

Abstract
Elongation factor P (EF-P) accelerates diprolyl synthesis and requires a posttranslational modification to maintain proteostasis. Two phylogenetically distinct EF-P modification pathways have been described and are encoded in the majority of Gram-negative bacteria, but neither is present in Gram-positive bacteria. Prior work suggested that the EF-P-encoding gene (efp) primarily supports Bacillus subtilis swarming differentiation, whereas EF-P in Gram-negative bacteria has a more global housekeeping role, prompting our investigation to determine whether EF-P is modified and how it impacts gene expression in motile cells. We identified a 5-aminopentanol moiety attached to Lys32 of B. subtilis EF-P that is required for swarming motility. A fluorescent in vivo B. subtilis reporter system identified peptide motifs whose efficient synthesis was most dependent on 5-aminopentanol EF-P. Examination of the B. subtilis genome sequence showed that these EF-P-dependent peptide motifs were represented in flagellar genes. Taken together, these data show that, in B. subtilis, a previously uncharacterized posttranslational modification of EF-P can modulate the synthesis of specific diprolyl motifs present in proteins required for swarming motility.
Keywords: Bacillus, cell motility, microbiology, posttranslational modification, protein synthesis, translation elongation factor
Introduction
Ribosomes, along with a set of translation factors, translate genetic information by polymerizing amino acids into proteins. Of the 20 amino acids common to all organisms, proline is the slowest to form a peptide bond, especially when consecutive proline residues are polymerized (1, 2). If not resolved, the slow synthesis of poly-proline sequences can lead to translational pausing (3, 4). Bacterial translation elongation factor P (EF-P)3 remedies proline-induced pausing by binding to the ribosome near the pepitdyl transfer site and makes synthesis of oligoprolines entropically favorable (5). The presence of EF-P alone is not sufficient to maintain efficient translation of oligoprolines because the active protein also requires posttranslational modification (6–9).
Although EF-P is conserved in all bacteria, EF-P posttranslational modification systems are seemingly diverse (6, 9, 10). In Gammaproteobacteria, the genes yjeK, yjeA, and yfcM coordinate the attachment of (R)-β-lysine onto Lys34 of EF-P (Salmonella enterica numbering), followed by a hydroxylation of Lys-β-Lys34 (7, 11). In contrast, both Gamma- and Betaproteobacteria harbor the gene earP, which posttranslationally glycosylates Arg32 of EF-P with L-rhamnose, which is produced by rmlABCD-encoded proteins (6, 9). Thus far, EF-P function and modification systems have only been studied in detail for a narrow set of Gram-negative bacteria. Gram-negative efp mutants exhibit severe pleiotropy because of an abundance of functionally diverse poly-proline-containing proteins (12, 13), prompting us to determine whether EF-P is similarly important in Gram-positive bacteria. A previous study carried out insertional mutagenesis and identified efp as necessary for swarming motility in the Gram-positive bacterium Bacillus subtilis, whereas vegetative growth and swimming motility were unimpaired (14). Another study reported defects in sporulation in B. subtilis efp mutants (13).
To ascertain the posttranslational modification state of B. subtilis EF-P, we characterized a missense Lys32-to-Ala mutant, the residue analogous to the modification site in Gamma- and Betaproteobacteria. Consistent with a role important for the function of EF-P, swarming motility was impaired in efpK32A to a similar extent as observed in Δefp mutants, whereas sporulation was unaffected in either mutant. Furthermore, use of a chromosomally inserted reporter system determined that Δefp and efpK32A strains were both unable to efficiently translate the canonical EF-P-dependent sequence of three consecutive proline residues. Bioinformatic analysis of the B. subtilis genome identified several swarming motility-associated genes with diprolyl motifs that were shown to be EF-P-dependent, as indicated by the in vivo reporter system. Finally, structural investigation by mass spectrometry elucidated a 5-aminopentanol moiety covalently linked to Lys32. Taken together, the data indicate B. subtilis requires EF-P to be posttranslationally modified to control the synthesis of a subset of proteins containing specific diprolyl motifs in the swarming motility regulon.
Experimental Procedures
Strains and Growth Conditions
Unless otherwise noted, B. subtilis and Escherichia coli strains were grown in Luria-Bertani (LB) broth (0.5% NaCl, 0.5% yeast extract, and 1% Tryptone) or LB agar plates fortified with 1.5% Bacto agar. When appropriate, antibiotics were included at the following concentrations: 5 μg/ml kanamycin, 100 μg/ml spectinomycin, 100 μg/ml ampicillin, or 1 μg/ml erythromycin plus 25 μg/ml lincomycin.
For swarm assays, strains were grown to mid-log phase at 37 °C in 3 ml of LB medium, and 1 ml was harvested by centrifugation. Cells were resuspended to an A600 of 10 in PBS buffer containing 0.5% India ink. 0.7% agar LB plates were dried for 10 min in a laminar flow hood, inoculated with 10 μl of cell resuspension, dried for an additional 10 min, and incubated at 37 °C. The swarm radius was recorded every 30 min for 6 h along the same axis for consistency.
Sporulation efficiency was determined as described previously (15). Overnight cultures were used to inoculate 3 ml of growth medium (0.01% casein hydrolysate, 25 mm l-glutamate, 15 mm l-alanine, 10 mm l-asparagine, 1 mm KH2PO4, 25 mm NH4CL2, 0.77 mm Na2SO4, 1.2 mm NH4NO3, 3.7 μm FeCl3, 0.4 mm MgSO4, 0.002% CaCl2, 10 μm MnSO4, and 1 μm l-tryptophan) and grown to mid-log phase at 37 °C. Cultures were harvested by centrifugation, resuspended in 3 ml of resuspension medium (3 μm FeCl3, 37 μm MgCl2, 90 mm MnCl2, 5 mm NH4Cl2, 0.67 mm Na2SO4, 0.45 mm KH2PO4, 1 mm NH4NO3, 0.002% l-glutamate, 1 mm CaCl2, and 40 mm MgSO4) and incubated overnight at 37 °C. The following morning, cultures were diluted in H2O and plated onto LB plates. Dilutions were subsequently incubated at 80 °C for 20 min to heat-kill vegetative cells and replated onto LB plates. Colony-forming units were counted following overnight incubation at 37 °C. Growth curves were performed by subculturing overnight cultures into 25 ml of LB to an A600 of 0.01. Cultures were incubated at 37 °C with shaking at 175 rpm, and A600 was monitored every 30 min for 7 h.
For analysis of colony morphology on LB agar, strains were grown to mid-log phase at 37 °C, diluted into H2O, and plated onto LB agar plates. After incubation at 37 °C overnight, colonies were imaged using a Leica EZ4D microscope. Pellicle assays were performed by growing strains to mid-log phase at 37 °C and harvesting 1 ml by centrifugation. Cells were resuspended to an A600 of 10 in LBGM (0.5% NaCl, 0.5% yeast extract, 1% Tryptone, 0.5% glycerol, and 0.05 mm MnSO4). A 10-μl cell resuspension was used to inoculate 10 ml of LBGM in 6-well plates (Corning). Following incubation at 22 °C for 2 days, pellicles were imaged against a black background with a tripod-mounted Canon PowerShot A620 camera. Colony architecture analysis was performed by stamping colonies grown overnight at 37 °C onto 1.5% agar LBGM plates. Following incubation at 22 °C for 5 days, colonies were imaged as described for the pellicle assay.
Strain Construction
All strain descriptions are detailed in Supplemental Tables 1 and 2, with primers used for the generation of plasmids and strains found in Supplemental Table 2. To generate Δefp, the upstream flanking region was amplified with primer pair 4031/4032, and the downstream flanking region was amplified with primer pair 4033/4034. A Gibson assembly was used to ligate the flanking regions into the SmaI restriction site of pMiniMAD, which contains a temperature-sensitive origin and mls resistance cassette (16–18). The resulting construct was transformed into DK1042, and mls resistance was selected at 37 °C. Plasmid eviction was subsequently induced by incubating 3 ml of LB cultures at room temperature overnight. Resulting mls-sensitive colonies were analyzed by PCR to ascertain whether eviction resulted in reversion to the wild-type efp allele or deletion.
To generate efpK32A at the native site, primer pair 4031/4039 was used to amplify the upstream flanking region, and primer pair 4034/4038 was used to amplify the downstream flanking region. Primers 4038 and 4039 are complementary to one another and encode the K32A mutation. A Gibson assembly was used to ligate the flanking regions into the SmaI site of pMiniMAD, and the resulting plasmid was transformed and evicted as described for Δefp construction. Mls-sensitive colonies were analyzed for the retention of the efpK32A allele through sequencing.
B. subtilis 168 strains deficient in spermidine biosynthesis were purchased from the Bacillus Genetic Stock Center (Ohio State University). Mutant strains were grown in minimal salt medium as described previously (19) and lysed in 25 mm Tris (pH 8) with 100 μg/ml of lysozyme for 30 min at 37 °C, followed by addition of 3 units of DNase and incubation for another 30 min at 37 °C. The lysate was clarified on a tabletop centrifuge spun at 20,000 × g for 15 min, decanted, and flash-frozen to be stored at −80 °C for further analysis.
EF-P-FLAG Mutant Construction
An IPTG-inducible efp construct was constructed at the amyE locus by amplification of the efp gene with primer pair 3575/3576. The resulting fragment was digested with NheI and SphI restriction enzymes and ligated into the corresponding restriction sites of pDRIII (a gift from David Rudner, Harvard Medical School). pDRIII contains the Physpank promoter, the lacI lactose repressor, and a spectinomycin resistance cassette. The resulting plasmid was used to transform DS2569. Genomic DNA harvested from a spectinomycin-resistant transformant (DK755) was used to amplify the amyE locus with primer pairs 3177/4250 and 3180/4251. Primers 4250 and 4251 are complementary and introduce a FLAG epitope to the C terminus of EF-P. The two fragments were ligated with a Gibson assembly and transformed into DK2050. The amyE locus of spectinomycin-resistant isolates was sequenced to verify the introduction of the FLAG tag. The efpK32A allele was introduced into this construct by amplifying the amyE locus of DK755 with primer pairs 3177/4250, 4251/4038, and 4039/3180. The resulting fragments were ligated by Gibson assembly, transformed into DK2050, and sequence-verified.
Construction of GFPPPX, fliP-GFP, flhP-GFP, and Recombinant EF-P Expression Plasmids
Using the pDR111 plasmid as described above, IPTG-inducible GFPPPX constructs were generated by modifying templates used in prior studies (4, 20). GFP and GFPPPP were amplified with the restriction sites NheI and SphI and cloned into pDR111 using primer pairs 4832/4834 and 6449/4834, respectively. XL1 Blue cells were transformed with the resulting plasmid and screened by colony PCR for the presence of a correct insert. To generate the PPX motifs of interest, site-directed mutagenesis was performed using primer pairs 4293/4294 (PPG), 4067/4068 (PPV), 4069/4070 (PPW), 1373/1374 (PPL), and 7825/7826 (PPR) on the GFPPPP plasmid using the QuikChange site-directed mutagenesis kit (Stratagene). Otherwise, a Gibson assembly was used to generate GFPPPN with primer pairs 7516/7515 and 7514/7517.
The fusion constructs flhP and flip were amplified from B. subtilis 3610 genomic DNA and cloned into pDR111 by Gibson assembly using primer pairs 7340/7341 and 7343/5272. The linker region GSGGG was inserted between gfp and the fusion proteins using the primer pair 3933/7342. All reporter constructs were sequenced to confirm that the correct clone was obtained. Each reporter construct was transformed into strains DS2569, DK3235, and DK2050 and assessed for the presence of GFP fluorescence. Chromosomal integration into the correct location was obtained with primer pairs 7122/7123 flanking the AmyE locus and LacI gene. Recombinant expression of B. subtilis EF-P was obtained by amplifying B. subtilis EF-P from genomic DNA of strain 3610 with primer pair 2712/1961. The amplified efp fragment was digested with SapI and XhoI, cloned into the intein expression vector pTYB11, and transformed into XJB BL21(DE3) for expression.
spoIIIE::kan Construction
To construct the spoIIIE::kan allele, upstream and downstream flanking regions of spoIIIE were amplified with primer pairs 3393/3394 and 3395/3396, respectively. The kanamycin resistance cassette from pDG780 was amplified with primer pair 3250/3251 (3) and ligated between the flanking regions with a Gibson assembly. The resulting fragment was transformed into DS2569, and kanamycin-resistant isolates were selected. The spoIIIE::kan allele was transduced into 3610 using the general transducing phage SPP1 (21).
Assessing PPX Motif Dependence in Vivo
Overnight cultures of the various GFPPPX reporter constructs were inoculated into fresh LB broth and grown to mid-log phase. When mid-log was reached, cells were induced with 1 mm IPTG for 1 h. After 1 h of induction, 1 ml of cells was collected, washed three times with 1× phosphate-buffered saline (22), and an A600 was taken for purposes of normalization. Wild-type B. subtilis transformed with pRD111 was used to account for background fluorescence. Fluorescent readings for GFP were measured on a Fluorolog-3 as described previously (4, 20).
In the case of the fusion constructs, colonies from plates were directly inoculated into minimal salt medium. After 16 h of growth, cells were induced for 7 h with 1 mm IPTG. Cells were diluted 5-fold, and an A600 was taken prior to GFP fluorescence measurements.
Western Blotting
2 mg of purified EF-P was sent to Cocalico Biologicals for polyclonal antiserum generation in a rabbit host. Anti-SigA antiserum was a gift from Masaya Fujita (University of Houston). 1 ml of mid-log cultures grown at 37 °C was harvested by centrifugation. Pellets were resuspended to an A600 of 10 in lysis buffer (20 mm Tris (pH 7.0), 10 mm EDTA, 1 mg/ml lysozyme, 10 μg/ml DNase I, 100 μg/ml RNase I, and 1 mm PMSF) and incubated at 37 °C for 30 min. The appropriate volume of 6× SDS loading dye was added, and samples were incubated at 95 °C for 5 min. 12-μl samples were separated on an 12% SDS-polyacrylamide gel and transferred onto nitrocellulose. Nitrocellulose blots were probed with 1:40,000 dilution anti-EF-P or 1:80,000 anti-SigA polyclonal antiserum and subsequently probed with horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G. Blots were developed with Pierce ECL substrate (Thermo Fisher Scientific).
Isoelectric Focusing
Isoelectric focusing gels were adapted from previous studies with minor alterations (8, 23). In brief, proteins were resolved on a native isoelectric focusing gel with a pH gradient range of 4.5–5.4. (Pharmalyte 4.5–5.4, GE Healthcare). Isoelectric focusing gels required refrigeration during the run and were prefocused for 5 min at 100 V. Prior to loading the samples, the wells were rinsed with cathode buffer (50 mm NaOH) to remove unreacted ammonium persufate. Gels were focused for 1 h at 200 V, 1 h at 300 V, and 30 min at 500 V. Isoelectric focusing gels were then incubated in Towbin buffer for 5 min and transferred onto a HybondTM-C Extra nitrocellulose membrane (GE Healthcare) according to protocols established previously (8).
Bioinformatics and Statistical Methods
The fasta file for the genome of Bacillus subtilis 168 was obtained from ftp://ftp.ncbi.nih.gov/genomes/Bacteria/. In-house scripts were written with biopython to search PPX motifs for each gene in B. subtilis and return the identity of the motif as well the gene name and total instances of PPX motifs (24). Microsoft Office software was used to view the data. Student's t test was performed for comparisons of growth rate doubling times as well for fluorescence ratios comparing wild-type/efpK32A and wild type/Δefp. The data for PPN were not normally distributed, and fluorescence ratios were instead compared using a Wilcoxon signed-rank test.
Purification of Recombinant and Native B. subtilis EF-P
Native B. subtilis EF-P purified for top-down mass spectrometry was adapted from a prior paper with the following changes (25). Wild-type B. subtilis 3610 cells were grown in 40 liters of LB medium. Cells were harvested by centrifugation at 7500 rpm for 10 min (JLA 16.250 rotor) and resuspended in 100 ml of buffer A (25 mm Tris-HCl (pH 8), 150 mm NaCl, 10% glycerol, 2 mm β-mercaptoethanol) and supplemented with a protease inhibitor mixture (Roche Molecular Biochemicals). Cell lysis was performed on a French press with three passages, and the lysate was clarified by centrifuging at 75,000 × g for 90 min at 4 °C (JA 25.50 rotor). B. subtilis EF-P was precipitated between 40–60% of ammonium sulfate, as determined by immunoblotting precipitated fractions. Fractions containing EF-P were pooled and dialyzed in buffer A without glycerol before being loaded onto a Hiprep XK26 Sepharose Q column (65 ml) using an AKTA Prime FPLC machine. EF-P-containing fractions, as determined by immunoblot analysis, were collected and concentrated to 200 μl using an Amicon Ultra centrifugal filter (molecular weight cut-off 10 kDA). A concentrated sample containing EF-P was injected onto a Hiload 26/600 Superdex 200 prep grade column (330 ml) and eluted off in buffer A without glycerol. Fractions containing EF-P were concentrated to 100 μl and dialyzed against buffer A.
Purification of native B. subtilis EF-P by multistep chromatography proved to be inefficient for further analysis. Therefore, FLAG-tagged B. subtilis EF-P was chromosomally integrated into a Δefp B. subtilis strain and purified with anti-FLAG M2 magnetic beads (Sigma-Aldrich) following the instructions of the manufacturer. Recombinant intein-tagged B. subtilis EF-P was expressed in XJB BL21(DE3) cells in an autoinduction medium as described before, with slight modifications (25). Lysate expressing recombinant EF-P was applied to an in-house packed intein column (New England Biolabs) and allowed to sit overnight at room temperature to increase cleaving efficiency. Protein was eluted in 50 mm DTT and dialyzed in buffer A.
High-resolution Top-Down Mass Spectrometry
Individual native and recombinant B. subtilis EF-P was initially analyzed as described before by tandem LC-MS+ using a triple-quadrupole mass spectrometer (API III+, Applied Biosystems) prior to being introduced to Fourier transform-ICR by direct infusion nanospray (6, 26, 27). Envelopes of multiply charged ions were measured for each sample on a hybrid linear ion-trap/Fourier transform-ICR mass spectrometer (7T, LTQ FT Ultra, Thermo Scientific) operated with a standard (up to m/z 2000) or extended mass range (up to m/z 4000). Spectra were derived from an average of 500 transient signals, and mass resolution was set to 100,000. Data were analyzed exactly as before using ProSight PC 2.0 software (Thermo Fisher) (6).
Bottom-up Proteomics and Structural Investigation
Tandem LC-MS2 and tandem LC-MS3 analyses were performed with an Easy-nLC 1000 (Thermo Scientific) coupled to an Orbitrap Fusion mass spectrometer (Thermo Scientific). The LC system consisted of a fused silica nanospray needle (PicoTipTM emitter, 75 μm internal diameter, New Objective) packed in-house with 40 cm of Magic C18 AQ 100-Å reverse-phase media (Michrom Bioresources Inc.). Native FLAG-tagged and recombinant EF-P were separated by SDS-PAGE and in-gel digested with chymotrypsin (Promega) as described in a previous study (28). Peptide samples were resuspended in 0.1% formic acid with 2% acetonitrile at a concentration of 400 ng/μl, and 2 μl was loaded onto the column and separated using a two-mobile-phase system consisting of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). The chromatographic separation was achieved over a 38-min gradient from 3–50% B (3–30% B for 30 min, 30–50% B for 5 min, and 50% B for 3 min) at a flow rate of 300 nl/min. The mass spectrometer was operated in a data-dependent MS/MS mode over the m/z range of 350–1200, with target m/z values of 380.5495 and 414.2393. The mass resolution was set to 120,000, and the mass tolerance was set to 25 ppm. For each cycle, up to four ions with target m/z values from the precursor scan were selected for MS2 and MS3 analysis. If the precursor m/z was 380.5495, then m/z 477.2566 was selected for MS3 analysis, and if the precursor m/z was 414.2393, then m/z 578.3403 was selected for MS3. The MS2 analysis was performed using electron transfer dissociation (ETD) with charge-dependent ETD parameters and analyzed with a Orbitrap mass analyzer with the resolution set to 30,000. The MS3 analysis was performed using higher-energy collision-induced dissociation (HCD) with 40% normalized collision energy and analyzed with the Orbitrap with the mass resolution set to 30,000. The isolation window for both MS2 and MS3 was 1.6 m/z.
Results
Lys32 of EF-P Is Critical for Function
In the majority of Gammaproteobacteria, EF-P is posttranslationally modified on a conserved lysine residue, whereas most Betaproteobacteria and a few Gammaproteobacteria modify EF-P on an analogous arginine residue. Mutation of efp at its posttranslational modification site in these organisms has highly pleiotropic effects similar to a null mutant, including a significant reduction in growth rate (29, 30). The homologous residue in B. subtilis EF-P is Lys32, and to determine its biological relevance, it was replaced with alanine at the native chromosomal locus (efpK32A). Only a slight reduction in growth rate compared with the wild type was observed in the B. subtilis Δefp or efpK32A missense mutation strains, in sharp contrast to the significant reductions in growth rates observed for efp mutants in other bacteria (Fig. 1, A and B) (6, 7). Furthermore, a sporulation defect was reported previously in a B. subtilis efp mutant in laboratory strains (13), but no sporulation defect was observed in the Δefp or efpK32A strains when compared with the wild type in our strains (Fig. 1C). Colonies of the efp mutant had a smooth morphology on LB medium, often indicative of a defect in biofilm formation (Fig. 1D). However, no defect in either complex colony morphology or floating pellicles was observed in solid or liquid biofilm-promoting media, respectively (Fig. 1D). We conclude that the phenotype of an efp mutant is less pleiotropic in B. subtilis than what has been reported for Gram-negative bacteria.
FIGURE 1.

Phenotypic effects of loss of EF-P or mutation of the conserved lysine 32 residue. A, growth curve of strains grown in LB at 37 °C. The symbols represent an average of three replicates, and error bars depict the standard deviation. B, exponential growth rate of strains averaged over three replicates. Error bars indicate standard deviation. *, p < 0.05. C, sporulation assay in which cells were grown in sporulation-promoting medium and colony-forming units determined before and after 20-min incubation at 80 °C. The data represent an average of five replicates. Error bars indicate standard deviation. D, colony morphology of strains grown on LB 1.5% agar at 37 °C for 1 day (top row), LBGM 1.5% agar at 22 °C for 5 days (center row), or pellicle formation in LBGM after 2 days of incubation at 22 °C (bottom row). E, swarm assays in which symbols represent the average of three replicates. F, SDS-PAGE Western blot of lysates probed with anti-EF-P or anti-SigA polyclonal antisera. The following strains were used: WT (DK1042), efp (DK2050), efpK32A (DK3235), spoIIIE (DK453), and epsH (DS6776).
EF-P has previously been shown to be required for a flagellum-mediated form of surface migration called swarming motility (14). Consistent with a role important for the function of EF-P in B. subtilis, the efpK32A mutant displayed an impaired ability to swarm, although not to the extent observed in an efp deletion mutant (Fig. 1E). Importantly, the EF-PK32A protein was synthesized to the same level as the wild type when assayed by Western blotting, indicating that the mutation affected EF-P function rather than protein stability (Fig. 1F). These data suggest a physiological role of EF-P in swarming motility.
Motility-related Genes Code EF-P-dependent Motifs
The absence of EF-P in B. subtilis is associated with a swarming defect, whereas vegetative growth is only slightly impaired, suggesting that EF-P may play a role in the synthesis of a subset of motility-related proteins. A bioinformatic search for the canonical pause sequence PPP in the ancestral strain B. subtilis 3610 identified 34 genes, with none linked to motility or swarming. When expanded to all diprolyl motifs, the search revealed 927 gene-encoded proteins with PPX motifs, of which 59 were essential and 14 were associated with motility (supplemental Table 3). From the essential genes, 19 of the 20 possible PPX motifs were represented, with PPR being absent and PPG the most abundant (supplemental Table 3). The motility genes contained 12 unique PPX motifs: PPD, PPN, PPI, PPT, PPR, PPK, PPN, PPV, PPW, PPG, PPE, and PPA, of which PPV had the highest number of occurrences (supplemental Table 3). Based on comparison to ribosome profiling studies of EF-P dependent motifs, we selected from the above motifs those previously shown to produce a strong pause in E. coli Δefp strains (PPW, PPN, PPG, and PPP) and motifs less likely to create a pause (PPV, PPR, and PPL) (3, 4) to test using an in vivo reporter.
A single-copy, IPTG-inducible, in vivo reporter to measure pause strength of PPX motifs was engineered to form a chromosomal insertion at the native amyE locus in B. subtilis. The reporter consisted of the Physpank IPTG-inducible promoter, a variable representative poly-proline motif inserted in-frame after the fourth codon, with the remainder of gfp fused translationally downstream. This allowed the use of GFP florescence to quantify the efficiency of synthesis of a particular PPX motif. The average ratio of GFPPPX fluorescence produced in the wild type compared with the Δefp mutant strain decreased in the following order: PPW (8.87 ± 2.46), PPP (4.41 ± 1.36), PPG (4.40 ± 1.29), PPN (3.15 ± 0.15), PPR (2.97 ± 1.06), PPV (2.24 ± 0.06), and PPL (2.15 ± 0.48) (Fig. 2). We conclude that B. subtilis EF-P improves the translation efficiency of poly-prolyl motifs at a level comparable with that reported for Gram-negative bacteria. Contribution of the putative modification site with respect to the in vivo function of EF-P was also assessed using the native efpK32A mutant, revealing no statistical difference compared with the corresponding wild-type/Δefp motif, except in the case of PPV (p < 0.05): PPW (11.85 ± 3.61), PPG (4.59 ± 0.42), PPR (3.91 ± 1.68), PPN (3.70 ± 0.76), PPP (3.35 ± 0.49), PPL (1.73 ± 0.35), and PPV (1.49 ± 0.18) (Fig. 2).
FIGURE 2.
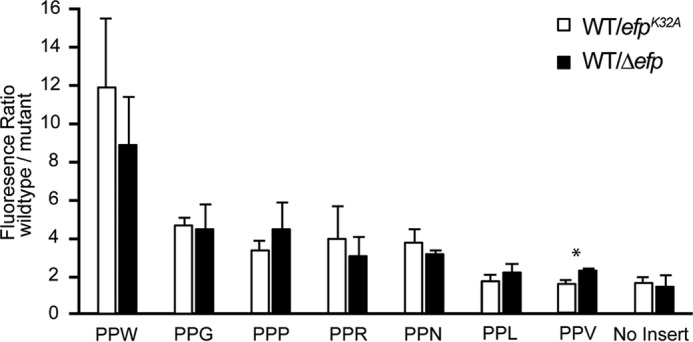
In vivo analysis of EF-P-dependent PPX motifs found in swarming genes. Translation efficiency of chromosomally inserted GFPPPX reporters were tested in the B. subtilis Δefp and efpK32A strains and compared with the wild type. GFPPPX expression was assayed in cells grown in LB in the presence of 1 mm IPTG and harvested during exponential phase. Fluorescence was normalized to A600, and error bars represent the mean ± S.D. from three biological replicates. A Student's t test was performed between Δefp and efpK32A strains within the same motif, indicating that PPV was significantly different (*, p = 0.0137).
The reporter-generated in vivo pause scores suggest that the diprolyl motif PPW has the greatest dependence on EF-P for translation, PPV and PPL have little if any dependence, and PPR, PPN, and PPG are about as dependent as PPP. Using the putative model of the B. subtilis flagellar machinery, the different diprolyl motifs were mapped to nine of the respective motility genes, revealing that the majority of PPX motifs were concentrated in the secretion machinery (supplemental Fig. 1, A and B). Other components containing poly-prolines included the stator, basal body, and filament. Mapping the in vivo pause scores to the corresponding flagellar components indicated that motifs with strong and moderate pauses were coded for in the secretion components flhA (PPW), fliP (PPN), and fliL (PPN) (supplemental Fig. 1C). In addition, the stator motB gene and the putative rod gene flhP encode the moderate pausing motifs PPR and PPG, respectively (supplemental Fig. 1C). In summary, the flagellar machinery contains a variety of EF-P-dependent motifs, with strong pauses found predominantly in the secretion components. Previous reports indicated that FliP expression is controlled by EF-P in E. coli. FliP has also been demonstrated to be required for swarming motility in B. subtilis. To validate whether EF-P facilitates the expression of genes involved with swarming, a fliP-gfp fusion was generated and expressed in wild-type, Δefp, and efpK32A backgrounds. Significant reductions in GFP fluorescence were measured in the Δefp and efpK32A strains, 1.7- and 3.8-fold compared with the wild type, respectively (Fig. 3A). To determine whether the trend extends to other proteins required for swarming, a flhP-gfp construct was produced and found to show similar results to FliP (Fig. 3B). These data suggest that EF-P is required for optimal production of multiple swarming motility-associated proteins.
FIGURE 3.

EF-P is required for optimal expression of fliP and flhP swarming motility genes. Chromosomally inserted fliP-gfp and flhP-gfp were expressed in the B. subtilis wild-type, Δefp, and efpK32A strains. Bacterial cells were grown in minimal salt medium to saturation, followed by a 7-h induction of the fusion protein construct with the addition of 1 mm IPTG. Fluorescence was normalized to A600, and errors bars represent the mean ± S.D. from four biological replicates.
B. subtilis EF-P Is Posttranslationally Modified at Lys32 with 5-Aminopentanol
Posttranslational modifications have been shown previously to be required for the activity of all characterized forms of EF-P and its eukaryotic and archaeal paralogs eIF5A and aIF5A, respectively. To determine whether the B. subtilis protein is also posttranslationally modified, B. subtilis EF-P was purified natively using multistep chromatography or purified recombinantly from E. coli as an unmodified control and then analyzed by high-resolution mass spectrometry. Recombinant EF-P yielded a measured monoisotopic mass of 20,455.603 Da (Fig. 4A and supplemental Table 4), consistent with that calculated from the gene sequence (20,455.374 Da, Δ mass 0.229 Da), and native EF-P yielded a measured monoisotopic mass of 20,556.624 Da (Fig. 4D and supplemental Table 5) that was 101.250 Da heavier. The spectra of both samples revealed adducts that were 113 Da heavier, attributed to TFA adducts formed in the mass spectrometer ion source.
FIGURE 4.

Structural characterization of the B. subtilis EF-P posttranslational modification 5-aminopentanol. A and D, high-resolution mass spectrum of intact recombinant and native BsEF-P measured on a 7T Fourier transform-ICR. Monoisotopic masses were deconvoluted based on the envelope of multiply charged ions. B and E, chymotrypsin-digested modified and unmodified peptide was sequenced by ETD and revealed Lys32 to harbor the modification. C and F, ETD-HCD MS3 of the z5+ ion and proposed charge-directed fragmentation pattern for the modification. Roman numerals and Greek letters represent ion fragments unique to the modification.
To obtain an elemental composition and determine whether the additional mass localized to a specific residue, bottom-up proteomics were carried out on in-gel chymotrypsin-digested native and recombinant FLAG-EF-P peptides. Fragmentation by ETD of the peptide QHVKPGKGAAF produced a series of z and c ions, sufficient to assign the additional mass to Lys32 from native EF-P (Fig. 4E). ETD fragmentation of the same peptide, but from recombinant EF-P, achieved complete backbone cleavage without any additional mass on any of the residues (Fig. 4B). The average mass difference between modified and unmodified fragments (Δ mass 101.0834 Da) was used to compute the elemental composition of the modification, yielding C5H11NO with a 6.92-ppm difference between measured and theoretical values (supplemental Table 6).
To add to the limited information available regarding the structure of the modification, ETD/HCD MS3 was performed to clarify the molecular arrangement of the atoms. The modified and unmodified z5+ ions (m/z 578.339, 477.256) generated during MS2 ETD were isolated and fragmented further with HCD. Based on the resulting spectra, Δ masses with respect to the precursor ion and measured ion fragments were compared to obtain a set of common and unique mass differences. Common to both modified and unmodified peptides were a series of a, b, and y ions (Fig. 4, C and F). From a set of Δ masses unique for the modified ion, 16.018 u, 74.057 u, 88.073 u, 132.062 u, 221.089 u, 265.116 u, 279.131 u, and 293.148 u, a charge directed fragmentation pattern of the modification was determined that suggested a five-carbon chain with a terminal amine group attached to the η amine of lysine (Fig. 4F). However, the fragmentation data were incomplete and did not provide the location of the hydroxyl group, which is postulated to be on either the third or fourth carbon, because fragment ions (ϵ, δ-a4) measured the addition of an OH on or before the fourth carbon, whereas ions before the second carbon did not measure the addition of an OH (Fig. 4F).
The structure of the 5-aminopentanol modification bears a striking resemblance to hypusine, the essential posttranslational modification of the eukaryotic EF-P paralog eIF5A (supplemental Fig. 2). Hypusine is derived from spermidine, suggesting that 5-aminopentanol could also be derived from a polyamine precursor substrate. Furthermore, ETD/HCD MS3 produced an ion (102.092 m/z) matching that of the modification, suggesting that the modification can carry a charge, analogous to β-Lys and hypusine (Fig. 4F). Based on the positive charge gained when EF-P is modified, isoelectric focusing followed by Western blotting was carried out to assess whether B. subtilis strains deficient in polyamine biosynthesis could modify EF-P. To rapidly test a variety of deletion strains, B. subtilis 168 mutants were purchased from the Bacillus Genetic Stock Center. B. subtilis EF-P recombinantly purified from E. coli served as an unmodified control, although partial modification with (R)-β-lysine was observed (Fig. 5). EF-PK32A was also run on the isoelectric focusing gel and migrated below recombinant B. subtilis EF-P because of the absence of a modification and the substitution of the lysine residue to an alanine (Fig. 5). EF-P remained modified in mutants disrupted for each step in spermidine biosynthesis when grown in minimal salt medium (Fig. 5). Therefore, it is likely that the modification does not originate from the polyamines putrescine or spermidine.
FIGURE 5.
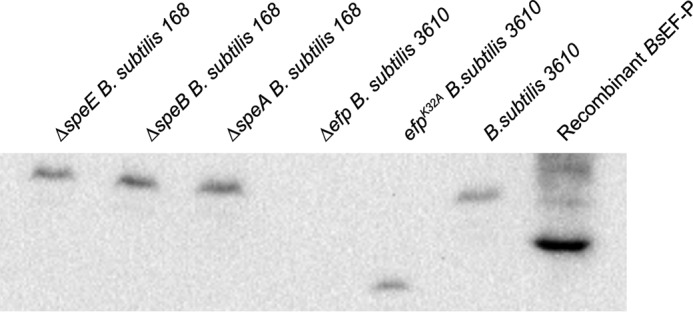
Isoelectric focusing gel resolves modified and unmodified BsEF-P. Samples were loaded onto a gel with a sharp pH gradient (4.5–5.4). The deletion strains ΔspeE, ΔspeB, and ΔspeA were acquired from the Bacillus Genetic Stock Center and have the B. subtilis 168 background, whereas Δefp, efpK32A, and the wild type were generated in a B. subtilis 3610 background strain. The isoelectric focusing gel was probed for EF-P and run with positively charged proteins migrating toward the top of the gel and negatively charged proteins migrating lower on the gel.
Discussion
The rapid translation of poly-proline residues in Gammaproteobacteria is dependent on a fully modified EF-P, and absence of the modification results in aberrant phenotypes such as impaired swimming motility, growth defects, and compromised membrane integrity (6, 33). In contrast, when efp is disrupted in B. subtilis, limited pleiotropy is observed, with swarming motility abolished and vegetative growth only mildly affected, calling into question the broad role of EF-P and its possible modification in B. subtilis. Here we investigated the importance of the putative modification site for efp in B. subtilis by generating a native missense variant, K32A. Sporulation was unaffected in the Δefp and efpK32A strains, contrary to results obtained in a prior study, a discrepancy perhaps attributed to differences in ancestral and laboratory strain backgrounds (13). Previous studies that replaced modification sites of EF-P with alanine also resulted in phenotypes resembling those where genes involved in modifying EF-P were knocked out (6). However, those mutant efp genes were overexpressed in trans from a vector that resulted in dominant-negative phenotypes. The efpK32A strain was shown to produce EF-P at comparable levels to the wild type, thus preventing the possibility of an overexpression artifact, further supporting a prominent role for Lys32 in the function of EF-P. The efpK32A mutation resulted in phenotypes similar to the Δefp strain, displaying a severe defect in swarming motility compared with the wild type. One possible reason for the observed swarming defect is that modified EF-P is required to accelerate the synthesis of diprolyl motifs in proteins required for swarming motility. An assortment of diproyl motifs encoded in motility-related genes were analyzed using an in vivo reporter in both the Δefp and efpK32A backgrounds. Of the motifs that contributed to pausing in both mutant strains, PPW, had the greatest effect. PPG, PPP, PPN, and PPR all had similar moderate effects, whereas PPV and PPL were not dependent on EF-P for translation. Each of these motifs that produced a significant pause in the mutant strains are found in several motility genes that are essential for swarming (34). For instance, fliP and flhP encode PPN and PPG motifs, respectively, and gfp fusion constructs revealed that their expression depended on EF-P for efficient translation. Although loss of either of these genes abolishes motility (35), the absence of efp could lead to a decrease in the levels of flagellar proteins, disrupting secretion machinery stoichiometry and overall flagellin output (36). The requirement for EF-P during the synthesis of particular diprolyl motifs matches the trend observed in Gram-negative bacteria, confirming that the function of EF-P remains the same in Gram-positive bacteria regardless of differences in poly-proline abundance (3, 4).
The PPP motif has a significant pausing effect, and 38% of genes encoding PPP are involved in sporulation, including the sporulation-essential transcription factor SigE, but no defect in sporulation was observed. By investigating the amino acids flanking the triple proline in SigE, we observed two leucine residues predicted to diminish the reliance on EF-P for efficient translation (3, 4). The rate of initiation is another factor that was shown recently to influence the dependence on EF-P of ribosomes translating poly-proline-containing proteins (32). Therefore, it is also possible that initiation is the rate-limiting step in translating the majority of B. subtilis genes with poly-prolines, whereas swarming motility may require a substantial increase in the production of certain flagellar components dependent on modified EF-P to facilitate this transition in motile cell behavior. Flagellar proteins, particularly structural proteins, may be needed in abundance because cells may require as many as 30 flagella per cell to swarm (31).
For B. subtilis to effectively synthesize certain poly-proline-containing proteins, posttranslational modification of Lys32 of EF-P is required. Mass spectrometry revealed a novel posttranslational modification, 5-aminopentanol, for EF-P from B. subtilis, which resembles the previously described modification of eIF5A with hypusine. Although modification of B. subtilis EF-P with 5-aminopentonal is reminiscent of the addition of positively charged molecules to other EF-Ps and IF5As, the mechanism by which modifications contribute to peptide bond formation remains unclear.
The modification pathway of EF-P 5-aminopentonylation has yet to be completely elucidated. Assuming that the hydroxyl moiety is a secondary posttranslational modification, spermidine, which is also an intermediate in eIF5A modification, is an attractive candidate for a precursor substrate during 5-aminopentanol addition. Disruption of spermidine biosynthesis did not result in the loss of modified B. subtilis EF-P, as determined by isoelectric focusing. Furthermore, spermidine would only provide four carbons and require a subsequent methylation to match the correct mass of the modification. Cadaverine, on the other hand, is a polyamine that could provide five carbons and match the structure of the modification more accurately than spermidine. Cavaderine biosynthesis has not been described to date in B. subtilis, and, as a result, there are no obvious candidate genes to test for its possible roles in EF-P modification (10, 19). To identify the gene(s) responsible for modifying EF-P in B. subtilis, forward genetic screens searching for mutants defective in swarming motility are now necessary. Alternatively, it may be possible to complement a ΔpoxA mutant with the modification machinery from B. subtilis using a genomic library screen because of the resemblance between B. subtilis and E. coli EF-P. Future endeavors will then be able to determine whether the roles of these modification systems are complementary or whether other non-canonical functions exist.
Author Contributions
M. I., A. R., and D. B. K. designed the experiments. A. W., A. R., S. E., P. R. G., and K. R. H. performed the experiments. P. R. G., A. R., A. W., J. P. W., K. F. F., and K. R. H. analyzed the data. A. R., K. R. H., M. I., D. B. K., A. W., and P. R. G. wrote the manuscript. All authors edited and approved the manuscript. Responsibility of data analysis integrity is assumed by all authors. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Yuko Ogata for assistance with collecting mass spectrometry data. We also thank Masaya Fujita, Rebecca Calvo, Eric Vanderpool, and Sampriti Mukherjee for strains and reagents. The Fred Hutchinson Cancer Research Center Proteomics Facility is funded by Cancer Center Support Grant P30 CA015704 from the National Institutes of Health, and the OrbiTrap Fusion used in this research was funded in part by the M. J. Murdock Charitable Trust.
This work was supported by National Institutes of Health Grant GM065183 (to M. I.), National Institutes of Health Training Grant T32 GM007757 and National Science Foundation Graduate Research Fellowship Grant 1342962 (to K. R. H.), and National Institutes of Health Grant GM 093030 (to D. B. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Figures 1 and 2 and Tables 1–6.
- EF-P
- elongation factor P
- LB
- Luria-Bertani
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- ETD
- electron transfer dissociation
- HCD
- higher-energy collision-induced dissociation
- mls
- macrolide-lincosamide-streptogramin B
- ICR
- ion cyclotron resonance.
References
- 1. Pavlov M. Y., Watts R. E., Tan Z., Cornish V. W., Ehrenberg M., and Forster A. C. (2009) Slow peptide bond formation by proline and other N-alkylamino acids in translation. Proc. Natl. Acad. Sci. U.S.A. 106, 50–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wohlgemuth I., Brenner S., Beringer M., and Rodnina M. V. (2008) Modulation of the Rate of peptidyl transfer on the ribosome by the nature of substrates. J. Biol. Chem. 283, 32229–32235 [DOI] [PubMed] [Google Scholar]
- 3. Woolstenhulme C. J., Guydosh N. R., Green R., and Buskirk A. R. (2015) High-precision analysis of translational pausing by ribosome profiling in bacteria lacking EFP. Cell Rep. 11, 13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Elgamal S., Katz A., Hersch S. J., Newsom D., White P., Navarre W. W., and Ibba M. (2014) EF-P dependent pauses integrate proximal and distal signals during translation. PLoS Genet. 10, e1004553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Doerfel L. K., Wohlgemuth I., Kubyshkin V., Starosta A. L., Wilson D. N., Budisa N., and Rodnina M. V. (2015) Entropic contribution of elongation factor P to proline positioning at the catalytic center of the ribosome. J. Am. Chem. Soc. 137, 12997–13006 [DOI] [PubMed] [Google Scholar]
- 6. Rajkovic A., Erickson S., Witzky A., Branson O. E., Seo J., Gafken P. R., Frietas M. A., Whitelegge J. P., Faull K. F., Navarre W., Darwin A. J., and Ibba M. (2015) Cyclic rhamnosylated elongation factor P establishes antibiotic resistance in Pseudomonas aeruginosa. mBio 6, e00823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Navarre W. W., Zou S. B., Roy H., Xie J. L., Savchenko A., Singer A., Edvokimova E., Prost L. R., Kumar R., Ibba M., and Fang F. C. (2010) PoxA, YjeK, and elongation factor P Coordinately modulate virulence and drug resistance in Salmonella enterica. Mol. Cell 39, 209–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Roy H., Zou S. B., Bullwinkle T. J., Wolfe B. S., Gilreath M. S., Forsyth C. J., Navarre W. W., and Ibba M. (2011) The tRNA synthetase paralog PoxA modifies elongation factor-P with (R)-β-lysine. Nat. Chem. Biol. 7, 667–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lassak J., Keilhauer E. C., Furst M., Wuichet K., Godeke J., Starosta A. L., Chen J. M., Sogaard-Andersen L., Rohr J., Wilson D. N., Haussler S., Mann M., and Jung K. (2015) Corrigendum: arginine-rhamnosylation as new strategy to activate translation elongation factor P. Nat. Chem. Biol. 11, 299. [DOI] [PubMed] [Google Scholar]
- 10. Bailly M., and de Crécy-Lagard V. (2010) Predicting the pathway involved in post-translational modification of elongation factor P in a subset of bacterial species. Biol. Direct 5, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peil L., Starosta A. L., Virumäe K., Atkinson G. C., Tenson T., Remme J., and Wilson D. N. (2012) Lys34 of translation elongation factor EF-P is hydroxylated by YfcM. Nat. Chem. Biol. 8, 695–697 [DOI] [PubMed] [Google Scholar]
- 12. Starosta A. L., Lassak J., Peil L., Atkinson G. C., Woolstenhulme C. J., Virumäe K., Buskirk A., Tenson T., Remme J., Jung K., and Wilson D. N. (2014) A conserved proline triplet in Val-tRNA synthetase and the origin of elongation factor P. Cell Rep. 9, 476–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ohashi Y., Inaoka T., Kasai K., Ito Y., Okamoto S., Satsu H., Tozawa Y., Kawamura F., and Ochi K. (2003) Expression profiling of translation-associated genes in sporulating Bacillus subtilis and consequence of sporulation by gene inactivation. Biosci. Biotechnol. Biochem. 67, 2245–2253 [DOI] [PubMed] [Google Scholar]
- 14. Kearns D. B., Chu F., Rudner R., and Losick R. (2004) Genes governing swarming in Bacillus subtilis and evidence for a phase variation mechanism controlling surface motility. Mol. Microbiol. 52, 357–369 [DOI] [PubMed] [Google Scholar]
- 15. Harwood C. R., and Cutting S. M. (1990) Molecular Biological Methods for Bacillus, Wiley, Chichester, UK [Google Scholar]
- 16. Arnaud M., Chastanet A., and Débarbouillé M. (2004) New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, Gram-positive bacteria. Appl. Environ. Microbiol. 70, 6887–6891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Patrick J. E., and Kearns D. B. (2008) MinJ (YvjD) is a topological determinant of cell division in Bacillus subtilis. Mol. Microbiol. 70, 1166–1179 [DOI] [PubMed] [Google Scholar]
- 18. Gibson D. G., Young L., Chuang R. Y., Venter J. C., Hutchison C. A. 3rd, and Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 [DOI] [PubMed] [Google Scholar]
- 19. Sekowska A., Bertin P., and Danchin A. (1998) Characterization of polyamine synthesis pathway in Bacillus subtilis 168. Mol. Microbiol. 29, 851–858 [DOI] [PubMed] [Google Scholar]
- 20. Hersch S. J., Wang M., Zou S. B., Moon K. M., Foster L. J., Ibba M., and Navarre W. W. (2013) Divergent protein motifs direct elongation factor P-mediated translational regulation in Salmonella enterica and Escherichia coli. mBio 4, e00180–00113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yasbin R. E., and Young F. E. (1974) Transduction in Bacillus subtilis by bacteriophage SPP1. J. Virol. 14, 1343–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Doherty G. P., Bailey K., and Lewis P. J. (2010) Stage-specific fluorescence intensity of GFP and mCherry during sporulation in Bacillus subtilis. BMC Res. Notes 3, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cooper H. L., Park M. H., Folk J. E., Safer B., and Braverman R. (1983) Identification of the hypusine-containing protein hy+ as translation initiation factor eIF-4D. Proc. Natl. Acad. Sci. U.S.A. 80, 1854–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cock P. J., Antao T., Chang J. T., Chapman B. A., Cox C. J., Dalke A., Friedberg I., Hamelryck T., Kauff F., Wilczynski B., and de Hoon M. J. (2009) Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bullwinkle T. J., Zou S. B., Rajkovic A., Hersch S. J., Elgamal S., Robinson N., Smil D., Bolshan Y., Navarre W. W., and Ibba M. (2013) (R)-β-lysine-modified elongation factor P functions in translation elongation. J. Biol. Chem. 288, 4416–4423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Whitelegge J. P., Zabrouskov V., Halgand F., Souda P., Bassilian S., Yan W., Wolinsky L., Loo J. A., Wong D. T., and Faull K. F. (2007) Protein-sequence polymorphisms and post-translational modifications in proteins from human saliva using top-down Fourier-transform ion cyclotron resonance mass spectrometry. Int. J. Mass Spectrom. 268, 190–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gómez S. M., Nishio J. N., Faull K. F., and Whitelegge J. P. (2002) The chloroplast grana proteome defined by intact mass measurements from liquid chromatography mass spectrometry. Mol. Cell Proteomics 1, 46–59 [DOI] [PubMed] [Google Scholar]
- 28. Zhang L., Xu H., Chen C. L., Green-Church K. B., Freitas M. A., and Chen Y. R. (2008) Mass spectrometry profiles superoxide-induced intramolecular disulfide in the FMN-binding subunit of mitochondrial Complex I. J. Am. Soc. Mass Spectrom. 19, 1875–1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Balibar C. J., Iwanowicz D., and Dean C. R. (2013) Elongation factor P is dispensable in Escherichia coli and Pseudomonas aeruginosa. Curr. Microbiol. 67, 293–299 [DOI] [PubMed] [Google Scholar]
- 30. Zou S. B., Hersch S. J., Roy H., Wiggers J. B., Leung A. S., Buranyi S., Xie J. L., Dare K., Ibba M., and Navarre W. W. (2012) Loss of elongation factor P disrupts bacterial outer membrane integrity. J. Bacteriol. 194, 413–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mukherjee S., and Kearns D. B. (2014) The structure and regulation of flagella in Bacillus subtilis. Annu. Rev. Genet. 48, 319–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hersch S. J., Elgamal S., Katz A., Ibba M., and Navarre W. W. (2014) Translation initiation rate determines the impact of ribosome stalling on bacterial protein synthesis. J. Biol. Chem. 289, 28160–28171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zou S. B., Roy H., Ibba M., and Navarre W. W. (2011) Elongation factor P mediates a novel post-transcriptional regulatory pathway critical for bacterial virulence. Virulence 2, 147–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Calvo R. A., and Kearns D. B. (2015) FlgM is secreted by the flagellar export apparatus in Bacillus subtilis. J. Bacteriol. 197, 81–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Carpenter P. B., and Ordal G. W. (1993) Bacillus subtilis FlhA: a flagellar protein related to a new family of signal-transducing receptors. Mol. Microbiol. 7, 735–743 [DOI] [PubMed] [Google Scholar]
- 36. Courtney C. R., Cozy L. M., and Kearns D. B. (2012) Molecular characterization of the flagellar hook in Bacillus subtilis. J. Bacteriol. 194, 4619–4629 [DOI] [PMC free article] [PubMed] [Google Scholar]