averaged from duplicates. Nicotine-treated pups are statistically significant from saline-treated pups, P < .05 (Dunnett multiple comparisons).
Progressive and Consistent Nicotine Exposure Causes Developmental Anomalies
Our results demonstrated that treatment of dams with increasing dosage of nicotine resulted in significant teratogenic outcomes compared to saline-treated control mice. These outcomes are (1) The pups of nicotine-treated dams were smaller in overall size and shape and weighed less than saline-treated mice; (2) The litter size of nicotine-treated groups was less than the control groups, and the number of still births was higher than the control group in a dose dependent ratio; and (3) The palates of nicotine-exposed pups were smaller that control pups and had persistent MES (P < .05).
The nicotine-treated newborns exhibited significantly lower median birth weight and body length in accordance with the elevated exposure to nicotine throughout gestation (Figure 1B and Figure 2B). We classified the litters for the occurrence of LBW (<1.3 gm) pups and stillbirths. While the saline-treated control group did not show any stillborn or LBW pups out of 83 newborns; all the nicotine-treated groups exhibited considerable number of stillbirth and LBW incidences (Figure 1B). The treatment group of 1.5mg/kg/d nicotine-administered pregnant mice gave birth to one stillborn (1.7%) and two LBW pups (3.4%) out of a total of 58 pups. As the nicotine dosage increased to 3.0mg/kg/d, three stillborn (6.4%) and 10 LBW pups (21.3%) were born out of 47 pups. Finally, the 4.5mg/kg/d nicotine-treated group gave birth to nine stillborn (29.0%) and six LBW pups (19.4%) out of 31 pups. These results clearly demonstrated the negative outcomes of nicotine as resulting in stillbirth and LBW cases on pregnant mice.
Figure 2.
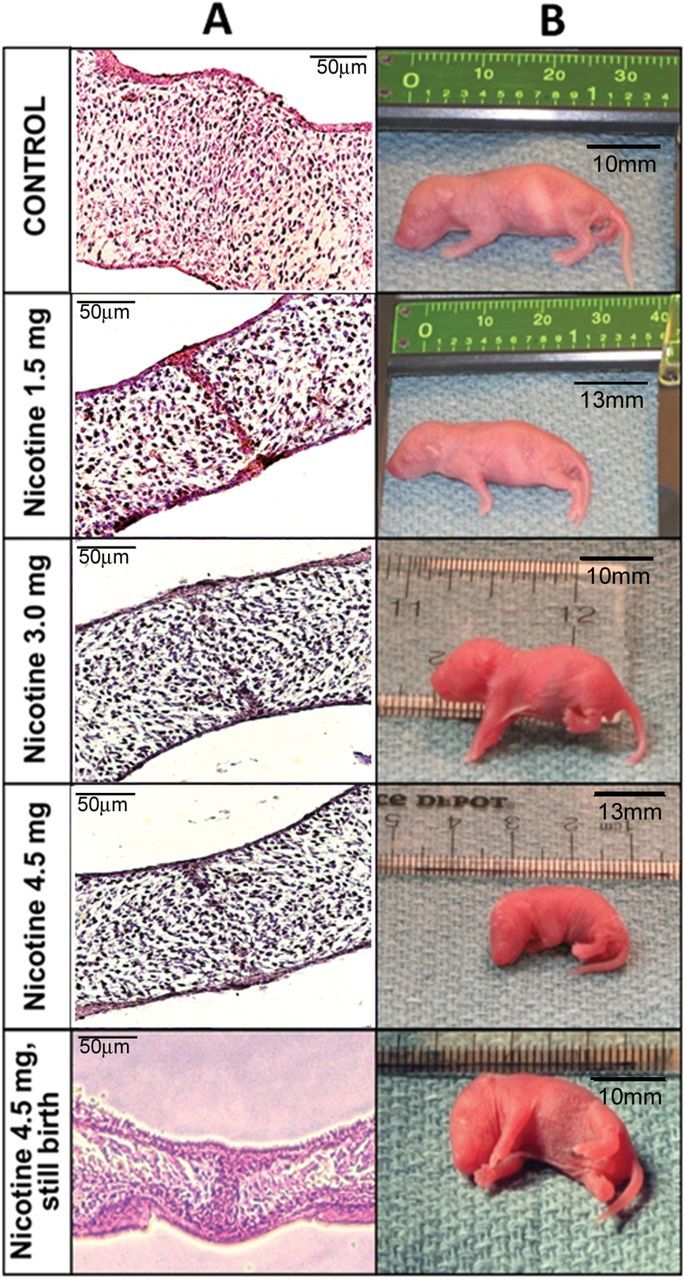
Morphology of the midline epithelial seam (MES) and size measurements of newborns. (A) Representative images of hematoxyline and eosin–stained sections of the palatal shelves. All newborn pups, including survived and stillborn, were evaluated. A detectable MES, which prevents palatal confluence, remained in the midline of all nicotine-treated pups. This persistent MES is classified as either type B or type C palatal fusion according to reference Nakajima et al (2014).37 (B) Size measurements of sample newborn pups from each treatment group. Bar represents relative size in mm.
The birth rate among the treated pregnant mice was also decreased in accordance with daily nicotine levels. While the saline-treated (control) group of mice had an average birth rate of 8.3 pups per litter, the group of mice treated with 1.5, 3.0, and 4.5mg/kg/d nicotine had average birth rates of 7.25, 5.88, and 3.88 pups per litter, respectively (Figure 1B).
The newborn pups of all treatment groups were examined by direct visual observation and under the dissecting microscope to detect the presence of any palatal deformities. Neither the pups of the saline-treated nor the nicotine-treated groups displayed any evidence of complete OFC, but they exhibited type B and C palatal fusion, in which the MES remained present with either on two layers. Meanwhile, there were significant difference with the size and shape of the palates of the newborn pups. According to dimensional analysis of hematoxyline and eosin–stained palate sections, both the height and width of palatal shelves of the nicotine-treated animals were considerably smaller than saline-treated pups (Figure 1C). We detected that the nicotine-administered newborns had a detectable MES remaining in the palate of the pups, which are either reached full term or stillborn (Figure 2A). During normal development of the palate, once the MES is formed at or around 14.0~15.0 dpc, it is destined to be disintegrated by epithelial-mesenchymal transition and/or apoptosis resulting in the palatal confluence. Based on our overall morphological analysis, we concluded that nicotine causes delays and alterations in fetal development, including palatogenesis, in the BALB/c mice. Surprisingly, the smaller palate size and interference of MES disintegration resulted in type B and C palatal fusion.
Nicotine Changes the Gene Profile of Developing Fetus
The microarray data analysis of control and nicotine-treated pups resulted in 6232 up-regulated and 6310 down-regulated genes in at least one of the nicotine-treated groups compared to control. Venn diagrams were generated to show the concurrent number of up- and down-regulated genes in response to different nicotine concentrations (Supplementary Figure 1). The number of genes individually affected in treatment groups is outlined in Table 1. Duplicate chips in each treatment group resembled each other with a correlation coefficient of 0.95±0.02 (average ± SD) using complete expression profiles showing high reproducibility. The unsupervised hierarchical clustering analysis of the differentially expressed genes based on the progressively up-regulated (n = 69) and down-regulated (n = 227) transcripts showed a clear separation of each group (Figure 3). The majority of subjects clustered accurately into the four groups. This unsupervised analysis approach, not using any pre-selected subset of genes, rendered a clear distinction between the control versus nicotine-treated pups.
Table 1.
Dataset for Number of Regulated Genes
| Comparison | # of genes | Comparison | # of genes | Comparison | # of genes | |||
|---|---|---|---|---|---|---|---|---|
| Up-regulated genes | N1.5 vs. C | 2253 | Down-regulated genes | N1.5 vs. C | 3461 | No change in gene expression | N1.5 vs. C | 34 007 |
| N3.0 vs. C | 3722 | N3.0 vs. C | 3552 | N3.0 vs. C | 31 930 | |||
| N4.5 vs. C | 2742 | N4.5 vs. C | 4376 | N4.5 vs. C | 32 699 | |||
| N3.0 vs. N1.5 | 3040 | N3.0 vs. N1.5 | 2587 | N3.0 vs. N1.5 | 33 203 | |||
| N4.5 vs. N1.5 | 1073 | N4.5 vs. N1.5 | 2067 | N4.5 vs. N1.5 | 37 312 | |||
| N4.5 vs. N3.0 | 971 | N4.5 vs. N3.0 | 1485 | N4.5 vs. N3.0 | 37 905 |
N = nicotine treatment; C = saline control.
Figure 3.
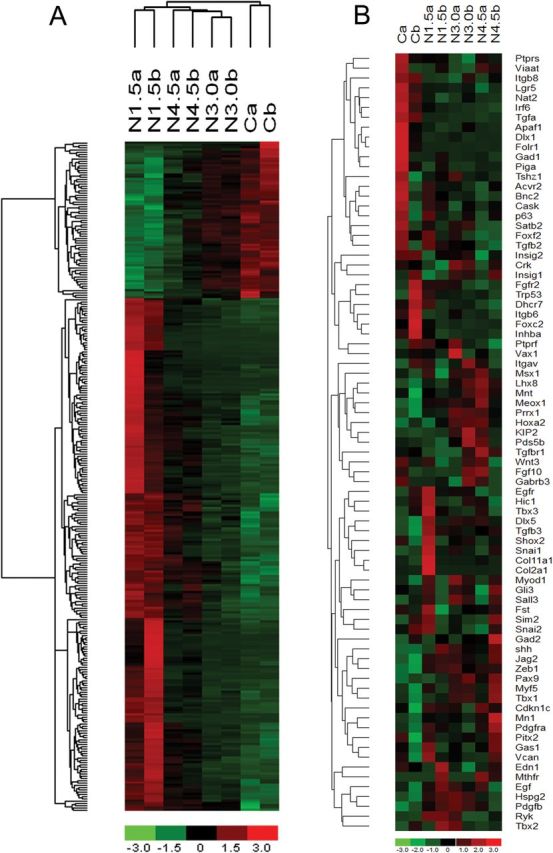
Hierarchical clustering (heatmap) analysis of differentially expressed transcripts. Unsupervised hierarchical clustering of the expression of probe sets differentially expressed in the nicotine-treated versus control pups. (A) Complete transcriptional profile of progressively up-regulated (n = 69) and down-regulated (n = 227) genes. (B) Significantly up-regulated (n = 18) and down-regulated (n = 20) genes known to be susceptible for induction of cleft palate (n = 84). Nicotine-treated pups cluster primarily into three distinct groups on a dose-dependent manner. Each column corresponds to the expression profile of a treatment (either nicotine [N] or saline [C]) in duplicates, and each row corresponds to an mRNA. Upper portion (green-to-red) represents down-regulated transcripts and lower portion (red-to-green) represents up-regulated transcripts in A and vice versa in B. The increasing intensities of red signify a higher expression in the given sample of a specific mRNA, whereas the increasing intensities of green indicate a lower expression of mRNA and black indicates mean level expression.
Several Genes Involved in Palate Development are Affected by Nicotine Exposure
According to several transgenic knockout mice models, currently, there are 84 genes, which their mutations exhibit cleft palate in newborns.21 The molecules involved in cleft palate formation in knockout models include cytokines, receptors, growth factors, ion channels, kinases, extracellular matrix proteins, homeobox genes, and transcription regulators22,23 (Supplementary Table 1). According to the findings of this study, 46% of the cleft palate-causing genes (39 out of 84) were found to be affected by nicotine exposure. The palatal genome of the nicotine-exposed pups are altered as follows: 19 of the cleft-palate related genes were up-regulated (Col11a1, Egfr, Gas1, Hoxa2, Hspg2, Jag2, Lhx8, Meox1, Mn1, Myf5, Myod1, Pdgf, Pdgfra, Pds5b, Prrx1, Shh, Tbx1, Tgf-β3, and Zeb10), and 20 of them were down-regulated (Bnc2, Crk, Dhcr7, Fgfr2, Foxf2, Inhba, Insig1, Irf6, Itg-αV, Itg-β6, Lgr5, Nat2, Piga, Ptprf, Sall3, Satb2, Snai2, Tbx3, Tgf-α, and Tgf-βr1) in response to consistent nicotine uptake throughout gestation (Supplementary Figure 2).
Additionally, unsupervised hierarchical cluster heatmap analysis of these genes also confirmed that these changes were not always dose-dependent (Figure 3B). Among these differentially expressed cleft palate-susceptible genes, Tgf-β3, Irf6, Snai2, Tgf-α, and Tgf-βr1 constitute the TGF-β family, which has been shown to play an essential role during palatogenesis.24 These results demonstrated that consistent nicotine exposure of the pregnant mice had considerable effect on expression levels of the genes affecting palate development.
In order to identify other developmental disorders related to cleft palate-susceptible genes, we performed a disease network analysis using the IPA. In nicotine-treated groups, 18 of these genes (Tgf-α, Tgf-β3, Gas1, Gabrb3, Ryk, Pdgfr-A, Gad1, Crk, Dhcr7, Insig1, Insig2, Cdkn1C, Tbx22, Sat-B2, Snai2, Snai1, and Tp63) were differentially expressed (false discovery rate criteria 0.05) and qualified to be analyzed for merged direct interaction network of the developmental disorders. More than 15 developmental disorders, involving several organs of the body such as the eyes, limbs, kidney, and oral cavity, were detected (Supplementary Figure 3). These disorders are caused by the genes of the cleft palate network and have known connectivity information from published literature according to the Ingenuity Knowledge Base. Types of molecules are annotated in the legend, subcellular localization of each molecule and its relationship with the particular genetic diseases are shown with connecting lines (Supplementary Figure 3).
Nicotine Alters Genes Involved in Cellular Function and Systems Development
Using the IPA, a total of 6473 nicotine-altered genes included information on functions and/or canonical pathways from the published literature. The most significant assignments included genetic disorders, cancer, neurological diseases, and gastrointestinal diseases. The most significantly affected molecular and cellular functions were cell death, cellular growth and proliferation, cell cycle, and gene expression. Within the development and functions of the physiological system, the changes occurred in tissue development, tissue morphology, skeletal and muscular system development, and organismal development. The alterations of these biological pathways are graphed in Supplementary Figure 4 and listed in detail in Supplementary Table 2, along with the P value of each assignment.
Our overall microarray analyses demonstrated that nicotine caused differential expression of more than 12 500 transcripts, which comprise roughly 30% of the whole mouse genome (39 000 transcripts on MG-430), with a fold change value of more than 1.2. Among the significantly up/down-regulated genes, at least 39 (19 up + 20 down) of them were involved in palatogenesis. Furthermore, according to pathway and network analysis using IPA, nicotine-exposure throughout pregnancy resulted in significant alterations in various biological functions and pathways.
Discussion
Although there are several studies analyzing the genetic and environmental causes of palatal deformities, this study primarily shows the morphological and large-scale genomic outcomes of gestational nicotine exposure in neonatal mice palate. Among the recent gene-environment interaction studies for OFC, significant interaction was detected between maternal cigarette smoking and the TaqI polymorphism in the gene for transforming growth factor alpha (TGFα).25 Meanwhile, polymorphisms of several genes, including MSX1, TGFB3, BCL3, CYP1A1, GSTP1, and GSTT1 have been examined in conjunction with maternal alcohol consumption, cigarette smoking, medication use, and multivitamin supplementation during pregnancy.25–27 It was shown that smoking by both parents interacts with a specific allelic variant of MSX1, which significantly increase the OFC risk for their offspring.28 Moreover, a novel approach was developed by Wehby et al.26 to study the role of maternal smoking and OFC, in which they found out that smoking before and during pregnancy increased the risk of OFC by about 4–5 times at the sample average smoking rate. Furthermore, in a recent genome-wide analysis study (GWAS), biases for maternally inherited genetic factors influencing the risk of OFC was suggested.29 On the other hand, in a recent study, the associations between maternal smoking and neonatal complications in humans were analyzed, however the palatal deformities were omitted.30
According to several studies, cigarette smoking induces developmental anomalies both in human and animal models.17,30–33 The objective of our study was to test the hypothesis that nicotine intake during pregnancy causes delays in fetal development and interferes with palate formation. Installing the micro-osmotic pump at 1 dpc and allowing the nicotine-treated dams for natural birth maintained consistent nicotine administration throughout the entire pregnancy. The mode of delivery using the micro-osmotic pump was continuous as 0.11µL/h throughout the pregnancy.19 This mode largely mimics the nicotine patch therapy, which is applied to the skin and continuously administer a stable dose of nicotine slowly over 16–24 hours.34 Meanwhile, it would be expected that physiological effects of nicotine may differ according to the mode of delivery, since the human smoking is more intermittent throughout the day. Moreover, although tobacco smoking would induce hypoxia, the scope of this study covers only the nicotine’s effect on morphological and genomic changes in neonatal mice. However, further studies related to comparison of intermittent versus continuous delivery of nicotine can be planned. The determination of blood nicotine concentration levels for each treatment group accurately confirmed delivery of nicotine into the dams and pups via micro-osmotic pump. The average nicotine levels of fetuses, treated as 1.5, 3.0, and 4.5mg/kg/d, were comparable to the nicotine levels found in their corresponding mother mice, which confirm that nicotine penetrates into the fetuses during gestation in a manner consistent with the dose of nicotine to which mothers are exposed.
In human newborns, smoking during pregnancy has been shown to result in increased rates of stillbirth and LBW.26,35 The pups exposed to nicotine had statistically significant LBW and shorter body length as compared to control group, confirming the momentous negative impact of maternal nicotine exposure on the size of the offspring. Additionally, frequency of stillborn pups was considerably higher in nicotine-treated dams, thus showing the deleterious effect of nicotine on the developing fetus. In terms of litter size, which also includes stillborn pups, all of the above-mentioned developmental anomalies are likely due to the effect of nicotine on morphogenesis and organ development as reviewed in Rogers36. It would be also noteworthy to analyze if the higher number of stillborn pups and lower litter size were due to failures during implantation, early development, or late development. Presence of persistent MES in the stillborn pups (Figure 2), shows that the death of pups was most likely happened in the late phase of pregnancy following palatal adherence at 14.5 dpc.
This study showed gestational treatment of mice with nicotine causes abnormalities in the size and shape of palates of newborn pups and generate persistent MES.37 The height and width of the palates in offspring of nicotine-treated dams were found to be significantly decreased (Figure 1C). The observed smaller palate size of nicotine-treated fetuses might simply be a reflection of the smaller size of the pups. There was an observed failure of the MES to disintegrate in the palates of nicotine-treated mice. In normal palate development, ultimate confluence of the fused palatal shelves requires disintegration of the MES through epithelial-mesenchymal transition and/or apoptosis between 14.5 to 16 dpc.38 Failure of epithelial cells within the MES to undergo these cellular changes results in a persistent MES and nonconfluent palate, thus the palatal shelves may separate with the growing face and result in cleft palate. The occurrence of partially disintegrated MES without cleft palate in nicotine-treated pups might be due to the alteration in the expression of the genes responsible for epithelial-mesenchymal transition and/or apoptosis, particularly the TGF-β family as shown in our microarray analysis (Supplementary Figure 2). These alterations may interfere with the signaling pathways involved in the final phase of palatogenesis and disrupt the MES disintegration, which may result in either failed epithelial-mesenchymal transition or apoptosis within the medial edge epithelium cells. Previous studies have shown especially Tgfβ-signaling plays a crucial role in palatal fusion, acting via the Alk5 and TgfβR2 receptors to activate Smad2/Smad4 and the p38 MAPK pathways, which together regulate p21 expression in the MES. These, in parallel with the transcription factors Snai1 and Snai2, promote MES apoptosis and disintegration.6,21,39–43 Our results demonstrated that not only the Tgfβ-pathway genes but also other pathway-related genes might interact each other to regulate medial edge epithelium disintegration and complete palatogenesis (Supplementary Figure 2)
In several studies, it has been suggested that there is a gene-environment interaction exists between smoking and TGFα expression for the induction of cleft palate.8,25,44–49 By using our microarray analysis and filtering cleft palate-related genes, we determined that nicotine is the highly susceptible element of smoking to induce down-regulation of TGFα, which may explain the palatal size abnormality observed in nicotine-treated pups. The presence of partial palatal seam in the newborn pups, which otherwise would have disintegrated at 14.5 to 16.5 dpc indicates: (1) the growth of the palate towards each other to form a seam, although in smaller size and shape, is not altered; (2) the genes that causes immaculate seam disintegration might have been effected by nicotine exposure.
According to our microarray data analysis, clustering of samples using the complete transcriptional profiling showed that nicotine treatment affects global gene expression. Venn diagrams allowed us straightforward evaluation of the comparative and concurrent numbers of genes significantly altered in response to multiple doses of nicotine (Supplementary Figure 1). Detection of differential expression of approximately 30% of the analyzed transcripts reflects the large-scale impact of nicotine on gene expression during development.
The normal development of the palate involves cell proliferation, differentiation, and matrix synthesis. These processes are coordinated by secreted proteins and their signaling pathways, ExtraCellular Matrix components, and cell surface-receptors.22 Gene targeting to knock-out specific genes in mice has generated, by the date, 84 loss-of-function mutants that exhibits cleft palate. Information about these knockout models was collected from a literature review4,21,23,50 and genes were clustered into families of cytokine/receptor, homeobox genes, and miscellaneous (Supplementary Table 1). Out of 84 cleft palate-susceptible genes, expression of 19 of them was up-regulated, and 20 of them were down-regulated significantly in response to consistent nicotine uptake during pregnancy. Discovering altered expression of 46% of cleft palate-related genes (39/84) with persistent MES in nicotine-treated mice is an important result of this study. This finding suggests that changes in the expression levels of genes involved in palate development can be compensated by other genes, thus the palatogenesis can be brought until MES formation and persistence. However, failure of MES disintegration demonstrates that genes required for final stages of palatal confluence are either not compensable by other genes or differential expression of them was not at the right direction (up/down).
Our graphical and heatmap analysis of cleft palate-susceptible molecules recommended that nicotine has negatively regulated several genes involved in palatogenesis. Interestingly some of these altered genes were expressed differentially, either up- or down-regulated. According to our disease network analysis, these genes are vulnerable to various other developmental disorders as displayed in Supplementary Figure 3. Albeit examination of all fetal disorders is beyond the scope of this study, alteration of cleft palate-susceptible genes and their relationship in response to nicotine exposure necessitates further investigation.
In order to identify gene ontology, specific pathways, and functional assignments involved in the response to nicotine treatment, we performed a series of formal pathway analyses using the IPA. According to our biological pathway analysis, it is obvious that nicotine alters the expression of genes involved in genetic disorders and cancer at the highest significance. It is likely that this observed effect of nicotine has substantial implications in the induction of various genetic disorders as an environmental risk factor. This treasured microarray data, which can be accessed at Gene Expression Omnibus, is worth further investigation and data mining to study the genotoxic role of nicotine on the developing fetus. Moreover, although nicotine itself is not classified as a carcinogen,51 by acting through nAChRs on nonneuronal cells, it facilitates tumor growth, angiogenesis, metastasis, survival, and chemoresistance by regulating diverse signaling pathways.51 Therefore, the highest significance of differential expression of genes involved in cancer in response to nicotine exposure presents valuable data to further study nicotine’s role in developmental toxicology.
In conclusion, our results suggest that consistent nicotine exposure during pregnancy interferes with normal growth and development of the fetus in the BALB/c mouse model. In terms of palatogenesis, nicotine exposure caused improper palatal fusion, which resulted in persistent MES with no or partial disintegration of the seam. Our results also suggest that nicotine does not induce complete cleft palate in the BALB/c mouse model; however, other constituents of tobacco such as tar, carbon monoxide, nitrogen oxides, cannot be ruled out for the induction of cleft palate. Studying these patterns of differential gene expression in response to nicotine exposure throughout pregnancy may provide the key to understanding the impact of nicotine in the genetic and morphological make-up of the developing fetus.
Supplementary Material
Supplementary Data S1–S5, Figures 1–4, and Tables 1 and 2 can be found online at http://www.ntr.oxfordjournals.org
Funding
This research is supported by NIDCR, NIH grant (R01DE017986) and Tobacco Settlement Biomedical Research Development Fund (LB692) to Dr. Ali Nawshad. This publication’s contents are the sole responsibility of the authors and do not necessarily represent the official views of the NIH.
Declaration of Interests
None declared.
Supplementary Material
Acknowledgments
We thank Dr Eric Fung for his significant contribution to reviewing the manuscript and nicotine analysis; Ms Kendra Kaldahl and Ms Namita Sakhuja for their contribution with the research project; and Ms Marian Schmidt for her help with animal handling and housing within the College of Dentistry of UNMC.
References
- 1. Lebby KD, Tan F, Brown CP. Maternal factors and disparities associated with oral clefts. Ethn Dis. 2010;20(1)(suppl 1):S1-146–149. [PMC free article] [PubMed] [Google Scholar]
- 2. Ferguson MW. Palate development. Development. 1988;103(suppl):41–60. [DOI] [PubMed] [Google Scholar]
- 3. Murray JC, Schutte BC. Cleft palate: players, pathways, and pursuits. J Clin Invest. 2004;113(12):1676–1678. doi:10.1172/JCI22154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dixon MJ, Marazita ML, Beaty TH, Murray JC. Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet. 2011;12(3):167–178. doi:10.1038/nrg2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Juriloff DM, Harris MJ. Mouse genetic models of cleft lip with or without cleft palate. Birth Defects Res A Clin Mol Teratol. 2008;82(2):63–77. doi:10.1002/bdra.20430. [DOI] [PubMed] [Google Scholar]
- 6. Kaartinen V, Voncken JW, Shuler C, et al. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. 1995;11(4):415–421. doi:10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- 7. Ingraham CR, Kinoshita A, Kondo S, et al. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6). Nat Genet. 2006;38(11):1335–1340. doi:10.1038/ng1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beaty TH, Ruczinski I, Murray JC, et al. Evidence for gene-environment interaction in a genome wide study of nonsyndromic cleft palate. Genet Epidemiol. 2011;35 (6):469–478. doi:10.1002/gepi.20595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ericson A, Kallen B, Westerholm P. Cigarette smoking as an etiologic factor in cleft lip and palate. Am J Obstet Gynecol. 1979;135(3):348–351. [DOI] [PubMed] [Google Scholar]
- 10. Leite IC, Paumgartten FJ, Koifman S. Chemical exposure during pregnancy and oral clefts in newborns. Cad Saude Publica. 2002;18(1):17–31. doi:10.1590/S0102-311X2002000100003. [DOI] [PubMed] [Google Scholar]
- 11. Wehby GL, Murray JC. Folic acid and orofacial clefts: a review of the evidence. Oral Dis. 2010;16(1):11–19. doi:10.1111/j.1601-0825.2009.01587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pauly JR, Slotkin TA. Maternal tobacco smoking, nicotine replacement and neurobehavioural development. Acta Paediatr. 2008;97(10):1331–1337. doi:10.1111/j.1651-2227.2008.00852.x. [DOI] [PubMed] [Google Scholar]
- 13. Ion R, Bernal AL. Smoking and preterm birth. Reprod Sci. 2015;22 (8):918–926. doi:10.1177/1933719114556486. [DOI] [PubMed] [Google Scholar]
- 14. Schneider T, Bizarro L, Asherson PJ, Stolerman IP. Gestational exposure to nicotine in drinking water: teratogenic effects and methodological issues. Behav Pharmacol. 2010;21(3):206–216. doi:10.1097/FBP.0b013e32833a5bb5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhao Z, Reece EA. Nicotine-induced embryonic malformations mediated by apoptosis from increasing intracellular calcium and oxidative stress. Birth Defects Res B Dev Reprod Toxicol. 2005;74(5):383–391. doi:10.1002/bdrb.20052. [DOI] [PubMed] [Google Scholar]
- 16. Brook DW, Zhang C, Rosenberg G, Brook JS. Maternal cigarette smoking during pregnancy and child aggressive behavior. Am J Addict. 2006;15(6):450–456. doi:10.1080/10550490600998559. [DOI] [PubMed] [Google Scholar]
- 17. Sabbagh HJ, Hassan MH, Innes NP, Elkodary HM, Little J, Mossey PA. Passive smoking in the etiology of non-syndromic orofacial clefts: a systematic review and meta-analysis. PLoS One. 2015;10(3):e0116963. doi:10.1371/journal.pone.0116963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frazer KA, Eskin E, Kang HM, et al. A sequence-based variation map of 8.27 million SNPs in inbred mouse strains. Nature. 2007;448(7157):1050–1053. doi:10.1038/nature06067. [DOI] [PubMed] [Google Scholar]
- 19. Corp. D. Micro-Osmotic Pump ALZET® - Model 1004 - Instruction and Specification Sheet 2008. www.alzet.com/downloads/1004specs.pdf Accessed August 12, 2015.
- 20. Fung YK, Lau YS. Effect of nicotine pretreatment on striatal dopaminergic system in rats. Pharmacol Biochem Behav. 1989;32(1):221–226. [DOI] [PubMed] [Google Scholar]
- 21. Ozturk F, Li Y, Zhu X, Guda C, Nawshad A. Systematic analysis of palatal transcriptome to identify cleft palate genes within TGFbeta3-knockout mice alleles: RNA-Seq analysis of TGFbeta3 Mice. BMC Genomics. 2013;14:113. doi:10.1186/1471-2164-14-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Meng L, Bian Z, Torensma R, Von den Hoff JW. Biological mechanisms in palatogenesis and cleft palate. J Dent Res. 2009;88(1):22–33. doi:10.1177/0022034508327868. [DOI] [PubMed] [Google Scholar]
- 23. Gritli-Linde A. Molecular control of secondary palate development. Dev Biol. 2007;301(2):309–326. doi:10.1016/j.ydbio.2006.07.042. [DOI] [PubMed] [Google Scholar]
- 24. Nawshad A, LaGamba D, Hay ED. Transforming growth factor beta (TGFbeta) signalling in palatal growth, apoptosis and epithelial mesenchymal transformation (EMT). Arch Oral Biol. 2004;49(9):675–689. doi:10.1016/j.archoralbio.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 25. Skare O, Jugessur A, Lie RT, et al. Application of a novel hybrid study design to explore gene-environment interactions in orofacial clefts. Ann Hum Genet. 2012;76(3):221–236. doi:10.1111/j.1469-1809.2012.00707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wehby G, Jugessur A, Murray JC, Moreno L, Wilcox A, Lie RT. Genes as instruments for studying risk behavior effects: an application to maternal smoking and orofacial clefts. Health Serv Outcomes Res Methodol. 2011;11(1–2):54–78. doi:10.1007/s10742-011-0071-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krapels IP, Raijmakers-Eichhorn J, Peters WH, et al. The I,105V polymorphism in glutathione S-transferase P1, parental smoking and the risk for nonsyndromic cleft lip with or without cleft palate. Eur J Hum Genet. 2008;16(3):358–366. doi:10.1038/sj.ejhg.5201973. [DOI] [PubMed] [Google Scholar]
- 28. van den Boogaard MJ, de Costa D, Krapels IP, et al. The MSX1 allele 4 homozygous child exposed to smoking at periconception is most sensitive in developing nonsyndromic orofacial clefts. Hum Genet. 2008;124(5):525–534. doi:10.1007/s00439-008-0569-6. [DOI] [PubMed] [Google Scholar]
- 29. Garg P, Ludwig KU, Böhmer AC, et al. Genome-wide analysis of parent-of-origin effects in non-syndromic orofacial clefts. Eur J Hum Genet. 2014;22(6):822–830. doi:10.1038/ejhg.2013.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bakker R, Kruithof C, Steegers EA, et al. Assessment of maternal smoking status during pregnancy and the associations with neonatal outcomes. Nicotine Tob Res. 2011;13(12):1250–1256. doi:10.1093/ntr/ntr117. [DOI] [PubMed] [Google Scholar]
- 31. Bnait KS, Seller MJ. Ultrastructural changes in 9-day old mouse embryos following maternal tobacco smoke inhalation. Exp Toxicol Pathol. 1995;47(6):453–461. doi:10.1016/S0940-2993(11)80327-1. [DOI] [PubMed] [Google Scholar]
- 32. Chung KC, Kowalski CP, Kim HM, Buchman SR. Maternal cigarette smoking during pregnancy and the risk of having a child with cleft lip/palate. Plast Reconstr Surg. 2000;105(2):485–491. [DOI] [PubMed] [Google Scholar]
- 33. Esposito ER, Horn KH, Greene RM, Pisano MM. An animal model of cigarette smoke-induced in utero growth retardation. Toxicology. 2008;246(2–3):193–202. doi:10.1016/j.tox.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rigotti NA. Clinical practice. Treatment of tobacco use and dependence. N Engl J Med. 2002;346(7):506–512. doi:10.1056/NEJMcp012279. [DOI] [PubMed] [Google Scholar]
- 35. Aliyu MH, Salihu HM, Wilson RE, Alio AP, Kirby RS. The risk of intrapartum stillbirth among smokers of advanced maternal age. Arch Gynecol Obstet. 2008;278(1):39–45. doi:10.1007/s00404-007-0529-8. [DOI] [PubMed] [Google Scholar]
- 36. Rogers JM. Tobacco and pregnancy: overview of exposures and effects. Birth Defects Res C Embryo Today. 2008;84(1):1–15. doi:10.1002/bdrc.20119. [DOI] [PubMed] [Google Scholar]
- 37. Nakajima A, Ito Y, Tanaka E, et al. Functional role of TGF-β receptors during palatal fusion in vitro. Arch Oral Biol. 2014;59(11):1192–1204. doi:10.1016/j.archoralbio.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nawshad A. Palatal seam disintegration: to die or not to die? That is no longer the question. Dev Dyn. 2008;237(10):2643–2656. doi:10.1002/dvdy.21599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kaartinen V, Cui XM, Heisterkamp N, Groffen J, Shuler CF. Transforming growth factor-beta3 regulates transdifferentiation of medial edge epithelium during palatal fusion and associated degradation of the basement membrane. Dev Dyn. 1997;209(3):255–260. doi:10.1002/(SICI)1097-0177(199707)209:3<255::AID-AJA1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 40. Dudas M, Kim J, Li WY, et al. Epithelial and ectomesenchymal role of the type I TGF-beta receptor ALK5 during facial morphogenesis and palatal fusion. Dev Biol. 2006;296(2):298–314. doi:10.1016/j.ydbio.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ahmed S, Liu CC, Nawshad A. Mechanisms of palatal epithelial seam disintegration by transforming growth factor (TGF) beta3. Dev Biol. 2007;309(2):193–207. doi:10.1016/j.ydbio.2007.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cui XM, Shiomi N, Chen J, et al. Overexpression of Smad2 in Tgf-beta3-null mutant mice rescues cleft palate. Dev Biol. 2005;278(1):193–202. doi:10.1016/j.ydbio.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 43. Xu X, Han J, Ito Y, Bringas P, Deng C, Chai Y. Ectodermal Smad4 and p38 MAPK are functionally redundant in mediating TGF-beta/BMP signaling during tooth and palate development. Dev Cell. 2008;15(2):322–329. doi:10.1016/j.devcel.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vieira AR. Association between the transforming growth factor alpha gene and nonsyndromic oral clefts: a HuGE review. Am J Epidemiol. 2006;163(9):790–810. doi:10.1093/aje/kwj103. [DOI] [PubMed] [Google Scholar]
- 45. Zeiger JS, Beaty TH, Liang KY. Oral clefts, maternal smoking, and TGFA: a meta-analysis of gene-environment interaction. Cleft Palate Craniofac J. 2005;42(1):58–63. [DOI] [PubMed] [Google Scholar]
- 46. Jugessur A, Lie RT, Wilcox AJ, et al. Cleft palate, transforming growth factor alpha gene variants, and maternal exposures: assessing gene-environment interactions in case-parent triads. Genet Epidemiol. 2003;25(4):367–374. doi:10.1002/gepi.10268. [DOI] [PubMed] [Google Scholar]
- 47. Shaw GM, Wasserman CR, Lammer EJ, et al. Orofacial clefts, parental cigarette smoking, and transforming growth factor-alpha gene variants. Am J Hum Genet. 1996;58(3):551–561. [PMC free article] [PubMed] [Google Scholar]
- 48. Chevrier C, Bahuau M, Perret C, et al. Genetic susceptibilities in the association between maternal exposure to tobacco smoke and the risk of nonsyndromic oral cleft. Am J Med Genet A. 2008;146A(18):2396–2406. doi:10.1002/ajmg.a.32505. [DOI] [PubMed] [Google Scholar]
- 49. Romitti PA, Lidral AC, Munger RG, Daack-Hirsch S, Burns TL, Murray JC. Candidate genes for nonsyndromic cleft lip and palate and maternal cigarette smoking and alcohol consumption: evaluation of genotype-environment interactions from a population-based case-control study of orofacial clefts. Teratology. 1999;59(1):39–50. doi:10.1002/(SICI)1096–9926(199901)59:1<39::AID-TERA9>3.0.CO;2–7. [DOI] [PubMed] [Google Scholar]
- 50. Wilkie AO, Morriss-Kay GM. Genetics of craniofacial development and malformation. Nat Rev Genet. 2001;2(6):458–468. doi:10.1038/35076601. [DOI] [PubMed] [Google Scholar]
- 51. Singh S, Pillai S, Chellappan S. Nicotinic acetylcholine receptor signaling in tumor growth and metastasis. J Oncol. 2011;2011:456–743. doi:10.1155/2011/456743. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.