

Abstract
Nav1.5, the pore-forming α subunit of the cardiac voltage-gated Na+ channel complex, is required for the initiation and propagation of the cardiac action potential. Mutations in Nav1.5 cause cardiac arrhythmias and sudden death. The cardiac Na+ channel functions as a protein complex; however, its complete components remain to be fully elucidated. A yeast two-hybrid screen identified a new candidate Nav1.5-interacting protein, αB-crystallin. GST pull-down, co-immunoprecipitation, and immunostaining analyses validated the interaction between Nav1.5 and αB-crystallin. Whole-cell patch clamping showed that overexpression of αB-crystallin significantly increased peak sodium current (INa) density, and the underlying molecular mechanism is the increased cell surface expression level of Nav1.5 via reduced internalization of cell surface Nav1.5 and ubiquitination of Nav1.5. Knock-out of αB-crystallin expression significantly decreased the cell surface expression level of Nav1.5. Co-immunoprecipitation analysis showed that αB-crystallin interacted with Nedd4-2; however, a catalytically inactive Nedd4-2-C801S mutant impaired the interaction and abolished the up-regulation of INa by αB-crystallin. Nav1.5 mutation V1980A at the interaction site for Nedd4-2 eliminated the effect of αB-crystallin on reduction of Nav1.5 ubiquitination and increases of INa density. Two disease-causing mutations in αB-crystallin, R109H and R151X (nonsense mutation), eliminated the effect of αB-crystallin on INa. This study identifies αB-crystallin as a new binding partner for Nav1.5. αB-Crystallin interacts with Nav1.5 and increases INa by modulating the expression level and internalization of cell surface Nav1.5 and ubiquitination of Nav1.5, which requires the protein-protein interactions between αB-crystallin and Nav1.5 and between αB-crystallin and functionally active Nedd4-2.
Keywords: cardiovascular disease, electrophysiology, patch clamp, sodium channel, ubiquitylation (ubiquitination), αB-crystallin, Nav1.5, SCN5A
Introduction
Nav1.5 is the pore-forming α subunit of the major cardiac voltage-gated Na+ channel complex. It generates the sodium current (INa)4 that plays an essential role in the initiation and propagation of the cardiac action potential (1–3). Mutations in the SCN5A gene (encoding Nav1.5) cause several inherited arrhythmias, including atrial fibrillation, Brugada syndrome, long QT syndrome, progressive cardiac conduction defect disease, sick sinus syndrome, and dilated cardiomyopathy (4).
Nav1.5 exists in vivo in a multiprotein complex, which interacts with the actin cytoskeleton and the extracellular matrix to provide an important functional link between channel complexes, cardiac structure, and electrical functioning (5, 6). Several proteins have been reported to bind to Nav1.5 (5–7). We have previously reported a small protein, MOG1, with a function in nucleocytoplasmic protein transport that interacts directly with Nav1.5, promotes trafficking of Nav1.5 to the cell surface, and increases peak INa density (4, 6). Specifically, MOG1 facilitates export of Nav1.5 from the endoplasmic reticulum as well as targeting of Nav1.5 to caveolae on plasma membranes (4). Other Nav1.5-interacting proteins include four β-subunits, fibroblast growth factor homologous factor 1B, calmodulin, Nedd4-like ubiquitin-protein ligases (Nedd4-2), ankyrin-G, the Src family tyrosine kinase Fyn, syntrophin (associated with dystrophin), the protein tyrosine phosphatase PTPH1, and 14-3-3 β (5, 7). The majority of these proteins bind to Nav1.5 and affect its biophysical properties. MOG1, 14-3-3 β and ankyrin-G regulate Nav1.5 localization to the cell surface in cardiomyocytes (4, 7). Nedd4-2 interacts with Nav1.5 and regulates Nav1.5 degradation (8). However, the Nav1.5 protein complex is highly sophisticated. Here, we aim to identify other important components of the Nav1.5 protein complex.
The CRYAB gene encodes αB-crystallin, a small molecular weight heat shock protein widely expressed in many tissues, including the heart, lens, and skeletal muscle (9). Mutations in CRYAB cause a number of inherited human disorders, including cataracts, skeletal muscle myopathy, and cardiomyopathy (9). In this study, using yeast two-hybrid screening and follow-up biochemical technologies, we have identified αB-crystallin as a new binding partner of Nav1.5. We further show that αB-crystallin interacts with Nav1.5 and enhances Nav1.5 cell surface expression by reducing ubiquitination of Nav1.5.
Experimental Procedures
Plasmids, Mutagenesis, Antibodies, and Animals
The cDNA for the human CRYAB gene encoding αB-crystallin (NM_001885) was amplified by PCR using plasmid pcDNA3.1-αB-crystallin (10) as a template and subcloned into the pIRES2-EGFP vector between the XhoI and EcoRI restriction sites, resulting in expression plasmid pIRES2-CRYAB for CRYAB or αB-crystallin. The disease-causing mutations in CRYAB, including R11H, P20S, R56W, D109H, R120G, D140N, G154S, R151X, R157H, and A171T, were created into pIRES2-CRYAB using a PCR-based mutagenesis method (11, 12).
The expression construct for the human cardiac sodium channel gene SCN5A (hH1a) in vector pcDNA3 (pcDNA3-Nav1.5) was previously described (13–15). The cDNAs for HA-tagged Nav1.5-Loop II (cytoplasmic Loop II between DII and DIII; amino acids 940–1200), HA-tagged Nav1.5-Loop III (cytoplasmic Loop III between DIII and DIV; amino acids 1471–1523), and HA-tagged Nav1.5-C terminus (C-terminal domain; amino acids 1773–2016) were amplified by PCR using plasmid pcDNA3-Nav1.5 as a template and subcloned into the pCMV-HA vector between the EcoRI and XhoI restriction sites. The expression construct for mutant SCN5A with mutation V1980A was generated using a PCR-based mutagenesis method (11, 12). The expression plasmids for wild type human Nedd4-2 (KIAA0439) and a catalytically inactive form of mutant Nedd4-2 with mutation C801S in pcDNA3.1(−) were described previously (8). All expression plasmids were verified by direct DNA sequencing analysis.
The mouse anti-ubiquitin monoclonal antibody FK2 (BML-PW8810, Enzo Life Science) was used at a dilution factor of 1:500. A mouse anti-αB-crystallin (Cryab) antibody (ADI-SPA-222, Enzo Life Science), a rabbit anti-Nav1.5 antibody (ASC-005, ASC-013, Alomone), and a rabbit anti-Na+K+-ATPase antibody (3010, Cell Signaling Technology) were all used at a dilution factor of 1:1000. A mouse anti-β-actin antibody (60008, PTGCN) was used at a dilution factor of 1:3000. The goat anti-rabbit IgG and goat anti-mouse IgG were purchased from Santa Cruz Biotechnology, Inc. A goat anti-rabbit HRP-conjugated secondary antibody and a goat anti-mouse HRP-conjugated secondary antibody were purchased from Millipore and used at a dilution factor of 1:20,000. All procedures performed on animals were approved by the ethics committee of Huazhong University of Science and Technology and conformed to the National Institutes of Health guidelines for the care and use of laboratory animals.
Yeast Two-hybrid Screening for Nav1.5-interacting Proteins
Yeast two-hybrid screening for Nav1.5-interacting proteins was reported by us previously (6).
GST Pull-down Analysis
GST pull-down assays were carried out as described by us previously (6). A cDNA fragment encoding Nav1.5-Loop II (amino acids 940 and 1200) was amplified using PCR and fused to GST in the pGEX-4T-1 vector (pGEX-4T-1-Nav1.5 LII) as described previously (6). The construct was verified by direct DNA sequencing analysis. Expression of GST and the GST-Nav1.5-Loop II fusion protein in Escherichia coli BL21 cells was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside for 8 h at 26 °C. Following induction, the cells were harvested, resuspended in lysis buffer (0.5% Nonidet P-40, 50 mm Tris/HCl, 150 mm NaCl, 1 mm EDTA supplemented with 1× protease inhibitor cOmplete Mini EDTA-free mixture from Roche Applied Science), and sonicated on ice. The lysate was then centrifuged at 13,000 × g for 30 min at 4 °C, and the supernatants were incubated with glutathione-Sepharose 4B beads to precipitate GST and GST-Nav1.5-Loop II with gentle agitation on a rotator overnight at 4 °C according to the manufacturer's instructions (GE Healthcare). After centrifugation, the beads were washed three times for 5 min in PBS containing 1% Triton X-100. An equal amount of soluble fractions from HEK293 cells transiently transfected with pIRES2-EGFP-αB-crystallin was incubated with the prewashed beads coupled with GST or GST-Nav1.5-Loop II with gentle agitation on a rotator for 1 h at room temperature. The beads were centrifuged and washed three times for 5 min in NETN buffer (0.5% Nonidet P-40, 50 mm Tris/HCl, 900 mm NaCl, 1 mm EDTA). The bound proteins were eluted with 1× SDS-PAGE sample buffer and separated on a 15% SDS-polyacrylamide gel and transferred to PVDF membranes. The membranes were subsequently probed with an anti-αB-crystallin antibody, and the rest of the procedures for Western blotting analysis were as described previously (6).
Co-immunoprecipitation (Co-IP) Analysis
Co-IP studies were performed as described previously (4, 6, 16, 17). A stable HEK293 cell line with constitutive expression of Nav1.5 (HEK/Nav1.5) was described previously (4, 6, 15). The HEK/Nav1.5 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), l-glutamine (2 mm), penicillin G (100 units/ml), streptomycin (10 mg/ml), and G418 (200 μg/ml) (Gibco) in a humidified incubator with 5% CO2 at 37 °C. HEK/Nav1.5 cells with 70–80% confluence in a 10-cm plate were transiently transfected with 10 μg of plasmid DNA (pIRES2-EGFP-αB-crystallin) using 20 μl of Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions and cultured for 48 h, harvested, and lysed in ice cold lysis buffer (50 mm Tris/HCl, pH 7.5, 150 mm NaCl, 2 mm EDTA, 1% Nonidet P-40, 1× protease inhibitor cOmplete Mini EDTA-free mixture from Roche Applied Science). The cell lysate was centrifuged at 13,000 × g for 30 min at 4 °C. Cell extracts in the supernatants (500 μg) were preabsorbed with 30 μl of Protein A/G PLUS-agarose (sc-2003, Santa Cruz Biotechnology) for 1 h at 4 °C and microcentrifuged at 4 °C. An equal volume of the supernatants was incubated with 1.5 μg of an anti-αB-crystallin antibody or the same amount of anti-mouse IgG on a rotator overnight at 4 °C and then mixed with 30 μl of Protein A/G PLUS-agarose. The antibody·protein A/G PLUS·agarose complex was incubated on a rocker for 2 h at 4 °C, centrifuged at 1000 × g for 5 min, and washed five times with washing buffer (lysis buffer with 0.1% Tween 20). The washed pellets were resuspended in 50 μl of 1× SDS loading buffer, incubated at 37 °C for 5 min, and electrophoresed through 8–15% SDS-polyacrylamide gel using the Bio-Rad minigel system. The proteins were transferred onto a PVDF membrane (Millipore) overnight at 100 mA at 4 °C. The membrane was blocked with blocking buffer (3% BSA and 0.05% Tween in PBS) for 2 h at room temperature with gentle agitation and incubated with an anti-Nav1.5 antibody overnight at 4 °C with gentle agitation. After three washes with PBST (0.05% Tween in PBS) for 15 min at room temperature, the membrane was incubated with goat anti-rabbit HRP-conjugated secondary antibody for 2 h at room temperature. After three washes with PBST for 15 min at room temperature, the proteins were visualized using a SuperSignal West Pico Chemiluminescent Substrate (Pierce).
For reciprocal co-IP analysis, 1.5 μg of anti-Nav1.5 antibody was used for immunoprecipitation, and the anti-αB-crystallin antibody was used for Western blotting analysis. For the negative control, 1.5 μg of anti-rabbit IgG antibody was substituted for the anti-Nav1.5 antibody.
For co-IP analysis of αB-crystallin and Nav1.5 in cardiac cells, Sprague-Dawley rats were euthanized with nembutal (intraperitoneal injection of 162.5 units/kg body weight), and hearts were excised and rinsed with ice-cold Hanks' buffer. The cardiac tissue samples were homogenized in lysis buffer (20 mm Tris/HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 1× protease inhibitor cOmplete Mini EDTA-free mixture from Roche Applied Science) on ice. The cardiac protein extracts were then used for co-IP analysis as described above.
Electrophysiological Studies
For patch clamping experiments, HEK/Nav1.5 cells were transfected with 0.8 μg of pIRES2-GFP-αB-crystallin DNA or pIRES2-GFP control DNA using 2 μl of GenJetTM in vitro DNA transfection reagent. After 48 h of transfection, the cells expressing an approximately equal amount of GFPs were selected for electrophysiological studies for recordings of INa (6, 18–21). The pipette was filled with a solution containing 20 mm NaCl, 130 mm CsCl, 10 mm EGTA, and 10 mm HEPES (pH 7.2-adjusted with CsOH). The bath solution contained 70 mm NaCl, 80 mm CsCl, 5.4 mm KCl, 2 mm CaCl2, 10 mm HEPES, 10 mm glucose, and 1 mm MgCl2 (pH 7.4-adjusted with CsOH). All reagents were obtained from Sigma-Aldrich. INa was recorded using a whole-cell voltage clamp recording method with an Axon multiclamp700B patch clamp amplifier using the Digidata1440A digitizer (Axon Instruments, Sunnyvale, CA) on a desktop computer at room temperature (22 °C). INa currents were filtered at 5 kHz with a 4-pole Bessel filter and sampled at 50 kHz. Pipette resistance ranged from 2 to 3 megaohms. The series resistance recorded in the whole cell configuration was compensated (80%) to minimize voltage errors. The holding potential for all pulse protocols was −120 mV, and the voltage dependence of the relative Na+ conductance activation, voltage-dependent inactivation, and recovery from inactivation were determined by means of custom voltage clamp protocols modified from those published previously. Details of each pulse protocol are given schematically in the related figures. INa density was normalized using the cell capacitance. The data were analyzed using a combination of Clampfit version 10.2 (Molecular Devices), Microsoft Excel, and Origin version 8.5 (Microcal Software, Northampton, MA). The steady-state activation and voltage dependence of inactivation curves were fitted with the Boltzmann equation, I/Imax = (1 + exp((V − V½)/k))−1 to determine the membrane potential for half-maximal (in) activation (V½) and the slope factor k. Recovery from inactivation was analyzed by fitting the data with a two-exponential equation, It/Imax = Af × exp(−t/τf) + As × exp(−t/τs), to determine the fractions of the fast (Af) and the slow (As) components of recovery from inactivation and the time constants for recovery from fast (τf) and slow (τs) inactivation, respectively.
Immunohistochemistry
The immunostaining analysis of neonatal rat cardiomyocytes and paraffin-embedded adult rat heart sections (8 μm) was performed with an anti-Nav1.5 antibody (ASC-013, Alomone) and an anti-αB-crystallin antibody (ADI-SPA-222, Enzo Life Science) according to standard protocols as described previously (6).
RNA Interference
The αB-crystallin siRNA targeting rat αB-crystallin (5′-GUGGAUCCUCUCACCAUUATT-3′) and scrambled control siRNA were designed and synthesized by GenePharma (Suzhou, Jiangsu, China). The scramble siRNA does not recognize any mRNA from H9C2 cells. H9C2 cells were cultured in DMEM supplemented with 15% fetal bovine serum and transfected with siRNA using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen). After 48 h of transfection, cells were harvested for assaying siRNA knockdown efficiency using quantitative real-time RT-PCR and Western blotting analysis as described (4, 22–25). Sequences of primers used for quantitative real-time PCR analysis are listed in Table 1.
TABLE 1.
Sequences of primers used for quantitative RT-PCR analysis and siRNA
| Primer | Sequence |
|---|---|
| αB-Crystallin (rat) | |
| Forward | 5′-TGCGGGCACCTAGCTGGATTGA-3′ |
| Reverse | 5′-GCGCTCTTCGTGCTTGCCGTG-3′ |
| β-Actin (rat) | |
| Forward | 5′-CCGTAAAGACCTCTATGCCAACA-3′ |
| Reverse | 5′-CGGACTCATCGTACTCCTGCT-3′ |
| αB-Crystallin siRNA (rat) | 5′-GUGGAUCCUCUCACCAUUATT-3′ |
Isolation of Cell Surface Proteins and Analysis of Expression Levels of Cell Surface Nav1.5
Transfected cells were used for preparation of plasma membrane protein extracts using EZ-Link Sulfo-NHS-SS-Biotin (Pierce) as described by us previously (4). HEK/Nav1.5 cells were transiently transfected with 10 μg of pIRES2-EGFP-αB-crystallin or pIRES2-EGFP using 20 μl of Lipofectamine 2000 (Invitrogen). H9C2 cells were transfected with αB-crystallin siRNA or control scramble siRNA, also using Lipofectamine 2000 (Invitrogen). After 48 h of transfection, cells were washed three times with ice-cold PBS and incubated for 30 min at 4 °C with 2 ml of biotin per 10-cm dish (1 mg/ml; EZ-Link Sulfo-NHS-SS-Biotin, Pierce) to label the cell surface proteins. After three washes with 100 mm glycine in PBS, the cells were incubated with 100 mm glycine for 15 min at 4 °C to quench the biotinylation reaction. After quenching, the cells were scraped and lysed in lysis buffer (20 mm Tris/HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, and 1× protease inhibitor cOmplete Mini EDTA-free mixture from Roche Applied Science). Cell lysates were centrifuged at 13,000 × g for 30 min at 4 °C, and the supernatants were incubated with NeutrAvidin-agarose resins (Pierce) overnight at 4 °C with gentle agitation. The protein·agarose complexes were washed three times with PBS and then resuspended in 1× SDS-PAGE sample buffer containing 50 mm DTT and analyzed by Western blotting analysis with an anti-Nav1.5 antibody as described above. The plasma membrane protein Na+K+-ATPase was used as loading control to calibrate the cell surface proteins.
Analysis of Stability and Internalization of Cell Surface Nav1.5
The assay for stability of cell surface Nav1.5 was performed as described previously (4). Cells were transfected and cultured in 10-cm culture dishes for 48 h. Cell surface proteins were tagged with biotin at 4 °C and subsequently quenched with 100 mm glycine as described above. The culture media were then added, and biotin-tagged cells were cultured in a 37 °C incubator to allow biotinylated cell surface proteins to traffic inside cells (internalization) for 9 h. The biotin-tagged proteins remained on cell surface were stripped off with 50 mm MESNA (1392807, Sigma) in 100 mm Tris/HCl (pH 8.6) containing 100 mm NaCl and 2.5 mm CaCl2 at 4 °C for 30 min and quenched with 5 mg/ml iodoacetamide in PBS at 4 °C for 15 min. The cells were then lysed in ice with cold lysis buffer, and the supernatants were incubated with 200 μl of precleared NeutrAvidin-agarose resins (Pierce) to precipitate the internalized biotinylated cell surface proteins for overnight at 4 °C with gentle agitation. The protein·agarose complexes were washed five times with lysis buffer and resuspended in 100 μl of 1× SDS-PAGE loading buffer (39001, Pierce) containing 50 mm DTT. The expression levels of internalized biotinylated cell surface proteins for 9 h were normalized to the total biotinylated cell surface proteins at the zero time point to calculate the percentage of internalization and the percentage of biotinylated proteins remaining on the cell surface.
Statistical Analysis
All data were from at least three independent experiments and expressed as means ± S.E. Statistical analysis was carried out using two-tailed paired or unpaired Student's t tests between two groups. A p value of ≤0.05 was considered to be statistically significant. The differences between groups over a time period were analyzed by two-way analysis of variance.
Results
Identification of αB-Crystallin as a New Protein That Interacts with Nav1.5
We have previously reported a yeast two-hybrid screen to identify candidate proteins that interact with Nav1.5 (6). MOG1 was identified as a Nav1.5-interacting protein during the study and previously reported (6). In the same screen, we also identified a cDNA clone that encodes human αB-crystallin (NM_001885). In this study, we validated the interaction between αB-crystallin and Nav1.5 by an in vitro GST pull-down assay first and then by a co-immunoprecipitation assay. We constructed an expression plasmid that will express a GST-Nav1.5-Loop II protein with GST fused to the cytoplasmic Loop II of Nav1.5 (between transmembrane domain II and III, amino acids 940–1200) in E. coli BL21 cells. The GST-Nav1.5-Loop II protein was purified using glutathione beads. The affinity-purified GST or GST-Nav1.5-Loop II was incubated with cell lysates from HEK293 cells transfected with a pIRES2-EGFP-αB-crystallin expression plasmid. Unbound proteins were washed off, and proteins pulled down were analyzed by Western blotting analysis using an anti-αB-crystallin antibody. As illustrated in Fig. 1A, GST-Nav1.5-Loop II successfully pulled αB-crystallin down, whereas no binding for αB-crystallin was observed with GST alone (negative control). These results suggest that αB-crystallin interacts with Nav1.5-Loop II in vitro.
FIGURE 1.
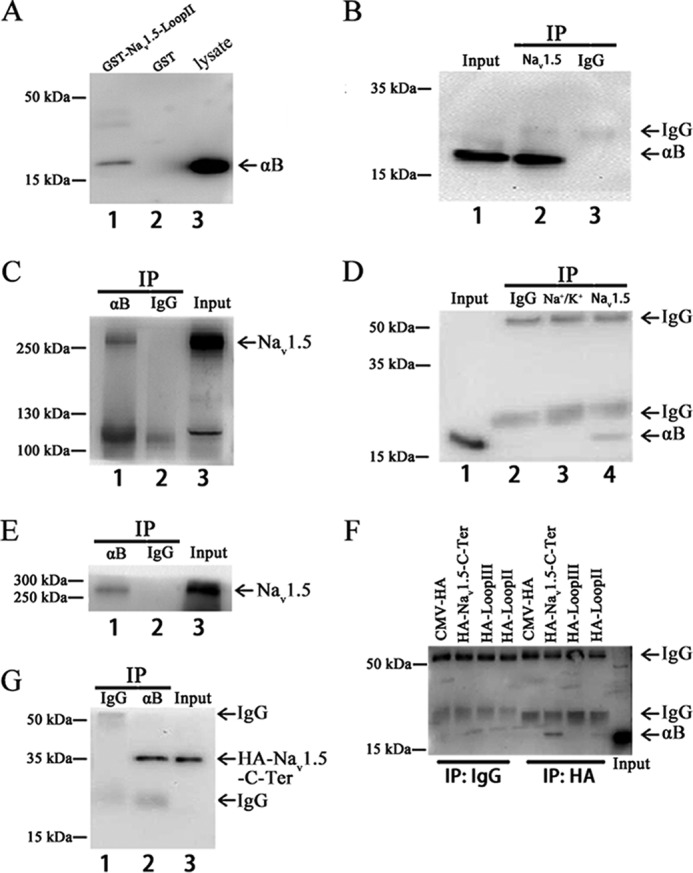
Interaction between αB-crystallin and Nav1.5 in vitro by GST pull-down assays and in vivo by co-IP assays). A, GST pull-down assays. The cell lysates extracted from HEK293 cells with overexpression of αB-crystallin were incubated with a purified GST fusion protein with the intracellular domain between transmembrane domains II and III of Nav1.5 (GST-Nav1.5-Loop II) or negative control GST alone. GST-Nav1.5-Loop II (lane 1), but not control GST (lane 2), successfully pulled down αB-crystallin (the lysate as a positive control). B and C, co-IP with HEK/Nav1.5 cell extracts. HEK/Nav1.5 cells were transiently transfected with pIRES2-EGFP-αB-crystallin. Cell lysates were incubated and immunoprecipitated with anti-Nav1.5 antibody and examined by immunoblotting with an anti-αB-crystallin antibody (B). C, reciprocal co-IP with anti-αB-crystallin for immunoprecipitation and anti-Nav1.5 for Western blotting. D and E, co-IP as in B and C but with protein extracts from adult rat hearts. An anti-Na+/K+-ATPase antibody and normal IgG were used as negative controls. F, co-IP with HEK293 cell extracts. Vector pCMV-HA (negative control), Nav1.5-Loop II (positive control), Nav1.5-Loop III, or Nav1.5-C terminus expression plasmids were each co-transfected with pIRES2-EGFP-αB-crystallin into HEK293 cells. Cell lysates were incubated and immunoprecipitated with anti-HA antibody and examined by immunoblotting with an anti-αB-crystallin antibody. G, co-IP with extracts from HEK293 cells co-transfected with pIRES2-EGFP-αB-crystallin and the Nav1.5-C terminus expression plasmid. An anti-αB-crystallin antibody was used for immunoprecipitation, and an anti-HA antibody was used for Western blotting. All studies were repeated at least three times.
To determine whether αB-crystallin interacts with Nav1.5 in vivo, a co-IP assay was performed using protein extracts from HEK293 cells and rat hearts. A HEK293 stable cell line that overexpresses Nav1.5 (HEK/Nav1.5) was transfected with pIRES2-EGFP-αB-crystallin for 48 h. The HEK/Nav1.5 cell lysates were immunoprecipitated using an anti-Nav1.5 antibody (Fig. 1B, lane 2) or anti-rabbit IgG as a negative control (Fig. 1B, lane 3). The precipitates were analyzed by Western blotting analysis with an anti-αB-crystallin antibody. The anti-Nav1.5 antibody successfully precipitated αB-crystallin (Fig. 1B, lane 2), but the anti-rabbit IgG failed to precipitate αB-crystallin (Fig. 1B, lane 3). Consistently, reciprocal co-IP showed that the anti-αB-crystallin antibody, but not the control anti-mouse IgG, precipitated Nav1.5 (Fig. 1C). These results demonstrated that αB-crystallin interacted with Nav1.5 in HEK293 cells.
Similar to studies with HEK/Nav1.5 cells, a co-IP assay was also performed using protein extracts from adult rat hearts. Fig. 1D shows that the anti-Nav1.5 antibody, but not the control IgG or an anti-Na+/K+-ATPase antibody, pulled αB-crystallin down. Reciprocally, the anti-αB-crystallin antibody, but not the control IgG, precipitated Nav1.5 protein (Fig. 1E). These results demonstrated that αB-crystallin interacted with Nav1.5 in the rat heart.
We also assessed whether αB-crystallin interacts with Loop III (between transmembrane domain III and IV) and the C terminus of Nav1.5. Interestingly, co-IP assays demonstrated that αB-crystallin interacted strongly with the C terminus of Nav1.5 but not with Loop III (Fig. 1, F and G).
Because αB-crystallin interacted with Nav1.5, we hypothesized that αB-crystallin co-localized with Nav1.5 in the cardiac tissue. To test the hypothesis, immunostaining was performed in adult rat hearts fixed in paraformaldehyde and embedded in paraffin. The paraffin sections were used for immunostaining using both anti-αB-crystallin and anti-Nav1.5 antibodies (Fig. 2A). Immunostaining without primary antibodies was used as negative controls (Fig. 2B). The anti-αB-crystallin produced red signals, whereas the anti-Nav1.5 produced green signals. When the two signals merged, a yellow signal was generated. The αB-crystallin protein showed strong expression in atrial and ventricular cardiomyocytes and co-localized with Nav1.5 (Fig. 2A). These results suggested the co-localization of αB-crystallin and Nav1.5 expression in cardiac tissue.
FIGURE 2.

Co-localization of αB-crystallin and Nav1.5 in adult rat cardiac muscle tissue. The paraffin-embedded adult rat heart sections were used for co-immunostaining with both anti-αB-crystallin and anti-Nav1.5 antibodies (Ab) (A) or without primary antibodies as negative controls (B). Overlay images show co-localization of green signals (Nav1.5) and red signals (αB-crystallin), which generated yellow signals in cardiac tissue in vivo. Nuclei were stained with DAPI (blue).
αB-Crystallin Modulates Sodium Current in HEK293 Cells
To investigate the functional role of αB-crystallin in Nav1.5 physiology, electrophysiological studies were performed using the whole-cell patch clamp method. Plasmid pIRES2-EGFP-αB-crystallin or control vector pIRES2-EGFP was transfected into HEK/Nav1.5 cells. Whole-cell sodium currents were then measured (Fig. 3A). Compared with the control, overexpression of αB-crystallin significantly increased sodium current densities (Fig. 3, B and C). Cells with αB-crystallin overexpression significantly increased the peak sodium current density (−297.22 ± 45.27 pA/picofarads) at −25 mV compared with the cells with control EGFP (−202.05 ± 23.82 pA/picofarads, p < 0.01) (Fig. 3C). The voltage dependence of the activation and inactivation of sodium currents was also investigated. However, no differences were observed in the voltage dependence kinetics of activation and inactivation of sodium currents between cells with αB-crystallin overexpression and those with expression of control EGFP (Fig. 3D). An investigation of time dependence of recovery from inactivation showed moderate deceleration of tf by 3.19 ms (from 12.88 to 9.69 ms for fast time constant of recovery) by αB-crystallin (Fig. 3E). The fitted data are shown in Table 2.
FIGURE 3.

Functional effects of αB-crystallin on INa in HEK/Nav1.5 cells. A, representative whole-cell sodium currents recorded from HEK/Nav1.5 cells transfected with the control vector or a αB-crystallin overexpression plasmid. The voltage clamp protocol is shown in the inset. B, the relationship of average current densities (current normalized to cell capacitance) and voltage. C, peak sodium current densities at −25 mV. D, steady-state activation and inactivation curves. The holding potential was −120 mV. The protocol for recording the steady-state activation and inactivation curves is shown in the inset. E, time course of recovery from inactivation was studied using a two-pulse protocol of −20 mV at the −120 mV holding potential as in the inset. F and G, representative late INa recorded at the −20 mV test potential from HEK293 cells with co-expression of wild type SCN5A with the control vector or the αB-crystallin expression plasmid. The ΔKPQ mutation associated with long QT syndrome, which is known to generate large late INa, was used as a positive control. Note that overexpression of αB-crystallin did not impact the generation of late INa; αB-crystallin slowed the rate of decay, although the effect did not reach a significant level. Data are shown as means ± S.E. (error bars). NS, not significant; *, p < 0.05. All studies were repeated at least three times. pF, picofarads.
TABLE 2.
Summary data on effects of overexpression of αB-crystallin on cardiac sodium current INa
| Sodium current parametera | Control vector EGFP | αB-Crystallin |
|---|---|---|
| Activation | n = 12 | n = 12 |
| V½ (mV) | −34.73 ± 0.79 | −34.72 ± 0.61 |
| k | 6.57 ± 0.16 | 6.7 ± 0.12 |
| Steady-state inactivation | n = 10 | n = 11 |
| V½ (mV) | −86.88 ± 1.09 | −88.03 ± 0.39 |
| k | 5.74 ± 0.12 | 5.29 ± 0.10 |
| Recovery from inactivation | n = 15 | n = 14 |
| Af | 0.120 ± 0.08 | 0.156 ± 0.01b |
| τf | 9.69 ± 0.40 | 12.88 ± 0.45b |
| As | 0.87 ± 0.01 | 0.831 ± 0.01b |
| τs | 158.033 ± 16.76 | 165.454 ± 14.95 |
a V½, voltage of half-maximal activation; k, slope factor of voltage dependence of (in)activation; τ, time constant for development of slow inactivation; τf, fast time constant of recovery from inactivation; τs, slow time constant of recovery from inactivation; Af, fraction of the fast component of recovery from inactivation; As, fraction of the slow component of recovery from inactivation. Data were shown as means ±⎭S.E.
b p < 0.5 versus vector EGFP (Student's t test). The experiment was repeated at least three times.
We also assessed whether αB-crystallin affects the late sodium currents generated by Nav1.5. Mutant ΔKPQ-Nav1.5 associated with long QT syndrome and known to generate large late sodium current was used as a control for easy detection of late sodium currents (26). Overexpression of αB-crystallin did not show an effect on late INa generated by wild type Nav1.5 compared with control cells transfected with pIRES2-EGFP (Fig. 3, F and G). The rate of decay of INa was slowed by overexpression of αB-crystallin (Fig. 3F), although it did not reach a significant level (Fig. 3G).
Overexpression of αB-Crystallin Increased the Amount of Nav1.5 on the Cell Surface
Given the significant role of the level of cell surface expression of Nav1.5 in sodium current densities, αB-crystallin overexpression may increase sodium current densities by increasing cell surface expression of Nav1.5. Forty-eight hours after transfection of HEK/Nav1.5 cells with pIRES2-EGFP-αB-crystallin or pIRES2-EGFP, cell surface biotinylation assays were performed. Because Na+K+-ATPase is expressed on the plasma membrane of HEK293, it was used as a loading control for cell surface proteins. Western blotting analysis showed that compared with the control EGFP vector, overexpression of αB-crystallin significantly increased the amount of Nav1.5 in plasma membranes (Fig. 4A). However, αB-crystallin overexpression did not affect the expression level of total Nav1.5 protein in cells (Fig. 4B). These data suggest that αB-crystallin regulates cell surface expression of Nav1.5 but not expression of total Nav1.5.
FIGURE 4.
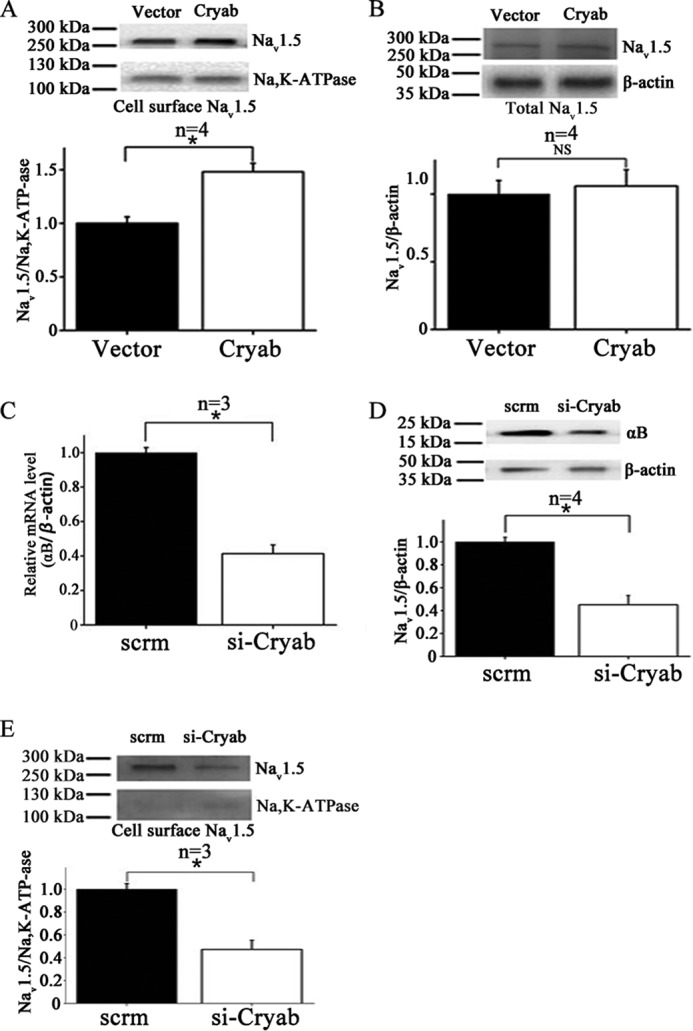
Effects of αB-crystallin on the expression level of Nav1.5 on the cell surface by Western blotting analysis. A, Western blotting analysis with biotinylated cell surface proteins from Nav1.5/HEK293 cells transfected with empty control vector pIRES2-EGFP or a αB-crystallin expression plasmid for 48 h. Na+K+-ATPase was used as a loading control for cell surface proteins. Quantification of Western blotting data showed that overexpression of αB-crystallin significantly increased the amount of Nav1.5 in plasma membranes by 1.48-fold. B, similar Western blotting analysis as in A but with total protein extracts (measuring the expression level of total Nav1.5 protein). C, real-time quantitative PCR analysis showing successful knockdown of CRYAB expression in H9C2 cells by siRNA against CRYAB as compared with control scramble siRNA. D and E, Western blotting analysis with biotinylated cell surface proteins from H9C2 cells transfected with siRNA against CRYAB or control scramble siRNA. D, successful knockdown of αB-crystallin expression by siRNA against CRYAB as compared with control scramble siRNA by Western blotting analysis. E, the effect of knockdown of αB-crystallin expression on the cell surface expression of Nav1.5 using cell surface biotinylation assays in combination with Western blotting analysis. Data were scanned, quantified, and plotted. Quantification of the cell surface expression of Nav1.5 showed a significant reduction by 52% in H9C2 cells transfected with siRNA against CRYAB or control scramble siRNA. Data were shown as means ± S.E. (error bars). NS, not significant; *, p < 0.05. All studies were repeated at least three times.
Knockdown of αB-Crystallin Decreased the Cell Surface Expression Level of Nav1.5 in H9C2 Cells
We next evaluated the role of endogenous αB-crystallin in regulation of Nav1.5 function. Because Western blotting analysis failed to detect αB-crystallin protein expression in HEK293 cells (data not shown), we studied H9C2 rat cardiac myoblasts. Small interfering RNA (siRNA) against αB-crystallin (si-Cryab) was transfected into H9C2 rat cardiac myoblasts to knock down the endogenous expression level of αB-crystallin. Quantitative real-time RT-PCR analysis of mRNA expression showed a significant reduction (about 57%) of αB-crystallin mRNA in cells transfected with si-Cryab as compared with control scrambled siRNA (Fig. 4C). Western blotting analysis showed that si-Cryab was able to significantly decrease the expression of αB-crystallin (Fig. 4D); therefore, it was used to determine the effects of knockdown of αB-crystallin on Nav1.5 cell surface expression.
Biotinylated cell surface protein extracts from H9C2 cells were analyzed by Western blotting analysis (Fig. 4E). Compared with control scrambled siRNA, si-Cryab significantly decreased the amount of Nav1.5 in the plasma membranes (Fig. 4E).
αB-Crystallin Overexpression Decreased Ubiquitination of Nav1.5 and Increased the Stability of Nav1.5 on the Cell Surface
The steady state level of Nav1.5 on the cell surface is determined by the rate of trafficking to the plasma membrane and the internalizing rate to intracellular organelles. Ubiquitination is one of the major processes that regulate the degradation of membrane proteins and their trafficking to other membrane compartments. Heat shock proteins, including Hsp70, Hsc70, and αA-crystallin have been shown to be involved in regulating the cell surface expression level of epithelial sodium channels through regulating ubiquitination (27). Thus, we hypothesized that αB-crystallin also regulated the cell surface expression level of Nav1.5 by modulating ubiquitination of Nav1.5. Western blotting analysis was performed with cell extracts from HEK/Nav1.5 cells with overexpression of αB-crystallin or transfected with the EGFP vector and treated with a 10 μm concentration of the proteasome inhibitor MG132 to detect the ubiquitination level of Nav1.5 by the FK2 mouse monoclonal anti-ubiquitination antibody. First, we used the anti-Nav1.5 antibody to precipitate Nav1.5 from HEK/Nav1.5 cell extracts. Then the precipitates were analyzed by Western blotting analysis. The anti-Nav1.5 antibody was able to precipitate an approximately equal amount of Nav1.5 from cells with EGFP and αB-crystallin (Fig. 5A, top). Western blotting analysis showed that overexpression of αB-crystallin decreased the ubiquitination level of wild type Nav1.5 as compared with EGFP but had no effect on the ubiquitination level of mutant V1980A-Nav1.5 (Fig. 5, A (bottom) and B). This experiment was repeated three times, and similar results were obtained (data not shown).
FIGURE 5.
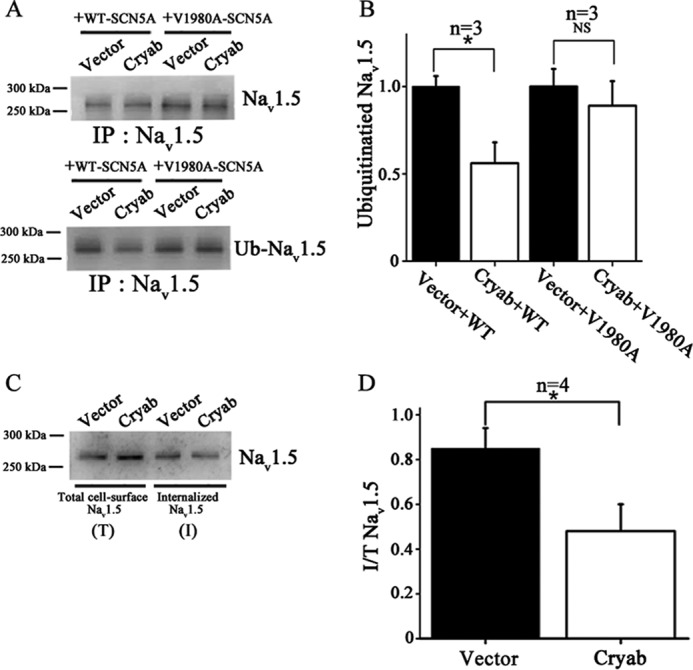
Functional effects of αB-crystallin on ubiquitination of Nav1.5 and internalization of Nav1.5 on cell surface. A and B, plasmid pIRES2-EGFP-αB-crystallin or control vector pIRES2-EGFP was co-transfected with expression plasmids for WT-SCN5A or mutant V1980A-SCN5A into HEK293 cells. All studies were repeated at least three times. A, Nav1.5 was immunoprecipitated using an anti-Nav1.5 antibody and immunoblotted against the Nav1.5 antibody to demonstrate that the same amount of Nav1.5 was precipitated in ubiquitination assays (top). Ubiquitination levels of Nav1.5 were detected by the FK2 antibody (bottom). B, the data from A were scanned, quantified, and plotted. Overexpression of αB-crystallin significantly decreased the ubiquitination level of wild type Nav1.5 but did not affect the ubiquitination level of mutant V1980A-Nav1.5. C and D, effects of αB-crystallin on Nav1.5 endocytosis at the time point of 9 h. All studies were repeated at least four times. C, Western blotting analysis showing the amount of biotinylated cell surface Nav1.5 internalized into the cytoplasm and total biotinylated cell surface Nav1.5. D, The data from C were scanned, quantified, and plotted. Overexpression of αB-crystallin significantly decreased the amount of biotinylated cell surface Nav1.5 internalized into the cytoplasm. Data were shown as means ± S.E. (error bars). NS, not significant; *, p < 0.05.
To determine whether αB-crystallin is involved in regulating Nav1.5 internalization from the plasma membranes, we tested whether αB-crystallin affects the stability of Nav1.5 on the cell surface. HEK/Nav1.5 cells transfected with pIRES2-EGFP-αB-crystallin or pIRES2-EGFP were labeled with biotin at 4 °C and returned to the culture incubator for 9 h at 37 °C. At the end of the incubation, the remaining cell surface biotin was stripped with MESNA, and the internalized biotin-labeled Nav1.5 was extracted and analyzed by Western blotting analysis with anti-Nav1.5 antibody. As shown in Fig. 5, C and D, compared with EGFP (vector), overexpression of αB-crystallin significantly decreased the amount of internalized Nav1.5.
αB-Crystallin Interacts with Nedd4-2 and Decreases the Interaction between Nedd4-2 and Nav1.5
Nedd4-2 was shown to interact with Nav1.5 and regulate degradation of Nav1.5 by regulating ubiquitination (8). Considering that αB-crystallin decreases ubiquitination of Nav1.5, we investigated whether αB-crystallin interacts with Nedd4-2. Co-IP analysis showed that an anti-αB-crystallin antibody was able to precipitate Nedd4-2 in HEK293 cells overexpressing αB-crystallin (Fig. 6A). Reciprocal co-IP analysis showed that an anti-Nedd4-2 antibody also was able to precipitate αB-crystallin (Fig. 6B). These data suggest that αB-crystallin forms a complex with Nedd4-2.
FIGURE 6.
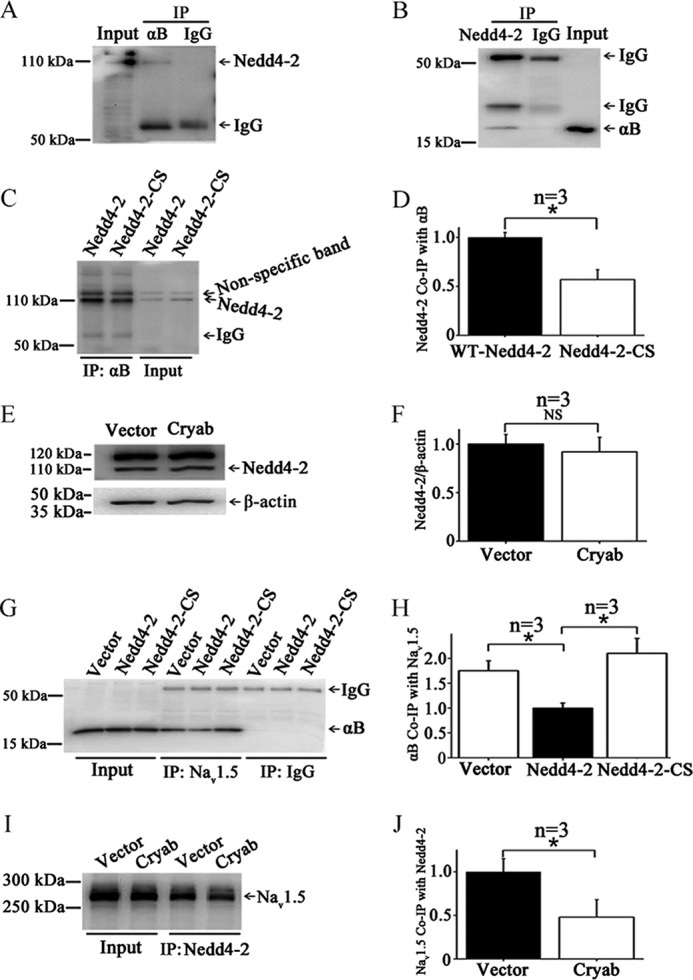
αB-Crystallin interacts with Nedd4-2. A, co-IP with an anti-αB-crystallin antibody for immunoprecipitation and an anti-Nedd4-2 for Western blotting analysis. HEK293 cells overexpressing αB-crystallin were used. Anti-αB-crystallin antibodies pulled down Nedd4-2. B, co-IP with an anti-Nedd4–2 antibody for immunoprecipitation and an anti-αB-crystallin for Western blotting analysis. Anti-Nedd4-2 antibodies pulled down αB-crystallin. C, comparison between wild type Nedd4-2 and mutant Nedd4-2 with a mutation at the catalytic site (C801S) in co-IP studies between αB-crystallin and Nedd4-2. D, the data from C were scanned, quantified, and plotted. E, Western blotting analysis for Nedd4-2. F, the data from E were scanned, quantified, and plotted. Overexpression of αB-crystallin did not affect the total expression level of Nedd4-2. G, co-IP analysis to assess the effect of wild type Nedd4-2 and mutant Nedd4-2-CS on interaction between αB-crystallin and Nav1.5. H, the data from G were scanned, quantified, and plotted. Overexpression of wild type Nedd4-2, but not mutant Nedd4-2-C801S, reduced the interaction between Nav1.5 and αB-crystallin. I, co-IP for interaction between Nav1.5 and Nedd4-2. J, the data from I were scanned, quantified, and plotted. Data were shown as means ± S.E. (error bars). NS, not significant; *, p < 0.05. All studies were repeated at least three times.
Mutation C801S (Nedd4-2-CS) is a dominant negative mutant that affects the catalytic activity of Nedd4-2 (8). Co-IP analysis showed that the interaction between Nedd4-2 and αB-crystallin was reduced by the C801S mutation in Nedd4-2 (Fig. 6, C and D). In a separate study, we showed that overexpression of αB-crystallin did not affect the expression level of Nedd4-2 (Fig. 6, E and F). Together, these data suggest that functionally active Nedd4-2 is required for interaction between αB-crystallin and Nedd4-2.
We also assessed whether Nedd4-2 affects the interaction between Nav1.5 and αB-crystallin. Overexpression of wild type Nedd4-2 significantly reduced the interaction between Nav1.5 and αB-crystallin (Fig. 6, G and H), which is consistent with the reported finding that wild type Nedd4-2 reduced sodium current density (8). However, mutant Nedd4-2-CS did not affect the interaction between Nav1.5 and αB-crystallin or slightly increased the interaction (Fig. 6, G and H), which is also consistent with the reported finding that Nedd4-2-CS did not affect sodium current density (8).
Next, we investigated whether αB-crystallin affects the interaction between Nedd4-2 and Nav1.5. Co-immunoprecipitation analysis showed that, compared with EGFP (vector), overexpression of αB-crystallin reduced the interaction between Nedd4-2 and Nav1.5 (Fig. 6, I and J).
αB-Crystallin Modulates INa by Affecting Its Interaction with Nedd4-2 and Nedd4-2-mediated Ubiquitination of Nav1.5
Electrophysiological studies were then used to further assess the effects of αB-crystallin on sodium currents by regulating Nedd4-2. In HEK/Nav1.5 cells with overexpression of wild type Nedd4-2, additional overexpression of αB-crystallin significantly increased the peak sodium current density (Fig. 7A). However, in HEK/Nav1.5 cells with overexpression of mutant Nedd4-2-CS, additional overexpression of αB-crystallin did not affect the peak sodium current density (Fig. 7B). Together with the co-immunoprecipitation data above, these data suggest that the interaction between αB-crystallin and functionally active Nedd4-2 is important for the function of Nav1.5.
FIGURE 7.
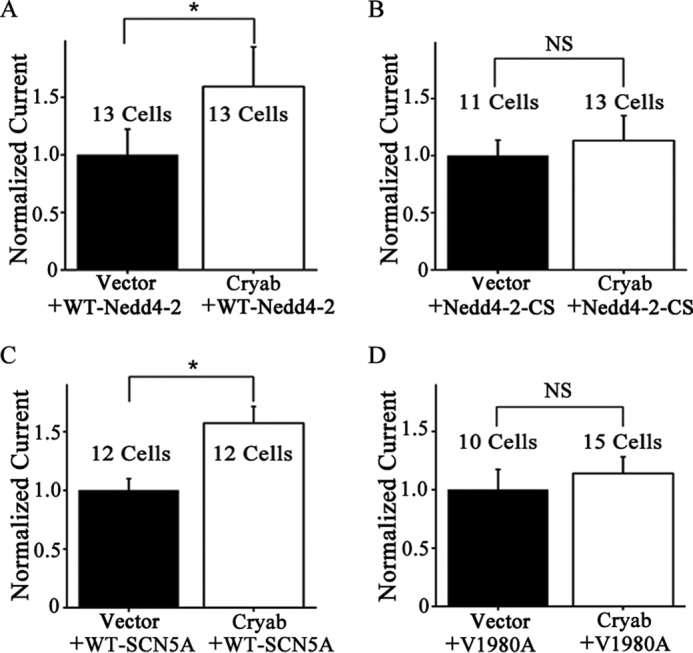
Analysis of peak INa. A and B, functional effects of overexpression of αB-crystallin on INa densities from wild type Nav1.5 in the presence of wild type Nedd4-2 or catalytically inactive mutant Nedd4-2 (Nedd4-2-C801S). Nedd4-2-C801S disrupts the activity of Nedd4-2 on ubiquitination of Nav1.5. HEK/Nav1.5 cells were co-transfected with wild type Nedd4-2 or mutant Nedd4-2-C801S as well as with the control vector and an expression plasmid for αB-crystallin. C and D, functional effects of overexpression of αB-crystallin on INa densities from wild type Nav1.5 or mutant Nav1.5 with mutation V1980A at the interaction site for Nedd4-2. HEK293 cells were co-transfected with an expression plasmid for either wild type SCN5A or mutant SCN5A with mutation V1980A together with an expression plasmid for αB-crystallin or control empty vector. Data were shown as means ± S.E. (error bars). NS, not significant; *, p < 0.05. All studies were repeated at least three times.
The valine residue at position 1980 in Nav1.5 (V1980A-SCN5A) has been shown to interact with Nedd4-2 and facilitate Nav1.5 ubiquitination (8). Mutating Val-1980 to Ala (V1980A) significantly increased the INa density (8). Interestingly, electrophysiological analysis showed that overexpression of αB-crystallin could increase the sodium current density only from the wild type Nav1.5 (Fig. 7C). It failed to increase the sodium current density from the mutant V1980A-Nav1.5 channel (Fig. 7D).
Effects of Disease-associated Mutations of αB-Crystallin on Sodium Currents
Multiple mutations in αB-crystallin have been associated with a variety of diseases. These mutations include R11H, P20S, R56W, R109H, R120G, D140N, R151X, G154S, R157H, and A171T. To determine whether these naturally occurring mutations can alter the physiological roles of αB-crystallin in Nav1.5, we analyzed these αB-crystallin mutations for their effects on sodium current densities.
Mutations R11H, P20S, R56W, R120G, G154S, R157H, and A171T in αB-crystallin did not affect the capability of αB-crystallin in enhancing the sodium current density (Fig. 8A). However, the R109H mutation, which is linked to multisystemic disease, including cataracts, myofibrillar myopathy, and cardiomyopathy, and the R151X mutation, which is linked to proximal and distal leg muscle weakness, abolished the function of αB-crystallin to increase the sodium current density (Fig. 8A). We selected one αB-crystallin mutation that reduced the sodium current density (R151X) and one mutation that did not reduce the density (R120G) and examined their effects on αB-crystallin interaction with Nav1.5 and Nedd4-2. Mutation R151X significantly reduced the interaction between αB-crystallin and Nav1.5, whereas mutation R120G had little effect on the interaction (Fig. 8B). Together, these data suggest that the interaction between αB-crystallin and Nav1.5 plays an important role in increasing the sodium current density by αB-crystallin. Mutation R151X, but not R120G, also significantly reduced αB-crystallin interaction with Nedd4-2, but the effect was small (Fig. 8C).
FIGURE 8.
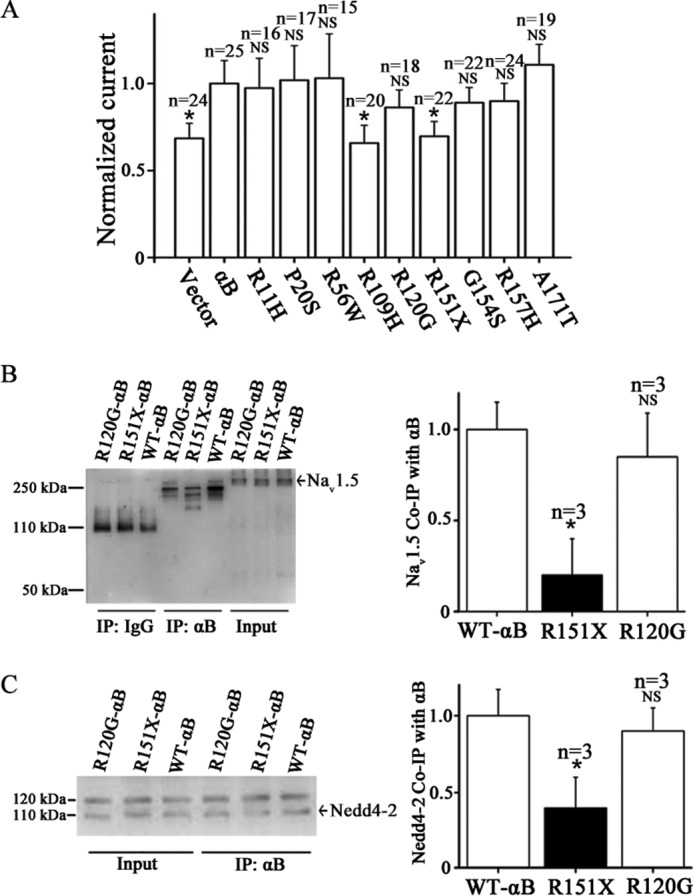
Functional effects of αB-crystallin mutations associated with human diseases on peak INa densities and the interaction of αB-crystallin with either Nav1.5 or Nedd4-2. A, HEK/Nav1.5 cells were transfected with an expression plasmid for wild type αB-crystallin or mutant αB-crystallin and then used for whole-cell patch clamp recordings of INa. The peak sodium current density was normalized to the sodium current density recorded from HEK/Nav1.5 cells transfected with wild type αB-crystallin and plotted. B, co-IP analysis for interaction between wild type αB-crystallin or two mutant αB-crystallin proteins and Nav1.5. C, co-IP analysis for interaction between wild type αB-crystallin or two mutant αB-crystallin proteins and Nedd4-2. NS, not significant; *, significant with p < 0.05. Data are shown as means ± S.E. (error bars) All studies were repeated at least three times.
Discussion
In this report, we describe the identification of a new protein factor, αB-crystallin, which interacts with Nav1.5 and increases peak INa density. The cardiac sodium channel is in a multiprotein complex; therefore, identification of a new key component of the complex will be important for full understanding of the function of the channel and the physiology of the cardiovascular system. The αB-crystallin was identified as a candidate protein that interacts with Nav1.5 by a yeast two-hybrid screen. The interaction between αB-crystallin and Nav1.5 was verified both by in vitro GST pull-down assays and by in vivo Co-IP studies in HEK/Nav1.5 cells (Fig. 1, A–C). Interestingly, the αB-crystallin was shown to interact with both Loop II and the C terminus of Nav1.5 but not with Loop III (Fig. 1, F and G). The interaction between αB-crystallin and Nav1.5 was also validated by Co-IP studies using protein extracts from rat hearts (Fig. 1, D and E) and co-immunostaining analysis of rat heart sections (Fig. 2). The interaction of αB-crystallin with Nav1.5 appeared to have selectivity because no interaction was found between αB-crystallin and another membrane protein Na+/K+ ATPase (Fig. 1D). On the other hand, as a chaperone, αB-crystallin is expected to interact with other ion channels or transporters. Indeed, we have found that αB-crystallin interacts with potassium channel KCNH2 (data not shown), although the functional effect of the interaction is under investigation.
The interaction between αB-crystallin and Nav1.5 results in increased sodium current densities. Whole-cell patch clamping revealed that overexpression of αB-crystallin significantly increased INa densities (Fig. 3). We further showed that the interaction between αB-crystallin and Nav1.5 played an important role in increasing the sodium current density by αB-crystallin. As shown in Fig. 8, αB-crystallin mutation R115X reduced the interaction between αB-crystallin and Nav1.5, which was accompanied by significantly reduced sodium current density. On the other hand, R120G did not affect the interaction between αB-crystallin and Nav1.5 and did not have any effect on the sodium current density. Previously, we reported that MOG1 interacts with Nav1.5 and increases INa densities by increasing expression of Nav1.5 on the cell surface (4, 6). Therefore, similar to MOG1, one molecular mechanism by which αB-crystallin increases INa densities is the increased expression of Nav1.5 on the cell surface. Western blotting analysis with biotinylated cell surface proteins showed that overexpression of αB-crystallin significantly increased cell surface expression of Nav1.5 (Fig. 4A), and knockdown of αB-crystallin expression significantly decreased the cell surface expression level of Nav1.5 (Fig. 4E). In contrast to MOG1, which increases the cell surface expression level of Nav1.5 by facilitating ER export of Nav1.5 (4), αB-crystallin reduces turnover of cell surface Nav1.5 by slowing down internalization of Nav1.5 for degradation (Fig. 5, C and D).
αB-crystallin belongs to the family of α-crystallins. The α-crystallin family is composed of αA-crystallin and αB-crystallin and belongs to the heat shock protein family, which exhibits important molecular chaperone activities. Recent studies showed that overexpression of αA-crystallin accelerated the degradation of misfolded membrane proteins, including a mutant cystic fibrosis transmembrane conductance regulator (CFTRΔ508) and epithelial Na+ channels via a process referred to as ER-associated degradation (27, 28). However, the effect of αB-crystallin on ion channels is unknown. In this study, for the first time, we demonstrated that αB-crystallin modulates the function of the cardiac sodium channel, Nav1.5. Because αB-crystallin decreased internalization of Nav1.5 for degradation, we investigated the effect of αB-crystallin on ubiquitination of Nav1.5. Interestingly, overexpression of αB-crystallin decreased ubiquitination of Nav1.5 (Fig. 5, A and B). This finding is consistent with the report showing that knock-out of αB-crystallin expression increased ubiquitination of VEGF-A in RPE cells (29).
Ubiquitination of Nav1.5 was previously reported to be mediated by the ubiquitin-protein ligase Nedd4-2, which binds to the PY-motif at the C terminus of Nav1.5 (8). Nedd4-2 can increase Nav1.5 ubiquitination and reduce INa densities (8). When the PY motif of Nav1.5 was mutated (V1980A-SCN5A), we found that the effect of overexpression of αB-crystallin on INa densities was blocked (Fig. 7B). These data suggest that the effect of αB-crystallin on Nav1.5 function is dependent on the Nedd4-2 binding site. Furthermore, we found that αB-crystallin interacted with Nedd4-2 (Fig. 6). Nedd4-2 was shown to decrease peak INa density by 65%, but its effect on Nav1.5 with the V1980A mutation in the PY motif was reduced (8). In contrast, the catalytically inactive mutation, Nedd4-2-C801S, failed to decrease peak INa density (8). We found that Nedd4-2-C801S reduced the interaction between αB-crystallin and Nedd4-2 (Fig. 6, C and D) and also blocked the increase of peak INa density by αB-crystallin (Fig. 7A). The data suggest that the interaction between αB-crystallin and Nedd4-2 is critical to increased INa density by αB-crystallin. As discussed earlier, the interaction between αB-crystallin and Nav1.5 is also critical to increased INa density by αB-crystallin (Fig. 8, A and B).
Genetic studies have linked mutations in αB-crystallin to cataracts, skeletal muscle myopathy, and cardiomyopathy (9). In particular, mutations G154S and R157H in αB-crystallin were identified in a Japanese patient and an Italian patient with dilated cardiomyopathy, respectively, but both patients did not present with cataracts or arrhythmias (30, 31). Later, mutation G154S was also reported in a 73-year-old German patient with late onset distal vacuolar myopathy but without cardiac involvement or cataracts (32). Due to the critical importance of Nav1.5 in cardiac physiology and human disease, including dilated cardiomyopathy, it would thus be interesting to determine whether these mutations affect function of Nav1.5. We studied the effects of mutations G154S and R157H on the function of αB-crystallin on peak INa density, but no significant difference was found when compared with wild type αB-crystallin (Fig. 8A). Similarly, mutations R11H, P20S, R56W, A171T, and R120G associated with cataracts or muscle myopathy did not have a significant effect on the function of Nav1.5 when compared with the wild type αB-crystallin (Fig. 8A). However, mutation R109H, linked to multisystemic disease, including cataract, myofibrillar myopathy, and cardiomyopathy, and mutation R151X, linked to proximal and distal leg muscle weakness, significantly inhibited the function of αB-crystallin on Nav1.5 and peak INa density (Fig. 8A). Because Nav1.5 mutations were found to be associated with cardiomyopathy (33), it should be interesting to further study how αB-crystallin mutation R109H causes cardiomyopathy by affecting the function of Nav1.5. Moreover, because reduced peak INa density has been linked to Brugada syndrome, progressive cardiac conduction disease, sinus node disease, and atrial fibrillation, carriers with mutations R109H and R151X of αB-crystallin are predicted to be at risk of these arrhythmic disorders. Although no arrhythmias were examined or detected in patients with mutations R109H and R151X, they should be monitored for a potential risk of arrhythmias. Moreover, mutational analysis may identify αB-crystallin mutations in patients with Brugada syndrome, ventricular arrhythmias, cardiac conduction disease, sick sinus syndrome, and atrial fibrillation.
There are limitations to the present study. Although overexpression of αB-crystallin significantly increased cell surface expression of Nav1.5 and INa densities in HEK293 cells, future studies are needed to explore αB-crystallin expression levels in pathological states and to assess the physiological role of αB-crystallin interaction with Nav1.5 in cardiomyocytes. The low transfection efficiency of HL-1 cells and cardiomyocytes with plasmid DNA makes it difficult to achieve a sufficiently high overexpression of αB-crystallin to observe the functional effects of αB-crystallin with Western blotting analysis and patch clamping studies. Moreover, we found that wild type Nedd4-2 reduced the interaction between Nav1.5 and αB-crystallin, whereas the mutant Nedd4-2-C801S did not have an effect on the interaction or slightly increased the interaction (Fig. 6, G and H). The data are consistent with the previous report that wild type Nedd4-2, but not mutant Nedd4-2-C801S, reduced INa density (8). However, we could not distinguish the two possible mechanisms by which wild type Nedd4-2 reduces the interaction between αB-crystallin and Nav1.5: 1) the competitions for the complex formation with Nav1.5 by αB-crystallin and Nedd4-2 or 2) the possibility that increased Nedd4-2 leads to increased ubiquitination and degradation of Nav1.5, resulting in less Nav1.5 and less αB·crystallin·Nav1.5 complex formation.
Conclusions
This study identifies a new binding partner, a small heat shock protein αB-crystallin, for Nav1.5. We show that αB-crystallin interacts with Nav1.5 and increases INa densities by increasing cell surface expression levels of Nav1.5 via inhibition of its internalization. Moreover, we show that αB-crystallin also interacts with functionally active Nedd4-2, which has been reported to interact with Nav1.5 to regulate ubiquitination of Nav1.5 and INa densities. Considering the critical roles of Nav1.5 in cardiac physiology, cardiac arrhythmias, and sudden death, our finding of a critical role of αB-crystallin in regulation of Nav1.5 predicts that αB-crystallin may play a role in cardiac physiology, cardiac arrhythmias, and sudden death, too. This study may also provide an interesting target for developing new therapeutic strategies to treat lethal arrhythmias associated with reduced Nav1.5 function and INa.
Author Contributions
Y. H., Q. C., and Q. K. W. designed the research; Y. H., Z. W., Y. L., H. X., Y. Z., L. Wu, C. Y., L. Wang, Y. H., G. Y., and Z. H. performed the research; Y. H., L. W., C. X., Q. C., and Q. K. W. analyzed the data; and Y. H., Q. C., and Q. K. W. wrote the paper.
Acknowledgments
We thank Hyunjin Rho for the initial yeast two-hybrid screen and other members of the Wang Laboratory for help, assistance, discussions, and advice. We thank Dr. Hughes Abriel for expression plasmids for wild type human Nedd4-2 and mutant Nedd4-2-C801S.
This work was supported by the Chinese National Basic Research Program (973 Program 2013CB531101); China National Natural Science Foundation Key Program Grant 31430047; 973 Program 2012CB517801; NHLBI, National Institutes of Health, Grants R01 HL121358 and R01 HL126729; Hubei Province's Outstanding Medical Academic Leader Program; Hubei Province Natural Science Key Program Grant 2014CFA074; China National Natural Science Foundation Grant 91439129, NSFC-J1103514; the Specialized Research Fund for the Doctoral Program of Higher Education from the Ministry of Education; and “Innovative Development of New Drugs” Key Scientific Project Grant 2011ZX09307-001-09. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- INa
- sodium current
- IP
- immunoprecipitation
- EGFP
- enhanced GFP
- MESNA
- 2-mercaptoethanesulfonic acid sodium salt.
References
- 1. Wang Q., Li Z., Shen J., and Keating M. T. (1996) Genomic organization of the human SCN5A gene encoding the cardiac sodium channel. Genomics 34, 9–16 [DOI] [PubMed] [Google Scholar]
- 2. Wang Q., Chen Q., Li H., and Towbin J. A. (1997) Molecular genetics of long QT syndrome from genes to patients. Curr. Opin. Cardiol. 12, 310–320 [DOI] [PubMed] [Google Scholar]
- 3. Wang Q., Chen Q., and Towbin J. A. (1998) Genetics, molecular mechanisms and management of long QT syndrome. Ann. Med. 30, 58–65 [DOI] [PubMed] [Google Scholar]
- 4. Chakrabarti S., Wu X., Yang Z., Wu L., Yong S. L., Zhang C., Hu K., Wang Q. K., and Chen Q. (2013) MOG1 rescues defective trafficking of Na(v)1.5 mutations in Brugada syndrome and sick sinus syndrome. Circ. Arrhythm. Electrophysiol. 6, 392–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abriel H., and Kass R. S. (2005) Regulation of the voltage-gated cardiac sodium channel Nav1.5 by interacting proteins. Trends Cardiovasc. Med. 15, 35–40 [DOI] [PubMed] [Google Scholar]
- 6. Wu L., Yong S. L., Fan C., Ni Y., Yoo S., Zhang T., Zhang X., Obejero-Paz C. A., Rho H. J., Ke T., Szafranski P., Jones S. W., Chen Q., and Wang Q. K. (2008) Identification of a new co-factor, MOG1, required for the full function of cardiac sodium channel Nav1.5. J. Biol. Chem. 283, 6968–6978 [DOI] [PubMed] [Google Scholar]
- 7. Detta N., Frisso G., and Salvatore F. (2015) The multi-faceted aspects of the complex cardiac Nav1.5 protein in membrane function and pathophysiology. Biochim. Biophys. Acta 1854, 1502–1509 [DOI] [PubMed] [Google Scholar]
- 8. van Bemmelen M. X., Rougier J. S., Gavillet B., Apothéloz F., Daidié D., Tateyama M., Rivolta I., Thomas M. A., Kass R. S., Staub O., and Abriel H. (2004) Cardiac voltage-gated sodium channel Nav1.5 is regulated by Nedd4-2 mediated ubiquitination. Circ. Res. 95, 284–291 [DOI] [PubMed] [Google Scholar]
- 9. Graw J. (2009) Genetics of crystallins: cataract and beyond. Exp. Eye Res. 88, 173–189 [DOI] [PubMed] [Google Scholar]
- 10. Li H., Li C., Lu Q., Su T., Ke T., Li D. W., Yuan M., Liu J., Ren X., Zhang Z., Zeng S., Wang Q. K., and Liu M. (2008) Cataract mutation P20S of αB-crystallin impairs chaperone activity of αA-crystallin and induces apoptosis of human lens epithelial cells. Biochim. Biophys. Acta 1782, 303–309 [DOI] [PubMed] [Google Scholar]
- 11. Du W, Bautista J. F., Yang H., Diez-Sampedro A., You S. A., Wang L., Kotagal P., Luders H. O., Shi J., Cui J., Richerson G. B., and Wang Q. K. (2005) Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat. Genet. 37, 733–738 [DOI] [PubMed] [Google Scholar]
- 12. Zhang X., Chen S., Yoo S., Chakrabarti S., Zhang T., Ke T., Oberti C., Yong S. L., Fang F., Li L., de la Fuente R., Wang L., Chen Q., and Wang Q. K. (2008) Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell 135, 1017–1027 [DOI] [PubMed] [Google Scholar]
- 13. Chen Q., Kirsch G. E., Zhang D., Brugada R., Brugada J., Brugada P., Potenza D., Moya A., Borggrefe M., Breithardt G., Ortiz-Lopez R., Wang Z., Antzelevitch C., O'Brien R. E., Schulze-Bahr E., et al. (1998) Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 392, 293–296 [DOI] [PubMed] [Google Scholar]
- 14. Wan X., Wang Q., and Kirsch G. E. (2000) Functional suppression of sodium channels by β1-subunits as a molecular mechanism of idiopathic ventricular fibrillation. J. Mol. Cell Cardiol. 32, 1873–1884 [DOI] [PubMed] [Google Scholar]
- 15. Wan X., Chen S., Sadeghpour A., Wang Q., and Kirsch G. E. (2001) Accelerated inactivation in a mutant Na+ channel associated with idiopathic ventricular fibrillation. Am. J. Physiol. Heart Circ. Physiol. 280, H354–H360 [DOI] [PubMed] [Google Scholar]
- 16. Fan C., Chen Q., and Wang Q. K. (2009) Functional role of transcriptional factor TBX5 in pre-mRNA splicing and Holt-Oram syndrome via association with SC35. J. Biol. Chem. 284, 25653–25663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tian X. L., Kadaba R., You S. A., Liu M., Timur A. A., Yang L., Chen Q., Szafranski P., Rao S., Wu L., Housman D. E., DiCorleto P. E., Driscoll D. J., Borrow J., and Wang Q. (2004) Identification of an angiogenic factor that when mutated causes susceptibility to Klippel-Trenaunay syndrome. Nature 427, 640–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tian X. L., Yong S. L., Wan X., Wu L., Chung M. K., Tchou P. J., Rosenbaum D. S., Van Wagoner D. R., Kirsch G. E., and Wang Q. (2004) Mechanisms by which SCN5A mutation N1325S causes cardiac arrhythmias and sudden death in vivo. Cardiovasc. Res. 61, 256–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yong S. L., Ni Y., Zhang T., Tester D. J., Ackerman M. J., and Wang Q. K. (2007) Characterization of the cardiac sodium channel SCN5A mutation, N1325S, in single murine ventricular myocytes. Biochem. Biophys. Res. Commun. 352, 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang T., Yong S. L., Tian X. L., and Wang Q. K. (2007) Cardiac-specific overexpression of SCN5A gene leads to shorter P wave duration and PR interval in transgenic mice. Biochem. Biophys. Res. Commun. 355, 444–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang T., Yong S. L., Drinko J. K., Popović Z. B., Shryock J. C., Belardinelli L., and Wang Q. K. (2011) LQTS mutation N1325S in cardiac sodium channel gene SCN5A causes cardiomyocyte apoptosis, cardiac fibrosis and contractile dysfunction in mice. Int. J. Cardiol. 147, 239–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang F., Xu C. Q., He Q., Cai J. P., Li X. C., Wang D., Xiong X., Liao Y. H., Zeng Q. T., Yang Y. Z., Cheng X., Li C., Yang R., Wang C. C., Wu G., et al. (2011) Genome-wide association identifies a susceptibility locus for coronary artery disease in the Chinese Han population. Nat. Genet. 43, 345–349 [DOI] [PubMed] [Google Scholar]
- 23. Xu Y., Zhou M., Wang J., Zhao Y., Li S., Zhou B., Su Z., Xu C., Xia Y., Qian H., Tu X., Xiao W., Chen X., Chen Q., and Wang Q. K. (2014) Role of microRNA-27a in down-regulation of angiogenic factor AGGF1 under hypoxia associated with high-grade bladder urothelial carcinoma. Biochim. Biophys. Acta 1842, 712–725 [DOI] [PubMed] [Google Scholar]
- 24. Zhou B., Si W., Su Z., Deng W., Tu X., and Wang Q. (2013) Transcriptional activation of the Prox1 gene by HIF-1α and HIF-2α in response to hypoxia. FEBS Lett. 587, 724–731 [DOI] [PubMed] [Google Scholar]
- 25. Zhou B., Ma R., Si W., Li S., Xu Y., Tu X., and Wang Q. (2013) MicroRNA-503 targets FGF2 and VEGFA and inhibits tumor angiogenesis and growth. Cancer Lett. 333, 159–169 [DOI] [PubMed] [Google Scholar]
- 26. Dumaine R., Wang Q., Keating M. T., Hartmann H. A., Schwartz P. J., Brown A. M., and Kirsch G. E. (1996) Multiple mechanisms of Na+ channel-linked long-QT syndrome. Circ. Res. 78, 916–924 [DOI] [PubMed] [Google Scholar]
- 27. Kashlan O. B., Mueller G. M., Qamar M. Z., Poland P. A., Ahner A., Rubenstein R. C., Hughey R. P., Brodsky J. L., and Kleyman T. R. (2007) Small heat shock protein αA-crystallin regulates epithelial sodium channel expression. J. Biol. Chem. 282, 28149–28156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ahner A., Nakatsukasa K., Zhang H., Frizzell R. A., and Brodsky J. L. (2007) Small heat-shock proteins select δF508-CFTR for endoplasmic reticulum-associated degradation. Mol. Biol. Cell 18, 806–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kase S., He S., Sonoda S., Kitamura M., Spee C., Wawrousek E., Ryan S. J., Kannan R., and Hinton D. R. (2010) αB-crystallin regulation of angiogenesis by modulation of VEGF. Blood 115, 3398–3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Inagaki N., Hayashi T., Arimura T., Koga Y., Takahashi M., Shibata H., Teraoka K., Chikamori T., Yamashina A., and Kimura A. (2006) αB-crystallin mutation in dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 342, 379–386 [DOI] [PubMed] [Google Scholar]
- 31. Pilotto A., Marziliano N., Pasotti M., Grasso M., Costante A. M., and Arbustini E. (2006) αB-crystallin mutation in dilated cardiomyopathies: low prevalence in a consecutive series of 200 unrelated probands. Biochem. Biophys. Res. Commun. 346, 1115–1117 [DOI] [PubMed] [Google Scholar]
- 32. Reilich P., Schoser B., Schramm N., Krause S., Schessl J., Kress W., Müller-Höcker J., Walter M. C., and Lochmuller H. (2010) The p.G154S mutation of the α-B crystallin gene (CRYAB) causes late-onset distal myopathy. Neuromuscul. Disord. 20, 255–259 [DOI] [PubMed] [Google Scholar]
- 33. Veerman C. C., Wilde A. A., and Lodder E. M. (2015) The cardiac sodium channel gene SCN5A and its gene product NaV1.5: role in physiology and pathophysiology. Gene 573, 177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]