

Abstract
The I-domain is a genetic insertion in the phage P22 coat protein that chaperones its folding and stability. Of 11 acidic residues in the I-domain, seven participate in stabilizing electrostatic interactions with basic residues across elements of secondary structure, fastening the β-barrel fold. A hydrogen-bonded salt bridge between Asp-302 and His-305 is particularly interesting as Asp-302 is the site of a temperature-sensitive-folding mutation. The pKa of His-305 is raised to 9.0, indicating the salt bridge stabilizes the I-domain by ∼4 kcal/mol. Consistently, urea denaturation experiments indicate the stability of the WT I-domain decreases by 4 kcal/mol between neutral and basic pH. The mutants D302A and H305A remove the pH dependence of stability. The D302A substitution destabilizes the I-domain by 4 kcal/mol, whereas H305A had smaller effects, on the order of 1–2 kcal/mol. The destabilizing effects of D302A are perpetuated in the full-length coat protein as shown by a higher sensitivity to protease digestion, decreased procapsid assembly rates, and impaired phage production in vivo. By contrast, the mutants have only minor effects on capsid expansion or stability in vitro. The effects of the Asp-302–His-305 salt bridge are thus complex and context-dependent. Substitutions that abolish the salt bridge destabilize coat protein monomers and impair capsid self-assembly, but once capsids are formed the effects of the substitutions are overcome by new quaternary interactions between subunits.
Keywords: bacteriophage, nuclear magnetic resonance (NMR), protein folding, protein stability, virus, ionization constant, nanoparticle, procapsid
Introduction
Virus and phage coat proteins requisitely encounter the dilemma of balancing counteracting forces during folding and assembly. For icosahedral particles built from multiple copies of a single coat protein and a triangulation number (T) >1, subunits have to adopt pseudo-symmetric conformations dependent on their position in the capsid (1). Counteracting the needs for flexibility and protection of interaction surfaces until assembly is complete is the requirement that capsid proteins be folded into assembly-competent structures. Despite the potential difficulties inherent in the folding of coat proteins, the resulting capsids are naturally extremely stable and provide excellent platforms for nanoengineering (2). The economical architecture of icosahedral symmetry has been applied to simultaneously display a multiplicity of cargo molecules on the surfaces of capsids, enabling the design of nanomaterials with unique properties (3, 4), including nanoparticle scaffolds based on phage P22 (5–7).
P22 coat protein first assembles into a metastable precursor capsid, known as a procapsid, in a process driven by its scaffolding protein, which serves as an assembly chaperone (8). In its simplest form, the P22 procapsid has 420 copies of coat protein arranged in a T = 7 (T = triangulation number) icosahedral shell with 100–300 copies of the scaffolding protein bound within its confines with unknown symmetry (9). The procapsid has an overall stability of ∼3000 kcal/mol, although individual coat protein subunits are only marginally stable (10, 11). The large stability of the procapsid is due to an extensive network of non-covalent contacts between coat protein subunits. During phage morphogenesis, procapsids mature concomitant with genome packaging (12). Maturation increases the particle diameter by 10% (13), stabilizes the icosahedral shell (14), and induces the release of scaffolding protein (15). The maturation from procapsid to capsid can be recapitulated in vitro by heating the particles (14, 16). Heat expansion can lead to the release of the penton subunits, yielding a “wiffle ball” form of the capsid (16, 17). Phage P22 is an extremely attractive platform for nanomaterial design (5–7) because it is a structurally characterized virus (18–25), with well understood genetics, assembly, and maturation (26). Moreover, procapsids and capsids can be manipulated in vivo and in vitro. To be able to fully exploit P22 capsids for the design of nanomaterials, an understanding the particle's stability properties is critical, as there are many examples where single amino acid substitutions in coat protein can affect self-assembly and/or disassembly (17, 27–29).
The P22 coat protein (430 amino acids) has a core structure based on the HK97 fold (30) but has an additional genetic insertion, the 123-aminoacyl I-domain (19, 20). The I-domain folds very rapidly and stabilizes full-length coat protein, suggesting it serves as an uncleaved intermolecular chaperone and the folding nucleus of the protein (31). Over half of the known temperature-sensitive-folding (tsf) coat protein mutants are localized in the I-domain, attesting to the importance of this module in modulating folding (32). The NMR structure of the I-domain (32) consists of a six-stranded β-barrel fold (strands β1-β6) and a smaller sub-domain (strands βi–βiii together with helix αi). Several ion-pair interactions fasten the β-strands in the I-domain structure (Fig. 1A, Table 1). Of these, the Asp-302–His-305 salt bridge is of particular interest because the D302G mutation has been identified as a tsf mutant (33), suggesting this site is particularly important for proper coat protein folding. Here we show the salt bridge between Asp-302 and His-305 contributes significantly to the stability of the I-domain, such that the unfolding free energy change for the I-domain decreases by ∼50% between neutral and basic pH, as His-305 becomes deprotonated.
FIGURE 1.

Ion-pair interactions in the I-domain. A, stereo diagram of the I-domain NMR structure showing charged residues. Acidic and basic residues that do not participate in ion pairs are colored orange and cyan, respectively. Residues that participate in ion pairs (Table 1) are labeled red and blue for acidic and basic amino acids, respectively. Hydrogen bonds that link ion pairs in a salt bridge (Asp-302–His-305, Asp-253–Lys-311, Asp-271–Lys-268, Asp-317–Lys-237) are indicated by dashed green lines. B, representative NMR pH titration data for aspartate Hβ protons. Asp-246 is in the disordered D-loop and has a random coil pKa. Asp-316 forms an ion pair with Arg-325 and has the lowest pKa measured in the I-domain. Asp-302 and Asp-317 form hydrogen-bonded salt bridges (Table 1) and do not shift with pH. C, NMR pH titration of histidine Hϵ1 protons. The unresolved resonances from the His6 tag used for purification gave a pKa of 6.7, in agreement with the random coil histidine value (43). By contrast His-305 had a pKa value shifted up by 2.5 pH units, consistent with the residue forming a stabilizing salt bridge with Asp-302.
TABLE 1.
I-domain ion-pairs and pKa values
| Residue | pKaa | Ion-pair partner | Salt bridgeb | d(O … HN)c | Structure context | Energeticsd |
|---|---|---|---|---|---|---|
| % | Å | |||||
| Asp-244e | NAf | None | 0 | NA | D-loop | NA |
| Asp-246 | 4.2 ± 0.1 | None | 0 | NA | D-loop | Null |
| Asp-253 | <2.8 | Lys-311 | 60 | 1.7 | Links β2-β5 | Stabilizing |
| Asp-271 | <2.8 | Lys-268 | 53 | 1.7 | Turn before β3 | Stabilizing |
| Asp-292 | <2.8 | Lys-279 | 37 | 3.4 | Links β4-βi | Stabilizing |
| Asp-302 | <2.8 | His-305 | 43 | 2.2 | β4-β5 hairpin | Stabilizing |
| His-305 | 9.05 ± 0.04 | Asp-302 | See Asp-302 | See Asp-302 | see Asp-302 | Stabilizing |
| Glu-307 | 3.9 ± 0.3 | Arg-299 | 10 | 5.3 | Links β5-β4 | Slight stabilizing |
| Asp-316 | 3.0 ± 0.3 | Arg-325 | 0 | 6.0 | Links βii-αi | Slight stabilizing |
| Asp-317 | < 2.8 | Lys-237 | 97 | 1.7 | Links βii-β1 | Stabilizing |
| Glu-323 | 3.6 ± 0.1 | Unknown | 0 | NA | Unknown | Slight stabilizing |
| Asp-336 | 4.3 ± 0.3 | None | 0 | NA | Surface β6 | Null |
a Uncertainties in pKa values are given as the S.E. of non-linear least square fits of the chemical shift data as a function of pH (e.g. Fig. 1, B and C). The minimum error in the pKa, however, is likely to be limited by the accuracy of the pH meter, which is 0.1 pH units.
b We use the terminology “salt bridge” for a hydrogen-bonded ion pair that has the closest approach distance between the side-chain NH hydrogen-bond donor and O carbonyl acceptor atoms shorter than 2.5 Å in the NMR structure closest to the ensemble mean (structure 1 in the ensemble of PDB code 2M5S). The column gives the fraction of structures in the NMR ensemble of 30 structures (32) in which the side chains of the ion-pair are hydrogen-bonded.
c Nearest approach NH to O side-chain distance in the NMR structure of the I-domain closest to the ensemble average.
d Based on differences of pKa values in the I-domain from the following random-coil values (43): histidine = 6.5, aspartate = 4.0; glutamate = 4.4. An interaction is considered stabilizing if the pKa shift is greater than 1 pH unit, slightly stabilizing if the shift is between 0.5 and 1 pH unit and negligible if it is <0.5 pH.
e Resonances for Asp-244 were not observed in the NMR spectrum due to conformational exchange line-broadening (19).
f NA, not applicable.
The role of the salt bridge in the I-domain, full-length coat protein, and assembled P22 procapsids is further examined by substituting alanine at both the Asp-302 and His-305 sites. Substitution of Asp-302 with alanine destabilizes the I-domain, increases the susceptibility of the full-length coat protein monomers to proteolytic digestion, and causes a tsf phenotype leading to impaired phage assembly in vivo. The H305A I-domain mutation has smaller effects than D302A, possibly due to partial compensation of the loss of the Asp-302–His-305 salt bridge by the substitution of the histidine with a smaller alanine side chain. Although the mutations destabilize coat protein monomers leading to assembly defects, they have only minor effects on the heat expansion and urea denaturation of assembled procapsids, as the contributions of the mutations are overcome by new inter-capsomer quaternary interactions.
Experimental Procedures
Materials
D2O (99.96%) for hydrogen exchange studies, DCl and NaOD were from Cambridge Isotope Laboratories (Tewksbury, MA). Urea (Molecular Biology grade, >99% pure) was from Fisher. Thermolysin from Bacillus thermoproteolyticus rokko was from Sigma.
Plasmids
The plasmid pID, which encodes the I-domain consisting of amino acids Ser-223–Val-345 of the full-length phage P22 coat protein, was previously described (19, 32, 34, 35). The D302A and H305A mutants were constructed by site-directed mutagenesis of the pID plasmid for the I-domain and the pPC plasmid for the full-length coat protein. The pPC plasmid has the genes for scaffolding (gene 8) and coat protein (gene 5) cloned into pET30b (a gift from Peter Prevelige) and is used for purification of procapsids. For complementation experiments to determine phage relative titers, the D302A and H305A mutations were also generated in the pMS11 plasmid (36).
Purification of Procapsids
Escherichia coli BL21 cells transformed with pPC encoding full-length WT, D302A, or H305A coat protein together with scaffolding protein were grown to mid-log phase at 37 °C with aeration. H305A and WT coat proteins were induced for 4 h at 37 °C, whereas D302A was induced overnight at 16 °C. Cells were harvested and resuspended in buffer B (50 mm Tris, 25 mm NaCl, 2 mm EDTA, pH 7.6). Lysozyme (200 μg/ml) and 0.1% (w/v) Triton X-100 were added before freezing the suspension at −20 °C. The cells were thawed, and Complete EDTA-free Protease Inhibitor Mixture (Roche Applied Science) was added according to the manufacturer's instructions together with 50 μg/ml DNase, 50 μg/ml RNase, 2 mm MgCl2, and 0.5 mm CaCl2 followed by incubation at room temperature for 30 min. Four freeze-thaw cycles were used to lyse the cells. The lysate was clarified by centrifugation at 32,000 × g and 4 °C for 15 min using a F-18 12X50 rotor (Thermo Scientific) followed by ultra-centrifugation at 206,000 × g in a T-865 rotor (Sorvall) for 40 min at 4 °C to pellet the procapsids. The procapsids were then resuspended by gentle shaking at 4 °C in buffer B and loaded onto a 150-ml Sephacryl S-1000 column (GE Healthcare) that was run at 4 °C with a flow rate of 0.2 ml/min. Fractions containing pure procapsids were pooled, sedimented by ultra-centrifugation, and resuspended in a small volume of buffer B.
Preparation of Full-length Coat Protein Monomers
To generate coat protein monomers, empty procapsid shells prepared as previously described (9) were denatured for 30 min at room temperature in 20 mm sodium phosphate buffer (pH 7.6) containing 6.75 m urea. To refold coat protein, an equal volume of phosphate buffer was added, and the sample was dialyzed three times against 1 liter of phosphate buffer at 4 °C using a 12-kDa cutoff dialysis membrane (Spectrum Labs) (24). The refolded protein was then centrifuged at 221,121 × g in a S120AT2 rotor (Sorvall) for 20 min at 4 °C to remove uncontrolled assembly product aggregates.
NMR Spectroscopy
Expression of the I-domain, purification, and 15N isotopic labeling for NMR studies were done as previously described (19, 31, 32). NMR spectra were collected on a Varian Inova 600 MHz spectrometer equipped with a cryogenic probe. All 1H chemical shifts were referenced to internal DSS (2,2-dimethyl-2-silapentane-5-sulfonate), whereas 15N chemical shifts were referenced indirectly as described in the literature (37). Assignments for the D302A and H305A I-domain mutants were obtained from 60-ms mixing time three-dimensional total correlation spectroscopy (TOCSY)-HSQC3 and 150-ms mixing time three-dimensional NOESY-HSQC spectra, starting from the published assignments for the WT I-domain, BMRB accession code 18566 (19). Fast ps-ns backbone dynamics were characterized using 1H,15N NOE experiments recorded at 600 MHz on a 0.1 mm sample of the D302A mutant and 0.5 mm samples of the WT and H305A mutant. The 1H,15N NOE values were obtained from interleaved spectra in which the proton signals were saturated for 3 s and control experiments replaced the saturation period with an equivalent preacquisition delay. Errors in the NOE values were calculated from the baseline noise in the saturated and control spectra as previously described (38). Hydrogen exchange (HX) experiments were performed on 15N-labeled I-domain samples in 20 mm sodium phosphate buffer. Protein concentrations were 0.2 mm for WT, 0.1 mm for D302A, and 0.5 mm for H305A. HX experiments for all three proteins were done at a temperature of 25 °C. Samples were lyophilized, dissolved in 99.96% D2O, and adjusted to pD 6.0 with DCl right before the experiments. Exchange rates were calculated with the program KaleidaGraph version 4.1 (Synergy Software) by fitting the amide proton cross-peak intensity decay for each residue to a three-parameter exponential decay function (39, 40),
where I0 is the initial peak intensity, kobs is the observed HX rate, and C is the baseline noise of the spectrum. Using the HX rates, ΔGHX was calculated from,
where R is the gas constant, T is absolute temperature, kobs is the observed HX rate constant, and kint is the intrinsic HX rate predicted for the I-domain amino acid sequence using the program SPHERE (41).
NMR pH Titrations of the I-domain
All pH titration experiments were done at a temperature of 25 °C. Titrations of aspartate and glutamate residues were monitored using 13 200-ms mixing-time three-dimensional 15N-edited NOESY-HSQC and two-dimensional 1H,15N HSQC spectra recorded on 1.6 mm I-domain samples between pH 2.9 and pH 8.6. Side-chain aspartate (Hβ1/Hβ2) protons and glutamate (Hγ1/Hγ2 or Hβ1/Hβ2) protons were identified from intra-residue NOEs to the backbone amide protons, and the pH dependence of these side-chain chemical shifts was used to obtain pKa values. The pH titration of the I-domain's unique histidine was monitored from the Hϵ1 aromatic-ring resonance of His-305 using one-dimensional 1H NMR spectra recorded on 0.33 mm samples in 99.96% D2O. The pH values were adjusted using 1 m DCl and 1 m NaOD as previously described to minimize drifts in pH (42). A total of 23 experiments distributed between pH 2.6 and 11.5 were used to characterize the titration of the histidine.
Solution pH values were measured with a MA235 pH meter equipped with a glass InLab Micro pH electrode from Mettler (Columbus, OH). The pH value was taken as the average of measurements before and after NMR experiments, which typically differed by <0.1 pH unit. To obtain pKa values, the chemical shift (δ) versus pH data were fit to a four-parameter modified Henderson-Hasselbalch equation,
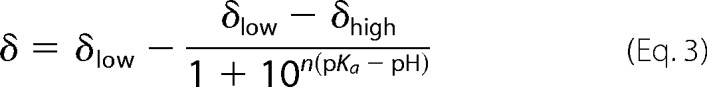 |
where pKa is the logarithmic acid ionization constant, δlow is the low pH chemical shift plateau, δhigh is the high pH chemical shift plateau, and n is the apparent Hill coefficient (42, 43).
Circular Dichroism (CD)
CD experiments were performed on a Pi-Star 180 spectropolarimeter from Applied Photophysics (Leatherhead, Surrey, UK). CD spectra were used to evaluate the effects of the substitutions on the secondary structures of WT, D302A, and H305A full-length coat protein monomers at concentrations of 0.2 mg/ml in 20 mm phosphate buffer (pH 7.6). Wavelength scans were done between 195 and 250 nm using a 1-nm increment, a bandwidth set to 3 nm, and a time-per-point averaging of 30 s (leading to a spectrum acquisition time of ∼25 min). The 1-mm path length cuvette used for the experiments was thermostated to 20 °C.
For equilibrium denaturation studies of the I-domain, samples were diluted to a final protein concentration of 0.4 mg/ml (28 μm) in 20 mm sodium phosphate buffer (pH 7.6) and mixed with 9 m urea using a Hamilton Microlab 50 titrator to generate a range of final urea concentrations between 0 m and 6 m in 0.1 m increments. Exact urea concentrations were determined by refractometry (44) using a Carl Zeiss refractometer (Oberkochen, Germany). The samples were incubated at 20 °C for 3 h to reach equilibrium followed by measurement of the CD signal at 220 nm (averaged over 20 s). ΔΔGu values were calculated from the CD denaturation data as a function of urea concentration using a six-parameter non-linear least-squares fit (45). To look at the pH dependence of stability, urea denaturation series for the WT, D302A, and H305A I-domains were performed at pH values of 4.5, 6.0, 7.6, 8.4, 9.0, 10.0, and 11.0.
Time Course of Coat Protein Thermolysin Digestion
Protease digestion experiments with WT, D302A, or H305A full-length monomeric coat protein were done as previously described (31). Briefly, thermolysin was prepared in 50 mm Tris (pH 8.0) sample buffer containing 0.5 mm CaCl2. Coat protein monomers were digested with an enzyme:substrate ratio of 1:200 at a temperature of 20 °C. For each time point, an aliquot of the sample was removed and quenched with sample buffer containing 4.4% SDS and 75 mm EDTA. The samples were then heated for 5 min at 95 °C before analysis by 16% Tricine-SDS-PAGE (46).
Procapsid Assembly Reactions
For assembly reactions, scaffolding protein in 20 mm phosphate buffer (pH 7.6) and potassium acetate were each pipetted into a cuvette to final concentrations of 0.5 mg/ml and 40 mm, respectively. The reaction was initiated by the addition of coat protein monomers (in 20 mm phosphate buffer, pH 7.6) to a final concentration of 0.5 mg/ml. The formation of procapsids was monitored by light scattering at 500 nm using an Amino-Bowman AB2 fluorimeter with the cuvette thermostatted to 20 °C.
Determination of Phage Relative Titer
For these experiments we used a P22 phage strain that carries an amber mutation in the coat protein gene 5 (5−am N114) and the clear plaque c1–7 allele to prevent lysogeny. The non-amber suppressing host strain Salmonella enterica serovar Typhimurium, DB7136 (leuA414am, hisC525am, su0), has been previously described (47, 48). DB7136 cells were transformed with pMS11 plasmid encoding WT gene 5 or gene 5 carrying the D302A or H305A mutations and grown on LB agar plates supplemented with 100 μg/ml ampicillin. Gene 5 amber phages plated on lawns of DB7136 cells carrying each of the plasmids were induced by the addition of 1 mm isopropyl 1-thio-β-d-galactopyranoside to the soft agar overlay. The experiments were repeated at temperatures ranging from 23 to 41 °C. The relative titer was calculated by dividing the titer at the experimental temperature by the titer of phage complemented with WT gene 5 at 30 °C.
Heat Expansion of Procapsids
Procapsids were diluted to 1 mg/ml in buffer B at pH 7.6. Aliquots of 20 μl were incubated at 23, 49, 51, 53, 58, 63, 67, 70, or 72 °C for 15 min in a thermal cycler (14, 27). The aliquots for each temperature were mixed with 3× agarose sample buffer containing 50% glycerol, 0.25% bromphenol blue, and TAE buffer (40 mm Tris, 20 mm acetic acid, 1 mm EDTA, pH 8.3). Aliquots of 5 μl were loaded on a 1% agarose gel that was run at 100 V for 45 min in TAE buffer. Coomassie Blue stain was used to visualize protein bands.
Urea Titration of Empty Procapsid Shells
Titrations of 0.1 mg/ml WT, D302A, and H305A empty procapsid shells were done in 20 mm phosphate buffer at pH 7.6 using a denaturant concentration range from 0 to 7.6 m urea. Samples were incubated for 16 h at room temperature before measuring light scattering at 500 nm to detect intact shells (49).
Results
Ion-pair Interactions in the I-domain
Because the coat protein I-domain is located on the surface of the P22 capsid (32), we reasoned ion pairs could be important for its stability, the stability of full-length coat protein monomers, and even the entire capsid. Considering ionizable amino acids, the I-domain has 11 acidic aspartate and glutamate residues, 11 basic arginine and lysine residues, 1 histidine, and 1 tyrosine. Although the precision of NMR structures is often too low to determine electrostatic interactions, the NMR structure of the I-domain is of high quality (32). The distribution of charged residues in the I-domain NMR structure is shown in Fig. 1A. The four residue pairs Asp-253–Lys-311, Asp-271–Lys-268, Asp-302–His-305, and Asp-317–Lys-237 come within hydrogen-bonded salt-bridging distances of each other (Table 1). Another three residue pairs, Asp-292–Lys-279, Glu-307–Arg-299, and Asp-316–Arg-235 (Table 1), although too far away to satisfy the requirements of a salt bridge, could form non-hydrogen-bonded ion-pair interactions given the long range nature of electrostatic interactions (50, 51).
To confirm the ion-pair interactions suggested by the NMR structure (Fig. 1A; Table 1), we characterized the pKa values of the acidic residues in the I-domain using a series of three-dimensional 1H,15N NOESY-HSQC spectra collected as a function of pH. Intra-residue NOEs between backbone amide and side-chain Hβ or Hγ protons were used to determine the pKa values of aspartate and glutamate residues in the I-domain. The opposite approach of characterizing basic residues was not used, as the amide protons used for NMR detection in 15N-edited spectra are lost at high pH due to fast exchange with solvent (52).
The pH titrations of the acidic residues in the I-domain showed three types of behavior illustrated by the representative data in Fig. 1B. First, residues in the disordered segments of the protein such as Asp-246 that do not participate in ion pairs have pKa parameters near random coil values (4.0 for aspartate, 4.4 for glutamate; Ref. 43). Second, acidic residues that are within ion-pair distance but do not form hydrogen-bonded salt bridges have pKa values shifted lower than random coil values by up to 1 pH unit. The lowest measurable pKa value was 3.0 for Asp-316 (Fig. 1B). The shift of pKa values to acidic pH for these residues indicates that they need a need a larger hydronium ion concentration (lower pH) to become protonated. They resist protonation as this would break the stabilizing ion-pair interactions in which they participate. The last class of residues, exemplified by Asp-317 (Fig. 1B), have side-chain chemical shifts that are invariant with pH. The pKa values of these residues are shifted lower than the midpoint of acid denaturation of the I-domain (estimated to be pH 2.8 based on NMR experiments) and cannot be measured quantitatively. All four of the aspartate residues that meet the distance criterion for a hydrogen-bonded salt bridge in the NMR structure, as well as Asp-292, belong to this latter group. As shown in Table 1 these acidic residues have the shortest ion-pair distances in the NMR structure closest to the ensemble average and the largest fractions of conformers that meet ion-pair criteria in the ensemble of NMR structures.
Of the ionizable residues that do not participate in salt bridges or ion pairs, the unique tyrosine 326 at the end of helix α1 is solvent-exposed, as are Glu-323 and Asp-336. These do not appear to form polar interactions with the rest of the protein. The acidic residues Asp-244 and Asp-246 are in the disordered D-loop and are not involved in ion pairs in the isolated I-domain (32). When coat protein assembles into procapsids, however, Asp-244 and Asp-246 form ion-pair interactions linking capsomer subunits (32, 36). Of the 11 basic residues, Lys-249, Arg-255, Arg-269, Lys-272, and Lys-286 are not involved in salt bridges or ion pairs in the I-domain. Residue Arg-269, however, may form an ion pair between coat protein subunits in capsids (32). Residues Lys-249 and Lys-286 are part of the disordered D- and S-loops, respectively. Based on a cryo-EM reconstruction of the phage P22 coat protein calculated using the I-domain NMR structure (32), residues Arg-255 and Lys-272 are part of a positively charged surface that docks the I-domain to a negatively charged patch on the core of coat protein and thus appear to be important in latching the I-domain to the rest of the coat protein core structure.
The Asp-302–His-305 Salt Bridge Stabilizes the I-domain by 4 kcal/mol
The Asp-302–His-305 salt bridge is of particular interest. The coat protein variant D302G is one of the tsf mutants identified from genetic studies of the P22 coat protein (33). Additionally, histidines have pKa values near neutral pH that are often physiologically important. The pH titration of His-305, monitored by the pH-induced changes in the chemical shift of its aromatic ring Hϵ1 resonance, is shown in Fig. 1C. Also shown in Fig. 1C is the pH titration of the unresolved Hϵ1 signals from the His6 tag used to purify the protein, which serves as an internal standard. The unresolved resonances from the His6-tag titrate with a pKa of 6.7, as expected for histidines in unstructured peptides (53). By contrast, His-305 titrates with a pKa of 9.0, which indicates that the charged state of His-305 is thermodynamically favorable.
The contribution of charging His-305 to the stability of the I-domain can be calculated through a thermodynamic linkage analysis (42, 54) using the equation,
where pKa,f is the value for His-305, and pKa,u is the value for a random coil histidine. At 25 °C, the charged state of His-305 is thus predicted to stabilize the I-domain by 3.4 kcal/mol compared with the uncharged histidine. This corresponds roughly to half of the global stability of the I-domain at neutral pH (ΔGu ∼ 6.9 kcal/mol). A corresponding shift of 2.5 pH units would lower the pKa of the salt-bridge partner residue Asp-302 to pH 1.5 compared with the random coil pKa of 4.0 for an aspartate. Consistently, Asp-302 is one of the residues we observed not to shift with pH (Table 1), indicating that its pKa must be below the pH 2.8 mid-point of acid denaturation for the I-domain.
Further insights into the stabilizing role of the Asp-302–His-305 salt bridge were obtained from equilibrium denaturation experiments on the WT I-domain as a function of pH (Fig. 2A). The maximum stability of 6.9 kcal/mol for the WT I-domain occurs at a pH of 7.6 (18) (Fig. 2B). In the range between pH 7.6 and 4.5 there is a shallow decrease in stability of ∼1.5 kcal/mol that corresponds to the onset of acid denaturation of the protein. Above pH 7.6 there is a steep decrease in the stability of the WT I-domain of ∼4.5 kcal/mol. The stability profile data for the WT I-domain were fit to the Henderson-Hasselbalch equation to obtain an apparent pKa value for the loss of stability at basic pH. For the fit we treated the stability of the protein as invariant in the range between pH 4.5 and 7.6 (e.g. neglecting acid denaturation) because of the rather limited number pH points examined. The apparent pKa of 9.0 ± 0.4 obtained from the fit matches the pKa of 9.0 obtained for His-305 by NMR. Moreover, the loss in stability of ∼4.5 kcal/mol for the WT I-domain (Fig. 2B) is consistent with the calculated stability loss of 3.4 kcal/mol from the thermodynamic linkage analysis of the pKa shift of His-305 (Equation 4). Taken together, these observations suggest that the decrease in stability of the WT I-domain of above pH 7.6 is due to the disruption of the Asp-302–His-305 salt bridge, as His-305 becomes uncharged at basic pH.
FIGURE 2.

Dependence of I-domain stability on pH. A, urea denaturation curves monitored by CD at 222 nm for the WT I-domain. The curves show fits to a 6-parameter equation (45) from which ΔGu0 and m values were obtained. The data for pH 7.6 (31) and 6.0 (57) were previously published. B, the pH dependence of I-domain stability obtained from equilibrium denaturation experiments as a function of urea (shown in panel A for the WT). The ΔGu0 versus pH data were fit to Equation 3. For WT we obtained a pKa value of 9.0 ± 0.4, consistent with the titration of His-305 (Fig. 1C). For D302A and H305A I-domains, ΔGu0 decreased with poorly defined pKa values >pH 10 and 11, respectively. The loss of stability for the mutants at high pH is likely due to the deprotonation of Arg and Lys residues.
The D302A Substitution Is More Destabilizing Than H305A
To further investigate the role of the Asp-302–His-305 salt bridge in stabilizing the I-domain, alanine substitutions were made for each residue. We first investigated the pH dependence of stability for the D302A and H305A I-domain mutants (Fig. 2B). Neither shows the apparent pKa of 9.0 seen in the stability profile of the WT I-domain. Rather, the D302A and H305A mutants showed stability losses at basic pH, but with apparent pKa values of >pH 10 and 11, respectively (Fig. 2B). These high pKa values for the mutants are likely due to deprotonation of Arg and Lys residues. At neutral pH the D302A mutant shows a 4 kcal/mol loss in stability compared with the WT I-domain (Fig. 2B), consistent with a stabilizing contribution of ∼4 kcal/mol for the Asp-302–His-305 salt bridge. The H305A mutant, however, shows only a slight ∼1.5 kcal/mol decrease in stability to unfolding compared with the WT at neutral pH (Fig. 2B).
Basis for the Stability Differences between the D302A and H305A Mutants
The data above present a conundrum; why is the D302A substitution more destabilizing than H305A? To address this question at residue-level resolution, we looked at the effects of the mutations on NMR chemical shifts, 1H,15N NOE relaxation parameters, and hydrogen exchange protection. The chemical shift of a nucleus in an NMR spectrum will be perturbed if its magnetic environment is altered due to either a structural change or proximity to the substituted site. In Fig. 3 we compare 1H,15N chemical shift differences between the WT I-domain and the mutants. The largest chemical shift differences occur for residues Val-300–Val-306 in the β4-β5 hairpin, which is the site of the substitutions (Fig. 3, A–D). The next largest differences are for the Thr-258–Gly-266 segment in the β2-β3 hairpin, which is adjacent to the β4-β5 hairpin. Additional smaller differences for the Ser-333–Ala-335 segment are conserved between the D302A and H305A mutants but are at a site in the I-domain distant from the substitutions (Fig. 3E). Because these perturbations are similar between D302A and H305A, however, it is unlikely that they can account for the stability differences between the mutants.
FIGURE 3.

Chemical shift changes in the I-domain accompanying the D302A and H305A substitutions at a temperature of 20 °C. A, superposition of the 1H,15N HSQC spectra for the WT I-domain (green) and the D302A mutant (red). (B) Superposed 1H,15N HSQC spectra for the WT I-domain (green) and the H305A mutant (red). In A and B large chemical shift changes are indicated by arrows, drawn from the position of the cross-peak in the WT to the position of the cross-peak in the mutant spectrum. Composite chemical shift differences were calculated as abs [ΔdHN + 0.1(ΔdN)] for D302A (C) and H305A (D). Errors in the composite chemical shift differences are estimated to be 0.08 ppm based on the acquired digital resolutions of the 1H and 15N dimensions. Segments with the largest chemical shift perturbations are labeled. E, mapping of the chemical shift differences between WT and D302A onto the I-domain structure: red, Val-300–His-305 reverse turn between strands β4-β5; orange, Thr-258–Gly-266 reverse turn between strands β2-β3; pink, long range effects for the Ser-333–Ala-335 segment. The corresponding structural mapping for H305A is nearly identical to that for D302A and is not shown.
An alternative possibility is that the mutations affect protein dynamics rather than the average structure in solution. Backbone flexibility can be investigated with the 1H,15N NOE, an NMR cross-relaxation parameter sensitive to dynamics on the ps-ns timescale. Rigid segments of a protein tend toward the maximum 1H,15N NOE value of 0.8, whereas flexible segments have lower values approaching the theoretical minimum of −3. Fig. 4A compares 1H,15N NOE data for the WT and mutant I-domains. The 1H,15N NOE sequence profile for the WT I-domain indicates flexibility in the N and C termini, the D-loop, and the S-loop as previously described (32). The dynamic character of the chain termini and two loops is conserved in the D302A and H305A mutants. A notable feature of the D302A profile (Fig. 4A) is that the site of the substitution experiences a large increase in dynamics, with residues Ala-302 and Gly-303 giving 1H,15N NOE values of 0.60 and −0.25, respectively. By contrast, the H305A substitution has small effects, showing 1H,15N NOE values of 0.82 and 0.68 for Ala-302 and Gly-303, respectively. In contrast to D302A, the H305A mutation does not appear to perturb the dynamics of the Asp-302–His-305 turn, suggesting the substituted alanine is accommodated in the structure as well as the histidine.
FIGURE 4.

Effects of mutations on I-domain dynamics. A, 1H,15N NOE values monitoring backbone dynamics on the ps-ns timescale. All 1H,15N NOE data were obtained at 20 °C. Because the least stable mutant D302A is prone to aggregation, data were collected using a 0.1 mm sample of D302A compared with 0.5 mm for WT and H305A. The lower protein concentration for D302A decreased NMR sensitivity; consequently the 1H,15N NOE data for D302A have larger experimental uncertainties. B, hydrogen exchange protection, expressed as ΔGHX values (Equation 2). All hydrogen exchange experiments were done at a pD of 6.0 and a temperature of 25 °C.
Finally, we examined the effects of the two substitutions on the microscopic stability of hydrogen bonds to structural perturbations that allow amide protons to exchange with deuterons (55–57). The largest ΔGHX values (calculated from Equations 1 and 2) in hydrogen exchange experiments (Fig. 4B) should correspond to the free energy differences for protein unfolding (ΔGu0) measured from equilibrium denaturation experiments followed by spectroscopic techniques such as circular dichroism (e.g. Fig. 2B). For all three proteins, the ΔGHX values are larger than the ΔGu0 values from equilibrium denaturation experiments by ∼2 kcal/mol. Reasons include that the ΔGHX values are not affected by the cis/trans isomerization of Pro-310 in the I-domain, the slightly larger stability of proteins in D2O compared with H2O, and the averaging of stabilities that occurs in spectroscopic experiments (40, 55–57). The differences in stabilities of ∼4 kcal/mol between WT and D302A and ∼2 kcal/mol between WT and H305A (Fig. 4B), however, are in agreement with the differences in ΔGu0 values obtained from equilibrium unfolding experiments measured by CD (Fig. 2B). In addition to the effects on global stability, the mutants appear to have more subtle effects on the stabilities of individual secondary structure elements. Thus, the D302A mutant shows a large selective decrease in protection of amide protons from the first three β-strands of the protein β1-β3, and the H305A mutant shows a smaller selective decrease in ΔGHX values for the βiii-β6 segment (Fig. 4B). Neither the β1-β3 nor βiii-β6 segments are close in the structure to the sites of the mutations, whose exchange protection appears to be unaffected. These long range effects on hydrogen exchange protection may be related to the long range chemical shift perturbations observed for the two variants (Fig. 3E). Non-cooperative unfolding of the mutants is also suggested by temperature melt experiments (data not shown). The midpoints for thermal unfolding of the three I-domains measured by CD at 220 nm were 50 °C for WT, 48 °C for H305A, and 34 °C for D302A. The order of the thermal melting points (WT > H305A ≫ D302A) is consistent with the stabilities of the I-domains in urea denaturation and hydrogen exchange experiments. In addition to reduced melting points, the D302A and H305A I-domain mutants showed shallower temperature denaturation transitions, suggesting that thermal unfolding of the mutants is less cooperative than WT. Thus, both mutations appear to increase the number of partially folded states accessible to the I-domain, with D302A having the larger effect as evinced by increased dynamics in 1H,15N NOE experiments together with decreased and less uniform protection in hydrogen exchange experiments.
The D302A and H305A Substitutions Destabilize Coat Protein, Impairing Phage Assembly
We next determined if the D302A and H305A substitutions affected the ability of full-length coat protein to fold and assemble in vivo. The far-UV CD spectra of the mutant coat proteins are similar to that of WT, indicating there are no major changes in secondary structure (Fig. 5A). This is consistent with the NMR chemical shift differences for the I-domain segment of coat protein, which show large differences only close to the sites of the substitutions (Fig. 3) and indicate that the I-domain structure is largely unaffected by the mutations. Consistent with the destabilizing effects on the I-domain, we observed that the D302A substitution also destabilizes the full-length coat protein as monitored by proteolysis. In previous experiments we showed the I-domain is resistant to thermolysin digestion of the full-length WT coat protein (31). If the substitutions in the I-domain affected the overall stability of coat protein monomers, we would expect an increase in the rate of digestion. Fig. 5B compared SDS gels for the time-course of thermolysin digestion of WT, D302A, and H305A coat protein monomers. As seen previously (31), the full-length WT coat protein was digested, but a thermolysin-resistant band corresponding to the I-domain was readily visible (marked by an arrow in Fig. 5B). By contrast, the D302A coat protein was completely digested within 30–60 min of incubation with thermolysin, as there were no bands consistent with the molecular mass of the I-domain on the gel. The H305A coat protein has a protease digestion time-course similar to the WT, consistent with its smaller effect on the stability of the I-domain.
FIGURE 5.

Effect of D302A and H305A substitutions on full-length coat protein stability and capsid assembly. A, CD spectra showing secondary structure conservation. B, stability of coat proteins monitored by the time-course of thermolysin proteolysis as analyzed by SDS-PAGE. The star marks the band of full-length coat protein, and the arrows indicate the protease-resistant I-domain fragment. MWM, molecular mass markers. C, comparison of procapsid assembly rates for WT, D302A, and H305A coat protein monomers upon the addition of scaffolding protein. Formation of procapsids with time was monitored by light scattering at 500 nm. D, plating efficiency of a P22 phage strain unable to synthesize coat protein (gene 5 amber) when complemented in vivo by the expression of WT, D302A, or H305A coat protein from a plasmid. The number of phages at each temperature is compared with the number when WT coat protein is expressed at a permissive temperature of 30 °C to calculate the relative titer. A decrease in relative titer occurred when the variant coat protein was less capable of complementing the gene 5 amber phage than WT coat protein expressed at 30 °C.
We next tested the ability of the WT and variant full-length coat proteins to assemble in vitro (Fig. 5C). When scaffolding protein was added to WT coat protein monomers, procapsids were rapidly produced in the typical time of ∼10 min (29, 58). The H305A coat protein was also able to assemble, albeit with slower kinetics that the WT coat protein. The D302A coat protein had the slowest assembly kinetics and the lowest yield. Thus, these data are also consistent with D302A causing a more severe defect of coat protein function than H305A in both the isolated I-domain and full-length coat protein monomers. Here, the lack of proper folding into assembly-competent monomers, as observed by the increase in the rate of thermolysin proteolysis and the absence of a stable I-domain fragment, led to a compromised capability to assemble.
Finally, we examined the ability of WT, D302A, or H305A coat proteins to support the production of phages in vivo. The coat proteins were expressed from a plasmid in Salmonella cells infected with a phage unable to make its own coat protein at temperatures from 23 to 41 °C as described under “Experimental Procedures.” If the plasmid-expressed coat protein were able to fold and assemble into phage at a particular temperature, we would expect to see clear plaques on the lawn of cells layered on the agar plate. The relative titer in the complementation experiments is defined as the titer generated at a particular temperature with plasmid-expressed WT or mutant coat protein divided by the titer from WT coat protein expressed from the plasmid at 30 °C (Fig. 5D). As expected, plasmid-encoded WT coat protein was able to support growth of the amber phage at all temperatures. By contrast, the D302A coat protein led to a temperature-sensitive phenotype at temperatures >37 °C. The H305A coat protein showed no temperature-sensitive or cold-sensitive phenotype in the complementation experiments. This again is consistent with the stability measurements on the isolated I-domain, where D302A is strongly destabilizing but H305A has only a small effect.
Effect of D302A and H305A Substitutions on Capsid Maturation and Denaturation Stability in Vitro
Some coat protein mutations drastically affect the ability of procapsids to undergo maturation to capsids. Maturation can be recapitulated in vitro by a process we call “heat expansion” (27). We tested the ability of procapsids assembled from coat proteins carrying the two salt-bridge mutations to undergo heat expansion (Fig. 6A). Capsid expansion is readily observable on an agarose gel, as the expanded heads have a larger diameter than procapsids and thus migrate through the gel more slowly. As expected, WT procapsids were completely expanded after incubation for 15 min at 70 °C. The D302A and H305A procapsids both show only a modest decrease of ∼3 °C in the temperature required to cause complete expansion in 15 min, consistent with their ability to support phage production.
FIGURE 6.

Effect of the D302A and H305A substitutions on procapsid stability. A, the ability of procapsid to undergo the expansion that occurs upon capsid maturation was assessed by heating procapsids for 15 min at different temperatures. Expanded heads (ExH) run more slowly on agarose gels because of their larger diameters compared with procapsids (PC). B, stability of capsids to urea denaturation. Apparent midpoints for urea denaturation (Cm) from fits of the pseudo-equilibrium data to a six-parameter model (45) were are as follows: WT, 5.2 ± 0.1 m; D302A, 5.0 ± 0.2 m; H305A, 5.4 ± 0.1 m. Based on duplicate experiments the uncertainties in the Cm values are ∼0.2 m urea.
We also tested the effect of the D302A and H305A substitutions on the overall stability of empty procapsid shells against urea denaturation (Fig. 6B). In this experiment we incubated shells with increasing concentrations of urea for a set time of 16 h and monitored the denaturation of the procapsid lattice by light scattering at 500 nm. The incubation time is important as this is a “pseudo-equilibrium” experiment, which is not reversible in the usual sense (e.g. scaffolding protein is needed for assembly, and coat protein monomers are much less stable to denaturant than shells). The data at denaturant concentrations <1.4 m urea are not shown, as the transition in this region is due to extraction of any remaining scaffolding protein and is not important for this analysis. In analogy to the experiments in Fig. 2A, we initially did experiments comparing the relative stability of WT shells at pH 7.6 and 10 (not shown). The midpoint for urea denaturation of procapsids was slightly lower at pH 10 (4.4 ± 0.1 m) compared with a physiological pH of 7.6 (5.1 ± 0.1 m). In contrast to the I-domain, which only has 1 histidine at position 305, the 430-aminoacyl coat protein has 2 histidines and 1 cysteine that could titrate near neutral pH as well as 8 tyrosines that could titrate near pH 10.1.
To get more precise information on the effects of the Asp-302–His-305 salt bridge on particle stability, we compared the urea titrations of WT, D302A, and H305A shells (Fig. 6B). For WT shells the denaturation midpoint occurred at 5.2 ± 0.1 m urea. The D302A shells had only a slightly lower midpoint of 5.0 ± 0.2 m urea, whereas the H305A shells show a slight but reproducible increase in stability with a midpoint of 5.4 ± 0.1 m urea. By comparison, other coat protein mutants have much larger effects on procapsid stability, with some decreasing the mid-point of urea denaturation by as much as 1–3 m urea (27, 28, 49).
In conclusion, neither the D302A nor H305A substitution had a large effect on the stability of the capsid lattice. Based on these data, we hypothesize that the Asp-302–His-305 salt bridge is critical for folding of the I-domain and full-length coat protein monomers leading to defects in the process of procapsid assembly. Once the procapsids are assembled, however, the new quaternary interactions stabilizing the procapsid lattices overcome the destabilizing effects of the substitutions on coat protein monomers (60) so that the integrities of the assembled capsid are sustained.
Discussion
The size of a virus genome is constrained by the size of its capsid. Thus, there is evolutionary pressure against genome size expansion. The insertion of the I-domain into gene 5 adds ∼375 base pairs (bp) to the 43,400-bp DNA of the P22 genome (61) or ∼1% of the total genome length. Athough P22 mutants have been found that over-package DNA by ∼2000 bp, their capsids are fragile compared with those of WT phage (62). Thus, we infer the addition of the I-domain must bestow evolutionary advantages that compensate for the higher DNA packing density resulting from its insertion. In previous work we showed the I-domain facilitates the folding of full-length coat protein by serving as the folding nucleus and also contributes about half of the ΔG of stabilization for monomeric coat protein (31). Additionally, the I-domain D-loop contributes to procapsid assembly by making critical inter-capsomer interactions across the icosahedral 2-fold symmetry-axis (32, 36). From these data the I-domain clearly has several critical roles in coat protein folding and assembly. Here, we asked how the I-domain itself is stabilized and the extent to which interactions that stabilize the I-domain module are important for the stability of the full-length coat protein and its resulting capsid assemblies.
Salt bridges and ion-pairs in the I-domain link secondary structure elements that form the six-stranded β-barrel structure of this module (Fig. 1A, Table 1). Based on the present work, the salt bridge between Asp-302 and His-305 is especially important for the stability of the I-domain. However, replacement of the histidine does not destabilize the I-domain as much as substituting the aspartate in the salt bridge. A possible explanation for the greater loss of stability with the D302A mutant is that aspartate is preferred compared with alanine at the first position in a β-turn (63). Thus, the alanine substitution would not be accommodated as well as an aspartate at position 302, perhaps accounting for the increased dynamics observed for the Asp-302–His-305 β-hairpin in the D302A mutant (Fig. 4A). This explanation, however, cannot account for the ∼4-kcal loss in stability experienced by the WT I-domain between physiological and basic pH. The loss of stability with the D302A substitution matches that when the WT salt bridge is disrupted by basic pH. Rather, the 1–2 kcal/mol decrease in stability for the H305A mutation is smaller than what would be expected if the mutation only disrupted the Asp-302–His-305 salt bridge. These observations suggest that the loss of the salt bridge in the H305A mutation is partially compensated by the alanine substitution. His-305 forms close contacts with Thr-258 and Thr-260 from strand β2 in the NMR structure of the I-domain. Unfavorable contacts such as steric clashes with the threonines or other residues in the I-domain may be relieved when the histidine is replaced by a smaller alanine side chain. Alternatively, the alanine substitution could be disfavored in the denatured state, raising its free energy and thereby conferring stability to the native state (64).
An analysis of coat protein sequences of phages related to P22 showed that in homologs with ∼60–70% sequence identity, position 302 is occupied by either an aspartate or asparagine.4 Both residues are favorable for the first position of a reverse turn. The histidine at position 305 is not conserved and is substituted by residues aspartate or asparagine. Thus, smaller side chains may be preferred at position 305 in lieu of a histidine ring. Nevertheless, in all cases the homologous residues have the potential for H-bonding. The fact that His-305 is not conserved suggests that its function is specific to phage P22. In this regard it is interesting to note, however, that recent NMR work on the I-domain from the distantly related phage CUS-3 identified a proton bound to an imidazole nitrogen atom on the aromatic ring of His-277 with an unusual shift of 11.75 ppm (59). For a histidine ring Nδ1/Nϵ2 proton to be protected and to have such an unusual shift strongly suggests that it is involved in an important stabilizing hydrogen-bonding interaction. His-277 in CUS-3 titrates with a pKa of 8.3, which from Equation 4 predicts a stabilizing contribution of ∼2.5 kcal/mol from its charged state, similar to that of His-305 in the I-domain from phage P22. Thus, although His-277 is not a sequence or structural homolog to His-305 in the P22 I-domain, it may nevertheless modulate the pH dependence of the CUS3 I-domain in a similar way, boosting its stability near physiological pH.
In summary, we have shown that the stability contributions of the Asp-302–His-305 salt bridge are context-dependent in the hierarchy of structural complexity going from the I-domain module to the coat protein monomer building block to the very stable capsid that protects the phage genome. Loss of the Asp-302–His-305 salt bridge at high pH as well as the D302A substitution destabilizes the I-domain module (Fig. 2B) and thereby the coat protein monomer (Fig. 5B), leading to impaired procapsid assembly (Fig. 5C) and a decrease in production of phage in vivo (Fig. 5D). By contrast, once icosahedral particles are assembled, loss of the salt bridge has relatively minor effects on the stability of procapsids to heat expansion (Fig. 6A) or urea denaturation (Fig. 6B). The position of the Asp-302–His-305 salt bridge in the context of the procapsid structure is illustrated in Fig. 7A, which shows the asymmetric unit of the phage P22 T = 7 (T = triangulation number) icosahedron. The salt bridge is located on the surface of the procapsid and is isolated from other coat-protein monomers related by the 6-fold symmetry axis. Interestingly, the salt bridges of adjacent monomers come in close proximity along the dyad 2-fold symmetry axis illustrated by the blue and brown coat protein monomers in Fig. 7B, with the Asp-302 and His-305 residues seemingly poised to form subunit-swapped salt bridges between coat protein monomers. The D-loops, which occur just below the Asp-302–His-305 salt bridge in the view of the I-domain structure shown in Fig. 1A, form intercapsomer salt bridges that stabilize the procapsid along the same 2-fold symmetry axis, shown in Fig. 7B (32, 36). Clearly the network of charged residues brought together in the assembled procapsids would lead to more complex electrostatic interactions than in coat protein monomers or the isolated I-domain. The stability of the capsid shell will depend on the totality of tertiary and quaternary interactions formed by its coat protein subunits. New interactions formed in the procapsid, including the intersubunit salt bridges formed by the D-loops (32, 36) as well as other types of polar and non-polar interactions, apparently override the loss in stability caused by disruption of the Asp-302–His-305 salt bridge. Thus the effects of the D302A mutation on procapsid denaturation are not commensurate with those on procapsid assembly. For the design of nanomaterials based on phage particles or other types of supramolecular assemblies, this implies that perturbations in the building blocks may not be additive in the assembled product, and that changes in subunits could have different consequences on assembly and disassembly pathways.
FIGURE 7.

The Asp-302–His-305 salt bridge in the context of the capsid structure. A, location of Asp-302–His-305 salt bridge on the capsid surface. The view on the left is from the outside looking at the surface of the capsid. The view on the right is along the width of the capsid shell. Subunits of the icosahedral T = 7 (T = triangulation number) capsid are shown in different colors, with Asp-302 in red and His-305 in blue space-filling spheres. B, view along the 2-fold of the asymmetric unit (corresponding to the blue and brown subunits in A). The Asp-302–His-305 salt bridge is poised to swap partners between subunits related by the dyad axis. The D-loops, which link monomers in the capsid through Asp-244–Arg-299, Asp-246–Arg-269, and Lys-249–Glu-81 salt bridges (32), are shown with thicker ribbons colored orange and turquoise.
Author Contributions
C. H., K. J. R., and A. T. A. conducted the NMR experiments, C. H. performed the I-domain urea denaturation experiments, and O. O. and T. M. did the phage and coat protein experiments. All authors contributed to the analysis of the data. C. M. T. and A. T A. wrote the manuscript and designed the project.
Acknowledgments
We thank Latasha C. R. Fraser and Dr. Margaret Suhanovsky for data on the pH dependence of ΔGu0 for the WT I-domain, Therese Tripler for an NMR sample of the CUS-3 I-domain, and the students of MCB5896, Practicum in NMR Spectroscopy, for performing the NMR pH titrations of histidines in the I-domains of phage P22 and CUS-3.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM076661 (to C. M. T. and A. T. A.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
S. Casjens, personal communication.
- HSQC
- heteronuclear single quantum correlation
- HX
- hydrogen exchange
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
References
- 1. Caspar D. L. D., and Klug A. (1962) Physical principles in the construction of regular viruses. Cold Spring Harbor Symp. Quant. Biol. 27, 1–24 [DOI] [PubMed] [Google Scholar]
- 2. Liu Z., Qiao J., Niu Z., and Wang Q. (2012) Natural supramolecular building blocks: from virus coat proteins to viral nanoparticles. Chem. Soc. Rev. 41, 6178–6194 [DOI] [PubMed] [Google Scholar]
- 3. Parent K. N., Deedas C. T., Egelman E. H., Casjens S. R., Baker T. S., and Teschke C. M. (2012) Stepwise molecular display utilizing icosahedral and helical complexes of phage coat and decoration proteins in the development of robust nanoscale display vehicles. Biomaterials 33, 5628–5637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Flenniken M. L., Uchida M., Liepold L. O., Kang S., Young M. J., and Douglas T. (2009) A library of protein cage architectures as nanomaterials. Curr. Top Microbiol. Immunol. 327, 71–93 [DOI] [PubMed] [Google Scholar]
- 5. Kang S., Uchida M., O'Neil A., Li R., Prevelige P. E., and Douglas T. (2010) Implementation of p22 viral capsids as nanoplatforms. Biomacromolecules 11, 2804–2809 [DOI] [PubMed] [Google Scholar]
- 6. Patterson D. P., Prevelige P. E., and Douglas T. (2012) Nanoreactors by programmed enzyme encapsulation inside the capsid of the bacteriophage P22. ACS Nano 6, 5000–5009 [DOI] [PubMed] [Google Scholar]
- 7. O'Neil A., Prevelige P. E., Basu G., and Douglas T. (2012) Coconfinement of fluorescent proteins: spatially enforced communication of GFP and mCherry encapsulated within the P22 capsid. Biomacromolecules 13, 3902–3907 [DOI] [PubMed] [Google Scholar]
- 8. King J., Lenk E. V., and Botstein D. (1973) Mechanism of head assembly and DNA encapsulation in Salmonella phage P22 II: morphogenetic pathway. J. Mol. Biol. 80, 697–731 [DOI] [PubMed] [Google Scholar]
- 9. Prevelige P. E. Jr., Thomas D., and King J. (1988) Scaffolding protein regulates the polymerization of P22 coat subunits into icosahedral shells in vitro. J. Mol. Biol. 202, 743–757 [DOI] [PubMed] [Google Scholar]
- 10. Zlotnick A., Suhanovsky M. M., and Teschke C. M. (2012) The energetic contributions of scaffolding and coat proteins to the assembly of bacteriophage procapsids. Virology 428, 64–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Parent K. N., Zlotnick A., and Teschke C. M. (2006) Quantitative analysis of multi-component spherical virus assembly: Scaffolding protein contributes to the global stability of phage P22 procapsids. J. Mol. Biol. 359, 1097–1106 [DOI] [PubMed] [Google Scholar]
- 12. King J., Botstein D., Casjens S., Earnshaw W., Harrison S., and Lenk E. (1976) Structure and assembly of the capsid of bacteriophage P22. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 276, 37–49 [DOI] [PubMed] [Google Scholar]
- 13. Zhang Z., Greene B., Thuman-Commike P. A., Jakana J., Prevelige P. E. Jr., King J., and Chiu W. (2000) Visualization of the maturation transition in bacteriophage P22 by electron cryomicroscopy. J. Mol. Biol. 297, 615–626 [DOI] [PubMed] [Google Scholar]
- 14. Galisteo M. L., and King J. (1993) Conformational transformations in the protein lattice of phage P22 procapsids. Biophys. J. 65, 227–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. King J., Hall C., and Casjens S. (1978) Control of the synthesis of phage P22 scaffolding protein is coupled to capsid assembly. Cell 15, 551–560 [DOI] [PubMed] [Google Scholar]
- 16. Teschke C. M., McGough A., and Thuman-Commike P. A. (2003) Penton release from P22 heat-expanded capsids suggests importance of stabilizing penton-hexon interactions during capsid maturation. Biophys. J. 84, 2585–2592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Y., Conway J. F., Cheng N., Steven A. C., Hendrix R. W., and Duda R. L. (2005) Control of virus assembly: HK97 “Whiffleball” mutant capsids without pentons. J. Mol. Biol. 348, 167–182 [DOI] [PubMed] [Google Scholar]
- 18. Suhanovsky M. M., and Teschke C. M. (2015) Nature's favorite building block: deciphering folding and capsid assembly of proteins with the HK97-fold. Virology 479, 487–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rizzo A. A., Fraser L. C., Sheftic S. R., Suhanovsky M. M., Teschke C. M., and Alexandrescu A. T. (2013) NMR assignments for the telokin-like domain of bacteriophage P22 coat protein. Biomol. NMR Assign. 7, 257–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Parent K. N., Sinkovits R. S., Suhanovsky M. M., Teschke C. M., Egelman E. H., and Baker T. S. (2010) Cryo-reconstructions of P22 polyheads suggest that phage assembly is nucleated by trimeric interactions among coat proteins. Phys. Biol. 7, 045004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Parent K. N., Khayat R., Tu L. H., Suhanovsky M. M., Cortines J. R., Teschke C. M., Johnson J. E., and Baker T. S. (2010) P22 coat protein structures reveal a novel mechanism for capsid maturation: stability without auxiliary proteins or chemical cross-links. Structure 18, 390–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cortines J. R., Motwani T., Vyas A. A., and Teschke C. M. (2014) Highly specific salt bridges govern bacteriophage P22 icosahedral capsid assembly: identification of the site in coat protein responsible for interaction with scaffolding protein. J. Virol. 88, 5287–5297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Padilla-Meier G. P., Gilcrease E. B., Weigele P. R., Cortines J. R., Siegel M., Leavitt J. C., Teschke C. M., and Casjens S. R. (2012) Unraveling the role of the C-terminal helix turn helix of the coat-binding domain of bacteriophage P22 scaffolding protein. J. Biol. Chem. 287, 33766–33780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Padilla-Meier G. P., and Teschke C. M. (2011) Conformational changes in bacteriophage P22 scaffolding protein induced by interaction with coat protein. J. Mol. Biol. 410, 226–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cortines J. R., Weigele P. R., Gilcrease E. B., Casjens S. R., and Teschke C. M. (2011) Decoding bacteriophage P22 assembly: identification of two charged residues in scaffolding protein responsible for coat protein interaction. Virology 421, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Teschke C. M., and Parent K. N. (2010) “Let the phage do the work”: using the phage P22 coat protein structures as a framework to understand its folding and assembly mutants. Virology 401, 119–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Capen C. M., and Teschke C. M. (2000) Folding defects caused by single amino acid substitutions in a subunit are not alleviated by assembly. Biochemistry 39, 1142–1151 [DOI] [PubMed] [Google Scholar]
- 28. Foguel D., Teschke C. M., Prevelige P. E. Jr., and Silva J. L. (1995) Role of entropic interactions in viral capsids: single amino acid substitutions in P22 bacteriophage coat protein resulting in loss of capsid stability. Biochemistry 34, 1120–1126 [DOI] [PubMed] [Google Scholar]
- 29. Parent K. N., Suhanovsky M. M., and Teschke C. M. (2007) Polyhead formation in phage P22 pinpoints a region in coat protein required for conformational switching. Mol. Microbiol. 65, 1300–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wikoff W. R., Liljas L., Duda R. L., Tsuruta H., Hendrix R. W., and Johnson J. E. (2000) Topologically linked protein rings in the bacteriophage HK97 capsid. Science 289, 2129–2133 [DOI] [PubMed] [Google Scholar]
- 31. Suhanovsky M. M., and Teschke C. M. (2013) An Intramolecular chaperone inserted in bacteriophage P22 coat protein mediates its chaperonin-independent folding. J. Biol. Chem. 288, 33772–33783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rizzo A. A., Suhanovsky M. M., Baker M. L., Fraser L. C., Jones L. M., Rempel D. L., Gross M. L., Chiu W., Alexandrescu A. T., and Teschke C. M. (2014) Multiple functional roles of the accessory I-domain of bacteriophage P22 coat protein revealed by NMR structure and CryoEM modeling. Structure 22, 830–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gordon C. L., and King J. (1994) Genetic properties of temperature-sensitive folding mutants of the coat protein of phage P22. Genetics 136, 427–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Anderson E., and Teschke C. M. (2003) Folding of phage P22 coat protein monomers: kinetic and thermodynamic properties. Virology 313, 184–197 [DOI] [PubMed] [Google Scholar]
- 35. Teschke C. M., and King J. (1993) Folding of the phage P22 coat protein in vitro. Biochemistry 32, 10839–10847 [DOI] [PubMed] [Google Scholar]
- 36. D'Lima N. G., and Teschke C. M. (2015) A molecular staple: D-loops in the I-domain of bacteriophage P22 coat protein make important intercapsomer contacts required for procapsid assembly. J. Virol. 89, 10569–10579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wishart D. S., Bigam C. G., Yao J., Abildgaard F., Dyson H. J., Oldfield E., Markley J. L., and Sykes B. D. (1995) 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 6, 135–140 [DOI] [PubMed] [Google Scholar]
- 38. Alexandrescu A. T., and Shortle D. (1994) Backbone dynamics of a highly disordered 131 residue fragment of staphylococcal nuclease. J. Mol. Biol. 242, 527–546 [DOI] [PubMed] [Google Scholar]
- 39. Watson E., Matousek W. M., Irimies E. L., and Alexandrescu A. T. (2007) Partially folded states of staphylococcal nuclease highlight the conserved structural hierarchy of OB-fold proteins. Biochemistry 46, 9484–9494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alexandrescu A. T., Jaravine V. A., Dames S. A., and Lamour F. P. (1999) NMR hydrogen exchange of the OB-fold protein LysN as a function of denaturant: the most conserved elements of structure are the most stable to unfolding. J. Mol. Biol. 289, 1041–1054 [DOI] [PubMed] [Google Scholar]
- 41. Zhang Y.-Z. (1995) Protein and peptide structure and interactions studied by hydrogen exchange and NMR, Ph.D. thesis, University of Pennsylvania [Google Scholar]
- 42. Sheftic S. R., Croke R. L., LaRochelle J. R., and Alexandrescu A. T. (2009) Electrostatic contributions to the stabilities of native proteins and amyloid complexes. Methods Enzymol. 466, 233–258 [DOI] [PubMed] [Google Scholar]
- 43. Croke R. L., Patil S. M., Quevreaux J., Kendall D. A., and Alexandrescu A. T. (2011) NMR determination of pKa values in α-synuclein. Protein Sci. 20, 256–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pace C. N. (1986) Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131, 266–280 [DOI] [PubMed] [Google Scholar]
- 45. Santoro M. M., and Bolen D. W. (1988) Unfolding free energy changes determined by linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl α-chymotrypsin using different denaturants. Biochemistry 27, 8063–8068 [DOI] [PubMed] [Google Scholar]
- 46. Schägger H., and von Jagow G. (1987) Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166, 368–379 [DOI] [PubMed] [Google Scholar]
- 47. Gordon C. L., Sather S. K., Casjens S., and King J. (1994) Selective in vivo rescue by GroEL/ES of thermolabile folding intermediates to phage P22 structural proteins. J. Biol. Chem. 269, 27941–27951 [PubMed] [Google Scholar]
- 48. Winston F., Botstein D., and Miller J. H. (1979) Characterization of amber and ochre suppressors in Salmonella typhimurium. J. Bacteriol. 137, 433–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Parent K. N., Suhanovsky M. M., and Teschke C. M. (2007) Phage P22 procapsids equilibrate with free coat protein subunits. J. Mol. Biol. 365, 513–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Barlow D. J., and Thornton J. M. (1983) Ion-pairs in proteins. J. Mol. Biol. 168, 867–885 [DOI] [PubMed] [Google Scholar]
- 51. Kumar S., and Nussinov R. (2002) Relationship between ion pair geometries and electrostatic strengths in proteins. Biophys. J. 83, 1595–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Croke R. L., Sallum C. O., Watson E., Watt E. D., and Alexandrescu A. T. (2008) Hydrogen exchange of monomeric α-synuclein shows unfolded structure persists at physiological temperature and is independent of molecular crowding in Escherichia coli. Protein Sci. 17, 1434–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Thurlkill R. L., Grimsley G. R., Scholtz J. M., and Pace C. N. (2006) pK values of the ionizable groups of proteins. Protein Sci. 15, 1214–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lumb K. J., and Kim P. S. (1995) Measurements of interhelical electrostatic interactions in the GCN4 leucine zipper. Science 268, 436–439 [DOI] [PubMed] [Google Scholar]
- 55. Bai Y., Milne J. S., Mayne L., and Englander S. W. (1994) Protein stability parameters measured by hydrogen exchange. Proteins 20, 4–14 [DOI] [PubMed] [Google Scholar]
- 56. Bai Y., Sosnick T. R., Mayne L., and Englander S. W. (1995) Protein folding intermediates: native-state hydrogen exchange. Science 269, 192–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Newcomer R. L., Fraser L. C., Teschke C. M., and Alexandrescu A. T. (2015) Partial unfolding of the phage P22 I-domain in native state hydrogen exchange experiments is promoted by urea binding. Biophys. J. 109, 2666–2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Prevelige P. E. Jr., and King J. (1993) Assembly of bacteriophage P22: a model for ds-DNA virus assembly. Prog. Med. Virol. 40, 206–221 [PubMed] [Google Scholar]
- 59. Tripler T. N., Maciejewski M. W., Teschke C. M., and Alexandrescu A. T. (2015) NMR assignments for the insertion domain of bacteriophage CUS-3 coat protein. Biomol. NMR Assign. 9, 333–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Parent K. N., Ranaghan M. J., and Teschke C. M. (2004) A second site suppressor of a folding defect functions via interactions with a chaperone network to improve folding and assembly in vivo. Mol. Microbiol. 54, 1036–1050 [DOI] [PubMed] [Google Scholar]
- 61. Casjens S., and Hayden M. (1988) Analysis in vivo of the bacteriophage P22 headful nuclease. J. Mol. Biol. 199, 467–474 [DOI] [PubMed] [Google Scholar]
- 62. Casjens S., Wyckoff E., Hayden M., Sampson L., Eppler K., Randall S., Moreno E. T., and Serwer P. (1992) Bacteriophage P22 portal protein is part of the gauge that regulates packing density of intravirion DNA. J. Mol. Biol. 224, 1055–1074 [DOI] [PubMed] [Google Scholar]
- 63. Creighton T. E. (1993) Proteins: Structures and Molecular Properties, 2nd Ed., pp. 225–227, W. H. Freeman and Co., New York [Google Scholar]
- 64. Shortle D. (1996) The denatured state (the other half of the folding equation) and its role in protein stability. FASEB J. 10, 27–34 [DOI] [PubMed] [Google Scholar]