

Abstract
The C1 domain represents the recognition module for diacylglycerol and phorbol esters in protein kinase C, Ras guanine nucleotide releasing protein (RasGRP), and related proteins. RasGRP2 is exceptional in that its C1 domain has very weak binding affinity (Kd = 2890 ± 240 nm for [3H]phorbol 12,13-dibutyrate. We have identified four amino acid residues responsible for this lack of sensitivity. Replacing Asn7, Ser8, Ala19, and Ile21 with the corresponding residues from RasGRP1/3 (Thr7, Tyr8, Gly19, and Leu21, respectively) conferred potent binding affinity (Kd = 1.47 ± 0.03 nm) in vitro and membrane translocation in response to phorbol 12-myristate 13-acetate in LNCaP cells. Mutant C1 domains incorporating one to three of the four residues showed intermediate behavior with S8Y making the greatest contribution. Binding activity for diacylglycerol was restored in parallel. The requirement for anionic phospholipid for [3H]phorbol 12,13-dibutyrate binding was determined; it decreased in going from the single S8Y mutant to the quadruple mutant. The full-length RasGRP2 protein with the mutated C1 domains also showed strong phorbol ester binding, albeit modestly weaker than that of the C1 domain alone (Kd = 8.2 ± 1.1 nm for the full-length protein containing all four mutations), and displayed translocation in response to phorbol ester. RasGRP2 is a guanyl exchange factor for Rap1. Consistent with the ability of phorbol ester to induce translocation of the full-length RasGRP2 with the mutated C1 domain, phorbol ester enhanced the ability of the mutated RasGRP2 to activate Rap1. Modeling confirmed that the four mutations helped the binding cleft maintain a stable conformation.
Keywords: cloning, guanine nucleotide exchange factor (GEF), phorbol ester, protein kinase C (PKC), Ras-related protein 1 (Rap1), C1 domain, RasGRP2
Introduction
Diacylglycerol (DAG),3 a lipid second messenger that is important in the transduction of signals from both receptor tyrosine kinases and G-protein-coupled receptors, is a product of the hydrolysis of phosphatidylinositol 4,5-bisphosphate by phospholipase C γ and β and is also indirectly generated from phosphatidylcholine via phospholipase D. Recognition of DAG is mediated by its binding to C1 domains, which are regulatory modules for protein kinase C (PKC), guanyl exchange factors (Ras guanine nucleotide releasing proteins (RasGRPs)), and other targets such as the chimaerins, munc-13, and some DAG kinase isoforms (1–3). The binding of DAG to the C1 domains translocates the target proteins to membranes, bringing these proteins into proximity with signaling complexes, substrates, and regulators (4). In addition, binding of DAG may lead to conformational change of the protein, as is the case with PKC, leading to enzyme activation.
C1 domains are small units of ∼50 amino acids that were originally discovered as the DAG binding modules in PKCs. Subsequent characterization revealed that C1 domains are functionally heterogeneous. Those that bound DAG and its ultrapotent analogs, the phorbol esters, were termed “typical.” C1 domains that possessed varying levels of sequence and structural homology but failed to bind were termed “atypical” (1). Examples of atypical C1 domains include those of PKCζ/ι, Vav, and Raf. The atypical C1 domains can be further subdivided depending on whether the geometry of the DAG/phorbol ester binding cleft is retained or lost. In the case of PKCζ/ι, lack of binding can be attributed to a ring of positively charged residues around the rim of the binding cleft (5). For Vav, a similar series of negatively charged residues is responsible for the lack of binding (6). For Raf-1, in contrast, deletion in one of the two loops of the binding cleft destroys its geometry (7).
Among RasGRP family members, RasGRP1, RasGRP3, and RasGRP4(α) have typical C1 domains (8, 9). Phorbol esters and DAG induce their translocation in intact cells and their binding to membrane vesicles in vitro (10). Direct [3H]PDBu binding to RasGRP1, RasGRP3, and RasGRP4 yielded Kd values of 0.58 (11), 1.5 (12), and 1.1 nm (13), respectively. In contrast, the C1 domain of RasGRP2 stands out as being atypical, neither evidencing [3H]PDBu binding (13) nor DAG/phorbol ester-driven membrane association, at least in vitro (10). Multiple methodological approaches including NMR, fluorescence, and molecular dynamics (14–19) as well as monolayer penetration (20) and vesicle binding (21) have shown that the hydrophobic and aromatic residues surrounding the DAG-binding pocket penetrate the membrane. Functional studies (22, 23) and comparisons with the sequences of other typical and atypical C1 domains have highlighted possibly significant differences between the C1 domain of RasGRP2 and typical C1 domains (1). An initial effort, however, using chemically synthesized variant C1 domains of RasGRP2 (S8Y, V12T/A13F, and R15D/K18A) failed to identify the residues responsible for the lack of PDBu binding ability (13).
RasGRP2 is an exchange factor for membrane-bound Ras or Rap GTPases. In vivo, RasGRP2 selectively catalyzes nucleotide exchange on N- and K-Ras but not Ha-Ras. RasGRP2 also catalyzes nucleotide exchange on Rap1 (24, 25). It displays a dispersed localization in the cytoplasm with occasional presence in membrane ruffles. Its membrane association can be induced by F-actin in response to regulators such as Vav and Rac1 (25). Conversely, its localization did not change when cells were treated with phorbol esters (10, 26). RasGRP2 has an important role in lymphocytes and neutrophils (27). For example, it plays a critical role in neutrophil chemotaxis in vitro (28) and recruitment of neutrophils into the inflamed peritoneal cavity in vivo (29). The most prominent known biological role of RasGRP2 is in platelet signaling (27, 30), and mutation in the RasGRP2 gene causes severe bleeding (31). RasGRP2 was also identified as a proto-oncogene in acute myeloid leukemia (32) and is elevated in trisomy12-associated chronic lymphocytic leukemia (33) where it is thought to contribute to the enhanced integrin signaling associated with drug resistance. In PC12D cells, RasGRP2 signals between the muscarinic acetylcholine receptor and Erk1/2 activation via B-Raf (34).
Here we describe that the C1 domain of RasGRP2 actually does bind phorbol ester although with very weak affinity. We then identify four residues in the C1 domain of RasGRP2 that, when replaced with the corresponding residues from RasGRP1, confer potent phorbol ester binding activity. We explore how these residues influence the behavior both of the isolated RasGRP2 C1 domain and the intact protein for membrane interaction and ligand-induced translocation. We demonstrate that the phorbol esters induce enhanced guanyl exchange activity for Rap1 in the presence of mutant RasGRP2 incorporating these residues. Finally, we use molecular dynamics simulation to show that the mutations stabilize the binding conformation of the C1 domain.
Experimental Procedures
Materials
[3H]Phorbol 12, 13-dibutyrate ([3H]PDBu) (13.5 Ci/mmol) was obtained from PerkinElmer Life Sciences. PDBu and phorbol 12-myristate 13-acetate (PMA) were purchased from LC Laboratories (Woburn, MA). Phosphatidyl-l-serine (PS), phosphatidylcholine, and 1,2-dioctanoylglycerol (DOG) were from Avanti Polar Lipids (Alabaster, AL). Dimethyl sulfoxide was purchased from Sigma-Aldrich. LNCaP human prostate cancer cells, human embryonic kidney 293 (HEK293) cells, fetal bovine serum (FBS), RPMI 1640 medium, l-glutamine, and Eagle's minimum essential medium were from the American Type Culture Collection (Manassas, VA). Reagents used for culturing bacteria (LB broth, LB agar plates with different selection of antibiotics, etc.) were from K-D Medical, Inc. (Columbia, MD). The oligonucleotide primers used for polymerase chain reaction (PCR) and for site-directed mutagenesis were obtained from Invitrogen. Rap1 activation assays were performed by using the Rap1 Activation Assay kit from Cell Biolabs, Inc. (San Diego, CA).
Construction of GFP-fused Full-length RasGRP2 and C1 Domains of RasGRP2
The full-length RasGRP2 cDNA (NCBI accession number NM_153819.1) was amplified by PCR using specific primers (forward primer, 5′-AATaagcttGCAGGCACCCTGGACCTGGAC-3′; reverse primer, 3′-AATggatccTTACAAGTGGATGTCAAACACCC-5′; HindIII and BamHI sites, indicated by lowercase letters, were incorporated to facilitate cloning) and subcloned into a pEGFP-C3 plasmid (BD Biosciences Clontech), generating pEGFP-C3-RasGRP2 with an N-terminal GFP tag. The full-length cDNA clone of RasGRP2 served as a template to generate the recombinant C1 domain. The C1 domain of RasGRP2 was subcloned into the pEGFP-C3 vector using the BamHI and HindIII sites (forward primer, 5′-CATaagcttCACAACTTCCAGGAGAGCAAC-3′; reverse primer, 3′-AATggatccTCAACACTCAACTGACAGGCG-5′). The DNA fragments of the PCR were purified with the QIAquick PCR purification kit (Qiagen, Inc., Valencia, CA) and afterward digested with HindIII and BamHI (New England Biolabs, Beverly, MA). After an additional step of purification, the fragments were finally ligated into the GFP-containing pEGFP-C3 plasmid using the restriction sites. The integrity of the inserts was verified by DNA sequencing, which was performed by the DNA Minicore (Center for Cancer Research, National Cancer Institute, National Institutes of Health).
Construction of the Glutathione S-Transferase (GST)-tagged C1 Domain of RasGRP2 and RasGRP1
To generate a recombinant RasGRP2/1 C1 domain fused to GST, PCR amplification of the appropriate sequence was performed. The full-length cDNA clone of RasGRP2/1 served as a template. The DNA fragments of the PCR were purified with the QIAquick PCR purification kit and ligated into the GST-containing pGEX-2T plasmid (GE Healthcare) using the BamHI restriction sites.
Site-directed Mutagenesis of the C1 Domain and the Full-length RasGRP2
Point mutations of the amino acid residues were introduced using the GeneTailor® and GeneArt® site-directed mutagenesis system (Invitrogen) according to the manufacturer's instructions. To generate the C1 domain and full-length mutants of RasGRP2, the above mentioned wild-type C1 and full-length constructs in pEGFP-C3 and pGEX-2T were used. Single mutations (S8Y) were introduced in one step, and double (S8Y/A19G), triple (N7T/S8Y/A19G and S8Y/A19G/I21L), and quadruple mutants (N7T/S8Y/A19G/I21L) were generated in a stepwise fashion using single and double mutants as templates. The presence of mutations was verified by DNA sequencing (DNA Minicore).
Construction of Halo-tagged Full-length Wild-type and Mutant RasGRP2
To generate Halo-tagged full-length wild-type and mutant RasGRP2, the cDNA clones of wild-type and mutant pEGFP-C3-RasGRP2 served as templates, and the following oligonucleotide primers were applied in the PCR: forward primer, 5′-AATcgatcgCTGCAGGCACCCTGGACCTG-3′, and reverse primer, 3′-AATtctagaTTACAAGTGGATGTCAAACAC-5′. The DNA fragments were ligated to the N-terminally Halo-tagged pHTN plasmid (Promega, Madison, WI) using the PvuI and XbaI restriction sites. The sequences of the Halo-tagged full-length wild-type and mutant constructs were verified by sequence analysis (DNA Minicore).
Expression in HEK293 Cells and Purification of Halo-tagged Full-length Wild-type and Mutant RasGRP2
For expression of wild-type and mutant RasGRP2, HEK293 cells were seeded on tissue culture dishes (100 mm) and grown in Eagle's minimum essential medium supplemented with 10% FBS. Cells were cultured at 37 °C with a humidified 5% CO2 atmosphere. Forty-eight hours later, cells were transfected with the pHTN-RasGRP2 wild-type, single, double, triple, and quadruple mutant constructs using Lipofectamine Plus reagent (Invitrogen) according to the manufacturer's instructions. The purification was performed 24 h posttransfection. Cells were subjected to sonication in 1× PBS supplemented with 0.005% IGEPAL CA-630 (Sigma) and 1 mm DTT (Thermo Scientific). The expressed Halo-tagged RasGRP2 proteins were purified using the HaloTag Mammalian Protein Purification System according to the manufacturer's instructions. Purification efficiency was evaluated by SDS-PAGE analysis. Purified proteins were stored in 20% glycerol at −80 °C.
Construction of Maltose-binding Protein (MBP)-tagged Full-length RasGRP1
The full-length RasGRP1 cDNA (NCBI accession number NM_005739.3) was amplified by PCR using specific primers (forward primer, 5′-AATcatatgGGCACCCTGGGCAAGGCGAGA-3′; reverse primer, 3′-AATggatccCTAAGAACAGTCACCCTGCTC-5′; NdeI and BamHI sites, indicated by lowercase letters, were incorporated to facilitate cloning) and subcloned into a pMAL-c5x plasmid (New England Biolabs, Beverly, MA), generating pMAL-c5x-RasGRP1 with an N-terminal MBP tag.
Expression in BL21 Cells and Purification of the MBP-tagged Full-length RasGRP1
The full-length RasGRP1 in the pMAL-c5x plasmid was transformed into BL21(DE3) One Shot chemically competent Escherichia coli (Invitrogen). Transformants were grown in LB broth medium (K-D Medical, Inc.) at 37 °C until the optical density of the bacterial suspension reached 0.5–0.6. Expression of the MBP fusion protein was induced with 0.3 mm isopropyl O-d-thiogalactopyranoside (Sigma) for 4 h at 37 °C. The expressed MBP-tagged RasGRP1 protein was purified using the pMALTM Protein Fusion and Purification System according to the manufacturer's instructions. Purification efficiency was evaluated by SDS-PAGE analysis. Purified proteins were stored in 20% glycerol at −80 °C.
Expression in BL21 Cells and Purification of the GST-tagged C1 Domain of Wild-type and Mutant RasGRP2/1
The C1 domains of wild-type and mutant RasGRP2/1 in the pGEX-2T plasmid were transformed into BL21(DE3) One Shot chemically competent E. coli. Transformants were grown in LB broth medium at 37 °C until the optical density of the bacterial suspension reached 0.6–0.8. Expression of the GST fusion proteins was induced with 0.3 mm isopropyl O-d-thiogalactopyranoside for 4 h at 37 °C. Bacterial cells were subjected to B-PER bacterial protein extraction reagent (Pierce). The expressed GST-tagged C1 proteins were purified using a GST Spin Purification kit (Pierce) according to the manufacturer's instructions. Purification efficiency was evaluated by SDS-PAGE analysis. Purified proteins were stored in 20% glycerol at −80 °C.
In Vitro [3H]PDBu Binding Assays
The dissociation constants (Kd values) of [3H]PDBu for the individual C1 domains and for the full-length RasGRP2/1 proteins were measured using the polyethylene glycol precipitation assay developed in our laboratory as described in detail previously (35). In competitive binding assays, we determined the Ki values of a competing ligand (DOG) using the same assay. Triton X-100, included in assays, did not exceed 0.003%. All values represent the mean ± S.E. of at least triplicate independent experiments where all points in each dose-response curve in each experiment were measured in triplicate.
Confocal Analysis of GFP-labeled RasGRP2 Proteins
LNCaP cells were plated at a density of 100,000 cells/plate on Ibidi μ-dishes (Ibidi, LLC, Verona, WI) and cultured at 37 °C in RPMI 1640 medium supplemented with 10% FBS and 2 mm l-glutamine. After 48 h in culture, cells were transfected with GFP-tagged recombinant constructs using Lipofectamine Plus reagent according to the manufacturer's recommendations. After 24 h, the cells were treated as indicated with 1000 nm and 10 μm PMA in confocal medium (Dulbecco's modified Eagle's medium without phenol red supplemented with 1% FBS), and time lapse images were collected every 30 s using the Zeiss AIM software. Imaging was with a Zeiss LSM 510 confocal microscopy system (Carl Zeiss, Inc.) with an Axiovert 100M inverted microscope operating with a 25-milliwatt argon laser tuned to 488 nm. A 63× 1.4 numerical aperture Zeiss Plan-Apochromat oil immersion objective was used together with varying zooms (1.4–2×). The imaging was performed using the resources of the Confocal Microscopy Core Facility, Center for Cancer Research, National Cancer Institute.
Quantification of Confocal Images
Two regions of 4 μm2 each were selected in each cell: one region in the cytoplasm and one in the cell membrane. Mean intensities of the GFP-tagged constructs in the selected regions were calculated using the Zeiss AIM software for the images at the different time points; the ratios of the intensities for membrane/cytoplasm were then calculated and normalized to the values at time 0. An increase in the membrane/cytoplasm ratio indicates translocation.
Rap1 Activation Assay
For Rap1 activation assays, HEK293 cells expressing the indicated RasGRP2 constructs were lysed in 1× assay buffer (Rap1 Activation Assay kit). Lysates were centrifuged at 14,000 × g for 10 min at 4 °C. The resulting supernatants were incubated for 60 min at 4 °C with 40 μl of RalGDS (where GDS is guanine nucleotide dissociation stimulator) Rap binding domain-agarose beads. After incubation, the beads were collected and washed three times with 1× assay buffer. Proteins were then eluted from the beads with Laemmli sample buffer, separated by electrophoresis, and analyzed by immunoblotting using anti-Rap1 antibody (Rap1 Activation Assay kit). The signal was developed by enhanced chemiluminescence (Amersham Biosciences) and imaged on Amersham Biosciences HyperfilmTM ECL (GE Healthcare).
Homology Modeling
The homology model for RasGRP2 was generated using the UniProt and ExPaSy SWISS-MODEL workspace, the web-based tools for automatic homology model generation. The sequence for the RasGRP2 zinc finger domain (residues 498–548) was from UniProt Q7LDG7. The crystal structure of RasGRP1 (Protein Databank code 4L9M) (36) showed 52% sequence identity homology with RasGRP2 and was used as the template. Using the Swiss-PdbViewer (37), residues at positions 7, 8, 19, and 21 of the RasGRP2 model were substituted respectively with the residues Thr, Tyr, Ala, and Ile that correspond to the residues at these positions present in RasGRP1 (Fig. 1A).
FIGURE 1.

Comparison of RasGRP isoforms. A, amino acid sequence alignment of the C1 domains of the RasGRP (human) isoforms. Amino acid residues in the C1 domain of RasGRP2 that were evaluated for their contributions to the weak PDBu binding activity of RasGRP2 are color-coded. Residues in RasGRP2 that, when substituted with the corresponding residues in RasGRP1, enhanced binding are shown in red. Those that failed to do so are shown in green. The C1 consensus sequence is highlighted in yellow. Numbers on the left and right indicate position in the protein sequence; the numbers on top indicate the position within the C1 domain consensus sequence, and the UniProt identification is on the right. B, space-filling models of RasGRP1 (Protein Data Bank code 4L9M) and RasGRP2 (based on homology model), both complexed with phorbol ester. Coloring of the C1 domains portrays the electrostatic potential according to Coulomb's law. Red represents hydrophobicity, blue represents hydrophilicity, and white is in between. The phorbol ester is colored dark gray for the carbon atoms and dark red for the oxygen atoms.
Molecular Docking
Docking of phorbol ester to the binding site of RasGRP2 or its mutated versions was performed using AutoDock4 (version 4.2) (38) with general protocols (39). The three-dimensional chemical structure of phorbol ester was taken from the structural data of the phorbol ester-PKCδ C1b complex (40). The protein data were prepared from the homology model described above. For AutoGrid4, grid volumes were set to cover the entire protein conformation with 0.375-Å spacing. 256 Lamarckian genetic algorithm local searches were performed with docking calculations consisting of a population size of 150 and 250,000 energy evaluations. The rest of the calculation parameters were left on default values for AutoDockTools4. Using the default cluster option in AutoDockTools4, the lowest energy conformation of the highest populated cluster was selected.
Molecular Dynamics Simulations
The molecular dynamics simulations were performed using the GROMACS 4.6.1 package of programs (41) using the GROMOS96 ffG53a6 force field (42). The initial structure used for the simulations was the three-dimensional model of RasGRP2 that was generated by the SWISS-MODEL server (43) as detailed above. The topology of the phorbol ester was generated with the PRODRG server (44). The bonds, angles, and dihedral angles were checked for compatibility with the ffG53a6 force field parameters. The net charge of the phorbol ester moiety was zero. The 100-ns molecular dynamics simulations were carried out starting with the initial structures for phorbol ester docked with the wild-type RasGRP2 or with the quadruple mutant of RasGRP2 (N7T/S8Y/S19G/I21L).
The protein-ligand complex was embedded in a box of simple point charge (SPC) water (45) that extended at least 12 Å beyond the protein to the edge of the box. Na+ and Cl− ions were added to neutralize the charges of the protein and to bring the ionic strength to 100 mm. Prior to the simulations, the structure was relaxed with energy minimization. Following the minimization, an equilibration run was conducted with restraints on the position of the heavy atoms (i.e. the non-hydrogen atoms) of the protein. The equilibration was conducted in two phases. The first phase (a 100-ps run) was conducted under NVT (number of particles, volume, and temperature) conditions, and the second phase (also 100 ps) was conducted under NPT (number of particles, pressure, and temperature) conditions to stabilize the pressure (and thus the density) of the system.
Following the above equilibration run, 100 ns of production molecular dynamics was carried out. The LINCS algorithm (46) was used to constrain the lengths of all bonds; the water molecules were restrained using the SETTLE algorithm (47). The time step for the simulations was 2 fs (0.002 ps). These simulations were conducted under NPT conditions using the Berendsen thermostat and Parrinello-Rahman barostat to keep the temperature and pressure constant (P = 1 bar; τp = 0.5 ps; T = 300 K; τT = 0.1 ps) (48). The van der Waals forces were treated using a 10-Å cutoff. Long range electrostatics were treated using the particle mesh Ewald method (49).
Visualization
The figures were generated with VMD (50) and Chimera (51). Hydrophobicity scale surfaces on the C1 domain of RasGRP1 and RasGRP2 were generated with Chimera.
Results
Four Critical Amino Acid Residues Are Responsible for the Lack of Phorbol Ester Sensitivity
Among RasGRP family members, RasGRP2 has been reported to stand out as not being responsive to phorbol esters or DAG. Comprehensive sequence alignment of the atypical RasGRP2 C1 versus the typical C1 domains of the other RasGRP family members revealed a number of differences in amino acid residues (Fig. 1A). Homology modeling of RasGRP2 and comparison with the structure of RasGRP1 revealed an overall similar structure (the C1 domains docked with phorbol ester are illustrated) (Fig. 1B). Comparison with the sequences of other typical C1 domains (for example, see Refs. 1, 6, and 23) as well as mutational studies exploring structure-function studies for other typical C1 domains provided guidance about which residues might contribute to the lack of phorbol ester binding of the RasGRP2 C1 domain (13, 22).
We used site-directed mutagenesis to prepare mutants of the C1 domain of RasGRP2 (Table 1). The binding properties of the purified recombinant proteins were then characterized by in vitro [3H]PDBu binding. The wild-type RasGRP2 C1 domain in fact proved not to be devoid of phorbol ester binding activity. Rather, if we were attentive to evaluate binding under conditions in which we could detect weak binding affinity, we in fact were able to demonstrate weak [3H]PDBu binding (Kd = 2890 ± 240 nm) (Fig. 2A). Just as the different behavior of the C1 domains of Vav and PKCζ/ι compared with that of Raf illustrated that atypical C1 domains could be further subdivided based on whether they retained the conformation of the phorbol ester/DAG binding cleft, so we conclude that the C1 domain of RasGRP2 represents a further subdivision in which the atypical C1 domain both retains an appropriate binding cleft and maintains a measurable, albeit perhaps not physiological, binding affinity.
TABLE 1.
Binding of [3H]PDBu to wild-type and mutants of RasGRP2 and to wild-type RasGRP1
Individual wild-type and mutant C1 domains were fused with GST, expressed in bacteria, and isolated. Individual full-length and mutant RasGRP2s were fused to HaloTag, and full-length RasGRP1 was fused with MBP. The constructs were expressed in HEK293 cells and bacteria, respectively, and then isolated. The binding affinities of the isolated proteins for [3H]PDBu were measured in the presence of 100 μg/ml PS. Values represent the mean ± S.E. (n = 3 independent experiments). NA, no measurable activity. The C1 domains containing the stepwise mutations that enhanced PDBu binding activity are indicated in bold.
| Receptor | Kd |
|---|---|
| nm | |
| hRasGRP2 C1 | |
| Wild type | 2890 ± 240 |
| S8Y | 50.4 ± 2.6 |
| S8Y/L22W | 207 ± 17 |
| S8Y/V12T | 174 ± 11 |
| S8Y/A13F | 32.8 ± 5.2 |
| S8Y/A19G | 12 ± 1 |
| S8Y/L22W/Y25I | 1230 ± 270 |
| S8Y/A19G/L20F | 84 ± 11 |
| S8Y/A19G/A33D | 100.2 ± 5.5 |
| S8Y/A19G/L29Y | 63 ± 12 |
| N7T/S8Y/A19G | 7.6 ± 1.8 |
| S8Y/A19G/I21L | 7.9 ± 5.5 |
| N7T/S8Y/A19G/I21L | 1.47 ± 0.03 |
| hRasGRP1 C1 | 0.26 ± 0.09 |
| hRasGRP2 full length | |
| Wild type | NA |
| S8Y | NA |
| S8Y/A19G | 63.6 ± 7.0 |
| N7T/S8Y/A19G | 24.5 ± 1.6 |
| S8Y/A19G/I21L | 20.14 ± 0.60 |
| N7T/S8Y/A19G/I21L | 8.2 ± 1.1 |
| hRasGRP1 full length | 0.40 ± 0.31 |
FIGURE 2.

Binding of [3H]PDBu to the wild-type and the quadruple mutant C1 domain of RasGRP2. Binding of [3H]PDBu to the wild-type (A) and the quadruple mutant (B) C1 domain of RasGRP2 was measured in the presence of 100 μg/ml PS. Curves shown are from representative individual experiments. Three independent experiments were performed for each construct. Points represent mean; error bars represent ±S.E. Where error bars are not visible they are within the size of the symbol.
In RasGRP1, the mutation Y8S was shown to abolish membrane translocation (5). The reverse mutation S8Y in the C1 domain of RasGRP2 had a major effect on binding, causing a 57-fold increase in binding affinity (Kd = 50.4 ± 2.6 nm) (Table 1). Although this substitution had previously not been found to restore binding in a chemically synthesized RasGRP2 C1 domain construct (13), a possible explanation was that the chemically synthesized protein might not have been properly folded. Starting with the S8Y mutant, we generated double, triple, and quadruple mutants to explore contributions to binding from other residues. Trp22 has been reported to be important for conferring binding activity, particularly for DAG (52), although it is clear that it makes a lesser contribution to phorbol ester binding at least in the case of the C1b domain of PKCδ (21, 53). In the present instance, L22W modestly diminished the binding affinity of the S8Y mutant (Table 1). It did not enhance it. Substituting Ala with Gly at position 19 (A19G) enhanced the binding affinity for PDBu. This enhancement may reflect the greater flexibility introduced by Gly in the second loop of the binding cleft. Among triple mutants, N7T and I21L individually conferred a modest improvement over the S8Y/A19G double mutant with further enhancement when both were included in the quadruple mutant. Together, replacement of the four amino acid residues Asn7, Ser8, Ala19, and Ile21 with the corresponding residues (Thr7, Tyr8, Gly19, and Leu21, respectively) of RasGRP1 resulted in potent [3H]PDBu binding affinity (Kd = 1.47 ± 0.03 nm) (Fig. 2B).
This binding affinity falls in the range reported for typical C1 domains. It is close to the value (0.58 nm) we reported previously for the C1 domain of RasGRP1 (11) and that reported for the C1 domain of RasGRP1 (Kd = 0.72 nm) by Irie et al. (13). Assayed under parallel conditions, the C1 domain of RasGRP1 yielded a Kd of 0.26 ± 0.09 nm. The locations within the C1 domain of the four mutations that conferred enhanced binding affinity to the RasGRP2 C1 domain are shown in Fig. 3. It is interesting to note that, unlike in the cases of PKCζ/ι and Vav1, most of the relevant residues do not occupy the tips of the binding loops or lead to marked changes in hydrophobic residues.
FIGURE 3.

Three-dimensional model structures of the wild-type (A and C) and quadruple mutated (B and D) RasGRP2 C1 domain. In the ribbon diagram of the structure of the quadruple mutated RasGRP2 C1 domain, the locations of the four residues that contributed to the enhanced binding are shown. In C and D, coloring portrays the hydrophobicity scale (Kyte-Doolittle scale). Red represents hydrophobicity, blue represents hydrophilicity, and white is in between.
Although the effects of the mutations in the C1 domain have been treated as independent of one another, it is important to emphasize that this is not fully true. For example, whereas the S8Y/A19G mutant had a Kd of 12 nm, conversion to the triple mutant with A33D, restoring the sequence in position 33 to that found in RasGRP1, caused 8-fold weaker binding, not a further improvement. Likewise, in our analysis of the residues accounting for the failure of Vav1 to bind phorbol ester, we had found that the introduction of combinations of these residues into the PKCδ C1b domain had a greater than multiplicative effect on the diminution of phorbol ester binding (6).
In an intact protein containing a C1 domain, binding affinities are modified from those of the isolated C1 domain. Other moieties in the protein such as the C2 domains of the classical PKC isoforms may help drive interaction with the lipid surface and enhance binding (55, 56). Indeed, Irie et al. (13) showed that simply including a short stretch of positively charged residues at the C terminus of their synthetic RasGRP1 C1 domain, by electrostatically enhancing interaction with the negatively charged phospholipids, improved binding affinity 7.5-fold. Conversely, the C1 domain may be involved in internal interactions with other portions of the protein as has been shown directly for PKCβII (57), β2-chimaerin (58), and RasGRP1 (36) and indeed appears to be the case for the PKC isoforms in general (59). The disruption of these interactions when the C1 domain binds to its ligand will necessarily result in a reduction in binding affinity, and conversely ligand binding affinity will be increased when these interactions are disrupted by mutation (60).
To test the binding affinities of full-length wild-type and mutant RasGRP2, we generated N-terminally Halo-tagged constructs, which we expressed in HEK293 cells and purified. We restricted our studies with the full-length proteins to variants in the four critical amino residues identified with the isolated C1 domain. The full-length RasGRP2 protein containing the quadruple mutant C1 domain showed strong phorbol ester binding (Kd = 8.2 ± 1.1 nm) (Table 1), albeit modestly weaker (5.6-fold relative to quadruple mutant C1 domain) than that of the purified quadruple mutant C1 domain. The double and triple mutants for the full-length RasGRP2 again reflected the binding affinities of the corresponding mutants in the isolated C1 domains but again with potencies reduced by 2.5–5-fold. Under our assay conditions, we were unable to measure binding to the full-length wild-type RasGRP2 and S8Y single mutant. Measurement of binding affinities requires concentrations of ligand in excess of the Kd. Because the assay background is proportional to the concentration of ligand, this requires a corresponding increase in the amount of protein, which was not achievable in these two instances. The full-length RasGRP1, which of course has multiple differences with RasGRP2 beyond those in the C1 domain, bound PDBu with 21-fold greater potency than did the full-length quadruple mutant RasGRP2.
Binding Affinities of DOG Showed a Pattern Similar to That for Phorbol Ester
Differences have been reported in the selectivities of C1 domains for diacylglycerol versus phorbol ester (2) as well as in the effects of mutations in the C1 domains on this selectivity (52, 53). We therefore examined the binding affinities of the synthetic diacylglycerol DOG for the more potent C1 domain mutants (S8Y and N7T/S8Y/A19G/I21L) and for the full-length protein containing the quadruple mutant C1 domain (Table 2). The mutations permitted DAG binding with values proportional to those seen for PDBu and with absolute values 40–100-fold weaker (Table 2). For comparison, DOG bound to the wild-type RasGRP1 C1 domain with an affinity of 51 ± 14 nm, which is 200-fold weaker than the affinity of PDBu assayed in parallel. Impressively, compared with the wild-type RasGRP1 C1 domain, the quadruple mutant of the RasGRP2 C1 domain bound DOG with an affinity only 1.2-fold weaker. Likewise, the full-length quadruple mutant RasGRP2 bound DOG with an affinity only 3.9-fold more weakly than did the full-length RasGRP1. We conclude that the mutations had generally similar effects on restoration of both phorbol ester and DAG binding, although in fact the quadruple mutant C1 domain, either by itself or in the intact RasGRP2, more closely approached the binding strength of that of RasGRP1 in the case of DOG than it did in the case of the phorbol ester.
TABLE 2.
Ki values of DOG for binding to the single and quadruple mutant RasGRP2 C1 domain and to the full-length quadruple mutant RasGRP2 as well as to the RasGRP1 C1 domain and the full-length RasGRP1
The single and quadruple C1 domains were fused with GST and expressed in a bacterial system, the quadruple full-length RasGRP2 was fused to HaloTag and expressed in HEK293 cells, and the full-length RasGRP1 was fused with MBP and expressed in bacteria. After isolation of the proteins, the binding of DOG was determined in a competitive binding assay with [3H]PDBu. Values represent the mean ± S.E. (n = 3 independent experiments).
| Receptor | Ki |
|---|---|
| nm | |
| hRasGRP2 C1 | |
| S8Y | 5240 ± 680 |
| N7T/S8Y/A19G/I21L | 60.7 ± 7.7 |
| hRasGRP1 C1 | 51 ± 14 |
| hRasGRP2 full length | |
| N7T/S8Y/A19G/I21L | 414 ± 88 |
| hRasGRP1 full length | 107.3 ± 9.3 |
The Effects of Lipid Content on the PDBu Binding Affinities of the RasGRP2 C1 Domain Mutants
We next addressed whether the phospholipid composition can influence the binding of PDBu to the C1 domain mutants. We examined the C1 mutants in the presence of PS/phosphatidylcholine mixtures with variable proportions of PS while total phospholipid was held constant at 0.1 mg/ml. For [3H]PDBu binding to the mutant C1 domains, the requirement for phosphatidylserine in the phosphatidylserine/phosphatidylcholine mixtures decreased in going from the single S8Y mutant to the double through quadruple mutants (Fig. 4). This shift fits with the stronger interaction of ligand with the binding cleft in the mutants diminishing the relative contribution of anionic membrane charge to formation of the C1 domain-ligand-membrane ternary complex.
FIGURE 4.
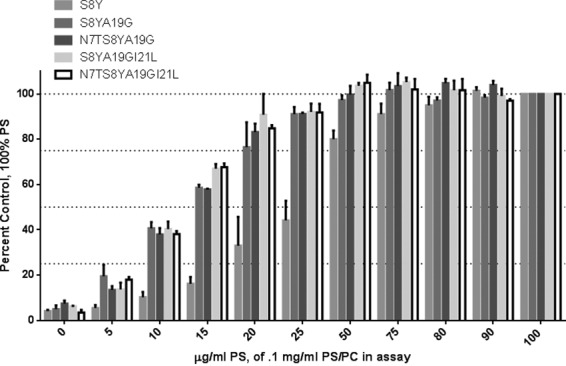
Requirement for anionic phospholipid for [3H]PDBu binding to the mutant C1 domains. The binding of [3H]PDBu to the mutant C1 domains was determined at constant total phospholipid (100 μg/ml) in which the proportion of phosphatidylserine in phosphatidylserine/phosphatidylcholine (PC) mixtures was varied as indicated. The concentration of [3H]PDBu for each mutant was adjusted to exceed the Kd for that mutant in 100% phosphatidylserine. Values were expressed relative to the binding in the presence of 100% phosphatidylserine. Data are the mean of triplicate experiments. Bars represent the mean; error bars represent ±S.E.
Substitution of the Critical Amino Acid Residues in the C1 Domain Significantly Increased Translocation of RasGRP2 to the Plasma Membrane
We examined the translocation patterns of the RasGRP2 mutants in live cells using confocal microscopy. We wished to address two questions. First, could the response to the phorbol esters be detected under physiological conditions with membranes of natural composition? Second, could we see selectivity for the plasma membrane because the plasma membrane is enriched in anionic lipids relative to internal membranes? We prepared fusion constructs between GFP and either the wild-type RasGRP2 C1 domain or the corresponding C1 domains with the single, double, triple, and quadruple mutations that enhanced phorbol ester binding in vitro. Translocation of the GFP-tagged full-length RasGRP2 with either the wild-type or mutated C1 domains was also tested. The constructs were transfected into the LNCaP human prostate cancer cell line, and the translocation of the overexpressed GFP-C1 and GFP-full-length RasGRP2 was visualized by confocal microscopy after the addition of PMA. Images were taken every 30 s. Time points at 0, 5, and 10/20 min after PMA addition are illustrated (Figs. 5A, 6A, and 7, A and C). Data at these time points as well as at 2 and 20 min were quantitated (Figs. 5B, 6B, and 7, B and D).
FIGURE 5.

Translocation in response to PMA of the GFP-tagged wild-type and multiple site mutants of the RasGRP2 C1 domain in living LNCaP cells. Cells expressing GFP-tagged wild-type and multiple site mutants of the C1 domain were treated with 1 μm PMA, and the living cells were imaged by confocal microscopy every 30 s as a function of time. A, images at 0, 5, and 10 min are illustrated. The images shown are representative of three to five independent experiments. Scale bars: 10 μm. B, the ratio of the intensities for membrane/cytoplasm (cyto) was calculated and normalized to the time 0 values. The increase in the membrane/cytoplasm ratio indicates translocation. Values are presented for time points at 0, 2, 5, 10, and 20 min. Values represent the mean of the independent experiments. Error bars represent ±S.E. The increased translocation of C1 domain mutants S8Y and S8Y/A19G/I21L was significant with p < 0.05; the increased translocation of mutants S8Y/A19G, N7T/S8Y/A19G, and N7T/S8Y/A19G/I21L was significant with p < 0.01 (Student's t test).
FIGURE 6.

Translocation in response to 1 μm PMA of the GFP-tagged wild-type and multiple site mutants of full-length RasGRP2 in living LNCaP cells. Cells expressing GFP-tagged wild-type and multiple site mutants of the full-length RasGRP2 were treated with 1 μm PMA, and the living cells were imaged by confocal microscopy every 30 s as a function of time. A, images at 0, 5, and 10 min are illustrated. The images shown are representative of three to six independent experiments except for wild-type, which is representative of nine experiments. Scale bars: 10 μm. B, the ratio of the intensities for membrane/cytoplasm (cyto) was calculated and normalized to the time 0 values. The increase in the membrane/cytoplasm ratio indicates translocation. Values are presented for time points at 0, 2, 5, 10, and 20 min. Values represent the mean of the independent experiments. Error bars represent ±S.E. The increased translocation of the wild-type and S8Y/A19G mutant of full-length RasGRP2 was significant with p < 0.05; the increased translocation of the other full-length RasGRP2 mutants S8Y/A19G/I21L, N7T/S8Y/A19G, and N7T/S8Y/A19G/I21L was significant with p < 0.001 (Student's t test).
FIGURE 7.

Translocation in response to 10 μm PMA of the GFP-tagged wild-type, S8Y, and S8Y/A19G C1 domains (A and B) and of the corresponding full-length RasGRP2 constructs (C and D) in living LNCaP cells. Cells expressing the GFP-tagged wild type and the indicated multiple site mutants were treated with 10 μm PMA to assess their response to an elevated level of PMA. The living cells were imaged by confocal microscopy every 30 s as a function of time after PMA addition. A and C, images taken at 0, 5, 10, and 20 min are illustrated. The images shown are representative of three to seven independent experiments. Scale bars: 10 μm. B and D, the ratio of the intensities for membrane/cytoplasm (cyto) was calculated and normalized to the time 0 values. The increase in the membrane/cytoplasm ratio indicates translocation. Values are presented for time points at 0, 2, 5, 10, and 20 min. Values represent the mean of the independent experiments. Error bars represent ±S.E. The increased translocation of full-length wild-type RasGRP2 was significant with p < 0.05; that of the full-length RasGRP2 mutants S8Y and S8Y/A19G was significant with p < 0.01. The increased translocation of the C1 domain mutant S8Y was significant with p < 0.01; that of the C1 domain mutant S8Y/A19G was significant with p < 0.05 (Student's t test).
Before PMA addition, the constructs displayed a mostly uniform expression throughout the membrane and cytoplasm. The wild-type C1 domain failed to translocate after addition of 1 μm PMA (Fig. 5, A, row 1, and B), consistent with its very weak binding activity in vitro. Substituting Ser with Tyr at position 8 (S8Y) yielded moderately strong [3H]PDBu binding in vitro, but in the live cell experiments the single mutant C1 domain gave only minimal translocation in response to 1 μm PMA (Fig. 5, A, row 2, and B; p = 0.02). In contrast, in response to 1 μm PMA, the double, triple, and quadruple C1 domain mutants, which all had shown a reduced phosphatidylserine requirement for phorbol ester binding in vitro, localized to the plasma membrane (Fig. 5, A, rows 3, 4, 5, and 6, and B). Translocation was first observed after 2 or 5 min of treatment and reached plateau levels at 10 min of treatment (Fig. 5B). The greatest translocation was seen for the quadruple mutant and for the N7T/S8Y/A19G triple mutant. Similar results were observed in confocal experiments with the wild-type and mutated C1 domain containing full-length RasGRP2 after 1 μm PMA treatment (Fig. 6, A and B) except that modestly more activity was observed for the S8Y single mutant. After translocation, there was some tendency for the constructs to transfer back from the plasma membrane to the interior of the cell. This behavior presumably reflects the changing distribution of the PMA, which first accumulates in the plasma membrane and then with time further equilibrates with internal membranes (61).
To assess whether a higher concentration of PMA would cause better translocation of the wild-type, S8Y, and S8Y/A19G C1 domains and of the corresponding full-length proteins, we used 10 μm PMA. After 10 μm PMA treatment of live LNCaP cells, we again saw weak translocation of the S8Y and S8Y/A19G GFP-C1 and GFP-full-length RasGRP2 (Fig. 7). Interestingly, very weak translocation of wild-type full-length RasGRP2 was observed here as was also the case in the presence of 1 μm PMA (Fig. 6; p = 0.01). This weak translocation might reflect either the weak binding of phorbol ester to the C1 domain or an indirect effect of phorbol ester activation of PKC, perhaps mediated by rearrangement of the actin cytoskeleton. Although we did not examine this issue in detail, inclusion of the PKC inhibitor Gö6983 did abolish the weak translocation of full-length wild-type RasGRP2, favoring the latter explanation. For the wild-type C1 domain, no significant effect was seen with 10 μm PMA, similar to the results for the C1 domain in the presence of 1 μm PMA (Fig. 5). The live cell imaging confirmed that substitution of four critical amino acid residues conferred phorbol ester responsiveness to the atypical RasGRP2 C1 domain. In the context of the full-length RasGRP2, the C1 mutants showed modestly more translocation than they did in isolation, whereas they had shown modestly less binding affinity in the in vitro binding assays. A plausible explanation is that other domains in the RasGRP2 also contribute to membrane binding as has been described for RasGRP1 (36, 62, 63).
The Quadruply Mutated RasGRP2 Caused Enhanced Rap1 Activation in the Presence of PMA
RasGRP2 is a guanyl exchange factor for Rap1. A Rap1 GTPase pulldown assay was used to compare the guanine nucleotide exchange activity of wild-type and mutated full-length RasGRP2 in HEK293 cells in the absence and presence of phorbol ester. The cells were transfected either with wild-type or mutated GFP-RasGRP2. At 24 h posttransfection, cells were treated with dimethyl sulfoxide or 1 μm PMA for 20 min. To control for possible indirect effects of the phorbol ester via activation of PKC, Gö6983, an inhibitor of PKC enzymatic activity, was included as indicated. Representative blots are illustrated in Fig. 8A; quantitation of the replicate experiments is presented in Fig. 8B. In the absence of PMA, the levels of activated Rap1 were similar for the cells transfected with either the wild-type or the mutant RasGRP2. In the presence of PMA, cells transfected with the mutated RasGRP2 yielded 2-fold higher levels of activated Rap1 than did the comparably treated cells transfected with wild-type RasGRP2 (p = 0.03). This stimulation cannot be explained as an indirect consequence of PMA activating PKC because the stimulation was not abrogated in the presence of the PKC inhibitor Gö6983. We cannot exclude an additional superimposed effect of PMA on Rap1 mediated by PKC in that a small stimulatory effect of PMA that could be inhibited by Gö6983 was observed for the wild-type RasGRP2, although this difference did not achieve statistical significance (p = 0.38).
FIGURE 8.
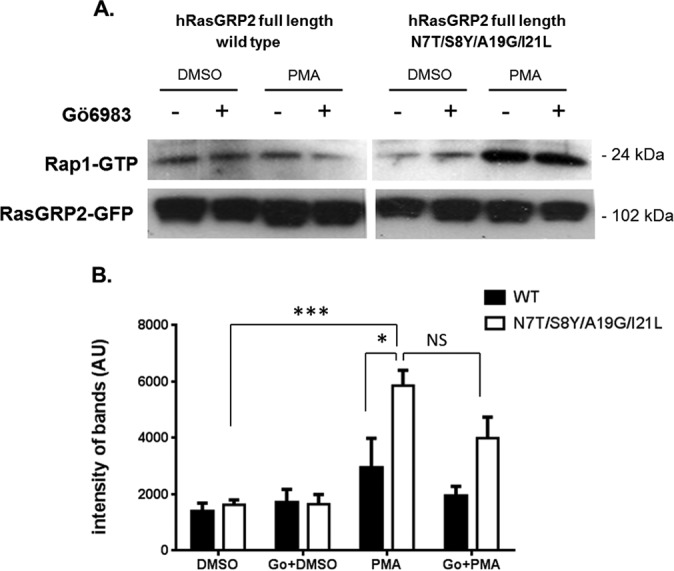
Activation of Rap1 by wild-type and quadruple mutant RasGRP2 after PMA treatment in HEK293 cells. HEK293 cells were transfected with wild-type and quadruple mutant RasGRP2. After 24 h, cells were treated with 1 μm PMA or dimethyl sulfoxide (DMSO) control (20 min). Where indicated, the cells were pretreated with the general PKC inhibitor Gö6983 (Go) (3 μm for 30 min) before addition of PMA or dimethyl sulfoxide. The cells were then lysed, and the levels of activation of the endogenous Rap1 protein were evaluated by pulldown of the activated Rap1 and detection by immunoblotting with anti-Rap1 antibody. The RasGRP2-GFP signal was used as a control for overexpression and loading. A, a representative immunoblot is illustrated. B, band intensities were quantitated for each of seven independent experiments. Values represent the mean. Error bars represent ±S.E. *, p < 0.05; ***, p < 0.001 by Student's t test. NS, not significant; AU, arbitrary units.
Modeling of the Wild-type and Quadruple Mutant C1 Domain of RasGRP2
To develop further insight into the structural effects of the four mutated residues on the function of the C1 domain of RasGRP2, we docked phorbol ester into the modeled wild-type and quadruple mutant C1 domains. We then subjected the complexes to molecular dynamics simulation for 100 ns. Snapshots at 10-ns intervals are illustrated (Fig. 9). In the case of the wild-type RasGRP2 C1 domain, the docked complex was unstable with the phorbol ester not being retained in the binding cleft (Fig. 9A). In contrast, the phorbol ester was retained in the binding cleft of the quadruple mutant (Fig. 9B). To facilitate visualization, the C20, C3, and C4 carbons of the phorbol ester are colored green, blue, and violet.
FIGURE 9.

Snapshots at 10-ns intervals of the molecular dynamics simulations of the complexes between phorbol ester and the RasGRP2 C1 domain. A, wild type. B, quadruple mutant. For easier visualization, the C20, C3, and C4 atoms of the phorbol ester are colored green, violet, and blue. The oxygens on these three carbons are the critical contributors to binding.
From the crystal structure of the PKCδ C1 domain-phorbol ester complex, the C20 primary hydroxyl group forms hydrogen bonds with Thr12 and Leu21, whereas the C4 tertiary hydroxyl and the C3 carbonyl provide hydrogen bonding to Gly23 (40). The distances of these same potential interactions with the corresponding residues in RasGRP2 wild type and quadruple mutant (Fig. 10A) were plotted over the 100-ns duration of the molecular dynamics simulation (Fig. 10B). For the quadruple mutant C1 domain, the distances for the C20 hydroxyl group with its interacting partners were well maintained over the molecular dynamics simulation. The distances were less stable for the C3 carbonyl and C4 hydroxyl over the earlier portion of the time course. In the case of the wild-type C1 domain, all of these distances showed marked increases as the phorbol ester moved away from its initially docked position.
FIGURE 10.

Interactions between the critical binding residues in the phorbol ester and the C1 domain. A, the distances for the critical binding interactions between phorbol ester and the C1 domain, as determined from the crystallographic structure of the binary binding complex with the C1b domain of PKCδ, were evaluated for the wild-type and quadruple mutant of RasGRP2. Ser/Tyr8 and Ile/Leu21 indicate the corresponding residues in the wild-type and mutant, respectively. The image is from LIGPLOT taken from the website PDBsum (54). Oxygen is red, nitrogen is blue, and carbon is black. The covalent bonds in the protein are in brown; those in the phorbol ester are in purple. Amino acid numbering is based on the position in the C1 domain or, in brackets, the position of the amino acid in PKCδ. B, distances calculated for the centers of mass of the interacting partners over the interval of the molecular dynamics simulations for the wild-type and quadruple mutant C1 domain of RasGRP2 as a function of stimulation time. Colors are for the interacting pairs as indicated in A.
Similar molecular dynamics simulations of another C1 domain, the second C1 domain of PKCα, showed that binding was associated with sufficient spreading of the loops forming the binding cleft of the C1 domain (15) where the loops are maintained by hydrogen bonding between residues at their ends (Fig. 11A). We therefore calculated the distances of these potential hydrogen bonds for the wild-type and mutant RasGRP2 C1 domain (Fig. 11A) over the duration of the molecular dynamics simulation (Fig. 11B). The quadruple mutant C1 domain shows a markedly wider spreading of the loops that was maintained over the simulation, whereas the loops of the wild-type C1 domain retain a more closed conformation. Another measure of the loop spreading was to calculate the distance between Pro11 and Ile24, the two residues that form the apices of the binding loops. Again, by this measure, the quadruple mutant shows a more spread conformation for the binding loops (Fig. 11C). Finally, using the SASA function in GROMACS, we calculated the solvent-accessible surface area of the loop region (residues 7–13 and 20–27) for the wild type and the quadruple mutant. The quadruple mutant displayed a consistently larger hydrophobic area (11.51 ± 0.39 nm2) than did the wild type (10.01 ± 0.37 nm2) (Fig. 11D).
FIGURE 11.

Comparison of the characteristics of the binding loops of the C1 domain of the wild-type and quadruple mutant RasGRP2. A, schematic of the residues in the binding cleft loops of the C1 domain of wild-type RasGRP2 and their potential hydrogen bonding interactions (the homologous interactions in the C1b domain of PKCα contribute to the stability of the loops (15)). B, distances measured from centers of mass of the interacting oxygen and nitrogen atoms over the time interval of the molecular dynamics simulations. Colors correspond to the interactions shown in A. C, distance between Pro11 and Ile24 on the wild-type (black) and quadruple mutant (red) C1 domains of RasGRP2 over the time interval of the molecular dynamics simulations. The distance (measured between the nitrogen of Pro11 and the oxygen of Ile24) provides one measure of the extent of spreading of the binding loops of the C1 domain. D, hydrophobic area within the solvent-accessible surface area defined by residues 7–13 (loop 1) and 20–27 (loop 2) over the time interval of the molecular dynamics simulations for the wild-type (black) and quadruple mutant (red) C1 domains of RasGRP2.
Similar behavior of the wild-type and quadruple mutant was observed upon a repetition of the molecular dynamics simulation. The single S8Y mutant, in contrast, gave intermediate but inconsistent results, suggesting that a contribution of the S8Y mutation was not fully captured in these simulations for the C1 domain-ligand binary complex. The functional studies of course were determined on the tertiary complex of C1 domain-ligand-lipid bilayer. The C1 domains with high phorbol ester binding affinity all have larger, hydrophobic residues at the 8 position (1, 18, 64) that interact with Leu21 and help stabilize the binding loops. This effect may not be captured in the simulations with the binary complex, which lacks the effect of the lipid environment on the ligand positioning and the binding loops.
Discussion
C1 domains have been subdivided into typical and atypical depending on whether they are able to bind phorbol ester. The RasGRP2 C1 domain occupies an intermediate position, being able to bind phorbol ester, albeit with an affinity 4 orders of magnitude less than that of the C1 domain of RasGRP1. The potencies of DAG for the mutant versions of the RasGRP2 C1 domain paralleled those for phorbol ester but with ∼40–100-fold lower affinity. It would thus seem most likely that the residual phorbol ester/DAG binding affinity of the RasGRP2 C1 domain would not be sufficient for regulation by physiological levels of DAG.
Our emerging understanding of the structure of C1 domain-containing proteins provides a plausible alternative function. The crystal structures of β2-chimaerin (58), PKCβII (57), and RasGRP1 (36) all indicate that the hydrophobic face of each of these typical C1 domains is not exposed in the absence of ligand. Rather, it is buried in intramolecular contacts with the binding cleft of the C1 domain involved in these contacts. In the case of the atypical C1 domain of Vav1, likewise, the C1 domain is again involved in intramolecular contacts. It would thus be plausible that the C1 domain of RasGRP2 performs a similar function in the closed conformation of the protein. When the protein is activated, these contacts would be broken, and the C1 domain would require an alternative hydrophobic site with which to interact. Kay and co-workers (10) have shown that the RasGRP2 C1 domain can provide a membrane binding motif even if not DAG-responsive. Thus, it is able to substitute for the C1 domain in RasGRP1 to support the ability of this family member to transform rat cells (10).
Mochly-Rosen and co-workers (65) have shown that peptides that can compete for a site of intramolecular interaction on PKC provide the potential of isoform-specific activation. Our emerging structural understanding of C1 domains suggests that the intramolecular domain interacting with the face of the C1 domain might represent a candidate structure. Moreover, to the degree that such a peptide would block the binding of the C1 domain to the membrane, it could function as an isoform-specific antagonist by impairing appropriate localization. This approach represents an alternative to the current strategy of developing hydrophilic variants of ligands such as the phorbol esters (66, 67), and indeed the approach of using peptides to interfere with the activity of specific PKC isoforms has proven to be successful (65).
The comparison of the abilities of the wild-type and mutant versions of the C1 domain of RasGRP2 as well as of the corresponding full-length RasGRP2 proteins further emphasizes that the C1 domain is not the only entity in the full-length protein responsible for membrane interaction. Thus, we observed greater translocation of the full-length RasGRP2 S8Y mutant than we did of the C1 domain S8Y mutant. The coordination of multiple lipid binding domains to enhance sensitivity to regulation is well recognized for PKC and AKT (49). In RasGRP1, for example, the plasma membrane targeter (PT) domain has been described as a key contributor along with the C1 domain (63).
Looking at functional activity, we demonstrated that mutation of the C1 domain of RasGRP2 so that it can translocate in response to phorbol ester led to elevated activity of Rap1 in the presence of phorbol ester. This would fit with our current understanding of RasGRP2 function in which stimulatory activity is affected by proximity with the typical activating element being actin organization (25). It should be noted that we saw suggestions of phorbol ester response in the wild-type RasGRP2 both for translocation and for Rap1 activation. In both cases, we showed that this effect could be blocked by the PKC inhibitor Gö6983, consistent with it being mediated by PKC. This may explain the occasional reports in the literature of in vivo effects of PMA on RasGRP2 (10). Possible mechanisms include direct phosphorylation and phorbol ester induction of actin rearrangement. Phosphorylation by PKC of RasGRP1 on Thr184 and of RasGRP3 on Thr133 is a critical element of their activation mechanism by phorbol ester (8). Conversely, although phosphorylation of RasGRP2 by PKC has not been reported, its phosphorylation by PKA was found to be inhibitory (68).
Among the residues in the C1 domain that made critical contributions to its very weak binding, the presence of Ser at position 8 of the C1 domain proved to be the most important. Similarly, a single residue at position 9 is largely responsible for the weak binding affinity of the C1a domain of PKCθ. PKCθ is unique among the typical PKC isoforms in that one of its two C1 domains, C1a, has been reported to have very weak phorbol ester binding affinity (69, 70). The C1a domain of PKCθ has Pro at position 9. Substitution of this Pro with Lys confers strong binding affinity (71). In RasGRP2, Ser is found at position 8 in place of Tyr, Phe, or Leu, which are present in C1 domains with potent binding activity (1). A potential explanation is that this residue normally packs in the C1 domain structure against the side chain of Leu21. The hydrophilicity and smaller size of the Ser would be less effective at stabilizing the binding structure. Molecular dynamics simulation indicated that the quadruple mutant of the RasGRP2 C1 domain showed stabilization of the binding conformation of the loops forming the binding cleft, whereas this stabilization was not reproducibly attained for the S8Y mutant. These findings fit with the combination of residues working together to maintain the active binding conformation.
We conclude that the C1 domain of RasGRP2 fills a gap in the continuum of properties of C1 domains (Fig. 12). The atypical C1 domains of proteins such as Raf show distorted geometry of their binding cleft. The atypical C1 domains of Vav and PKCζ/ι retain the binding cleft but incorporate residues along the binding cleft and elsewhere that disrupt binding sufficiently so that it is no longer measurable. The C1 domain of RasGRP2 represents a bridge situation between atypical and typical. It shows measurable binding, but the magnitude of binding is sufficiently weak so that this C1 domain is probably not a recognition module for physiological DAG levels. Finally, the typical C1 domains respond to phorbol ester and physiological DAG levels, often in cooperation with other lipid recognition elements in the intact protein. The differences between C1 domains that retain the binding cleft geometry provide the opportunity for future design of selective ligands. For both the atypical C1 domains of PKCζ/ι and of Vav1, we have described DAG lactones to begin to exploit such structural differences (6, 72).
FIGURE 12.

Continuum of characteristics for C1 domains. The traditional division of C1 domains into typical and atypical is now recognized to represent a simplification as characterization reveals a continuum of properties.
Author Contributions
A. C., D. J. L., D. C. B., N. K., and P. M. B. designed the study and wrote the paper. A. C., R. C. M., and E. A. C. performed the cloning of different constructs and purification of the proteins. N. E. L. and X. Z. measured the radioactive binding. A. C. conducted the confocal measurements and western blot assays. A. A. and M. L. P. did the modeling.
This work was supported in part by the Intramural Research Program, Center for Cancer Research, NCI, National Institutes of Health Project Z1A BC 005270 and in part with federal funds from the NCI, National Institutes of Health under Contract HHSN261200800001E. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- DAG
- diacylglycerol
- PDBu
- phorbol 12,13-dibutyrate
- PS
- phosphatidyl-l-serine
- DOG
- 1,2-dioctanoylglycerol
- PMA
- phorbol 12-myristate 13-acetate
- RasGRP
- Ras guanine nucleotide releasing protein
- MBP
- maltose-binding protein
- NPT
- number of particles, pressure, and temperature.
References
- 1. Hurley J. H., Newton A. C., Parker P. J., Blumberg P. M., and Nishizuka Y. (1997) Taxonomy and function of C1 protein kinase C homology domains. Protein Sci. 6, 477–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Colón-González F., and Kazanietz M. G. (2006) C1 domains exposed: from diacylglycerol binding to protein-protein interactions. Biochim. Biophys. Acta 1761, 827–837 [DOI] [PubMed] [Google Scholar]
- 3. Kazanietz M. G. (2002) Novel “nonkinase” phorbol ester receptors: the C1 domain connection. Mol. Pharmacol. 61, 759–767 [DOI] [PubMed] [Google Scholar]
- 4. Cho W., and Stahelin R. V. (2005) Membrane-protein interactions in cell signaling and membrane trafficking. Annu. Rev. Biophys. Biomol. Struct. 34, 119–151 [DOI] [PubMed] [Google Scholar]
- 5. Caloca M. J., Zugaza J. L., and Bustelo X. R. (2003) Exchange factors of the RasGRP family mediate Ras activation in the Golgi. J. Biol. Chem. 278, 33465–33473 [DOI] [PubMed] [Google Scholar]
- 6. Geczy T., Peach M. L., El Kazzouli S., Sigano D. M., Kang J. H., Valle C. J., Selezneva J., Woo W., Kedei N., Lewin N. E., Garfield S. H., Lim L., Mannan P., Marquez V. E., and Blumberg P. M. (2012) Molecular basis for failure of “atypical” C1 domain of Vav1 to bind diacylglycerol/phorbol ester. J. Biol. Chem. 287, 13137–13158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mott H. R., Carpenter J. W., Zhong S., Ghosh S., Bell R. M., and Campbell S. L. (1996) The solution structure of the Raf-1 cysteine-rich domain: a novel Ras and phospholipid binding site. Proc. Natl. Acad. Sci. U.S.A. 93, 8312–8317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stone J. C. (2011) Regulation and function of the RasGRP family of Ras activators in blood cells. Genes Cancer 2, 320–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stone J. C. (2006) Regulation of Ras in lymphocytes: get a GRP. Biochem. Soc. Trans. 34, 858–861 [DOI] [PubMed] [Google Scholar]
- 10. Johnson J. E., Goulding R. E., Ding Z., Partovi A., Anthony K. V., Beaulieu N., Tazmini G., Cornell R. B., and Kay R. J. (2007) Differential membrane binding and diacylglycerol recognition by C1 domains of RasGRPs. Biochem. J. 406, 223–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lorenzo P. S., Beheshti M., Pettit G. R., Stone J. C., and Blumberg P. M. (2000) The guanine nucleotide exchange factor RasGRP is a high-affinity target for diacylglycerol and phorbol esters. Mol. Pharmacol. 57, 840–846 [PubMed] [Google Scholar]
- 12. Lorenzo P. S., Kung J. W., Bottorff D. A., Garfield S. H., Stone J. C., and Blumberg P. M. (2001) Phorbol esters modulate the Ras exchange factor RasGRP3. Cancer Res. 61, 943–949 [PubMed] [Google Scholar]
- 13. Irie K., Masuda A., Shindo M., Nakagawa Y., and Ohigashi H. (2004) Tumor promoter binding of the protein kinase C C1 homology domain peptides of RasGRPs, chimaerins, and Unc13s. Bioorg. Med. Chem. 12, 4575–4583 [DOI] [PubMed] [Google Scholar]
- 14. Xu R. X., Pawelczyk T., Xia T. H., and Brown S. C. (1997) NMR structure of a protein kinase C-γ phorbol-binding domain and study of protein-lipid micelle interactions. Biochemistry 36, 10709–10717 [DOI] [PubMed] [Google Scholar]
- 15. Stewart M. D., Morgan B., Massi F., and Igumenova T. I. (2011) Probing the determinants of diacylglycerol binding affinity in the C1b domain of protein kinase Cα. J. Mol. Biol. 408, 949–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stewart M. D., Cole T. R., and Igumenova T. I. (2014) Interfacial partitioning of a loop hinge residue contributes to diacylglycerol affinity of conserved region 1 domains. J. Biol. Chem. 289, 27653–27664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Igumenova T. I. (2015) Dynamics and membrane interactions of protein kinase C. Biochemistry 54, 4953–4968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li J., Ziemba B. P., Falke J. J., and Voth G. A. (2014) Interactions of protein kinase C-α C1A and C1B domains with membranes: a combined computational and experimental study. J. Am. Chem. Soc. 136, 11757–11766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ziemba B. P., Li J., Landgraf K. E., Knight J. D., Voth G. A., and Falke J. J. (2014) Single-molecule studies reveal a hidden key step in the activation mechanism of membrane-bound protein kinase C-α. Biochemistry 53, 1697–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Medkova M., and Cho W. (1999) Interplay of C1 and C2 domains of protein kinase C-α in its membrane binding and activation. J. Biol. Chem. 274, 19852–19861 [DOI] [PubMed] [Google Scholar]
- 21. Wang Q. J., Fang T. W., Nacro K., Marquez V. E., Wang S., and Blumberg P. M. (2001) Role of hydrophobic residues in the C1b domain of protein kinase C δ on ligand and phospholipid interactions. J. Biol. Chem. 276, 19580–19587 [DOI] [PubMed] [Google Scholar]
- 22. Kazanietz M. G., Wang S., Milne G. W., Lewin N. E., Liu H. L., and Blumberg P. M. (1995) Residues in the second cysteine-rich region of protein kinase C δ relevant to phorbol ester binding as revealed by site-directed mutagenesis. J. Biol. Chem. 270, 21852–21859 [DOI] [PubMed] [Google Scholar]
- 23. Shindo M., Irie K., Masuda A., Ohigashi H., Shirai Y., Miyasaka K., and Saito N. (2003) Synthesis and phorbol ester binding of the cysteine-rich domains of diacylglycerol kinase (DGK) isozymes. DGKγ and DGKβ are new targets of tumor-promoting phorbol esters. J. Biol. Chem. 278, 18448–18454 [DOI] [PubMed] [Google Scholar]
- 24. Clyde-Smith J., Silins G., Gartside M., Grimmond S., Etheridge M., Apolloni A., Hayward N., and Hancock J. F. (2000) Characterization of RasGRP2, a plasma membrane-targeted, dual specificity Ras/Rap exchange factor. J. Biol. Chem. 275, 32260–32267 [DOI] [PubMed] [Google Scholar]
- 25. Caloca M. J., Zugaza J. L., Vicente-Manzanares M., Sánchez-Madrid F., and Bustelo X. R. (2004) F-actin dependent translocation of the Rap1 GDP/GTP exchange factor RasGRP2. J. Biol. Chem. 279, 20435–20446 [DOI] [PubMed] [Google Scholar]
- 26. Pasvolsky R., Feigelson S. W., Kilic S. S., Simon A. J., Tal-Lapidot G., Grabovsky V., Crittenden J. R., Amariglio N., Safran M., Graybiel A. M., Rechavi G., Ben-Dor S., Etzioni A., and Alon R. (2007) A LAD-III syndrome is associated with defective expression of the Rap-1 activator CalDAG-GEFI in lymphocytes, neutrophils, and platelets. J. Exp. Med. 204, 1571–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Canault M., Ghalloussi D., Grosdidier C., Guinier M., Perret C., Chelghoum N., Germain M., Raslova H., Peiretti F., Morange P. E., Saut N., Pillois X., Nurden A. T., Cambien F., Pierres A., van den Berg T. K., Kuijpers T. W., Alessi M. C., and Tregouet D. A. (2014) Human CalDAG-GEFI gene (RASGRP2) mutation affects platelet function and causes severe bleeding. J. Exp. Med. 211, 1349–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Carbo C., Duerschmied D., Goerge T., Hattori H., Sakai J., Cifuni S. M., White G. C. 2nd, Chrzanowska-Wodnicka M., Luo H. R., and Wagner D. D. (2010) Integrin-independent role of CalDAG-GEF1 in neutrophil chemotaxis. J. Leukoc. Biol. 88, 313–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stadtmann A., Brinkhaus L., Mueller H., Rossaint J., Bolomini-Vittori M., Bergmeier W., Van Aken H., Wagner D. D., Laudanna C., Ley K., and Zarbock A. (2011) Rap1a activation by CalDAG-GEFI and p38 MAPK is involved in E-selectin-dependent slow leukocyte rolling. Eur. J. Immunol. 41, 2074–2085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stefanini L., and Bergmeier W. (2010) CalDAG-GEF1 and platelet activation. Platelets 21, 239–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alon R., Aker M., Feigelson S., Sokolovsky-Eisenberg M., Staunton D. E., Cinamon G., Grabovsky V., Shamri R., and Etzioni A. (2003) A novel genetic leukocyte adhesion deficiency in subsecond triggering of integrin avidity by endothelial chemokines results in impaired leukocyte arrest on vascular endothelium under shear flow. Blood 101, 4437–4445 [DOI] [PubMed] [Google Scholar]
- 32. Dupuy A. J., Morgan K., von Lintig F. C., Shen H., Acar H., Hasz D. E., Jenkins N. A., Copeland N. G., Boss G. R., and Largaespada D. A. (2001) Activation of the Rap1 guanine nucleotide exchange gene, CalDAG-GEF1, in BXH-2 murine myeloid leukemia. J. Biol. Chem. 276, 11804–11811 [DOI] [PubMed] [Google Scholar]
- 33. Riches J. C., O'Donovan C. J., Kingdon S. J., McClanahan F., Clear A. J., Neuberg D. S., Werner L., Croce C. M., Ramsay A. G., Rassenti L. Z., Kipps T. J., and Gribben J. G. (2014) Trisomy 12 chronic lymphocytic leukemia cells exhibit upregulation of integrin signaling that is modulated by NOTCH1 mutations. Blood 123, 4101–4110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guo F. F., Kumahara E., and Saffen D. (2001) A CalDAG-GEF1/Rap1/B-Raf cassette couples M1 muscarinic acetylcholine receptors to the activation of ERK1/2. J. Biol. Chem. 276, 25568–25581 [DOI] [PubMed] [Google Scholar]
- 35. Lewin N. E., and Blumberg P. M. (2003) [3H]Phorbol 12,13-dibutyrate binding assay for protein kinase C and related proteins. Methods Mol. Biol. 233, 129–156 [DOI] [PubMed] [Google Scholar]
- 36. Iwig J. S., Vercoulen Y., Das R., Barros T., Limnander A., Che Y., Pelton J. G., Wemmer D. E., Roose J. P., and Kuriyan J. (2013) Structural analysis of autoinhibition in the Ras-specific exchange factor RasGRP1. eLife 2, e00813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Guex N., and Peitsch M. C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723 [DOI] [PubMed] [Google Scholar]
- 38. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., and Olson A. J. (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Morris G. M., Huey R., and Olson A. J. (2008) Using AutoDock for ligand-receptor docking. Curr. Protoc. Bioinformatics Chapter 8, Unit 8.14 [DOI] [PubMed] [Google Scholar]
- 40. Zhang G., Kazanietz M. G., Blumberg P. M., and Hurley J. H. (1995) Crystal structure of the cys2 activator-binding domain of protein kinase C δ in complex with phorbol ester. Cell 81, 917–924 [DOI] [PubMed] [Google Scholar]
- 41. Berendsen H. J., van der Spoel D., and van Drunen R. (1995) GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91, 43–56 [Google Scholar]
- 42. Oostenbrink C., Villa A., Mark A. E., and van Gunsteren W. F. (2004) A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 25, 1656–1676 [DOI] [PubMed] [Google Scholar]
- 43. Schwede T., Kopp J., Guex N., and Peitsch M. C. (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31, 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schüttelkopf A. W., and van Aalten D. M. (2004) PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60, 1355–1363 [DOI] [PubMed] [Google Scholar]
- 45. Hermans J. Jr., Lohr D., and Ferro D. (1969) Unfolding and hydrogen exchange of proteins: the three-dimensional Ising lattice as a model. Nature 224, 175–177 [DOI] [PubMed] [Google Scholar]
- 46. Hess B., Bekker H., Berendsen H. J., and Fraaije G. E. (1997) LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 [Google Scholar]
- 47. Miyamoto S., and Kollman P. (1992) Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 13, 952–962 [Google Scholar]
- 48. Berendsen H. J., Postma J. P., van Gunsteren W. F., DiNola A., and Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 [Google Scholar]
- 49. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., and Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys 103, 8577–8593 [Google Scholar]
- 50. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 51. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 52. Dries D. R., Gallegos L. L., and Newton A. C. (2007) A single residue in the C1 domain sensitizes novel protein kinase C isoforms to cellular diacylglycerol production. J. Biol. Chem. 282, 826–830 [DOI] [PubMed] [Google Scholar]
- 53. Kelsey J. S., Geczy T., Lewin N. E., Kedei N., Hill C. S., Selezneva J. S., Valle C. J., Woo W., Gorshkova I., and Blumberg P. M. (2014) Charge density influences C1 domain ligand affinity and membrane interactions. ChemBioChem 15, 1131–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Laskowski R. A., Hutchinson E. G., Michie A. D., Wallace A. C., Jones M. L., and Thornton J. M. (1997) PDBsum: a web-based database of summaries and analyses of all PDB structures. Trends Biochem. Sci. 22, 488–490 [DOI] [PubMed] [Google Scholar]
- 55. Newton A. C. (2009) Lipid activation of protein kinases. J. Lipid Res. 50, (suppl.) S266–S271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stahelin R. V. (2009) Lipid binding domains: more than simple lipid effectors. J. Lipid Res. 50, (suppl.) S299–S304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Leonard T. A., Różycki B., Saidi L. F., Hummer G., and Hurley J. H. (2011) Crystal structure and allosteric activation of protein kinase C βII. Cell 144, 55–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Canagarajah B., Leskow F. C., Ho J. Y., Mischak H., Saidi L. F., Kazanietz M. G., and Hurley J. H. (2004) Structural mechanism for lipid activation of the Ras-specific GAP, β2-chimaerin. Cell 119, 407–418 [DOI] [PubMed] [Google Scholar]
- 59. Lučić I., Truebestein L., and Leonard T. A. (2016) Novel features of DAG-activated PKC isozymes reveal a conserved 3-D architecture. J. Mol. Biol. 428, 121–141 [DOI] [PubMed] [Google Scholar]
- 60. Sosa M. S., Lewin N. E., Choi S. H., Blumberg P. M., and Kazanietz M. G. (2009) Biochemical characterization of hyperactive β2-chimaerin mutants revealed an enhanced exposure of C1 and Rac-GAP domains. Biochemistry 48, 8171–8178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Braun D. C., Cao Y., Wang S., Garfield S. H., Hur G. M., and Blumberg P. M. (2005) Role of phorbol ester localization in determining protein kinase C or RasGRP3 translocation: real-time analysis using fluorescent ligands and proteins. Mol. Cancer Ther. 4, 141–150 [PubMed] [Google Scholar]
- 62. Fuller D. M., Zhu M., Song X., Ou-Yang C. W., Sullivan S. A., Stone J. C., and Zhang W. (2012) Regulation of RasGRP1 function in T cell development and activation by its unique tail domain. PLoS One 7, e38796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zahedi B., Goo H. J., Beaulieu N., Tazmini G., Kay R. J., and Cornell R. B. (2011) Phosphoinositide 3-kinase regulates plasma membrane targeting of the Ras-specific exchange factor RasGRP1. J. Biol. Chem. 286, 12712–12723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Das J., and Rahman G. M. (2014) C1 domains: structure and ligand-binding properties. Chem. Rev. 114, 12108–12131 [DOI] [PubMed] [Google Scholar]
- 65. Churchill E. N., Qvit N., and Mochly-Rosen D. (2009) Rationally designed peptide regulators of protein kinase C. Trends Endocrinol. Metab. 20, 25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bertolini T. M., Giorgione J., Harvey D. F., and Newton A. C. (2003) Protein kinase C translocation by modified phorbol esters with functionalized lipophilic regions. J. Org. Chem. 68, 5028–5036 [DOI] [PubMed] [Google Scholar]
- 67. Blumberg P. M., Kedei N., Lewin N. E., Yang D., Czifra G., Pu Y., Peach M. L., and Marquez V. E. (2008) Wealth of opportunity—the C1 domain as a target for drug development. Curr. Drug Targets 9, 641–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Subramanian H., Zahedi R. P., Sickmann A., Walter U., and Gambaryan S. (2013) Phosphorylation of CalDAG-GEFI by protein kinase A prevents Rap1b activation. J. Thromb. Haemost. 11, 1574–1582 [DOI] [PubMed] [Google Scholar]
- 69. Irie K., Nakahara A., Nakagawa Y., Ohigashi H., Shindo M., Fukuda H., Konishi H., Kikkawa U., Kashiwagi K., and Saito N. (2002) Establishment of a binding assay for protein kinase C isozymes using synthetic C1 peptides and development of new medicinal leads with protein kinase C isozyme and C1 domain selectivity. Pharmacol. Ther. 93, 271–281 [DOI] [PubMed] [Google Scholar]
- 70. Melowic H. R., Stahelin R. V., Blatner N. R., Tian W., Hayashi K., Altman A., and Cho W. (2007) Mechanism of diacylglycerol-induced membrane targeting and activation of protein kinase Cθ. J. Biol. Chem. 282, 21467–21476 [DOI] [PubMed] [Google Scholar]
- 71. Shindo M., Irie K., Nakahara A., Ohigashi H., Konishi H., Kikkawa U., Fukuda H., and Wender P. A. (2001) Toward the identification of selective modulators of protein kinase C (PKC) isozymes: establishment of a binding assay for PKC isozymes using synthetic C1 peptide receptors and identification of the critical residues involved in the phorbol ester binding. Bioorg. Med. Chem. 9, 2073–2081 [DOI] [PubMed] [Google Scholar]
- 72. Pu Y., Kang J. H., Sigano D. M., Peach M. L., Lewin N. E., Marquez V. E., and Blumberg P. M. (2014) Diacylglycerol lactones targeting the structural features that distinguish the atypical C1 domains of protein kinase C ζ and ι from typical C1 domains. J. Med. Chem. 57, 3835–3844 [DOI] [PMC free article] [PubMed] [Google Scholar]