

Abstract
The AlkB repair enzymes, including Escherichia coli AlkB and two human homologues, ALKBH2 and ALKBH3, are iron(II)- and 2-oxoglutarate-dependent dioxygenases that efficiently repair N1-methyladenine and N3-methylcytosine methylated DNA damages. The development of small molecule inhibitors of these enzymes has seen less success. Here we have characterized a previously discovered natural product rhein and tested its ability to inhibit AlkB repair enzymes in vitro and to sensitize cells to methyl methane sulfonate that mainly produces N1-methyladenine and N3-methylcytosine lesions. Our investigation of the mechanism of rhein inhibition reveals that rhein binds to AlkB repair enzymes in vitro and promotes thermal stability in vivo. In addition, we have determined a new structural complex of rhein bound to AlkB, which shows that rhein binds to a different part of the active site in AlkB than it binds to in fat mass and obesity-associated protein (FTO). With the support of these observations, we put forth the hypothesis that AlkB repair enzymes would be effective pharmacological targets for cancer treatment.
Keywords: bacteria, cancer therapy, chemical biology, DNA repair, DNA-protein interaction, enzyme inhibitor, enzyme structure
Introduction
The nucleic acids in living cells are subject to modification by both endogenous and environmental agents (1). Direct-acting chemicals constantly damage nucleic acids and generate various methyl lesions with mutagenic and/or cytotoxic consequences (2, 3). O6-Methylguanine (O6mG)2 and N3-methyladenine lesions have the highest potential for methylating damage by an SN1 agent such as N-methyl-N′-nitro-N-nitrosoguanidine (MNNG), which block replication and are thought to be toxic (4, 5). For the most part, the SN2 agent such as methyl methane sulfonate (MMS) produces N1-methyladenine (m1A) and N3-methylcytosine (m3C) lesions in single-stranded DNA (ssDNA). Accumulation of these adducts can lead to cell death (6, 7). Organisms have evolved several mechanisms to efficiently remove various methyl lesions, including suicidal methyltransferases, DNA glycosylases, and the AlkB family dioxygenases (see Fig. 1A) (8, 9).
FIGURE 1.

Rhein selectively inhibits AlkB in vitro. A, the three major repair pathways in E. coli. DNA glycosylase is colored in cyan, DNA methyltransferase repair is in blue, and the AlkB repair is in green, respectively. B, scheme of AlkB repair methylated DNA. The structures of the inhibitor rhein and negative control BA are shown. C, rhein inhibits AlkB repair of m1A in ssDNA (left panel) and dsDNA (right panel) by using the DpnII digestion assay. The upper band is 49-nucleotide dsDNA, which contains an m1A lesion, and the lower band represents the mixture of 22- and 27-nucleotide dsDNA products after DpnII digestion. The 2OG concentration is 50 μm. D, quantitative determination of inhibitory activity using HPLC-based assay. The fitted IC50 is 12.7 μm assayed at 50 μm 2OG. The error bars are the means ± S.E. (n = 3). E, BA fails to inhibit m1A-dsDNA repair by AlkB. The 39-nucleotide m1A-containing DNA substrate was tested. F, C-Ada repair of O6mG is unimpaired in the presence of rhein. The upper band is 39-nucleotide O6mG-containg dsDNA, and the lower band is the digested fragments by PvuII. G, rhein is inactive to inhibit AlkA glycosylase. The 25-nucleotide mismatched dsDNA substrate is tested. All reactions were assayed in triplicate. nt, nucleotide.
To date, AlkB repair appears to be the major natural defense mechanism with the power to restore the canonical base structure in vivo. Escherichia coli AlkB and its human homologues, ALKBH2 and ALKBH3, utilize iron(II) and 2-oxoglutarate (2OG) to achieve oxidative demethylation of m1A and m3C (see Fig. 1B) (10–12). The lack of AlkB repair results in increased sensitivity to MMS, elevated level of mutations, and reduced cell proliferation (13–16). Furthermore, the accumulation of m1A and m3C lesions could also occur on RNA. Research suggests that the oxidative demethylation in mRNA and tRNA acts as a part of AlkB or ALKBH3 repair to protect cells against MMS (17, 18). The scope of substrates for AlkB repair has been largely extended to all simple N-alkyl lesions at the Watson-Crick base-pairing interface on the four bases (19), thus indicating the importance of oxidative demethylation for cell survival. In addition, human enzymes have been broadly linked to cancer. The housekeeping enzyme in mammalian cells, ALKBH2, plays a crucial role in pediatric brain tumors during chemotherapy treatment (20). Essential for prostate cancer progression, ALKBH3 presents a potential target for effective therapy in prostate cancer (21, 22).
Structural characterizations of AlkB repair enzymes have provided insights into the understanding of demethylation mechanism and substrate recognition (23–26). Similar to members of the 2OG-dioxygenase family, 2OG could inhibit AlkB at high concentrations (27). Recently, the method dynamic combinatorial chemistry, which is linked to mass spectrometric analyses, was used to identify AlkB inhibitors; these inhibitors showed improved potency and selectivity (28). A set of inhibitors of ALKBH3 were obtained from a random screening, and some of them significantly suppress tumor formation in a mice xenograft model (29). These data demonstrate that AlkB repair enzymes are amenable to potent inhibition by small molecules. What these studies lack, however, is 2-fold: one, a profile of the target engagement of inhibitor and two, a full elucidation of the inhibitor mode of action, which should provide a better understanding of the biological consequences of the replication-blocking m1A and m3C lesions (30).
As we reported in previous studies, the natural product rhein inhibits FTO demethylation of N6-methyladenine in vitro and elevates the level of N6-methyladenine within mRNA in HeLa cells (31, 32). In this paper, we provide significant new data that describe the in vitro and in vivo effects of rhein as an inhibitor of AlkB repair enzymes.
Experimental Procedures
Chemicals, DNA Oligonucleotides, Antibodies, E. coli Strains, and Cell Culture
Rhein, MMS, MNNG, and temozolomide (TMZ) in cell biology grade were purchased from Sigma-Aldrich. Bromaminic acid (BA) and JIB-04 were purchased from Tokyo Chemical Industry Co. and Selleck Chemicals, respectively. The DNA oligonucleotide were synthesized on an Expedite DNA synthesizer (PerSeptive Biosystems). Antibodies anti-m3C, anti-ALKBH2, anti-ALKBH3, anti-H3K9me3, and anti-β-actin were produced by Active Motif (61180), Sigma (SAB3500534), Millipore (09882), ABCAM (ab8898), and CWBIO (0096a), respectively. The rabbit polyclonal anti-AlkB antibody was generated by Shanghai Immune Biotech using the purified AlkB protein as the antigen. The E. coli AB1157 and HK82 strains were kind gifts from Dr. H. Krokan's lab (Norwegian University of Science and Technology). While this paper was in revision, the HK82 strain that has been widely used was reported to contain additional mutations affecting MMS sensitivity (33). U87 cells were purchased from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences and cultured in minimum essential medium (41500034; Gibco) supplemented with 10% FBS.
Restriction Endonuclease Digestion Assay
The AlkB, ALKBH2, and ALKBH3 proteins were expressed and purified as described (34–36). We cloned the expression plasmids of the C-terminal domain of Ada (C-Ada) and AlkA by incorporating the two genes into pET28a vector, and then the expression and purification of the two recombinant proteins followed known procedures (37, 38). The DNA oligonucleotides used in the enzymatic assays were 5′-TAGACATTGCCATTCTCGATAGG(m1A)TCCGGTCAAACCTAGACGAATTCCA-3′ or 5′-ATTGCCATTCTCGATAGG(m1A)TCCGGTCAAACCTAGACGAA-3′ for AlkB or ALKBH3 repair, 5′-TGGAATTCGTCTAGGTTTGACCGGATCCTATCGAGAATGGCAATGTCTA-3′ or 5′-TTCGTCTAGGTTTGACCGGATCCTATCGAGAATGGCAAT-3′ as the complementary DNA sequence for the duplex substrates of AlkB or ALKBH2 repair, 5′-GCCATTCTCGATAGGCGCA(O6mG)CTGAGCTCGCGTCCGGTCA-3′ complementary to 5′-TGACCGGACGCGAGCTCAGCTGCGCCTATCGAGAATGGC-3′ for Ada repair, and 5′-CGATAGCATCCTGCCTTCTCTCCAT-3′ complementary to 5′-ATGGAGAGAAGGAAGGATGCTATCG-3′ for AlkA repair, respectively. All reactions were run in 100-μl scale. The conditions were 50 mm Tris-HCl (pH 7.5), 1 μm DNA, 1 μm enzyme, 0.28 mΜ (NH4)2Fe(SO4)2, 2 mm l-ascorbic acid, and 50 μm 2OG in AlkB repair, 50 mm Tris-HCl (pH 8.0), 1 mm EDTA, 1 mm DTT, 1 μm DNA, and 1 μm C-Ada in Ada repair, and 50 mm Tris-HCl (pH 8.5), 1 mm EDTA, 1 mm DTT, 0.1 mg/ml bovine serum albumin, 5 μm AlkA, 1 μm DNA, and ionic strength adjusted to 100 mm with sodium chloride in AlkA glycosylase assay, respectively. The oxidative demethylation and C-Ada repair assays were run at room temperature for 1 h and quenched by heating at 50 °C for 10 min, and then a DpnII or PvuII digestion was performed at 37 °C. The AlkA assay was continually executed at 37 °C for 24 h and heated at 70 °C for 30 min in the presence of 0.2 m NaOH. All reactions were detected on 20% nonreducing PAGE. The gel was stained in the presence of GelRed (41003; Biotium) at room temperature for 0.5 h and then was photographed by UV light in a gel imaging system.
HPLC-based Quantification Assay
The reactions were run under the same conditions as described in the DpnII digestion assay in which a 15-mer ssDNA (5′-ATTGTCA(m1A)CAGCAGA-3′) was assayed at 5 μm. After the reaction was terminated, ssDNA was digested by nuclease P1 (N8630; Sigma) and alkaline phosphatase (P4252; Sigma), and then the nucleosides were analyzed by HPLC using an Agilent Eclipse XDB-C18 column (4.6 mm × 250 mm). The program was run with the following settings: mobile phase A (50 mm CH3COONH4, pH 5.0) and B (acetonitrile) at a flow rate of 1 ml/min. The IC50 values were then calculated from the variation as a percentage of demethylation at different inhibitor concentrations using nonlinear regression, with a normalized dose-response fit on GraphPad Prim 5.0 using the equation,
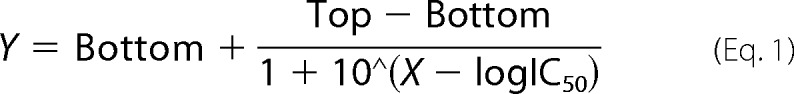 |
where Bottom and Top are the minimal and maximal inhibition percentages, respectively. All reactions were repeated in triplicate.
Plate Killing Assay
Overnight culture of E. coli was diluted to A600 0.005 or 0.008 (transformed with pET28a). After serial dilution by 10-fold, each volume of 10 μl of bacteria was spotted onto LB solid medium in the presence of rhein or MMS at varying concentrations, and isopropyl β-d-1-thiogalactopyranoside at 50 μm was supplemented to induce AlkB overexpression. After 10 h, static cultures at 37 °C, the plates were photographed with the gel imaging system (Tanon 2500R).
E. coli Colony-forming Unit Count
E. coli Gold, AB1157, or HK82 was cultured overnight and diluted 1:100 to fresh LB medium and grown to logarithmic phase. Then the bacteria were separated equally and grown for 5 h with MMS or rhein or both. The bacteria were diluted serially and cultured overnight on LB agar. The E. coli colonies were counted relative to the DMSO control group. All tests were performed in triplicate.
Western Blot and Dot Blot Analysis
Overnight E. coli culture was diluted 1:100 to fresh LB and incubated to the logarithmic phase. Compounds were added, and cultures were grown at 37 °C with constant shaking at 230 rpm for 5 h. The cells were collected and divided into two aliquots. One aliquot was used to isolate genomic DNA by means of a PureLink® genomic DNA mini kit (Invitrogen). 2-fold serial dilutions were loaded onto the nylon membrane and cross-linked by exposure to UV, and then m3C antibody was probed to the genomic DNA. The other aliquot was sonicated, and the supernatant was quantified by BCA assay. The equal 25-μg cell extracts were separated by 15% SDS-PAGE. Both AlkB and m3C were visualized by SuperSignal West Femto kit (Thermo).
Cellular Thermal Shift Assay (CETSA)
After exposure to 500 μm MMS with (for intact cell assay) or without rhein (for cell lysate assay), E. coli cells were collected and washed several times to avoid any excess compound residue. Then the cells were sonicated and centrifuged at 12,000 rpm for 20 min at 4 °C to fractionize the supernatant. For the cell lysate assay, rhein was added to the supernatant and incubated at 20 °C for 20 min. After denaturing at various temperatures for 5 min on Mastercycler® pro PCR instrument (Eppendorf), samples were centrifuged to eradicate the precipitate. The supernatants were analyzed by Western blot, and the density of AlkB protein bands was plotted using GraphPad Prism 5.0TM. All performances were repeated in triplicate (39, 40).
Isothermal Titration Calorimetry
Microcal iTC200 (GE Healthcare) was operated to measure the binding affinity at 25 °C (41). The titration buffer contained 50 mm Tris (pH 8.0), 150 mm NaCl, and 10 mm β-mercaptoethanol. Each sample was thoroughly degassed before titration. The first titration into the sample cell was a 0.5-μl injection, followed by 24 successive 1.5-μl injections at 180-s intervals. The Experimental data were analyzed with the Microcal ORIGIN V7.0 software (Microcal Software, MA).
Differential Scanning Fluorimetry
The experiments were performed using a RT-PCR detection system (ABI 7500 Fast) (42, 43). Each well consisted of a 30-μl solution containing 50 mm HEPES (pH 7.5), 50 μm MnCl2, and 1.25 μm protein, 1× SYPRO orange (Invitrogen), and tested compound. The unfolded protein was monitored by SYPRO orange using the wavelength 492 nm for excitation and 610 nm for emission, respectively. Fluorescence intensities were obtained every 0.4 °C in the range of 25 °C to 95 °C, which was heated at 1% ramp rate. The data were processed using Protein Thermal ShiftTM software (Applied Biosystems). The Tm was calculated by fitting the Boltzmann equation to the sigmoidal curve. All conditions were tested in triplicate.
Kinetics Analysis
To obtain the initial rate of inhibition, 20 nm AlkB was incubated with 2OG (5, 10, and 20 μm) and rhein at 0, 2, 4, and 8 μm, respectively. Another analysis was assayed in the presence of 50 nm AlkB and 20 μm 2OG with 15-mer ssDNA (2.5, 5, and 10 μm) and rhein at 0, 3, 6, and 12 μm, respectively. The consumption of m1A-containing ssDNA was adjusted to less than 20%. After digestion by nuclease P1 and alkaline phosphatase, the nucleosides were separated by HPLC. The kinetics parameters were determined with the Michaelis-Menten equation fits and Lineweaver-Burk plot in GraphPad Prism 5.0.
Cell Viability Assay
100 μl U87 cells (8 × 103) were seeded on a 96-well plate and cultured overnight. For ALKBH2/3 knockdown, cells were seeded as 60% confluent of each well. After additional culture with compounds for 24–48 h, cells in each well were mixed with 10 μl of MTT (5 mg/ml; Amresco). The precipitate was resuspended in 150 μl of DMSO, and the absorbance was detected at the wavelength of 570 nm. All assays were performed in triplicate.
RNA Interfering of ALKBH2 and ALKBH3
The assays were carried out using Lipofectamine RNAiMAX transfection reagent (Invitrogen). The target mRNA sequence of ALKBH2 is 5′-CACGGGAGCUUACUAAUGA-3′, and that of ALKBH3 is 5′-UGCCCAAAGAAUACCACUCUA-3′. Cells were collected for Western blot analysis after 24 h of culture.
Crystal Structure of AlkB-Rhein Complex
The purified AlkB was incubated with 20-fold excess rhein in 50 mm HEPES (pH 7.5), 500 μm MnCl2 at room temperature for 20 min. The crystallization was set up as 1:1 hanging drops at 20 °C. Crystals were grown in the reservoir solution containing 200 mm ammonium sulfate, 100 mm HEPES (pH 7.5), and 25% (w/v) polyethylene glycol 3,350. Crystals were flash cooled in liquid N2 with a well solution diluted to 20% (v/v) glycerol. All x-ray data were collected at the Shanghai Synchrotron Radiation Facility Beamline 17U and processed in HKL2000 (44). The structures were solved by molecular replacement using the AlkB-2OG complex (PDB code 3I3Q) as a search model in the CCP4 suite (45). The model building was manually performed in COOT (46), and iterative refinements were carried out in REFMAC5 (47).
Results
Rhein Inhibits AlkB in Vitro
AlkB and FTO share the homologous catalytic domain for oxidative demethylation. Therefore, we wondered whether the FTO inhibitor rhein could inhibit AlkB in vitro (Fig. 1B). The restriction endonuclease digestion assay was run to evaluate the inhibition of AlkB by rhein (Fig. 1C). We observed full activity of AlkB repair of m1A on ssDNA or dsDNA in the control experiment, whereas AlkB was inhibited in the presence of rhein in a concentration-dependent manner. The IC50 was quantitatively determined to be 12.7 μm in the HPLC-based detection when assayed at 50 μm 2OG (Fig. 1D) (48). BA is a moderate inhibitor of FTO (Fig. 1B) (31, 49). AlkB repair remained intact, however, even in the presence of 100-fold excess BA (Fig. 1E); thus BA was employed as an inactive control. Taken together, these results show that rhein effectively inhibits E. coli AlkB in vitro.
Rhein Does Not Inhibit Ada or AlkA
In addition to AlkB, Ada and AlkA were also induced in E. coli in response to MMS. The C-Ada specifically repairs O6mG by transferring the methyl group to an active Cys (50). AlkA glycosylase recognizes N3-methyladenine and mismatch pairs (51). We performed selectivity analysis to rule out the potential phenotypic consequences of inhibiting C-Ada or AlkA repair by rhein in E. coli. As expected, rhein does not inhibit the demethylation activity proceeded by either C-Ada methyltransferase or AlkA glycosylase in vitro (Fig. 1, F and G). These results clearly show that rhein selectively inhibits AlkB rather than Ada or AlkA of E. coli.
Rhein Sensitizes E. coli to MMS
The alkB mutant E. coli strain becomes more sensitive to MMS compared with the wild-type or AlkB-overexpressed strain. This is due to the lack of AlkB repair, which accumulates methyl lesions on nucleic acids (17). We wondered whether the AlkB inhibitor rhein would similarly result in the impaired viability of E. coli when exposed to MMS threats. The growth of E. coli Gold was minimally impaired in the presence of either 50–75 μm MMS or 100 μg/ml rhein (Fig. 2A). Interestingly, the combination of 50 μm MMS and 100 μg/ml rhein reduces bacterial survival on agar medium. Moreover, the reduction in bacterial survival is reinforced with elevated MMS (75 μm). In addition, we performed a measurement of cfu assay to quantitatively determine the sensitization of E. coli to MMS by rhein. As shown in Fig. 2B, rhein significantly makes E. coli sensitive to MMS threats when grown in liquid medium. In the control experiment, BA failed to sensitize the growth of E. coli in the presence of MMS (Fig. 2C). These data show that rhein sensitizes E. coli to MMS and ultimately impairs bacterial growth.
FIGURE 2.
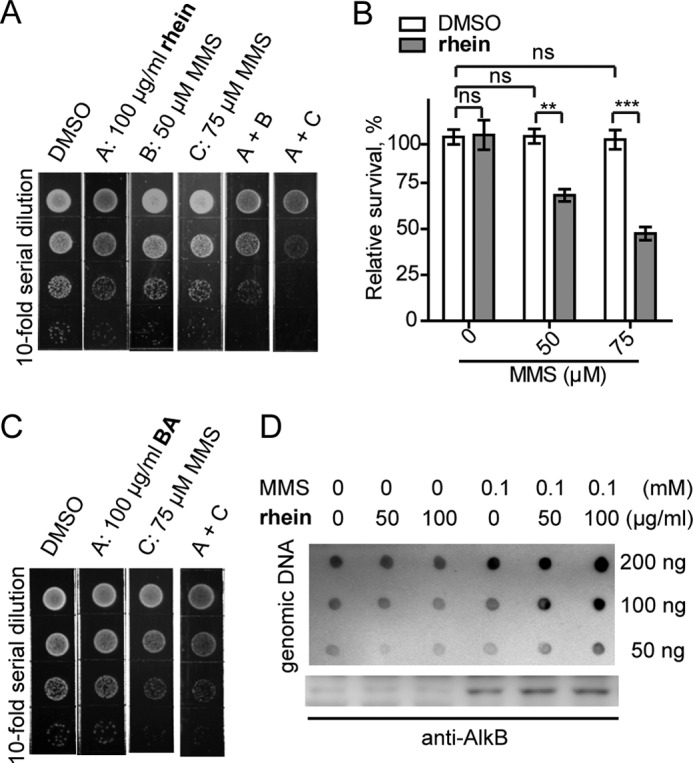
Rhein sensitizes E. coli to MMS by enhancing the m3C accumulation. A, plate killing assay to show growth of E. coli Gold during exposure to 50 or 75 μm MMS with 100 μg/ml rhein, respectively. The density in the top line is A600 0.005. B, cfu count assay to show resistance of E. coli Gold to MMS in the presence of rhein. The error bars are means ± S.E. (n = 6). **, p < 0.1; ***, p < 0.001. C, growth of E. coli Gold in the presence of MMS and compound BA. D, the quantification of m3C (upper blot) and AlkB protein (lower blot) in E. coli Gold using blot assays. ns, not significant.
Rhein Promotes the Accumulation of DNA Methyl Lesions
To test whether rhein sensitization of E. coli to MMS is a result of the inhibitory effects of AlkB repair, we sought to quantify the cellular m3C lesions on genomic DNA (52). Performance of Western blot analysis confirms that MMS indeed induces AlkB expression in the time course and that rhein had no effect on AlkB abundance (Fig. 2D). The content of genomic m3C was quantified in dot blot analysis. As expected, rhein left the cellular level of m3C unimpaired but did promote the accumulation of m3C lesions in a dose-dependent manner during continuous MMS exposure (Fig. 2D), which should be a result of the inhibition of AlkB by rhein in E. coli.
In Vivo Target Engagement of Rhein
To probe the cellular target of rhein, we explored the impact on the potency of rhein when AlkB is overexpressed or mutated. The growth of E. coli Gold complemented with pET28a empty vector was minimally impaired in the presence of either 75 μm MMS or 100 μg/ml rhein but significantly inhibited in the presence of both 75 μm MMS and 100 μg/ml rhein (Fig. 3A, left panel). However, the AlkB-overexpressed E. coli strain became more resistant to the combined treatment (Fig. 3A, right panel). Obviously, the overexpressed AlkB could be able to repair methyl lesions even in the presence of the inhibitor rhein. Rhein also sensitizes wild-type AB1157 to MMS (Fig. 3B, left panel). The E. coli HK82, which is a typical alkB mutant strain derived from wild-type AB1157, became more sensitive to MMS because of deficient AlkB repair. In addition, rhein did not further increase the toxicity of MMS toward HK82 strain (Fig. 3B, middle panel). However, the HK82 strain, when transformed with a low copy pJB658 for AlkB complement, regained resistance to 750 μm MMS. As expected, rhein obviously reduced the viability of AlkB-complement HK82 during MMS exposure (Fig. 3B, right panel). These results show that the inhibitory activity of rhein is dependent on the abundance and function of the cellular AlkB enzyme.
FIGURE 3.

Cellular target engagement of rhein. A, plate killing assay to show the resistance of E. coli Gold that overexpresses AlkB to MMS in the presence of rhein. The starting density is A600 0.008. B, cfu count assay to show resistance of E. coli AB1157 (wild type), HK82 (AlkB mutant), and AlkB complemented HK82 to MMS by rhein. The error bars are means ± S.E. (n = 6). **, p < 0.01; ***, p < 0.001. C, rhein could not sensitize E. coli Gold to MNNG. The density of bacteria in the top line is A600 0.005. D, rhein could not sensitize E. coli growth to other DNA-damaging agents such as the oxidizing agent (H2O2). E, CETSA showing that rhein increases the thermal stability of AlkB in E. coli cell lysate. The data are presented as means ± S.E., and experiments were performed in triplicate. F, CETSA shows that rhein stabilizes AlkB in intact bacterial cells. ns, not significant.
Next, we compared the cellular sensitivity of E. coli to SN2 agent versus SN1 agent in the presence of rhein. MMS is an SN2-alkylating agent that induces the formation of m1A and m3C lesions repaired specifically by AlkB (53). In contrast, SN1 agents, MNNG for example, induce predominantly O6mG lesions that are repaired by O6-methyltransferases (54). MNNG at 5 μm displayed minimal cytotoxic effects on E. coli growth. As expected, rhein could not enhance bacterial sensitivity to MNNG (Fig. 3C). In addition, rhein could not sensitize E. coli growth to other DNA-damaging agents such as H2O2 (Fig. 3D). These studies would also provide strong evidence that rhein is specifically targeting AlkB in vivo.
To further probe the in vivo target engagement of rhein, we performed a CETSA. AlkB in E. coli lysate shows a distinct shift in the melting curve (Fig. 3E), indicating that rhein directly binds to and stabilizes AlkB in the cell lysate. Next, the CETSA experiment in intact cells was also performed. E. coli were exposed to 500 μm MMS in the presence of 100 μg/ml rhein. The quantization of soluble AlkB reveals an obvious thermal shift in rhein-treated cells compared with that in the control experiment (Fig. 3F). The observations in CETSAs clearly show that rhein stabilizes AlkB in E. coli, perhaps through a direct binding.
Mechanistic Study of Rhein Inhibition of AlkB
We next investigated the mechanism of the inhibition of AlkB by rhein. Rhein binds tightly to the AlkB-Mn2+ complex (Kd = 0.29 ± 0.03 μm) as observed in the isothermal titration calorimetry assay (Fig. 4A), which is an enthalpically driven process (ΔH = −5.8 kcal mol−1, ΔS = 10.4 cal/mol/deg). As expected, no binding response was detected when compound BA was titrated into the AlkB-Mn2+ complex (Fig. 4B), which explains why BA is an inactive inhibitor of AlkB (Fig. 1E). In addition, the performance of a fluorescence-based thermal shift (differential scanning fluorimetry) assay showed that rhein significantly stabilizes AlkB as a reflection of the increasing melting temperature (Tm) of AlkB over 8 °C in the presence of 20-fold excess molar of rhein (Fig. 4C). Furthermore, we have performed an enzyme kinetics analysis. The initial rates linear fits by HPLC quantification were of good quality (Fig. 4D, left panel). Rhein acts as a 2OG competitive inhibitor (Ki = 1.3 μm; Fig. 4D, right panel) but a noncompetitive inhibitor toward substrate DNA (Ki = 2.1 μm; Fig. 4E). Taken together, we conclude that rhein competes on 2OG binding to AlkB and acts as a competitive inhibitor of AlkB.
FIGURE 4.

Mechanistic study for inhibition of AlkB by rhein. A, isothermal titration calorimetry of rhein binding to AlkB-Mn2+ complex. Binding curves were fitted as a single binding event, and the constant is the average of two measurements. The fitted Kd is 0.29 μm. B, compound BA could not bind to AlkB-Mn2+ complex. C, differential scanning fluorimetry assay shows that rhein stabilizes AlkB by increasing Tm over 8 °C. Also shown are graphs of unfolding transition of 1.25 μm AlkB in the presence of rhein at 6.25 and 25 μm, respectively. The experiments were performed in triplicate. D, kinetics analyses of the mode of AlkB inhibition by rhein with respect to 2OG. Some of the initial rates linear fits are shown (left panel). E, kinetics analyses of AlkB inhibition by rhein with respect to m1A.
Structural Insights into the Inhibitor Binding
We have determined the x-ray crystal structure of AlkB-rhein complex at a resolution of 1.5 Å (Table 1). The final Rwork and Rfree were 11.7 and 15.0%, respectively. The AlkB structure is not changed upon binding by rhein or 2OG, because the RMSD observed in the superimposition of our AlkB-rhein complex, and the known AlkB/2OG (PDB code 3I3Q) is only 0.28 Å (Fig. 5A). The Fo − Fc OMIT density contoured to 3.0 σ confirm that rhein is indeed bound (Fig. 5B). The binding site of rhein is partially overlapping with the 2OG binding site. This explains the competitive property of rhein inhibition of AlkB. The side chain of the carboxyl acid in rhein is positioned to form a salt bridge or hydrogen bonding with the side chains of Arg204 and Tyr122. Rhein is positioned to chelate Mn2+ in a bidentate manner by means of the carbonyl and hydroxyl groups. In addition to these major interactions, which are similar to 2OG binding to AlkB, rhein utilizes more interactions for AlkB binding, which take their origin from hydrogen-bonding between the other carbonyl in rhein and the side chains of Trp178 and Ser145 in AlkB.
TABLE 1.
Data collection and refinement statistics
| AlkB/rhein | |
|---|---|
| Data collection | |
| Space group | P1 |
| Cell dimensions | |
| a, b, c (Å) | 36.95, 39.20, 40.52 |
| α, β, γ (°) | 77.4, 74.7, 65.5 |
| Resolution (Å) | 50.0–1.50 (1.55–1.50)a |
| No. of observations | 118,619 (11,912) |
| No. unique | 30,221 (2,978) |
| Rsymb | 0.041 (0.146) |
| I/σ(I) | 31.0 (13.3) |
| Completeness (%) | 95.9 (94.6) |
| Redundancy | 3.9 (4.0) |
| Data refinement | |
| Resolution (Å) | 30.0–1.50 (1.54–1.50) |
| No. reflections | 28,702 (2,062) |
| Rwork/Rfree | 11.7/15.0 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.006 |
| Bond angles (°) | 1.176 |
| Ramachandran plotc | |
| Most favored (%) | 99.5 |
| Allowed (%) | 0.5 |
a Highest resolution shell is shown in parenthes2s.
b Rsym = Σ|(I − <I>) |/Σ(I), where I is the observed intensity.
c Values calculated in CCP4 suite using Procheck.
FIGURE 5.

Structural insights into the mode of rhein binding to AlkB. A, structure alignment of the AlkB-rhein (PDB code 4RFR) and 2OG-bound AlkB (PDB code 3I3Q) performed in PyMOL with RMSD = 0.28 Å. The AlkB-rhein structure is colored cyan, AlkB/2OG is magenta, and the oxygen atom is red, respectively. Mn2+ is shown as a sphere and colored orange. Rhein and 2OG are shown as sticks. B, an m|Fo| − D|Fc| map was calculated within the PHENIX program suite after omission of rhein from the complex model and subsequent simulated annealing. The map density is contoured to 3.0 σ. The coordination of Mn2+ by ligands and hydrogen bonding are denoted by dotted dark lines. The map is shown in blue. The superimposition of rhein and 2OG is presented. C, structural superimposition of AlkB-rhein and FTO-rhein complexes performed in PyMOL. The FTO/rhein (PDB code 4IE7) is colored orange. Rhein is shown as sticks. D, zoomed-in view token from C to show the pocket for rhein binding to AlkB and FTO, respectively. Rhein could not bind to FTO similarly to AlkB because of the steric clashes by Tyr295 and Met297 (left panel). A likely binding pocket is observed in AlkB for rhein binding similarly in FTO (right panel).
Rhein is a competitive inhibitor of both AlkB and FTO (31); however, the mode through which rhein binds to AlkB is different from the mode through which the inhibitor binds to FTO (Fig. 5C). Rhein mainly occupies the 2OG-binding site to bind to AlkB (Fig. 5B). In contrast, rhein fully occupies the methylated DNA binding site in FTO (49). The structural superimposition of FTO and the complex of rhein bound to AlkB clearly shows that rhein would not bind to the 2OG pocket of FTO because of steric clashes to Tyr295 and Met297 (Fig. 5D, left panel). On the other hand, a second site for rhein binding to AlkB appears likely in the structural alignment of AlkB to the FTO-rhein complex, which is similar to that observed in FTO-rhein complex (Fig. 5D, right panel). However, the functional relevance of this putative binding site of rhein in AlkB needs to be investigated.
Rhein Inhibition of ALKBH2 and ALKBH3 Sensitizes U87 Cells to MMS
The AlkB human homologues ALKBH2 and ALKBH3 are crucial to maintaining the integrity of the genome (55, 56). In glioblastoma multiforme, knockdown of ALKBH2 reduces alkylation resistance to MMS (57). Silence of the ALKBH3 sensitizes the prostate cancer cells to MMS threats (15). We wondered whether the inhibitor of ALKBH repair would similarly reduce the methylation resistance of mammalian cells. We confirmed the inhibitory activity of rhein toward ALKBH2 and ALKBH3 demethylation by using the DpnII digestion assay (Fig. 6A). Then we performed HPLC-based assay to quantitatively measure the inhibitory activity of rhein. The IC50 is 9.1 and 5.3 μm for the inhibition of ALKBH2 repair of m1A in dsDNA and ALKBH3 repair of m1A in ssDNA, respectively, assayed at 50 μm 2OG (Fig. 6B). Rhein promotes the thermal stability of ALKBH2 and ALKBH3 by enhancing Tm values in a concentration-dependent manner (Fig. 6C), which indicates that rhein also binds to ALKBH2 and ALKBH3 enzymes. Next, we evaluated whether rhein sensitizes U87, a glioblastoma multiforme cell line, to SN2-alkylating agents. As demonstrated in the MTT assay, the proliferation of U87 cells was minimally affected by either 80 μm rhein or 500 μm MMS after 48 h (Fig. 6D). Strikingly, rhein significantly sensitized U87 cells to MMS in a dose-dependent manner. The combined treatment of 80 μm rhein with 250 or 500 μm MMS caused a severe struggle for the U87 cells to survive, indicating that rhein and MMS inhibit U87 cell proliferation in a synergistic way.
FIGURE 6.

Rhein inhibits ALKBH2 and ALKBH3 in vitro and sensitizes U87 cells to MMS. A, DpnII digestion assay to show rhein inhibits ALKBH2 and ALKBH3 repair of m1A in 39 nucleotide dsDNA and ssDNA, respectively. The 2OG concentration is 50 μm. B, quantitative determination of rhein inhibition of ALKBH repair using HPLC-based assay. The IC50 is fitted at 9.1 μm for ALKBH2 repair of dsDNA and 5.3 μm for ALKBH3 repair of ssDNA, respectively. This is assayed at 50 μm 2OG. The error bars are means ± S.E. (n = 3). C, Tm shifts of ALKBH2 and ALKBH3 by rhein. D, assessment of the growth of U87 cells in the presence of rhein (left panel) and under the combined treatment of MMS and rhein (right panel) using MTT assay. All the t tests were carried out between combination-treated groups and those adding MMS alone. E, the expression of ALKBH2 and ALKBH3 were silenced in U87 cells. Rhein sensitization of the proliferation of U87 cells to MMS is dependent on ALKBH2 and ALKBH3 enzymes. F, rhein is inactive to sensitizing U87 cells to MNNG or TMZ by MTT assay. G, Western blot analyses to monitor the amount of H3K9me3 in the presence of rhein and MMS (upper panel) and under the treatment of JIB-04 (lower panel). The error bars are means ± S.E. (n = 6). *, p < 0.05; **, p < 0.01; ***, p < 0.001. All tests were performed in triplicate. ns, not significant; nt, nucleotide.
Target Selectivity of Rhein in U87 Cells
To investigate whether the rhein-dependent sensitization of U87 cells to the MMS threat requires the expression and oxidative demethylation of cellular ALKBH2 and ALKBH3, we silenced both ALKBH2 and ALKBH3 by RNAi (Fig. 6E). After 24 h of exposure to MMS, rhein showed enhanced cytotoxicity to U87 cells in the siRNA control group. Of note, knockdown of ALKBH2 and ALKBH3 removed the synergistic effect of MMS and rhein. In addition, rhein and the SN1-alkylating agents such as MNNG and TMZ, a common chemotherapy drug, failed to produce a synergistic effect on the proliferation of U87 cells (Fig. 6F). These data indicate the rhein-dependent sensitization of human cells to MMS alkylation damage, which is a result of the inhibition of ALKBH2- and ALKBH3 repair of m1A and m3C lesions by rhein.
To further address target selectivity in vivo, we tested whether rhein inhibits other demethylation dioxygenases. Rhein is inactive for the inhibition of prolyl-4-hydroxylase in the in vitro screens, indicating that rhein is not a broad spectrum inhibitor of the 2OG-dependent hydroxylases (58). Rhein could moderately inhibit JMJD2A and JMJD2E in vitro, two iron(II) and 2OG-dependent histone demethylases. We monitored the abundance of trimethylated H3K9me3 that is the downstream target of JMJD2A and JMJD2E, when U87 cell is treated with rhein or MMS. Rhein or MMS or the combination of rhein and MMS did not alter the cellular abundance of H3K9me3, indicating that rhein fails to inhibit JMJD2A or JMJD2E at the tested concentration (Fig. 6G, top panel). In the control experiment, we observed that JIB-04, a known selective inhibitor of JMJD family enzymes (59), increased the abundance of H3K9me3 in a concentration-dependent manner (Fig. 6G, bottom panel). These data indicate that rhein-dependent sensitization of human cells to MMS alkylation damage is due to ALKBH2 and ALKBH3 inhibition rather than perturbation of other dioxygenase-dependent cellular pathways.
Discussion
DNA-methylating agents constantly damage nucleic acids in cells, frequently with mutagenic and/or cytotoxic consequences. The accumulation of m1A and m3C lesions is generally very cytotoxic and even leads to cell death. In E. coli, AlkB repair of m1A and m3C appears to be the major natural defense mechanism in protecting bacteria survival on methylation threats. To our knowledge, several inhibitors of AlkB repair enzymes have been developed, but the cellular tractability by small molecules has not been explored (27, 28, 60). The validation of biological targets and profiling of cellular pathway remains a major challenge for those inhibitors.
We have carried out a number of assays to investigate the mechanism by which rhein inhibition of E. coli AlkB sensitizes bacterial cells to MMS methylation. Rhein efficiently inhibits AlkB without inhibiting the glycosylase AlkA or the methyltransferase Ada in vitro (Fig. 1). Rhein promotes E. coli death during continuous MMS exposure, which could be a result of rhein inhibition of AlkB repair of the major methyl lesion, such as m3C (Fig. 2). Indeed, we detected an increased amount of m3C lesion in genomic DNA, which could be responsible for bacterial death. Rhein binds to AlkB enzyme and promotes its stability in vitro. Structural features observed within the AlkB-rhein complex have further demonstrated the inhibition mechanism of rhein, which is consistent with the enzyme kinetics result that rhein is a competitive inhibitor of AlkB (Figs. 4 and 5). Taken together, we conclude that rhein inhibition of AlkB repair sensitizes E. coli to MMS-induced methylation damage by increasing the accumulation of cellular m3C lesions.
The cellular target engagement of rhein has been adequately addressed (Fig. 3). Either overexpression of AlkB or deletion of AlkB demethylation makes E. coli resistant to rhein during MMS exposure, suggesting that the inhibitory activity of rhein is dependent on the abundance and the function of the cellular AlkB enzyme. Furthermore, rhein fails to sensitize E. coli to MNNG, a SN1-alkylating agent, which indicates that rhein specifically inhibits AlkB-catalyzed repair in E. coli. Rhein increases the thermal stability of AlkB in vivo, thus revealing its ability for binding to the AlkB enzyme. All these data have pointed toward a direct inhibition of AlkB by rhein in vivo.
Human oxidative repair enzymes have been broadly linked to cancer. DNA damage created by alkylation agents would lead to apoptosis and is one of the major mechanisms of cytotoxic anticancer drugs such as TMZ (61, 62). We have demonstrated that rhein inhibits ALKBH2 and ALKBH3 in vitro and synergistically suppresses the proliferation of U87 cells during exposure to MMS (Fig. 6). Strikingly, this synergistic effect vanished in the absence of ALKBH2 and ALKBH3 enzymes. In addition, rhein enhances the sensitivity of U87 cells to an SN2 agent but not an SN1 agent, which also provides strong evidence that rhein is specifically targeting ALKBH-catalyzed DNA repair. Together, rhein could efficiently target the ALKBH-catalyzed repair, thus acting to sensitize mammalian cells to MMS threats.
In summary, we have characterized that the natural product rhein inhibits AlkB repair enzymes (AlkB, ALKBH2, and ALKBH3) in vitro and decreases cellular resistance to MMS. All data of the mechanism investigations point to rhein as the direct inhibitor of AlkB repair enzymes. Our proof of principle study supports the findings that ALKBH2 and ALKBH3 enzymes would be effective pharmacological targets to overcome tumor resistance to methylating anticancer drugs.
Author Contributions
C.-G. Y. designed the project and wrote the paper with Q. L., and Q. L. performed most of the experiments. Y. H. provided technical assistance in protein purification and biochemical experiments. X. L. and H. C. assisted in the isothermal titration calorimetry experiment. J. G. processed the x-ray data and solved the AlkB-rhein structure. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. H. Krokan (Norwegian University of Science and Technology) for gifts of E. coli AB1157 strains, all beamline staff at the 17U of Shanghai Synchrotron Radiation Facility, and S. F. Reichard for editing the manuscript.
This work was supported by National Natural Science Foundation of China Grants 21372237 and 91313303, National Basic Research Program Grant 2015CB910603, and National Science and Technology Major Project “Key New Drug Creation and Manufacturing Program” Grant 2014ZX09507009-01. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 4RFR) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- O6mG
- O6-methylguanine
- m1A
- N1-methyladenine
- m3C
- N3-methylcytosine
- MNNG
- N-methyl-N′-nitro-N-nitrosoguanidine
- MMS
- methyl methane sulfonate
- TMZ
- temozolomide
- ssDNA
- single-stranded DNA
- 2OG
- 2-oxoglutarate
- BA
- bromaminic acid
- CETSA
- cellular thermal shift assay
- C-Ada
- C-terminal domain of Ada
- PDB
- Protein Data Bank.
References
- 1. Sedgwick B. (2004) Repairing DNA-methylation damage. Nat. Rev. Mol. Cell Biol. 5, 148–157 [DOI] [PubMed] [Google Scholar]
- 2. Rose N. R., McDonough M. A., King O. N., Kawamura A., and Schofield C. J. (2011) Inhibition of 2-oxoglutarate dependent oxygenases. Chem. Soc. Rev. 40, 4364–4397 [DOI] [PubMed] [Google Scholar]
- 3. Zhang Y., Chen F. X., Mehta P., and Gold B. (1993) Groove- and sequence-selective alkylation of DNA by sulfonate esters tethered to lexitropsins. Biochemistry 32, 7954–7965 [DOI] [PubMed] [Google Scholar]
- 4. Loechler E. L., Green C. L., and Essigmann J. M. (1984) In vivo mutagenesis by O6-methylguanine built into a unique site in a viral genome. Proc. Natl. Acad. Sci. U.S.A. 81, 6271–6275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shah D., and Gold B. (2003) Evidence in Escherichia coli that N3-methyladenine lesions and cytotoxicity induced by a minor groove binding methyl sulfonate ester can be modulated in vivo by netropsin. Biochemistry 42, 12610–12616 [DOI] [PubMed] [Google Scholar]
- 6. van den Born E., Bekkelund A., Moen M. N., Omelchenko M. V., Klungland A., and Falnes P. Ø. (2009) Bioinformatics and functional analysis define four distinct groups of AlkB DNA-dioxygenases in bacteria. Nucleic Acids Res. 37, 7124–7136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dietlein F., Thelen L., and Reinhardt H. C. (2014) Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet. 30, 326–339 [DOI] [PubMed] [Google Scholar]
- 8. Mishina Y., Duguid E. M., and He C. (2006) Direct reversal of DNA alkylation damage. Chem. Rev. 106, 215–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sedgwick B., Bates P. A., Paik J., Jacobs S. C., and Lindahl T. (2007) Repair of alkylated DNA: recent advances. DNA Repair 6, 429–442 [DOI] [PubMed] [Google Scholar]
- 10. Falnes P. Ø., Johansen R. F., and Seeberg E. (2002) AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature 419, 178–182 [DOI] [PubMed] [Google Scholar]
- 11. Trewick S. C., Henshaw T. F., Hausinger R. P., Lindahl T., and Sedgwick B. (2002) Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature 419, 174–178 [DOI] [PubMed] [Google Scholar]
- 12. Aas P. A., Otterlei M., Falnes P. O., Vågbø C. B., Skorpen F., Akbari M., Sundheim O., Bjørås M., Slupphaug G., Seeberg E., and Krokan H. E. (2003) Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature 421, 859–863 [DOI] [PubMed] [Google Scholar]
- 13. Sikora A., Mielecki D., Chojnacka A., Nieminuszczy J., Wrzesinski M., and Grzesiuk E. (2010) Lethal and mutagenic properties of MMS-generated DNA lesions in Escherichia coli cells deficient in BER and AlkB-directed DNA repair. Mutagenesis 25, 139–147 [DOI] [PubMed] [Google Scholar]
- 14. Ringvoll J., Nordstrand L. M., Vågbø C. B., Talstad V., Reite K., Aas P. A., Lauritzen K. H., Liabakk N. B., Bjørk A., Doughty R. W., Falnes P. Ø., Krokan H. E., and Klungland A. (2006) Repair deficient mice reveal mABH2 as the primary oxidative demethylase for repairing 1meA and 3meC lesions in DNA. EMBO J. 25, 2189–2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dango S., Mosammaparast N., Sowa M. E., Xiong L. J., Wu F., Park K., Rubin M., Gygi S., Harper J. W., and Shi Y. (2011) DNA unwinding by ASCC3 helicase is coupled to ALKBH3-dependent DNA alkylation repair and cancer cell proliferation. Mol. Cell 44, 373–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liefke R., Windhof-Jaidhauser I. M., Gaedcke J., Salinas-Riester G., Wu F., Ghadimi M., and Dango S. (2015) The oxidative demethylase ALKBH3 marks hyperactive gene promoters in human cancer cells. Genome Med. 7, 66–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vågbø C. B., Svaasand E. K., Aas P. A., and Krokan H. E. (2013) Methylation damage to RNA induced in vivo in Escherichia coli is repaired by endogenous AlkB as part of the adaptive response. DNA Repair 12, 188–195 [DOI] [PubMed] [Google Scholar]
- 18. Ougland R., Zhang C. M., Liiv A., Johansen R. F., Seeberg E., Hou Y. M., Remme J., and Falnes P. Ø. (2004) AlkB restores the biological function of mRNA and tRNA inactivated by chemical methylation. Mol. Cell 16, 107–116 [DOI] [PubMed] [Google Scholar]
- 19. Li D., Fedeles B. I., Shrivastav N., Delaney J. C., Yang X., Wong C., Drennan C. L., and Essigmann J. M. (2013) Removal of N-alkyl modifications from N2-alkylguanine and N4-alkylcytosine in DNA by the adaptive response protein AlkB. Chem. Res. Toxicol. 26, 1182–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cetica V., Genitori L., Giunti L., Sanzo M., Bernini G., Massimino M., and Sardi I. (2009) Pediatric brain tumors: mutations of two dioxygenases (hABH2 and hABH3) that directly repair alkylation damage. J. Neurooncol. 94, 195–201 [DOI] [PubMed] [Google Scholar]
- 21. Konishi N., Nakamura M., Ishida E., Shimada K., Mitsui E., Yoshikawa R., Yamamoto H., and Tsujikawa K. (2005) High expression of a new marker PCA-1 in human prostate carcinoma. Clin. Cancer Res. 11, 5090–5097 [DOI] [PubMed] [Google Scholar]
- 22. Shimada K., Nakamura M., Ishida E., Higuchi T., Yamamoto H., Tsujikawa K., and Konishi N. (2008) Prostate cancer antigen-1 contributes to cell survival and invasion though discoidin receptor 1 in human prostate cancer. Cancer Sci. 99, 39–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yu B., Edstrom W. C., Benach J., Hamuro Y., Weber P. C., Gibney B. R., and Hunt J. F. (2006) Crystal structures of catalytic complexes of the oxidative DNA/RNA repair enzyme AlkB. Nature 439, 879–884 [DOI] [PubMed] [Google Scholar]
- 24. Yang C. G., Yi C., Duguid E. M., Sullivan C. T., Jian X., Rice P. A., and He C. (2008) Crystal structures of DNA/RNA repair enzymes AlkB and ABH2 bound to dsDNA. Nature 452, 961–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sundheim O., Vågbø C. B., Bjørås M., Sousa M. M., Talstad V., Aas P. A., Drabløs F., Krokan H. E., Tainer J. A., and Slupphaug G. (2006) Human ABH3 structure and key residues for oxidative demethylation to reverse DNA/RNA damage. EMBO J. 25, 3389–3397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yi C., Chen B., Qi B., Zhang W., Jia G., Zhang L., Li C. J., Dinner A. R., Yang C. G., and He C. (2012) Duplex interrogation by a direct DNA repair protein in search of base damage. Nat. Struct. Mol. Biol. 19, 671–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Welford R. W., Schlemminger I., McNeill L. A., Hewitson K. S., and Schofield C. J. (2003) The selectivity and inhibition of AlkB. J. Biol. Chem. 278, 10157–10161 [DOI] [PubMed] [Google Scholar]
- 28. Woon E. C., Demetriades M., Bagg E. A., Aik W., Krylova S. M., Ma J. H., Chan M., Walport L. J., Wegman D. W., Dack K. N., McDonough M. A., Krylov S. N., and Schofield C. J. (2012) Dynamic combinatorial mass spectrometry leads to inhibitors of a 2-oxoglutarate-dependent nucleic acid demethylase. J. Med. Chem. 55, 2173–2184 [DOI] [PubMed] [Google Scholar]
- 29. Nakao S., Mabuchi M., Shimizu T., Itoh Y., Takeuchi Y., Ueda M., Mizuno H., Shigi N., Ohshio I., Jinguji K., Ueda Y., Yamamoto M., Furukawa T., Aoki S., Tsujikawa K., and Tanaka A. (2014) Design and synthesis of prostate cancer antigen-1 (PCA-1/ALKBH3) inhibitors as anti-prostate cancer drugs. Bioorg. Med. Chem. Lett. 24, 1071–1074 [DOI] [PubMed] [Google Scholar]
- 30. Bunnage M. E., Chekler E. L., and Jones L. H. (2013) Target validation using chemical probes. Nat. Chem. Biol. 9, 195–199 [DOI] [PubMed] [Google Scholar]
- 31. Chen B., Ye F., Yu L., Jia G., Huang X., Zhang X., Peng S., Chen K., Wang M., Gong S., Zhang R., Yin J., Li H., Yang Y., Liu H., et al. (2012) Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J. Am. Chem. Soc. 134, 17963–17971 [DOI] [PubMed] [Google Scholar]
- 32. Wang T., Hong T., Huang Y., Su H., Wu F., Chen Y., Wei L., Huang W., Hua X., Xia Y., Xu J., Gan J., Yuan B., Feng Y., Zhang X., Yang C. G., and Zhou X. (2015) Fluorescein derivatives as bifunctional molecules for the simultaneous inhibiting and labeling of FTO protein. J. Am. Chem. Soc. 137, 13736–13739 [DOI] [PubMed] [Google Scholar]
- 33. Mielecki D., Sikora A., Wrzesiński M., Nieminuszczy J., Detman A., Zuchniewicz K., Gromadka R., and Grzesiuk E. (2016) Evaluation of the Escherichia coli HK82 and BS87 strains as tools for AlkB studies. DNA Repair 39, 34–40 [DOI] [PubMed] [Google Scholar]
- 34. Zhu C., and Yi C. (2014) Switching demethylation activities between AlkB family RNA/DNA demethylases through exchange of active-site residues. Angew. Chem. Int. Ed. Engl. 53, 3659–3662 [DOI] [PubMed] [Google Scholar]
- 35. Chen B., Gan J., and Yang C. (2013) The complex structures of ALKBH2 mutants cross-linked to dsDNA reveal the conformational swing of β-hairpin. Sci. China Chem. 57, 307–313 [Google Scholar]
- 36. Chen B., Liu H., Sun X., and Yang C. G. (2010) Mechanistic insight into the recognition of single-stranded and double-stranded DNA substrates by ABH2 and ABH3. Mol. Biosyst. 6, 2143–2149 [DOI] [PubMed] [Google Scholar]
- 37. Verdemato P. E., Brannigan J. A., Damblon C., Zuccotto F., Moody P. C., and Lian L. Y. (2000) DNA-binding mechanism of the Escherichia coli Ada O6-alkylguanine-DNA alkyltransferase. Nucleic Acids Res. 28, 3710–3718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Labahn J., Schärer O. D., Long A., Ezaz-Nikpay K., Verdine G. L., and Ellenberger T. E. (1996) Structural basis for the excision repair of alkylation-damaged DNA. Cell 86, 321–329 [DOI] [PubMed] [Google Scholar]
- 39. Martinez Molina D., Jafari R., Ignatushchenko M., Seki T., Larsson E. A., Dan C., Sreekumar L., Cao Y., and Nordlund P. (2013) Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341, 84–87 [DOI] [PubMed] [Google Scholar]
- 40. Jafari R., Almqvist H., Axelsson H., Ignatushchenko M., Lundbäck T., Nordlund P., and Martinez Molina D. (2014) The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 9, 2100–2122 [DOI] [PubMed] [Google Scholar]
- 41. Bleijlevens B., Shivarattan T., Flashman E., Yang Y., Simpson P. J., Koivisto P., Sedgwick B., Schofield C. J., and Matthews S. J. (2008) Dynamic states of the DNA repair enzyme AlkB regulate product release. EMBO Rep. 9, 872–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vedadi M., Niesen F. H., Allali-Hassani A., Fedorov O. Y., Finerty P. J. Jr., Wasney G. A., Yeung R., Arrowsmith C., Ball L. J., Berglund H., Hui R., Marsden B. D., Nordlund P., Sundstrom M., Weigelt J., and Edwards A. M. (2006) Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc. Natl. Acad. Sci. U.S.A. 103, 15835–15840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Niesen F. H., Berglund H., and Vedadi M. (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221 [DOI] [PubMed] [Google Scholar]
- 44. Otwinowski Z., and Minor W. (1997) Processing of X-ray Diffraction Data Collected in Oscillation Mode, pp. 307–326, Elsevier Science Publishing Co., Inc., New York: [DOI] [PubMed] [Google Scholar]
- 45. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 46. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 47. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 48. Huang Y., Yan J., Li Q., Li J., Gong S., Zhou H., Gan J., Jiang H., Jia G. F., Luo C., and Yang C. G. (2015) Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 43, 373–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Aik W., Demetriades M., Hamdan M. K., Bagg E. A., Yeoh K. K., Lejeune C., Zhang Z., McDonough M. A., and Schofield C. J. (2013) Structural basis for inhibition of the fat mass and obesity associated protein (FTO). J. Med. Chem. 56, 3680–3688 [DOI] [PubMed] [Google Scholar]
- 50. Mitra S. (2007) MGMT: a personal perspective. DNA Repair 6, 1064–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. O'Brien P. J., and Ellenberger T. (2004) The Escherichia coli 3-methyladenine DNA glycosylase AlkA has a remarkably versatile active site. J. Biol. Chem. 279, 26876–26884 [DOI] [PubMed] [Google Scholar]
- 52. Li P., Gao S., Wang L., Yu F., Li J., Wang C., Li J., and Wong J. (2013) ABH2 couples regulation of ribosomal DNA transcription with DNA alkylation repair. Cell Rep. 4, 817–829 [DOI] [PubMed] [Google Scholar]
- 53. Dinglay S., Trewick S. C., Lindahl T., and Sedgwick B. (2000) Defective processing of methylated single-stranded DNA by E. coli AlkB mutants. Genes Dev. 14, 2097–2105 [PMC free article] [PubMed] [Google Scholar]
- 54. Day R. S. 3rd, Babich M. A., Yarosh D. B., and Scudiero D. A. (1987) The role of O6-methylguanine in human cell killing, sister chromatid exchange induction and mutagenesis: a review. J. Cell Sci. Suppl. 6, 333–353 [DOI] [PubMed] [Google Scholar]
- 55. Zhao Y., Majid M. C., Soll J. M., Brickner J. R., Dango S., and Mosammaparast N. (2015) Noncanonical regulation of alkylation damage resistance by the OTUD4 deubiquitinase. EMBO J. 34, 1687–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang P., Wu J., Ma S., Zhang L., Yao J., Hoadley K. A., Wilkerson M. D., Perou C. M., Guan K. L., Ye D., and Xiong Y. (2015) Oncometabolite d-2-Hydroxyglutarate inhibits ALKBH DNA repair enzymes and sensitizes IDH mutant cells to alkylating agents. Cell Rep. 13, 2353–2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Johannessen T. C., Prestegarden L., Grudic A., Hegi M. E., Tysnes B. B., and Bjerkvig R. (2013) The DNA repair protein ALKBH2 mediates temozolomide resistance in human glioblastoma cells. Neuro Oncol. 15, 269–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhou Y. X., Xia W., Yue W., Peng C., Rahman K., and Zhang H. (2015) Rhein: a review of pharmacological activities. Evid. Based Complement. Alternat. Med. 2015, 578107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang L., Chang J., Varghese D., Dellinger M., Kumar S., Best A. M., Ruiz J., Bruick R., Peña-Llopis S., Xu J., Babinski D. J., Frantz D. E., Brekken R. A., Quinn A. M., Simeonov A., Easmon J., and Martinez E. D. (2013) A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat. Commun. 4, 2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hopkinson R. J., Tumber A., Yapp C., Chowdhury R., Aik W., Che K. H., Li X. S., Kristensen J. B., King O. N., Chan M. C., Yeoh K. K., Choi H., Walport L. J., Thinnes C. C., Bush J. T., et al. (2013) 5-Carboxy-8-hydroxyquinoline is a broad spectrum 2-oxoglutarate oxygenase inhibitor which causes iron translocation. Chem. Sci. 4, 3110–3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Drabløs F., Feyzi E., Aas P. A., Vaagbø C. B., Kavli B., Bratlie M. S., Peña-Diaz J., Otterlei M., Slupphaug G., and Krokan H. E. (2004) Alkylation damage in DNA and RNA-repair mechanisms and medical significance. DNA Repair 3, 1389–1407 [DOI] [PubMed] [Google Scholar]
- 62. Srinivasan A., and Gold B. (2012) Small-molecule inhibitors of DNA damage repair pathways: an approach to overcome tumor resistance to alkylating anticancer drugs. Future Med. Chem. 4, 1093–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]