

Abstract
Purpose of review
The present review will highlight some of the recent findings regarding the capacity of HIV-1 to replicate during antiretroviral therapy (ART).
Recent findings
Although ART is highly effective at inhibiting HIV replication, it is not curative. Several mechanisms contribute to HIV persistence during ART, including HIV latency, immune dysfunction, and perhaps persistent low-level spread of the virus to uninfected cells (replication). The success in curing HIV will depend on efficiently targeting these three aspects. The degree to which HIV replicates during ART remains controversial. Most studies have failed to find any evidence of HIV evolution in blood, even with samples collected over many years, although a recent very intensive study of three individuals suggested that the virus population does shift, at least during the first few months of therapy. Stronger but still not definitive evidence for replication comes from a series of studies in which standard regimens were intensified with an integration inhibitor, resulting in changes in episomal DNA (blood) and cell-associated RNA (tissue). Limited drug penetration within tissues and the presence of immune sanctuaries have been argued as potential mechanisms allowing HIV to spread during ART. Mathematical models suggest that HIV replication and evolution is possible even without the selection of fully drug-resistant variants. As persistent HIV replication could have clinical consequences and might limit the efficacy of curative interventions, determining if HIV replicates during ART and why, should remain a key focus of the HIV research community.
Summary
Residual viral replication likely persists in lymphoid tissues, at least in a subset of individuals. Abnormal levels of immune activation might contribute to sustain virus replication.
Keywords: abnormal levels of immune activation and inflammation; cell-to-cell infection; HIV residual replication during ART; limited drug penetration within tissues, presence of immune sanctuaries
INTRODUCTION
In a motivated HIV-1-infected person who can adhere to modern regimens indefinitely, a combination of optimally selected antiretroviral drugs can suppress HIV-1 replication to levels below which drug resistance cannot accumulate. As a consequence, these drugs work indefinitely. It is now widely assumed that a modern combination regimen used optimally will work for decades and that the life-time risk of treatment failure because of drug resistance is negligible. Indeed, as drug resistance is now so rarely encountered in the clinic, most have assumed that ART is able to completely and indefinitely suppress all replication. This assumption is increasingly being challenged, however, and the degree to which ART is fully suppressive is the focus of ongoing debate and investigation.
All antiretroviral drugs work by preventing the capacity of HIV-1 to replicate, which can be defined as the spread of infectious virus from one cell to another cell. These drugs do not target integrated HIV-1 DNA nor are they able to eliminate long-lived cells that harbor these integrants. Accordingly, even if ART is fully suppressive, the virus will persist for decades, and be the source of recrudescent virus if therapy is stopped. ART is not curative.
We believe that there are three major pathways that should be investigated and targeted if we aim to find a cure or, more precisely, a remission: residual low-level viral replication, immune dysfunction, and latency. The prevailing consensus is that ART is highly effective at preventing HIV-1 replication, moderately effective at reversing immune dysfunction, and mostly ineffective at clearing cells harboring latent HIV-1. The most stubborn barrier in developing a cure will almost certainly prove to be HIV-1 latency; most active cure programs are understandably focused on reversing latency and eliminating the virus producing cells (‘shock and kill’). However, this approach assumes that all HIV-1 replication is completely inhibited and that ART is so effective that it will inhibit even a resurgent virus population that emerges in the context of a latency reversing therapy. It is hence critically important to determine the degree to which HIV-1 replication persists during ART.
There is an open controversy as to whether HIV-1 continues to replicate during ART. Some of this debate is because of a lack of consensus regarding the term replication. Some have argued that the continued production of HIV-1 from an infected cell is evidence of replication. As viremia persists at low levels during ART, it is abundantly clear that virus production persists indefinitely. We and others have argued that the presence of viral particles in cells or plasma is not proof that the virus is actively spreading to new target cells. As we discuss in this review, there are ample data to support the thesis that ART is fully effective at blocking replication. There is also ample and we argue more persuasive data that the virus does indeed continue to replicate, albeit at low levels and perhaps only in a subset of individuals.
Box 1.

no caption available
MEASURING HIV-1 REPLICATION DURING ANTIRETROVIRAL THERAPY
Although there are multiple approaches to measure the reservoir of HIV-1 in plasma, cells, and tissues, none is able to directly determine if the virus is actively replicating. In the absence of ART, the standard way to estimate the total body rate of HIV-1 replication is to determine the plasma HIV-1 RNA level. At steady state, the level of viremia is a reasonable estimate of the number of virus-producing cells and presumably the level of replication [1]. This is assumed to be true even in states of immunologic control. Among those controlling HIV-1 in absence of therapy (‘elite controllers’), there exists a steady-state level of viremia [2] and a persistent reservoir of replication-competent virus that enables continued replication [3–5]. Among controllers with low or undetectable plasma HIV-1 RNA levels, the presence of persistent viremia almost certainly reflects, in part, active replication, as shown by recent studies in which controllers were given ART [5].
A steady-state level of very-low viremia has also been described among those on ART [6,7], but it is far less clear if active HIV-1 replication contributes to viremia during ART. The molecular nature of such techniques does not allow us to determine whether residual virus are infectious or not, but their long-term persistence (over 7 years) suggests that they are at least being produced over time, and not just released from trapped stages in certain cells. The genetic structure of virus population during ART is generally stable [8–10,11▪,12▪], arguing against ongoing cycles of HIV-1 replication, although recent data have challenged this assumption [13▪▪], as discussed below.
Given the lack of any direct methods to quantify replication in vivo, we and others have used treatment intensification of standard regimens to determine if more potent ART can alter the size of the reservoir. With regard to plasma HIV-1 RNA, the results of these studies have been very consistent. Adding a potent antiretroviral drug to a stable regimen has no measurable impact on plasma HIV-1 RNA levels [14–21], strongly suggesting that ongoing cycles of HIV-1 replication are not a major mechanism for persistent viremia during ART.
The unique mechanism of action of the integrase inhibitor drug class allowed us, and others, to indirectly assess replication during ART. These drugs inhibit the integration of episomal HIV-1 DNA strands– including two-long terminal repeat (2-LTR) circles – into host genome. In two randomized clinical trials of HIV-1-infected adults on apparently effective ART, the addition of an integrase inhibitor (raltegravir) to a stable regimen resulted in a transient increase in 2-LTR-circles, suggesting that HIV-1 integration had been blocked [22,23]. A transient increase in episomal DNA with inhibition of integration suggests that at baseline (preintensification), there existed a steady-state in which there were going cycles of HIV-1 DNA integration; this in turn implies that at steady-state, HIV-1 was being successfully spread from one cell to the next, with RNA being successfully reversed transcribed (despite the presence of reverse transcriptase inhibitors) and the newly produced DNA eventually being successfully integrated (i.e. a complete round of replication). Interrupting this cycle at steady state would be expected to result in a transient and early increase in the preintegrated episomes, as observed in both studies. Of note, this phenomenon was only observed in about one-third of subjects, with most of the activity observed in those who had been on a protease inhibitor-based regimen.
Whether transiently produced 2-LTR circles are the result of ongoing cycles of viral replication or just persistent viral release from productively infected cells is debatable. However, mathematical modeling of the data suggests that the transient dynamics of 2-LTR after intensification with integrase inhibitors would be more compatible with the former hypothesis [24]. Of note, similar 2-LTR dynamics are also observed in patients who initiate ART with integrase inhibitors [25]. Notably, over time, integrase inhibitor intensification results in lower levels of CD8+ T-cell activation [18] and D-dimers [23], an outcome which strongly suggests that low-level replication had been occurring. Moreover, when integrase inhibitor intensification was discontinued, CD8+ T-cell activation levels increased in patients who had shown increases in 2-LTR circles [26].
Preliminary viral genetic analysis suggested that cells sequestered in tissues rather than circulating T cells were supporting virus replication [27]. Subsequent spatial modeling suggests that conditions for the formation of an observed transient peak in 2-LTR formation following raltegravir intensification include a sanctuary site diameter larger than 0.2 mm, a viral basic reproductive ratio within the site larger than 1, and a total volume of active sanctuary sites above 20 ml [28]. However, treatment intensification therapy has not contributed to reduce the total number of HIV-1-infected cells [22].
The exploration of candidate molecules targeting viral reactivation has promoted the use of cell-associated HIV-1 RNA as a way to measure the pharmacological impact of those molecules to flush HIV-1 out of the host genome. Interestingly, the levels of HIV-1 transcription has been greater than expected, even in resting memory CD4+ T cells, which were considered to be transcriptionally silent [12▪]. Although it is clear that viral transcription does not imply generation of mature infectious virions [29], it is intriguing to observed such a basal production of unspliced viral RNA. In fact, it has been reported that higher levels of HIV-1 expression while on ART are associated with shorter time to HIV-1 rebound after treatment interruption, suggesting that quantification of the active HIV-1 reservoir may provide a biomarker of efficacy for therapies that aim to achieve ART-free HIV-1 remission [30▪▪]. Raltegravir-containing ART intensification has been shown to reduce the detection of unspliced HIV-1 RNA in CD4+ T cells in the terminal ileum, suggesting that this gut site might support ongoing productive infection in some patients on ART, even if the contribution to plasma RNA is not discernible [31].
MEASURING HIV-1 EVOLUTION DURING ANTIRETROVIRAL THERAPY
If, as suggested by the aforementioned studies, HIV-1 is replicating during ART, then it is reasonable to assume that the virus would continue to evolve. Among individuals with natural control of viral replication (‘elite controllers’), virus evolution can be readily detected [3,32]. Detecting HIV-1 evolution during ART has been difficult. Indeed, the characterization of HIV-1 genetic sequences in the majority of patients treated for up to 15 years with suppressive ART has shown no detectable HIV-1 molecular evolution, strongly arguing against the presence of active HIV-1 replication [11▪,12▪]. These studies relied largely on blood-based measurements, a major limitation as most of the so-called active reservoir is likely in tissues, in which cell-to-cell transfer is theoretically more efficient [33].
Some teams, however, have reported evidence of HIV-1 evolution during ART. Sequence evolution consisting of new drug resistance mutations and novel amino acid changes within a relevant HLA-restricted allele were described during recombinant HIV-1 poxvirus immunizations in patients with clinically undetectable viral loads on durable suppressive ART [34]. In a separate study, ultra-deep sequencing of V3 env regions of viruses in peripheral blood mononuclear cells of patients on long-term suppressive ART revealed evolution of HIV-1 quasispecies in cell reservoirs and genetic diversification of the virus in patients infected with CXCR4-tropic viruses [35].
A recent intensive study of three HIV-infected adults provides additional evidence for residual evolution (and, by extension, replication) during ART [13▪▪]. Deep sequencing methods were applied to HIV-1 DNA obtained from lymph nodes at baseline, month 3, and month 6 of ART. Although drug resistance mutations did not accumulate, a phylogenetic analysis revealed a temporal structure consistent with ongoing evolution.
Mathematical modeling has been used to better understand the mechanisms underlying viral persistence in patients on suppressive ART. Models that include stochastic population switch may reconcile the coexistence of low viral load persistence, emergence of intermittent viral blips and stability of the latent reservoir [36▪]. Similarly, mathematical models of latently and productively infected cells and virus predict that, in patients on suppressive ART, the contribution of viral replication to residual virus, whereas small, yields short-term evolution [37▪▪]. But even if the contribution is large (e.g. poor adherence to therapy), long-term evolution can still be limited, and de novo emergence of drug resistance remain rare. Therefore, this theoretical approach attempts to reconcile the seemingly contradictory experimental observations on residual viremia in patients on therapy.
MECHANISMS ENABLING HIV-1 REPLICATION DURING ANTIRETROVIRAL THERAPY
The collective data strongly suggest that low-levels of HIV-1 replication persist during ART, at least in some individuals. Given that current antiretroviral drugs are potent and should be able to fully block HIV-1 spread [38–40], why would HIV-1 replication persist?
It has also been proposed that cell-to-cell spread of HIV-1 contributes to ongoing replication despite ART [33]. In an in-vitro experimental model, infections involving cell-to-cell spread were markedly less sensitive to the drugs than infections originating from cell-free virus, without requiring drug-resistant mutations. Cell-to-cell viral transfer has also been reported to contribute to HIV-1 infection and persistence in astrocytes [41▪], suggesting that decreased drug sensitivity in these contacts might contribute to provide additional explanations for the HIV-1 persistence in the central nervous system where the access of antiretroviral drugs may be limited. It remains to be proved, however, whether cell-to-cell spread has the same properties in vivo. It should be noted that cell-to-cell transmission of HIV-1 efficiently triggers pyroptotic death of lymphoid-tissue-derived CD4+ T cells [42▪▪]. If antiretroviral drugs suboptimally inhibit cell-to-cell HIV-1 transmission, inflammation and progressive depletion of CD4+ T cells will still happen in spite of suppressive plasma viremia (Fig. 1).
FIGURE 1.
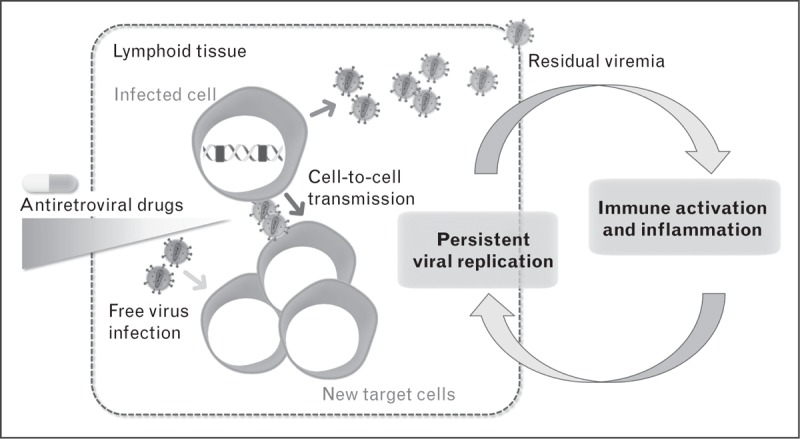
Suggested mechanisms of HIV residual replication during antiretroviral therapy (ART). Limited drug penetration within tissues, the presence of immune sanctuaries, cell-to-cell viral transmission, and abnormal levels of immune activation and inflammation have been argued as potential mechanisms allowing HIV to spread and evolve during ART.
Theoretical considerations and some emerging data suggest that the actively replication viral population resides in lymphoid tissues. The density of CD4+ T cells in these tissues likely makes successful infection events more likely [33]. The distribution of antiretroviral drugs within lymphoid tissues may also be a factor. In a recent study, intracellular concentrations of the most antiretroviral drugs in peripheral blood mononuclear cells did not predict concentrations in lymph node, ileal, and rectal mononuclear cells. In fact, tissue concentrations of several of the most frequently used antiretroviral drugs (including tenofovir disoproxil fumarate, emtricitabine, efavirenz, atazanavir, and darunavir) were lower than in peripheral blood [43▪▪]. These lower concentrations correlated with slower decay (and even possible increases) in the follicular dendritic cell network pool of virions and with detection of viral RNA in productively infected cells. Other groups have failed to find reduced concentrations of some of these and other drugs within gut tissue [44,45], however, and the potential mechanism accounting for reduced antiretroviral drug penetration into lymphoid tissues has not been identified.
The B-cell follicle within lymph nodes appears to be a major site for active viral replication in untreated simian immunodeficiency virus (SIV) and HIV infection [46,47,48▪▪]. This is particularly true in the context of strong host-mediated immune control (‘elite’ control) and perhaps during ART [48▪▪]. CD8+ T cells – the best characterized pathway by which the immune system can effectively control SIV/HIV – are excluded from the B-cell follicle (presumably to allow more efficient interactions between B cells and those cells which enable them to mature, including a T follicular helper cells). The lack of efficient CTL responses in these regions may allow SIV/HIV replication in lymphoid tissue [48▪▪,49,50], accounting for the low levels of viral evolution, which has recently been observed in lymphoid tissues [13▪▪].
Infected macrophages and resident microglia in the central nervous system serve as long-lived cellular reservoirs of HIV-1. These cells are found to harbor productive HIV-1, with perivascular macrophages constituting the principle reservoir of productive virus in the brain. Beyond the contribution of these long-lived cells to the clinical development and progression of HIV-1-associated neurocognitive disorders, they also represent a major obstacle in virus eradication that needs to be therapeutically targeted [51].
CLINICAL IMPLICATIONS OF RESIDUAL REPLICATION DURING ANTIRETROVIRAL THERAPY
Residual viremia has also been linked with development of blips and low-level viremia (50–1000 HIV-1 RNA copies/ml) in treated patients with HIV-1 [52]. In fact, replication-competent residual HIV-1 has been recovered from the plasma of a patient receiving long-term suppressive ART, suggesting that residual viruses produced in the absence of therapy could initiate fresh cycles of infection and spread in host cells [53]. Individuals with ultrasensitive viremia above 2.5 HIV-1 RNA copies/ml had greater plasma levels of lipopolysaccharide than those with viremia below 2.5 copies/ml, indicating an association between microbial translocation and residual viral production if not replication [54]. Many studies have found a positive association between the levels of HIV-1 persistence and inflammation [55–59], again suggesting these two phenomena are linked.
There likely exists a complex bi-directional association between residual inflammation and residual HIV-1 replication. Residual inflammation may contribute to HIV-1 persistence by inducing de-novo infection in activated CD4+ T cells and by upregulating the expression of immune checkpoint blockers and other immunoregulatory pathways that blunt HIV-1-specific immune responses [60]. Persistent HIV-1 replication may in turn contribute to an inflammatory environment [22,23]. Therefore, it has been proposed that compounds addressed to reduce immune activation and inflammation might contribute to limit residual viremia and reservoir replenishment [61,62▪▪].
FUTURE DIRECTIONS
The evidence of persistent replication in many if not all HIV-1-infected individuals on ART argues for the development and evaluation of novel therapeutic strategies that will fully suppress viral replication. Several approaches involve the implementation of improved drugs on existing targets, new formulations and delivery systems [63], new viral and cell targets [64], the use of therapies based on neutralizing monoclonal antibodies [65–67], or chimeric proteins as eCD4-Ig (a fusion of CD4-Ig with a small CCR5-mimetic sulfopeptide) expressed in adeno-associated virus vectors [68]. The goal of these new approaches is not only to achieve excellent potency but also improved drug penetration and reduced toxicity. Moreover, these strategies could also avert the long-term clinical consequences of chronic immune activation driven directly or indirectly by low-level viral replication to thereby improve immune reconstitution.
Cutting-edge research is being performed in the field of HIV-1 remission, including the development of new technologies that should facilitate the detection of viral dynamics and localization in lymphoid tissues. A new real-time, in-vivo viral imaging method to capture total-body SIV replication using antibody-targeted positron emission tomography (immunoPET) has been applied to the detection and localization of sites of SIV infection in antiretroviral-treated macaques [69▪▪]. Upon additional refinements to improve contrast and uptake, this approach should be easily translatable to humans because of the availability of anti-Env HIV-1 antibodies and because the imaging approach is based on technologies already used in the clinic. It also provides the ability to identify novel areas of virus replication that may otherwise be difficult to sample during studies investigating the eradication of HIV infection. Comprehensive characterization of HIV-1 antibody profiles has also been suggested as a method to monitor curative interventions [70].
CONCLUSION
In summary, complete block of residual viral replication is a prerequisite to successfully eliminating viral reservoir. Unfortunately, we lack a robust assay that ensures the identification of an active viral reservoir. Persistent levels of HIV-1 RNA in plasma, below the limit of detection of clinical assays, are difficult to interpret but suggestive of, at least, sustained viral leaking. Viral replication might also be confined to specific tissues and cell types where drug concentrations might be suboptimal. There is an association between residual viral replication and persistent immune activation and inflammation; however, we lack a clear immunological mechanism. The future implementation of new diagnostic tools and new treatment strategies with improved profiles of potency and tissue penetrability should facilitate the final remission of HIV-1 infection.
Acknowledgements
We appreciate the help of M.C. Puertas with graphics. Work in JMP group is supported by the Spanish Secretariat of Research (grant SAF2013–49042-R), the Spanish AIDS network ‘Red Temática Cooperativa de Investigación en SIDA’ (RD12/0017), the American Foundation for AIDS Research (amfAR), the FP7-HEALTH-2013-INNOVATION-1 through the iHIVARNA project, and the Agence Nationale de recherches sur la SIDA et les hépatites virales (ANRS).
The funders had no role in the design and preparation of this review, decision to publish, or preparation of the manuscript.
Financial support and sponsorship
None.
Conflicts of interest
Through his institution J. Martinez-Picado has received unrestricted research grant support from CHEMO, Gilead, Merck, and ViiV. He has received financial compensation for consultancies, lecturing or developing educational activities from Bristol-Myers Squibb, Gilead, Innovex, Merck, and Plasmia Biotech. S.G. Deeks has received research support from Merck and consulted for Merck.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1.Hockett RD, Kilby JM, et al. Constant mean viral copy number per infected cell in tissues regardless of high, low, or undetectable plasma HIV RNA. J Exp Med 1999; 189:1545–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hatano H, Delwart EL, et al. Evidence for persistent low-level viremia in individuals who control human immunodeficiency virus in the absence of antiretroviral therapy. J Virol 2009; 83:329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mens H, Kearney M, et al. HIV-1 continues to replicate and evolve in patients with natural control of HIV infection. J Virol 2010; 84:12971–12981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chun TW, Shawn Justement J, et al. Effect of antiretroviral therapy on HIV reservoirs in elite controllers. J Infect Dis 2013; 208:1443–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hatano H, Yukl SA, et al. Prospective antiretroviral treatment of asymptomatic, HIV-1 infected controllers. PLoS Pathog 2013; 9:e1003691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maldarelli F, Palmer S, et al. ART suppresses plasma HIV-1 RNA to a stable set point predicted by pretherapy viremia. PLoS Pathog 2007; 3:e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palmer S, Maldarelli F, et al. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc Natl Acad Sci U S A 2008; 105:3879–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joos B, Fischer M, et al. HIV rebounds from latently infected cells, rather than from continuing low-level replication. Proc Natl Acad Sci U S A 2008; 105:16725–16730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evering TH, Mehandru S, et al. Absence of HIV-1 evolution in the gut-associated lymphoid tissue from patients on combination antiviral therapy initiated during primary infection. PLoS Pathog 2012; 8:e1002506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Josefsson L, von Stockenstrom S, et al. The HIV-1 reservoir in eight patients on long-term suppressive antiretroviral therapy is stable with few genetic changes over time. Proc Natl Acad Sci U S A 2013; 110:E4987–E4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11▪.Kearney MF, Spindler J, et al. Lack of detectable HIV-1 molecular evolution during suppressive antiretroviral therapy. PLoS Pathog 2014; 10:e1004010. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article reports that low levels of virus detected in the blood of treated patients did not result from newly infected cells but originated from cellular populations that were infected before treatment was initiated.
- 12▪.Kearney MF, Wiegand A, et al. Origin of rebound plasma HIV includes cells with identical proviruses that are transcriptionally active before stopping of antiretroviral therapy. J Virol 2015; 90:1369–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]; This report shows several incidences where plasma HIV variants matched that of multiple proviral DNA copies from infected blood cells sampled before treatment interruption. In addition, it suggests that these cells were not dormant, but were generating unspliced RNA transcripts before treatment was interrupted.
- 13▪▪.Lorenzo-Redondo R, Fryer HR, et al. Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature 2016; 530:51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article describes shifts in the viral population within lymphoid tissues in three individuals receiving short-term ART. It also presents a mathematical model showing that development of drug resistance is not a foregone conclusion even if HIV continues to replicate at low levels.
- 14.Dinoso JB, Kim SY, et al. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc Natl Acad Sci U S A 2009; 106:9403–9408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McMahon D, Jones J, et al. Short-course raltegravir intensification does not reduce persistent low-level viremia in patients with HIV-1 suppression during receipt of combination antiretroviral therapy. Clin Infect Dis 2010; 50:912–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hatano H, Hayes TL, et al. A randomized, controlled trial of raltegravir intensification in antiretroviral-treated, HIV-infected patients with a suboptimal CD4+ T cell response. J Infect Dis 2011; 203:960–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Byakwaga H, Kelly M, et al. Intensification of antiretroviral therapy with raltegravir or addition of hyperimmune bovine colostrum in HIV-infected patients with suboptimal CD4+ T-cell response: a randomized controlled trial. J Infect Dis 2011; 204:1532–1540. [DOI] [PubMed] [Google Scholar]
- 18.Llibre JM, Buzón MJ, et al. Treatment intensification with raltegravir in subjects with sustained HIV-1 viraemia suppression: a randomized 48-week study. Antivir Ther 2012; 17:355–364. [DOI] [PubMed] [Google Scholar]
- 19.Gandhi RT, Coombs RW, et al. No effect of raltegravir intensification on viral replication markers in the blood of HIV-1-infected patients receiving antiretroviral therapy. J Acquir Immune Defic Syndr 2012; 59:229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vallejo A, Gutierrez C, et al. The effect of intensification with raltegravir on the HIV-1 reservoir of latently infected memory CD4 T cells in suppressed patients. AIDS 2012; 26:1885–1894. [DOI] [PubMed] [Google Scholar]
- 21.Hunt PW, Shulman NS, et al. The immunologic effects of maraviroc intensification in treated HIV-infected individuals with incomplete CD4+ T-cell recovery: a randomized trial. Blood 2013; 121:4635–4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buzón MJ, Massanella M, et al. HIV-1 replication and immune dynamics are affected by raltegravir intensification of HAART-suppressed subjects. Nat Med 2010; 16:460–465. [DOI] [PubMed] [Google Scholar]
- 23.Hatano H, Strain MC, et al. Increase in 2-long terminal repeat circles and decrease in D-dimer after raltegravir intensification in patients with treated HIV infection: a randomized, placebo-controlled trial. J Infect Dis 2013; 208:1436–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo R, Cardozo EF, et al. Modelling HIV-1 2-LTR dynamics following raltegravir intensification. J R Soc Interface 2013; 10:20130186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Puertas MC, Massanella M, et al. Intensification of a raltegravir-based regimen with maraviroc in early HIV-1 infection. AIDS 2014; 28:325–334. [DOI] [PubMed] [Google Scholar]
- 26.Massanella M, Esteve A, et al. Dynamics of CD8 T-cell activation after discontinuation of HIV treatment intensification. J Acquir Immune Defic Syndr 2013; 63:152–160. [DOI] [PubMed] [Google Scholar]
- 27.Buzón MJ, Codoñer FM, et al. Deep molecular characterization of HIV-1 dynamics under suppressive HAART. PLoS Pathog 2011; 7:e1002314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cardozo EF, Luo R, Piovoso MJ, Zurakowski R. Spatial modeling of HIV cryptic viremia and 2-LTR formation during raltegravir intensification. J Theor Biol 2014; 345:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barton KM, Rasmussen TA, Tolstrup M, et al. Panobinostat Broadly Activates Latent HIV-1 proviruses in patients. Proceedings of the 22nd Conference on Retroviruses and Opportunistic Infections Seattle, Washington, USA 2015; abstract 109. [Google Scholar]
- 30▪▪.Li JZ, Etemad B, et al. The size of the expressed HIV reservoir predicts timing of viral rebound after treatment interruption. AIDS 2016; 30:343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]; This report shows that higher levels of HIV expression whereas on ART are associated with shorter time to HIV rebound after treatment interruption.
- 31.Yukl SA, Shergill AK, et al. Effect of raltegravir-containing intensification on HIV burden and T-cell activation in multiple gut sites of HIV-positive adults on suppressive antiretroviral therapy. AIDS 2010; 24:2451–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Connell KA, Brennan TP, et al. Control of HIV-1 in elite suppressors despite ongoing replication and evolution in plasma virus. J Virol 2010; 84:7018–7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sigal A, Kim JT, et al. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature 2011; 477:95–98. [DOI] [PubMed] [Google Scholar]
- 34.Shiu C, Cunningham CK, et al. Identification of ongoing human immunodeficiency virus type 1 (HIV-1) replication in residual viremia during recombinant HIV-1 poxvirus immunizations in patients with clinically undetectable viral loads on durable suppressive highly active antiretroviral therapy. J Virol 2009; 83:9731–9742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raymond S, Saliou A, et al. Evolution of HIV-1 quasispecies and coreceptor use in cell reservoirs of patients on suppressive antiretroviral therapy. J Antimicrob Chemother 2014; 69:2527–2530. [DOI] [PubMed] [Google Scholar]
- 36▪.Wang S, Rong L. Stochastic population switch may explain the latent reservoir stability and intermittent viral blips in HIV patients on suppressive therapy. J Theor Biol 2014; 360:137–148. [DOI] [PubMed] [Google Scholar]; This article explores, through mathematical models, the contribution of ongoing viral replication to the persistence of the latent reservoir and virus in patients on long-term suppressive therapy.
- 37▪▪.Conway JM, Perelson AS. Residual viremia in treated HIV+ individuals. PLoS Comput Biol 2016; 12:e1004677. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article mathematically predicts that the contribution of viral replication to HIV persistence during ART may be small and could result in short-term evolution but not necessarily the generation of drug resistance and treatment failure.
- 38.Shen L, Peterson S, et al. Dose-response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat Med 2008; 14:762–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shen L, Rabi SA, et al. A critical subset model provides a conceptual basis for the high antiviral activity of major HIV drugs. Sci Transl Med 2011; 3:91ra63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jilek BL, Zarr M, et al. A quantitative basis for antiretroviral therapy for HIV-1 infection. Nat Med 2012; 18:446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41▪.Luo X, He JJ. Cell-cell contact viral transfer contributes to HIV infection and persistence in astrocytes. J Neurovirol 2015; 21:66–80. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study highlights the importance of the cell-to-cell contact as a mechanism for HIV persistence within astrocytes and provides evidence suggesting that HIV latency exists within astrocytes and the central nervous system.
- 42▪▪.Galloway NL, Doitsh G, et al. Cell-to-cell transmission of HIV-1 is required to trigger pyroptotic death of lymphoid-tissue-derived CD4 T cells. Cell Rep 2015; 12:1555–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article demonstrates that cell-to-cell spread of HIV-1 is required to deplete nonpermissive cells via caspase-1-dependent pyroptosis, whereas free HIV-1 particles, even in large quantities, are unable to trigger this form of cell death.
- 43▪▪.Fletcher CV, Staskus K, et al. Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc Natl Acad Sci U S A 2014; 111:2307–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article shows that measuring drug concentrations in plasma or in PBMCs does not predict those in lymphoid compartments, and that HIV persists at stable levels within lymphatic tissues in some individuals.
- 44.Patterson KB, Prince HA, et al. Penetration of tenofovir and emtricitabine in mucosal tissues: implications for prevention of HIV-1 transmission. Sci Transl Med 2011; 3:112re4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patterson KB, Prince HA, et al. Differential penetration of raltegravir throughout gastrointestinal tissue: implications for eradication and cure. AIDS 2013; 27:1413–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hufert FT, van Lunzen J, et al. Germinal centre CD4+ T cells are an important site of HIV replication in vivo. AIDS 1997; 11:849–857. [DOI] [PubMed] [Google Scholar]
- 47.Perreau M, Savoye AL, et al. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J Exp Med 2013; 210:143–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48▪▪.Fukazawa Y, Lum R, et al. B cell follicle sanctuary permits persistent productive simian immunodeficiency virus infection in elite controllers. Nat Med 2015; 21:132–139. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article demonstrates that SIV can take advantage of the relative exclusion of CD8+ effector T cells from B-cell follicles to persist and preferentially replicate within these regions.
- 49.Connick E, Mattila T, et al. CTL fail to accumulate at sites of HIV-1 replication in lymphoid tissue. J Immunol 2007; 178:6975–6983. [DOI] [PubMed] [Google Scholar]
- 50.Connick E, Folkvord JM, et al. Compartmentalization of simian immunodeficiency virus replication within secondary lymphoid tissues of rhesus macaques is linked to disease stage and inversely related to localization of virus-specific CTL. J Immunol 2014; 193:5613–5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gerngross L, Fischer T. Evidence for cFMS signaling in HIV production by brain macrophages and microglia. J Neurovirol 2015; 21:249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hofstra LM, Mudrikova T, et al. Residual viremia is preceding viral blips and persistent low-level viremia in treated HIV-1 patients. PLoS One 2014; 9:e110749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sahu GK, Sarria JC, Cloyd MW. Recovery of replication-competent residual HIV-1 from plasma of a patient receiving prolonged, suppressive highly active antiretroviral therapy. J Virol 2010; 84:8348–8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baroncelli S, Galluzzo CM, et al. Microbial translocation is associated with residual viral replication in HAART-treated HIV+ subjects with <50 copies/ml HIV-1 RNA. J Clin Virol 2009; 46:367–370. [DOI] [PubMed] [Google Scholar]
- 55.Chomont N, El-Far M, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med 2009; 15:893–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hatano H, Jain V, et al. Cell-based measures of viral persistence are associated with immune activation and programmed cell death protein 1 (PD-1)-expressing CD4+ T cells. J Infect Dis 2013; 208:50–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Klatt NR, Chomont N, Douek DC, Deeks SG. Immune activation and HIV persistence: implications for curative approaches to HIV infection. Immunol Rev 2013; 254:326–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vandergeeten C, Fromentin R, et al. Interleukin-7 promotes HIV persistence during antiretroviral therapy. Blood 2013; 121:4321–4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murray JM, Zaunders JJ, et al. HIV DNA subspecies persist in both activated and resting memory CD4+ T cells during antiretroviral therapy. J Virol 2014; 88:3516–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barouch DH, Deeks SG. Immunologic strategies for HIV-1 remission and eradication. Science 2014; 345:169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Katlama C, Deeks SG, et al. Barriers to a cure for HIV: new ways to target and eradicate HIV-1 reservoirs. Lancet 2013; 381:2109–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62▪▪.Micci L, Ryan ES, et al. Interleukin-21 combined with ART reduces inflammation and viral reservoir in SIV-infected macaques. J Clin Invest 2015; 125:4497–4513. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study indicates that reducing chronic inflammation within tissues during ART can reduce the burden of SIV, arguing that immune dysfunction contributes to SIV and perhaps HIV persistence during ART.
- 63.Trezza C, Ford SL, et al. Formulation and pharmacology of long-acting cabotegravir. Curr Opin HIV AIDS 2015; 10:239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sloan RD, Wainberg MA. Harnessing the therapeutic potential of host antiviral restriction factors that target HIV. Expert Rev Anti Infect Ther 2013; 11:1–4. [DOI] [PubMed] [Google Scholar]
- 65.Barouch DH, Whitney JB, et al. Therapeutic efficacy of potent neutralizing HIV-1-specific monoclonal antibodies in SHIV-infected rhesus monkeys. Nature 2013; 503:224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shingai M, Nishimura Y, et al. Antibody-mediated immunotherapy of macaques chronically infected with SHIV suppresses viraemia. Nature 2013; 503:277–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barouch DH, Alter G, et al. Protective efficacy of adenovirus/protein vaccines against SIV challenges in rhesus monkeys. Science 2015; 349:320–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gardner MR, Kattenhorn LM, et al. AAV-expressed eCD4-Ig provides durable protection from multiple SHIV challenges. Nature 2015; 519:87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69▪▪.Santangelo PJ, Rogers KA, et al. Whole-body immunoPET reveals active SIV dynamics in viremic and antiretroviral therapy-treated macaques. Nat Methods 2015; 12:427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes a real-time, in vivo, whole body viral imaging method for the detection and localization of sites of SIV production in chronically infected, ART-treated, and elite controller macaques.
- 70.Burbelo PD, Bayat A, et al. HIV antibody characterization as a method to quantify reservoir size during curative interventions. J Infect Dis 2014; 209:1613–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]