

Abstract
Purpose of review
The CARMA1/BCL10/MALT1 (CBM) complex is a multimeric signaling complex controlling several important aspects of lymphocyte activation. Gain-of-function mutations in the genes encoding CBM proteins or their upstream regulators are associated with lymphoid malignancies, whereas loss-of-function mutations lead to immunodeficiency. This review reports on recent findings advancing our understanding of how CBM proteins contribute to malignant and nonmalignant hematological diseases in humans.
Recent findings
Somatic gain-of-function mutations of CARMA1 (also known as CARD11), originally described for patients with diffuse large B-cell lymphoma, have recently been identified in patients with acute T-cell leukemia/lymphoma or Sézary syndrome, and in patients with a B-cell lymphoproliferative disorder known as BENTA. Loss-of-function mutations of CARMA1 and MALT1, on the other hand, have been reported to underlie human immunodeficiency. Lately, it has become clear that CBM-dependent signaling promotes lymphomagenesis not only via NF-κB activation, but also via the AP-1 family of transcription factors. The identification of new substrates of the protease MALT1 and the characterization of mice expressing catalytically inactive MALT1 have deepened our understanding of how the CBM complex controls lymphocyte proliferation through promoting MALT1's protease activity.
Summary
The discovery of CARMA1 gain-of-function mutations in T-cell malignancies and BENTA patients, as well as the association of CARMA1 and MALT1 mutations with human immunodeficiency highlight the importance of CBM proteins in the regulation of lymphocyte functions, and suggest that the protease activity of MALT1 might be targeted to treat specific lymphoid malignancies.
Keywords: AP-1, CARD11, immunodeficiency, lymphoma, NF-κB
INTRODUCTION
The multimeric CARMA1/BCL10/MALT1 (CBM) complex, composed of the scaffold protein CARMA1 (also known as CARD11), the adaptor protein BCL10, and the protease MALT1, plays an important role in signal transmission after antigen receptor stimulation [1]. Triggering of the antigen receptor induces a protein kinase C (PKC)-dependent phosphorylation of CARMA1, which leads to a conformational change in CARMA1. This nucleates the formation of a fibrillar, high molecular weight complex containing CARMA1 together with oligomerized BCL10 and MALT1 [2,3]. CBM complex formation is required for the activation of the transcription factors NF-κB and AP-1, which regulate various aspects of lymphocyte proliferation, differentiation, and survival [4]. The CBM complex also controls transcription-independent aspects of lymphocyte activation, such as regulation of transcript stability, cellular adhesion, and metabolic changes [4]. Genetic studies using mice deficient in CARMA1, BCL10, or MALT1 have revealed an essential role for these CBM proteins in immunoreceptor-induced cellular activation, as mice lacking these proteins are immunodeficient [4]. Moreover, inactivating germline mutations of CARMA1 and MALT1 have recently been identified in a small number of common immunodeficiency patients [5]. CBM hyperactivity, on the other hand, has emerged as a hallmark of lymphomagenesis. Originally, chromosomal translocations of BCL10 or MALT1 had been identified in lymphoma of the mucosa-associated lymphoid tissue (MALT lymphoma) [1]. Subsequently, gain-of-function mutations in CARMA1 or its upstream regulator, the B-cell receptor (BCR)-associated CD79 chains, have been described in diffuse large B-cell lymphoma (DLBCL) of the activated B-cell (ABC) subtype [6]. The purpose of this review is to update on recent findings describing novel gain-of-function mutations in CARMA1 and other CBM signaling components in an increasing number of B- and T-cell malignancies [7,8,9▪▪–11▪▪] and in patients with a lymphoproliferation disorder known as BENTA disease [12,13▪,14▪]. We also highlight novel molecular insights into aspects of lymphocyte activation that are controlled by CBM-dependent AP-1 activation [15▪▪–17▪▪] and by the MALT1-dependent cleavage of specific cellular substrates [18–20,21▪,22▪,23–26].
Box 1.

no caption available
CONSTITUTIVE CARMA1/BCL10/MALT1 SIGNALING CHARACTERIZES DIFFUSE LARGE B-CELL LYMPHOMA AND MANTLE CELL LYMPHOMA SUBSETS
Over the last few years, constitutive activation of CBM signaling has been recognized as a common feature of an increasing number of B- and T-cell malignancies and B-cell proliferative diseases (Fig. 1). Generally, this has been linked to gain-of-function mutations of CARMA1 or its upstream regulators, and/or to self-antigen-driven, constitutive BCR signaling [1,6].
FIGURE 1.
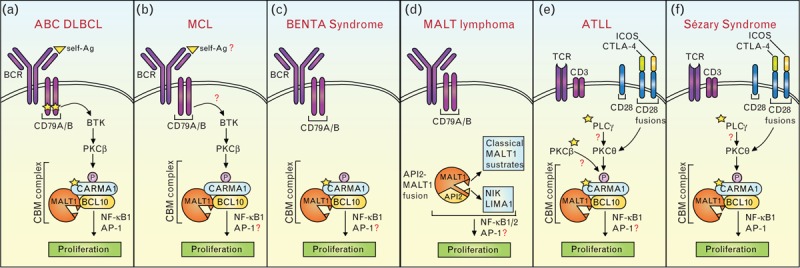
Constitutive CBM signaling in B- and T-cell malignancies. Underlying mechanisms include (a) mutations in CD79A or CD79B and CARMA1/CARD11, and self-antigen recognition, (b) self-antigen recognition or mutations upstream of BTK, (c) germline mutations in CARMA1, (d) generation of a MALT1-API2 fusion protein that activates the classical (NF-κB1) and nonclassical (NF-κB2) pathway, (e, f) gain-of function mutations in PLCγ1, PKCβ, or CARMA1, and in frame mutations of the T-cell co-receptor CD28 with ICOS or CTLA-4. In all figure panels, recurrent mutations are indicated with a yellow star. ABC, activated B-cell; ATLL, acute T-cell leukemia/lymphoma; BENTA, B-cell expansion with NF-κB and T-cell anergy; BCR, B-cell receptor; CBM, CARMA1/BCL10/MALT1; CTLA-4, cytotoxic T lymphocyte-associated protein 4; DLBCL, diffuse large B-cell lymphoma; ICOS, inducible costimulator; MALT, mucosa-associated lymphoid tissue; MCL, mantle cell lymphomas; PKC, protein kinase C; TCR, T-cell receptor.
A pathogenic role for CBM signaling was originally discovered in ABC DLBCL, in which this pathway can be activated or sustained by gain-of-function mutations in CARMA1 [27], or its upstream regulator, the BCR-associated CD79A/B complex [28], and by self-antigen driven BCR stimulation [29] (Fig. 1a). Mutations in CARMA1, which were present in 9.6% of the cases, were exclusively located in proximity of or within the coiled-coil region of CARMA1, which is required for the induction of an active conformation and oligomerization of this protein [27] (Fig. 2a). Functional characterization of these point mutants identified for the first time CARMA1 as a ‘bona fide’ oncogene [27]. CARMA1 mutations were independently identified in DLBCLs in several other studies [8,30–33]. Most of these mutations resided in the CARD-CC hot spot region, and are thus likely gain-of-function mutations.
FIGURE 2.
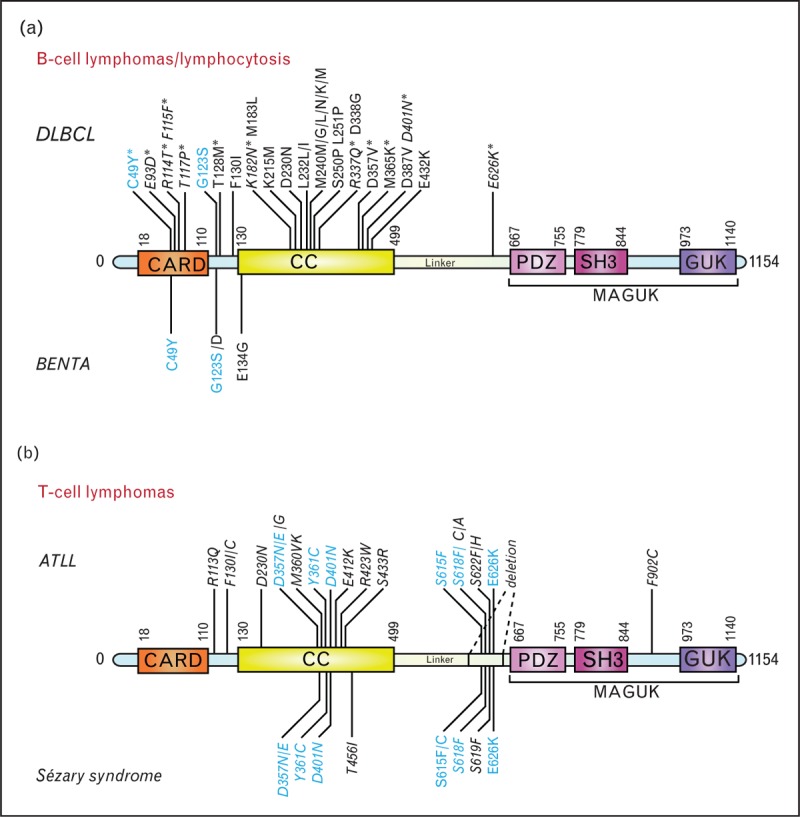
Described CARMA1/CARD11 mutations (a) Mutations found in ABC DLBCL and BENTA. An asterisk indicates mutations in DLBCL that were not clearly identified as ABC DLBCL. Amino acid numbers of some CARMA1 mutations [7] have been adjusted to the UniProt sequence. (b) Mutations identified in ATLL and Sézary syndrome; 8% of ATLL cases have a CARMA1 deletion within the linker domain. Mutations that were not functionally tested are indicated in italics. Mutations common to DLBCL and BENTA (a) or common to Sézary syndrome and ATLL (b) are highlighted in blue. ABC, activated B-cell; ATLL, acute T-cell leukemia/lymphoma; BENTA, B-cell expansion with NF-κB and T-cell anergy; CARD, caspase recruitment domain; CC, coiled coil; DLBCL, diffuse large B-cell lymphoma; GUK, guanylate kinase domain; MAGUK, membrane-associated guanylate kinase; PDZ, domain found in the proteins PSD95, Dlg1 and ZO-1; SH3, Src homology-3 domain.
Other recurrent alterations found in ABC DLBCL are mutations in the BCR-associated CD79A/B chains [28]. In contrast to the CARMA1 mutations, CD79 mutations are not sufficient to activate NF-κB signaling, but increase the surface expression of the BCR [28]. A recent study proposes that self-antigen stimulation is required in addition to CD79 mutations to sustain CBM signaling in ABC DLBCL [29]. Self-antigens may also drive the growth of other ABC DLBCL patients without detectable CD79 mutations [34] (Fig. 1a).
In addition to ABC DLBCL lymphomas, a subset of mantle cell lymphomas (MCL) has recently been shown to depend on chronic BCR-dependent signaling, and to present constitutive MALT1 and NF-κB activation [35]. Genome sequencing revealed no mutations in the BCR pathway that could explain why this subset is BCR addicted [35]. It is possible that these MCL cases are driven by yet unknown gene mutations and/or by nongenetic mechanisms such as self-antigen recognition, since the repertoire of the immunoglobulin heavy variable genes of MCL is biased [36] (Fig. 1b).
CONSTITUTIVE CARMA1 ACTIVATION CAN CAUSE BENTA SYNDROME
Recently, germline gain-of-function mutations in CARMA1 were identified in patients with a B-cell proliferative syndrome known as B-cell expansion with NF-κB and T-cell anergy (BENTA) [12,13▪,14▪] (Fig. 1c). The resulting point mutations were localized either between the CARD and the coiled-coil domain or in the CARD motif of CARMA1. As for the mutants described in ABC DLBCL, this induced enhanced NF-κB responses and CARMA1 oligomerization without the need for PKC-mediated CARMA1 phosphorylation [7,12,13▪,14▪] (Fig. 2a). Interestingly, the B-cell expansion phenotype of BENTA patients is associated with an increased risk to develop lymphoma, suggesting that CARMA1 mutations are predisposing but not by themselves sufficient for lymphoma development.
MALT LYMPHOMAS ARE DRIVEN BY BCL10 AND MALT1 TRANSLOCATIONS
Alterations in BCR signaling downstream of CARMA1 are a prominent feature of MALT lymphoma. Early stages of MALT lymphomas are driven by chronic infection with Helicobacter pylori, while advanced stages, which do not respond to H. pylori eradication, typically have characteristic gene translocations that lead to BCL10 or MALT1 overexpression, or to the expression of a constitutively active API2-MALT1 fusion protein [1]. Interestingly, transgenic overexpression of MALT1 or BCL10 alone in mice promotes lymphomagenesis [37,38], and MALT1 amplification together with p53 deletion can transform MALT lymphoma into DLBCL [37], showing a molecular link between the two subtypes of lymphomas. The API2-MALT1 fusion protein is constitutively active without the need for BCR triggering, and activates not only the canonical (NF-κB1) pathway but also the noncanonical or alternative (NF-κB2) pathway (Fig. 1d).
CONSTITUTIVE CARMA1/BCL10/MALT1 SIGNALING IS PRESENT IN SUBSETS OF ATLL AND SÉZARY SYNDROME
The role of CBM signaling in lymphoid malignancies has been recently extended to T-cell malignancies (Fig. 1e and f). Indeed, gain-of-function mutations in CARMA1, or upstream T-cell receptor (TCR)-signaling components, were identified in adult T-cell leukemia/lymphoma (ATLL) [11▪▪] and in a form of cutaneous T-cell lymphoma (CTCL) named Sézary syndrome [9▪▪,10▪▪] (Fig. 2b).
ATLL is known to be caused by human T-lymphotropic virus type I (HTLV-1) infection [39]. Since HTLV-1 has a long phase of latency, genomic alterations were suspected for a long time to be involved in the full transformation of HTLV-I-infected T cells into ATLL. Indeed, such mutations were recently found in components of the TCR-signaling pathway, such as PLCγ1, CARMA1, and PKCβ, and in the T-cell co-receptor CD28 (Fig. 1e). PLCγ1, which acts downstream of the TCR and upstream of CARMA1, was the most frequently mutated protein, and two of the identified PLCγ1 point mutations had already been characterized as gain-of-function mutations in CTCL [40]. CARMA1 was frequently mutated (24%) or amplified (12%) [11▪▪]. Some of the CARMA1 mutations were situated in the coiled-coil domain, similarly to ABC DLBCL, but others were present in the linker domain between the coiled-coil and PDZ domains (Fig. 2b) [11▪▪]. In addition, whole-genome sequencing identified a deletion in the linker region in 8% (4/48) of the cases [11▪▪]. Since this linker region has been shown to be auto-inhibitory [41], mutations in this region most likely activate CARMA1 by blocking auto-repression. An additional unexpected recurrent mutation was found in PKCβ [11▪▪], which is known as the direct upstream regulator of CARMA1 in B cells (Fig. 1). Interestingly, ATLL showed a high frequency of co-mutations in PKCβ and CARMA1, and synergism between PKCβ and CARMA1 mutations could be observed in 293T cells [11▪▪]. Finally, recurrent genetic alterations in ATLL patients were in-frame fusions between the gene encoding cytotoxic T lymphocyte associated protein 4 (CTLA-4) or inducible costimulator (ICOS) and CD28, which are predicted to lead to enhanced CD28 signaling [39]. Since CD28 co-stimulation is required for optimal TCR-induced CBM activation, these findings suggest that co-stimulatory signals can also boost CBM-dependent signaling events in some ATLL (Fig. 1e).
Sézary syndrome is a leukemic form of CTCL, which can arise de novo or can be derived from a chronic mycosis fungoides. Recently, two studies using genomic approaches have revealed mutations in CARMA1 [9▪▪,10▪▪], overexpression of CARMA1 mRNA [10▪▪], or presence of a CARMA1 fusion to PIK3R3 [10▪▪] as recurrent features of Sézary patients. CARMA1 mutations were present both in the coiled-coil region and in the linker domain [9▪▪,10▪▪] (Fig. 2b), and those in the linker region were identical to mutations found in ATLL [11▪▪]. Similarly to ATLL, Sézary syndrome patients had mutations in PLCγ1 and in-frame fusions between CTLA-4 or ICOS and CD28, suggesting that the development of these two T-cell malignancies is promoted by similar gain-of-function mutations [9▪▪,10▪▪].
Collectively, these findings suggest that the CBM signaling pathway is a highly relevant oncogenic pathway not only in B-cell lymphomas, but also in an increasing spectrum of T-cell lymphomas.
CARMA1/BCL10/MALT1-DEPENDENT AP-1 ACTIVATION CONTRIBUTES TO LYMPHOMAGENESIS
Most of the lymphomagenic activity of the CBM pathway has been ascribed to its capacity to activate the transcription factor NF-κB, which promotes cellular survival and proliferation. However, in ABC DLBCL, the CBM complex also activates transcription factors of the AP-1 family, in particular those of the JUN and ATF-type [15▪▪–17▪▪]. CBM-dependent NF-κB and AP-1 activation likely regulates gene expression in both synergistic and separate manners. Crosstalk between the NF-κB and AP-1 pathways can be mediated by synergistic promotor binding and activation [42]. IL-6, which promotes ABC DLBCL proliferation, is an example of a gene that is synergistically upregulated by both transcription factors [43]. Support for the existence of separate target genes, on the other hand, comes, for example, from experiments in which either the NF-κB or AP-1 pathways are specifically inhibited with dominant negative IκB, or A-Fos constructs, respectively. This impairs the viability of ABC DLBCL cell lines with distinct (fast versus slow) kinetics [15▪▪]. However, the full range of AP-1 target genes controlling lymphoma cell viability remains to be identified. A recent study has revealed an important role for AP-1 (and in particular c-Jun and JunB) in the control of genes required for the interaction of DLBCL with the tumor microenvironment [17▪▪]. Indeed, the expression of such c-Jun-regulated genes is significantly enriched in patients with disseminated, extranodal DLBCL [17▪▪].
The regulation and composition of AP-1 complexes in CBM-driven lymphomas has recently gained considerable attention. The JUN subfamily member c-Jun is known to be activated and stabilized by a c-Jun N-terminal kinase (JNK)-dependent phosphorylation [44]. BCR-induced JNK activation requires CARMA1 [45,46], and increased JNK activity and c-Jun expression is detectable in B-cells from mice expressing an oncogenic CARMA1 variant, which promotes aggressive B-cell lymphoproliferation [16▪▪]. JNK activation is also promoted by a human CARMA1 mutant that was identified in Sézary syndrome [9▪▪]. The exact role of JNK in CBM-driven AP-1 activation and lymphomagenesis remains unclear, however, since pharmacological JNK inhibition does not systematically affect c-Jun levels in CBM-driven ABC DLBCL cell lines [15▪▪]. Biochemical studies reveal that in these cell lines, c-Jun preferentially forms heterodimers with AP-1 subunits of the ATF subfamily, such as ATF2, ATF3, and ATF7 [15▪▪]. The resulting JUN/ATF complexes are essential for the CBM-driven proliferation of ABC DLBCL cell lines, and ATF3 expression is a hallmark of human ABC DLBCL [15▪▪]. Collectively, these studies reveal a critical role for CBM-dependent activation of the AP-1 family of transcription factors in lymphomagenesis, but the identity and contribution of individual AP-1 target genes remain to be further explored.
CARMA1/BCL10/MALT1-DEPENDENT LYMPHOMAGENESIS REQUIRES THE PROTEASE ACTIVITY OF MALT1
Over the last 8 years, our understanding of CBM-driven lymphomagenesis has considerably deepened with the discovery that MALT1, initially believed to act mainly as a scaffold protein, functions as an active protease cleaving multiple substrates to promote lymphocyte proliferation (Table 1) [18,23]. The MALT1 protease activity sustains NF-κB activation by the cleavage of RelB [19], which forms transcriptionally inactive complexes with RelA and c-Rel, and by the cleavage of the deubiquitinating enzyme A20 [18], which deubiquitinates and thereby inactivates several regulators of NF-κB [49]. MALT1 also undergoes auto-processing; this contributes to NF-κB activation in a manner that is not yet understood at the molecular level [20].
Table 1.
Overview of MALT1 and API2-MALT1 substrates and functional consequences of their cleavage
| Substrate | Cleavage site | Consequence | References |
| MALT1 | |||
| A20 | GASR439G | NF-κB1 activation | [18] |
| RelB | LVSR85G | NF-κB1 activation | [19] |
| MALT1 | LCCR149A | NF-κB1 activation | [20] |
| HOIL | LQPR165G | NF-κB1 termination | [21▪,22▪] |
| BCL10 | LRSR228T | Adhesion | [23] |
| CYLD | FMSR324G | AP-1 activation | [24] |
| Regnase-1 | LVPR111G | mRNA stabilization | [25] |
| Roquin-1 | LIPR510G/MVPR579G | mRNA stabilization | [26] |
| Roquin-2 | LISR509T | mRNA stabilization | [26] |
| API2–MALT1 | |||
| NIK | CLSR325G | NF-κB2 activation | [47] |
| LIMA1 | PDSR206A / FKSK289G | Proliferation and migration | [48▪] |
| Consensus | L-X-P/S-R-G | ||
An interesting new twist to MALT1's enzymatic role in NF-κB regulation comes from the recent identification of HOIL-1 as a novel MALT1 substrate in lymphocytes and lymphoma cells [21▪,22▪]. HOIL-1 is a component of the linear ubiquitin chain assembly complex (LUBAC), which comprises HOIL-1, HOIL-1 interacting protein (HOIP), and Sharpin, and promotes NF-κB activation by addition of linear (aminoterminally-linked) polyubiquitin chains on its substrates [50]. Interestingly, rare germline polymorphisms in the gene encoding HOIP (also known as RNF31), which lead to an increased HOIL-1/HOIP interaction and NF-κB activation, are enriched in ABC DLBCL [51]. MALT1-dependent HOIL-1 cleavage reduces linear ubiquitination of cellular proteins, and has thus been proposed to provide negative feedback on the NF-κB pathway [21▪,22▪,52].
Controlling NF-κB activation is so far the best-studied function of the protease activity of MALT1. However, MALT1 cleaves additional substrates that promote other aspects of lymphocyte activation, including AP-1 activation [via the cleavage of the deubiquitinating enzyme cylindromatosis (CYLD) [24]], regulation of transcript stability (via cleavage of RNA stability regulators Regnase-1 and Roquin-1/-2 [25,26]) and cellular adhesion (via cleavage of BCL10 [23]). It is likely that the cleavage of all reported MALT1 substrates contributes in one way or another to lymphomagenesis by promoting cellular proliferation, survival, adhesion and possibly migration.
A distinct and unique profile of MALT1 substrates have been identified in cells derived from MALT lymphoma expressing the API2 (also known as c-IAP2-MALT1 fusion protein [1] (Fig. 1d). This fusion protein is constitutively active and cleaves specific substrates that are recruited by the API2 moiety. One such substrate is the Ser/Thr kinase NIK [47], which has a known role as an activator of the noncanonical/NF-κB2 pathway. Recently, the tumor suppressor protein LIMA1α has been identified as an additional API2-MALT1-specific substrate [48▪]. LIMA1α cleavage disrupts the tumor suppressor function of LIMA1α, but it also results in the release of its Lim domain, an LMO-like oncogenic protein, to promote B-cell proliferation and adhesion [48▪].
PROTEASE ACTIVITY OF MALT1 COULD BE TARGETED TO TREAT LYMPHOMAS
The discovery and understanding of MALT1's protease activity has rapidly fostered efforts to develop MALT1 inhibitors for the therapy of lymphomas addicted to MALT1 signaling. So far, two types of small molecule MALT1 inhibitors have been identified by high throughput screening: phenothiazine-derivatives such as mepazine and thioridazine, which act as reversible allosteric MALT1 inhibitors [53], and MI-2, an active site-directed irreversible MALT1 inhibitor [54]. Both types of inhibitors were shown to efficiently slow down the growth of ABC DLBCL in xenograft models [53,54]. Whether MALT1 inhibitors show similar activity against other types of hematological malignancies remains to be explored. Lymphoma/leukemia types that should be considered as potential candidates for MALT1 inhibitor treatments include MALT lymphomas and MCL, chronic lymphocytic leukemia with stereotyped and potentially auto-reactive BCRs [6], and T-cell lymphomas with gain-of function mutations in CBM signaling, such as ATLL [11▪▪] and Sézary syndrome [9▪▪,10▪▪]. Therapeutic MALT1 inhibition should be particularly useful in patients who develop Ibrutinib-resistant mutations in BTK or who have mutations in signaling components downstream of BTK or ITK, such as PLCγ, PKCβ and CARMA1. In the future, the identification of lymphomas with constitutive MALT1 activity may be facilitated by MALT1 activity probes that are under development [55,56]. Other future developments may include the design/discovery of inhibitors that specifically target the scaffold (rather than the protease) function of MALT1. This may, alone or in combination with MALT1 protease inhibitors, open additional avenues for the treatment of CBM-dependent lymphomas.
CONCLUSION
Constitutive activation of the CBM complex, and/or of its protease component MALT1, is now identified in subsets of an increasing number of lymphoid malignancies including DLBCL, MALT lymphomas, MCL, ATLL, and Sézary syndrome, and is also likely to underlie disorders associated with milder lymphoproliferation, such as BENTA. Targeting the BCR/TCR–BTK/ITK–CBM–NF-κB pathway with small molecule drugs therefore presents a promising therapeutic strategy. Ongoing efforts to develop MALT1 inhibitors should in particular benefit patients who develop ibrutinib resistance or who have gain-of-function mutations in signaling components acting downstream of BTK. Further exploration of CBM activation in a broader range of hematological malignancies will certainly reveal additional lymphoma/leukemia subsets with MALT1 addiction.
Note added in proof
A new report identifying HOIL-1 as a MALT1 substrate has recently been published [57].
Acknowledgements
The authors would like to thank Stephan Hailfinger, Luca Bonsignore, Mikhail Kuravsky, Laurence de Leval, and David Vallois for critical comments on the manuscript.
Financial support and sponsorship
The work was supported by grants from the Swiss National Science Foundation (Sinergia programme) and the Swiss Cancer League.
Conflicts of interest
M.T. has a collaboration agreement with the Medical Research Council, UK, and had been supported in the past by a collaboration agreement with Ono Pharmaceuticals, Japan.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1. Rosebeck S, Rehman AO, Lucas PC, McAllister-Lucas LM. From MALT lymphoma to the CBM signalosome: three decades of discovery. Cell Cycle 2011; 10:2485–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rueda D, Thome M. Phosphorylation of CARMA1: the link(er) to NF-kappaB activation. Immunity 2005; 23:551–553. [DOI] [PubMed] [Google Scholar]
- 3. Qiao Q, Yang C, Zheng C, et al. Structural architecture of the CARMA1/Bcl10/MALT1 signalosome: nucleation-induced filamentous assembly. Mol Cell 2013; 51:766–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jaworski M, Thome M. The paracaspase MALT1: biological function and potential for therapeutic inhibition. Cell Mol Life Sci 2016; 73:459–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Turvey SE, Durandy A, Fischer A, et al. The CARD11-BCL10-MALT1 (CBM) signalosome complex: stepping into the limelight of human primary immunodeficiency. J Allergy Clin Immunol 2014; 134:276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat Rev Drug Discov 2013; 12:229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lenz G, Davis RE, Ngo VN, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 2008; 319:1676–1679. [DOI] [PubMed] [Google Scholar]
- 8. Compagno M, Lim WK, Grunn A, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 2009; 459:717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9▪▪. da Silva Almeida AC, Abate F, Khiabanian H, et al. The mutational landscape of cutaneous T cell lymphoma and Sezary syndrome. Nat Genet 2015; 47:1465–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]; Identifies genetic alterations in TCR signaling in Sézary syndrome, including CARMA1 (CARD11) gain-of-function mutations.
- 10▪▪. Wang L, Ni X, Covington KR, et al. Genomic profiling of Sezary syndrome identifies alterations of key T cell signaling and differentiation genes. Nat Genet 2015; 47:1426–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]; Identifies genetic alterations in TCR signaling in Sézary syndrome, including CARMA1 (CARD11) gain-of-function mutations.
- 11▪▪. Kataoka K, Nagata Y, Kitanaka A, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet 2015; 47:1304–1315. [DOI] [PubMed] [Google Scholar]; Identifies genetic alteration associated with ATLL. Frequent mutations were identified in TCR signaling components, including CARMA1 (CARD11), PLCγ1, and PKCβ.
- 12. Snow AL, Xiao W, Stinson JR, et al. Congenital B cell lymphocytosis explained by novel germline CARD11 mutations. J Exp Med 2012; 209:2247–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13▪. Brohl AS, Stinson JR, Su HC, et al. Germline CARD11 mutation in a patient with severe congenital B cell lymphocytosis. J Clin Immunol 2014; 35:32–46. [DOI] [PMC free article] [PubMed] [Google Scholar]; Identifies germline mutations in the CARD motif of CARMA1 (CARD11) in BENTA patients.
- 14▪. Buchbinder D, Stinson JR, Nugent DJ, et al. Mild B-cell lymphocytosis in patients with a CARD11 C49Y mutation. J Allergy Clin Immunol 2015; 136:819–821. [DOI] [PMC free article] [PubMed] [Google Scholar]; Identifies germline mutations in the CARD motif of CARMA1 (CARD11) in BENTA patients.
- 15▪▪. Juilland M, Gonzalez M, Erdmann T, et al. CARMA1- and MyD88-dependent activation of Jun/ATF-type AP-1 complexes is a hallmark of ABC diffuse large B-cell lymphomas. Blood 2016; 127:1780–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]; Reveals a critical role for CBM-dependent activation of the AP-1 family of transcription factors in lymphomagenesis.
- 16▪▪. Knies N, Alankus B, Weilemann A, et al. Lymphomagenic CARD11/BCL10/MALT1 signaling drives malignant B-cell proliferation via cooperative NF-kappaB and JNK activation. Proc Natl Acad Sci U S A 2015; 112:E7230–E7238. [DOI] [PMC free article] [PubMed] [Google Scholar]; Reveals a critical role for CBM-dependent activation of the AP-1 family of transcription factors in lymphomagenesis.
- 17▪▪. Blonska M, Zhu Y, Chuang HH, et al. Jun-regulated genes promote interaction of diffuse large B-cell lymphoma with the microenvironment. Blood 2015; 125:981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]; Reveals a critical role for CBM-dependent activation of the AP-1 family of transcription factors in lymphomagenesis.
- 18. Coornaert B, Baens M, Heyninck K, et al. T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-kappaB inhibitor A20. Nat Immunol 2008; 9:263–271. [DOI] [PubMed] [Google Scholar]
- 19. Hailfinger S, Nogai H, Pelzer C, et al. Malt1-dependent RelB cleavage promotes canonical NF-kappaB activation in lymphocytes and lymphoma cell lines. Proc Natl Acad Sci U S A 2011; 108:14596–14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baens M, Bonsignore L, Somers R, et al. MALT1 auto-proteolysis is essential for NF-kappaB-dependent gene transcription in activated lymphocytes. PLoS One 2014; 9:e103774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21▪. Klein T, Fung SY, Renner F, et al. The paracaspase MALT1 cleaves HOIL1 reducing linear ubiquitination by LUBAC to dampen lymphocyte NF-kappaB signalling. Nat Commun 2015; 6:8777. [DOI] [PMC free article] [PubMed] [Google Scholar]; Identifies the LUBAC component HOIL-1 as a novel MALT1 substrate.
- 22▪. Elton L, Carpentier I, Staal J, et al. MALT1 cleaves the E3 ubiquitin ligase HOIL-1 in activated T cells, generating a dominant negative inhibitor of LUBAC-induced NF-kappaB signaling. FEBS J 2015; 283:403–412. [DOI] [PubMed] [Google Scholar]; Identifies the LUBAC component HOIL-1 as a novel MALT1 substrate.
- 23. Rebeaud F, Hailfinger S, Posevitz-Fejfar A, et al. The proteolytic activity of the paracaspase MALT1 is key in T cell activation. Nat Immunol 2008; 9:272–281. [DOI] [PubMed] [Google Scholar]
- 24. Staal J, Driege Y, Bekaert T, et al. T-cell receptor-induced JNK activation requires proteolytic inactivation of CYLD by MALT1. EMBO J 2011; 30:1742–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Uehata T, Iwasaki H, Vandenbon A, et al. Malt1-induced cleavage of regnase-1 in CD4(+) helper T cells regulates immune activation. Cell 2013; 153:1036–1049. [DOI] [PubMed] [Google Scholar]
- 26. Jeltsch KM, Hu D, Brenner S, et al. Cleavage of roquin and regnase-1 by the paracaspase MALT1 releases their cooperatively repressed targets to promote T(H)17 differentiation. Nat Immunol 2014; 15:1079–1089. [DOI] [PubMed] [Google Scholar]
- 27. Lenz G, Davis RE, Ngo VN, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 2008; 319:1676–1679. [DOI] [PubMed] [Google Scholar]
- 28. Davis RE, Ngo VN, Lenz G, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010; 463:88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Young RM, Wu T, Schmitz R, et al. Survival of human lymphoma cells requires B-cell receptor engagement by self-antigens. Proc Natl Acad Sci U S A 2015; 112:13447–13454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang J, Grubor V, Love CL, et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A 2013; 110:1398–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Morin RD, Mendez-Lago M, Mungall AJ, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011; 476:298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lohr JG, Stojanov P, Lawrence MS, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A 2012; 109:3879–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pasqualucci L, Trifonov V, Fabbri G, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet 2011; 43:830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wilson WH, Young RM, Schmitz R, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med 2015; 21:922–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rahal R, Frick M, Romero R, et al. Pharmacological and genomic profiling identifies NF-kappaB-targeted treatment strategies for mantle cell lymphoma. Nat Med 2014; 20:87–92. [DOI] [PubMed] [Google Scholar]
- 36. Hadzidimitriou A, Agathangelidis A, Darzentas N, et al. Is there a role for antigen selection in mantle cell lymphoma? Immunogenetic support from a series of 807 cases. Blood 2011; 118:3088–3095. [DOI] [PubMed] [Google Scholar]
- 37. Vicente-Duenas C, Fontan L, Gonzalez-Herrero I, et al. Expression of MALT1 oncogene in hematopoietic stem/progenitor cells recapitulates the pathogenesis of human lymphoma in mice. Proc Natl Acad Sci U S A 2012; 109:10534–10539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li Z, Wang H, Xue L, et al. Emu-BCL10 mice exhibit constitutive activation of both canonical and noncanonical NF-kappaB pathways generating marginal zone (MZ) B-cell expansion as a precursor to splenic MZ lymphoma. Blood 2009; 114:4158–4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ishitsuka K, Tamura K. Human T-cell leukaemia virus type I and adult T-cell leukaemia-lymphoma. Lancet Oncol 2014; 15:e517–e526. [DOI] [PubMed] [Google Scholar]
- 40. Vaque JP, Gomez-Lopez G, Monsalvez V, et al. PLCG1 mutations in cutaneous T-cell lymphomas. Blood 2014; 123:2034–2043. [DOI] [PubMed] [Google Scholar]
- 41. Sommer K, Guo B, Pomerantz JL, et al. Phosphorylation of the CARMA1 linker controls NF-kappaB activation. Immunity 2005; 23:561–574. [DOI] [PubMed] [Google Scholar]
- 42. Stein B, Baldwin AS, Jr, Ballard DW, et al. Cross-coupling of the NF-kappa B p65 and Fos/Jun transcription factors produces potentiated biological function. EMBO J 1993; 12:3879–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Keller ET, Wanagat J, Ershler WB. Molecular and cellular biology of interleukin-6 and its receptor. Front Biosci 1996; 1:d340–d357. [DOI] [PubMed] [Google Scholar]
- 44. Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer 2003; 3:859–868. [DOI] [PubMed] [Google Scholar]
- 45. Gaide O, Favier B, Legler DF, et al. CARMA1 is a critical lipid raft-associated regulator of TCR-induced NF-kappa B activation. Nat Immunol 2002; 3:836–843. [DOI] [PubMed] [Google Scholar]
- 46. Blonska M, Pappu BP, Matsumoto R, et al. The CARMA1-Bcl10 signaling complex selectively regulates JNK2 kinase in the T cell receptor-signaling pathway. Immunity 2007; 26:55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rosebeck S, Madden L, Jin X, et al. Cleavage of NIK by the API2-MALT1 fusion oncoprotein leads to noncanonical NF-kappaB activation. Science 2011; 331:468–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48▪. Nie Z, Du MQ, McAllister-Lucas LM, et al. Conversion of the LIMA1 tumour suppressor into an oncogenic LMO-like protein by API2-MALT1 in MALT lymphoma. Nat Commun 2015; 6:5908. [DOI] [PubMed] [Google Scholar]; The study shows that the oncogenic API2-MALT1 fusion protein cleaves the tumor suppressor LIMA1a to generate a new oncogenic LIMA1a fragment.
- 49. Ma A, Malynn BA. A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nat Rev Immunol 2012; 12:774–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shimizu Y, Taraborrelli L, Walczak H. Linear ubiquitination in immunity. Immunol Rev 2015; 266:190–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang Y, Schmitz R, Mitala J, et al. Essential role of the linear ubiquitin chain assembly complex in lymphoma revealed by rare germline polymorphisms. Cancer Disc 2014; 4:480–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hailfinger S, Schmitt A, Schulze-Osthoff K. The paracaspase MALT1 dampens NF-kappaB signaling by cleaving the LUBAC subunit HOIL-1. FEBS J 2015; 283:400–402. [DOI] [PubMed] [Google Scholar]
- 53. Nagel D, Spranger S, Vincendeau M, et al. Pharmacologic inhibition of MALT1 protease by phenothiazines as a therapeutic approach for the treatment of aggressive ABC-DLBCL. Cancer Cell 2012; 22:825–837. [DOI] [PubMed] [Google Scholar]
- 54. Fontan L, Yang C, Kabaleeswaran V, et al. MALT1 small molecule inhibitors specifically suppress ABC-DLBCL in vitro and in vivo. Cancer Cell 2012; 22:812–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Eitelhuber AC, Vosyka O, Nagel D, et al. Activity-based probes for detection of active MALT1 paracaspase in immune cells and lymphomas. Chem Biol 2015; 22:129–138. [DOI] [PubMed] [Google Scholar]
- 56. Hachmann J, Edgington-Mitchell LE, Poreba M, et al. Probes to monitor activity of the paracaspase MALT1. Chem Biol 2015; 22:139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Douanne T, Gavard J, Bidère N. The paracaspase MALT1 cleaves the LUBAC subunit HOIL1 during antigen receptor signaling. J Cell Sci 2016; [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]