

Abstract
Matrix metalloproteinases (MMPs) are extracellularly acting enzymes that have long been known to have deleterious roles in brain injury and disease. In particular, widespread and protracted MMP activity can contribute to neuronal loss and synaptic dysfunction. However, recent studies show that rapid and focal MMP-mediated proteolysis proactively drives synaptic structural and functional remodelling that is crucial for ongoing cognitive processes. Deficits in synaptic remodelling are associated with psychiatric and neurological disorders, and aberrant MMP expression or function may contribute to the molecular mechanisms underlying these deficits. This Review explores the paradigm shift in our understanding of the contribution of MMPs to normal and abnormal synaptic plasticity and function.
Matrix metalloproteinases (MMPs), which are members of the metzincin clan of metalloproteinases, compose a large subgroup of zinc-binding, multi-domain endopeptidases that are present in most tissues of the body. In humans, there are 23 distinct MMPs comprising secreted and transmembrane proteins (BOX 1). Much of their biological activity is exerted extracellularly, where they critically influence cellular behaviour through the targeted degradation or the proteolytic processing of various extracellular matrix (ECM) molecules, peptide growth factors, cytokines, chemokines, cell adhesion molecules, and many other types of receptors and glycoproteins that reside on the cell surface. The collective effects of pericellular MMP-mediated proteolysis on cell behaviour can be permissive (they can degrade chemical or physical barriers) and instructive (they can proactively initiate or terminate signaling cascades through the processing of latent bioactive molecules)1, 2. MMP-mediated remodelling of the pericellular microenvironment is therefore essential for many physiological processes. However, MMP activity can also have deleterious effects, such as in cancer, rheumatoid arthritis and other disease states1, 3.
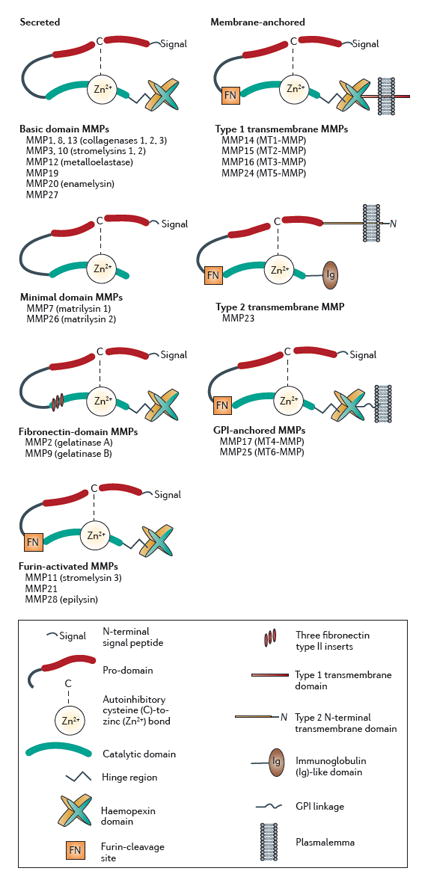
Box 1. Classification and domain structure of the MMPs.
Matrix metalloproteinases (MMPs) are multi-domain proteins that are named according to a sequential numbering scheme and organized into subgroups on the basis of common domains, inserts and other motifs and on shared canonical substrate preferences1, 136, 137. In humans, 24 MMP genes encode 23 distinct MMPs (two identical genes located on chromosome 1 encode MMP23). MMPs all possess an amino-terminal signal peptide that targets them to the secretory pathway, an autoinhibitory pro-domain and a catalytic domain (see the figure). Most MMPs also possess a carboxy-terminal hemopexin domain, which is coupled to the catalytic domain by a flexible hinge region and is an important mediator of protein–protein interactions. In cooperation with other exosites, the hemopexin domain contributes to the target specificity of MMP proteolysis by coordinating interactions with substrates. The hemopexin domain can anchor MMPs to other cell-surface proteins, thereby positioning or stabilizing MMPs at the membrane surface, which in turn markedly influences how and when MMPs become activated as well as regulates their accessibility to substrate targets. Additionally, MMPs, through their hemopexin domain, can act as ligands, activating downstream signal cascades by binding to receptors (for example, the low-density lipoprotein receptor-related protein)138, 139.
Most MMPs are secreted into the extracellular environment. However, a minority (seven) of MMPs are membrane-associated, anchored by a type 1 transmembrane domain (MMP14, MMP15, MMP16 and MMP24), a type II transmembrane domain (MMP23), or a glycosylphosphatidylinositol (GPI) linkage (MMP17 and MMP25). A key feature of all MMPs is that they are synthesized as proteolytically inactive zymogens containing a pro-domain (pro-MMPs). A conserved cysteine in the pro-domain controls proteolytic activity of the enzyme by acting as a fourth zinc-coordinating ligand, rendering the catalytic site masked and inoperative when bound. Activation of MMPs requires unmasking of the catalytic site by disruption of this cysteine–zinc bond (the ‘cysteine switch’)140. For most MMPs, the cysteine switch occurs extracellularly, either through physical removal of the pro-domain via proteolytic cleavage by other extracellular proteases (for example, other MMPs or serine proteases) or by conformationally destabilizing the pro-domain via modifications of the thiol group on the inhibitory cysteine (for example, via oxidation or S-nitrosylation69), which activates the MMP prior to subsequent proteolytic cleavage of the pro-domain. A minority of MMPs, including three secreted MMPs (MMP11, MMP21 and MMP28) and the membrane-associated ones, contain a unique sequence at the C-terminal end of the prodomain that is recognized by the convertase furin. Thus, for most of these MMPs, the cysteine switch can occur intracellularly, allowing the release or insertion of an already proteolytically active MMP into the extracellular environment. Although MMP activity is mostly extracellular, some active MMPs can be found in neuronal or glial nuclei141, 142, where they may have transcription factor-like or DNA repair-like activity143, 144.
MMP activity is terminated in brain mostly by four, small (~22 kD), secreted protein inhibitors called TIMPs (tissue inhibitor of metalloproteinases). The TIMPs bind reversibly to MMPs in a 1:1 stochiometric ratio, indicating that the balance of MMP:TIMP levels determines overall proteolytic activity. Each of the four TIMP proteins displays preferential MMP-binding specificities, and also possess biological activity independent of their role in regulating MMP activity.
In the CNS, MMPs are synthesized and secreted by neurons and glia and, historically, are associated with roles in early CNS morphogenesis4, 5 and protracted pathophysiological cellular remodelling that is triggered by injury or disease (BOX 2). Recent studies, however, show that rapid and focal MMP-mediated proteolysis proactively drives structural and functional synaptic circuit remodelling that is critical for ongoing cognitive processes.
Box 2. Well-established roles of MMPs in CNS injury and disease.
Excessive and protracted matrix metalloproteinase (MMP)-mediated proteolysis is a major pathological component of many CNS injuries and diseases such as neuroinflammation, ischaemia and hypoxia, demyelination, traumatic brain and spinal cord injuries, and neuronal degeneration5, 129, 145. A cascade of aberrant MMP activity is often initiated by MMP-mediated proteolytic degradation of the blood–brain barrier (BBB), where, for example, an ischaemic episode triggers upregulation of MMP2, MMP3, MMP7, MMP9 and MMP14 by endothelial cells, pericytes, leukocytes, and other components of the neurovascular epithelium. The MMP-mediated breakdown of the BBB, which occurs over the course of 24–48 h, allows large numbers of inflammatory and other cell types to invade the brain parenchyma, further stimulating excessive MMP expression and secretion by microglia, astrocytes and neurons. MMPs are activated by each other as well as by free radicals (such as nitric oxide), which are produced by hypoxia, thereby exacerbating a cascade of excessive MMP activity. In time, excessive MMP activity can trigger neuronal apoptosis and synapse loss. Such a cascade of excessive and essentially unregulated MMP activity within the brain parenchyma can last weeks, depending on the injury or particular insult. After this protracted destructive phase, further MMP expression can contribute to subsequent reparative processes of cell replacement, remyelination, and re-establishment of connectivity and neurovascular integrity.
Synaptic circuit remodelling, which includes structural alterations in the number or the morphology of dendritic spines and their synapses coupled with functional changes in the strength of synaptic signalling, occurs throughout life in naturally occurring contexts (for example, in learning6–9). It has become clear, largely based on studies of experimentally induced forms of synaptic plasticity such as long-term potentiation (LTP) and long-term depression (LTD), that synaptic remodelling depends crucially on a variety of ECM proteins and their receptors, cell adhesion molecules, growth factors and other targets of MMP proteolysis10, 11. These findings have led to a fresh outlook on the role, timecourse of activation, and regulation of MMPs in the fine-scale modulation of adaptive, non-pathophysiological synaptic function and plasticity.
The goal of this Review is to capture our rapidly evolving understanding of where, when and how MMPs contribute to normal synaptic functional and structural remodelling, and to speculate on the impact of perturbation of these processes in diseases that are characterized by synaptic plasticity deficits].
Expression and localization in brain
Most MMPs are synthesized and secreted as inactive pro-enzymes (zymogens) that are converted to proteolytically active molecules following several regulatory steps (BOX 1). As regulation of MMP activity can occur at the levels of transcription, translation and post-translation1, the levels of pro- and active forms of MMP and MMP proteolytic activity cannot always be inferred from MMP mRNA levels.
Biochemical experiments have shown that mRNA levels of MMP2, MMP3, MMP8, MMP9, MMP11, MMP12, MMP13, MMP14, MMP15 and MMP24 are low under basal conditions in adult human and rodent brains, but are much higher during early postnatal development12–17. The mRNA levels of other MMPs lie below detectable limits or have not been examined.
For some MMPs, their low transcript levels in total brain samples belie regionally higher levels of MMP mRNA expression and higher constitutive levels of protein. In adult mice and rats, elevated levels of constitutive Mmp9 and Mmp24 mRNA expression are found in the hippocampus compared with other brain regions16–19. Moreover, western blot analyses using MMP-specific antisera that recognize both the inactive pro-forms and the active forms of the enzymes, which can be distinguished by differences between the two forms in their molecular mass (the pro-form is heavier than the the active form), revealed that hippocampal lysates contain high levels of the pro-forms of MMP2, MMP3, MMP9 and MMP24, but lower levels of the active forms of these MMPs16, 18–24. High-resolution microscopy of immunolabelled hippocampal tissue sections showed that MMP9 is concentrated throughout the synaptic neuropil in discrete puncta that colocalize with presynaptic and postsynaptic molecular markers (FIG. 1A), in non-synaptic dendritic regions and in some glial processes18, 19, 21, 25. Similarly, MMP24 has a punctate, synaptic-like distribution in perfused tissue sections of adult mouse hippocampus and cerebellum16. Moreover, it colocalizes with synaptic molecular markers in cultured rat hippocampal neurons and is enriched in a synaptosome fraction prepared from adult rat forebrain23, 26. The cellular or synaptic distributions of MMP2 and MMP3 within specific brain regions are currently unknown.
FIGURE 1. MMP9 localization and activity in hippocampus before and after LTP.

a Under basal conditions in adult hippocampal area CA1, numerous matrix metalloproteinase 9 (MMP9) immunofluorescent puncta are present within the synaptic neuropil of stratum radiatum (a). Many such puncta codistribute with labelling for other synaptic molecular markers, for example, vGluts (vesicular glutamate transporters), which is a marker of excitatory presynaptic terminals (insets), indicating perisynaptic localization. Much of this perisynaptic immunoreactivity is probably the inactive pro-form, as there are few gelatinolytic puncta (hotspots of proteolytic activity) present under control conditions (b). Following induction of long-term potentiation (LTP) (c, image taken 75 minutes after LTP induction), there is a significant increase in numbers of gelatinolytic puncta. In the absence of LTP because of prior administration of the NMDA receptor (NMDAR) antagonist 3-(2-carboxypiperazin-4-yl)propyl-1-phosphonic acid (CPP), there is no increase in proteolysis (d), thus, MMP activation is related specifically to LTP and is not a non-specific effect of trains of electrical stimuli. At higher-power, many of the gelatinolytic puncta (e, gel’lysis) codistribute with MMP9 immunoreactivity (f,g, arrows), which consistent with the activation of MMP9 with LTP induction. Images B–G are adapted from REF 18. Bars represent 50 μm (A); 10 μm (B–D); 3 μm (E–G).
A key question raised by the data above is how much of the MMP protein is proteolytically active under basal conditions? This is a question that cannot be determined definitively by immunocytochemistry, western blotting or — for the gelatinases (that is, MMP2 and MMP9) — in gel gelatin-substrate zymography (BOX 3). However, the combination of in vivo or in situ gelatin-substrate zymography (BOX 3) with immunolocalization has been used to infer the status of endogenous MMP9-mediated proteolytic activity within the synaptic neuropil18, 27. Through these combined approaches, a small number of the MMP9-immunopositive puncta colocalize with discrete hotspots of gelatinolytic activity that are located along hippocampal dendrites and dendritic spines. This pattern is consistent with the idea that much of the MMP9 protein detected immunocytochemically under basal conditions is in the inactive pro-form. It should be noted that these data do not rule out the possibility that other proteases that are capable of cleaving gelatin also contribute to the gelatinolytic activity observed in these experiments21, 25. In any event, such focal gelatinolytic puncta colocalize with molecular markers of excitatory synapses, including AMPA- and NMDA-type glutamate receptors (AMPARs and NMDARs), postsynaptic scaffolding and cytoskeletal proteins, and presynaptic vesicle proteins18, 25, but they seem to be largely absent from GABAergic (inhibitory) synapses25. In the case of MMP24, western blotting and cell-surface biotinylation assays involving cultured rat cortical neurons together indicate that although both pro- and active forms reach the cell surface, the inactive pro-form is more abundant23. The active form of MMP24 is only visualized on the cell surface when proteolytic activity is blocked by the broad-spectrum MMP inhibitor GM6001 (also known as Ilomastat). This suggests that the duration of the active form of MMP24 on the cell surface is probably short because of proteolytic degradation, although the identity of the proteolytic enzyme responsible for surface degradation of MMP24 is unknown. Together, these data indicate that under basal conditions, high levels of MMP9 and MMP24, predominantly in their inactive pro-forms, are distributed perisynaptically at excitatory synapses that are located on dendritic spines. The observation that pro-forms of MMPs are located perisynaptically has important implications for where they exert their proteolytic effects when activated and which proteins they target (see below). Of note, various proteins have been identified that are involved in the targeting of some MMPs to synapses (BOX 4).
Box 3. Gelatin-substrate zymography techniques.
In gel gelatin-substrate zymography
An SDS-PAGE-based technique for the detection of enzyme activity based on the ability of the enzyme to cleave a substrate (gelatin) that is co-polymerized with the polyacrylamide gel. The advantages of this technique are that it is sensitive enough to detect picogram levels of enzyme and it can resolve pro-forms and active forms of gelatin-cleaving matrix metalloproteinases (MMPs) (for example, MMP2 and MMP9) on the basis of molecular mass. The disadvantages are that it lacks cellular resolution, is not applicable to non-gelatinolytic proteases, and does not accurately report on endogenous enzyme activity because of some auto-activation within the gel and the loss of any endogenous tissue inhibitor of metalloproteinase (TIMP) binding during preparation.
In vivo gelatin substrate zymography
An extension of in situ gelatin-substrate zymography, but in this case, the dye-quenched (DQ)-gelatin is injected directly into the brain of an intact animal in vivo. The animal is subsequently perfused with aldehydes and the fluorescent gelatinolytic puncta can be visualized by fluorescent microscopy. The advantage of this approach, in addition to those of in situ zymography, is that brains are not sliced prior to the gelatinolytic reaction, thereby eliminating the potentially confounding effects of the processing steps on enzymatic activity.
In situ gelatin substrate zymography
A technique for visualizing gelatinolytic enzyme activity within semi-intact tissues or cells. The approach is to expose acute tissue slices or cell cultures to dye-quenched (DQ)-gelatin, which is gelatin-coupled to a fluorescent molecule (fluorescein) that is masked until cleaved. Upon cleavage by endogenous gelatinases, the fluorescein becomes visible, and after aldehyde fixation, can be detected by fluorescent microscopy. The advantages of this approach are that it provides cellular and synaptic anatomical resolution of endogenase gelatinolytic enzymes, and can be combined with immunolocalization of other proteins of interest. The disadvantage is that it requires numerous controls to show that enzyme activity is not altered as a result of tissue slicing or other processing steps prior to visualization.
Box 4. How are MMPs directed to or near synapses?
Recent studies show that ABP (AMPA-receptor (AMPAR) binding protein) and GRIP1 (glutamate receptor interacting protein 1), two postsynaptic scaffolding proteins that bind to and direct AMPAR subunits to the synaptic membrane146, also bind to the carboxy-terminal tail of matrix metalloproteinase 24 (MMP24), which is a transmembrane protein, and direct it to synaptic sites23. In the case of secreted MMPs, the basis for such synaptic targeting is less well understood. Image analysis of intracellular trafficking of MMP2, MMP9 or the endogenous MMP inhibitor TIMP1 (tissue inhibitor of metalloproteinase 1) fused to green fluorescent protein expressed in cultured hippocampal neurons147 and glia141 shows that these MMPs and TIMP1 are transported in distinct 160–200 nm-wide vesicles that, for neurons, are concentrated in the somatodendritic domain. Interestingly, although TIMP1 is colocalized in vesicles carrying either MMP2 or MMP9, these two MMPs are distributed into separate vesicle populations in both neurons and astrocytes, which suggests that there is differential control of their trafficking and release. Such vesicles associate with cytoskeletal components and the molecular motors dynein, kinesin and myosin Va, are found in dendritic spines, and can be secreted, as exosomes, from these sites147. In non-neuronal cells, release of such MMP-containing vesicles depends on proteins of the SNARE (soluble NSF attachment protein receptor) family148, although it is unclear at present whether regulated release in neurons occurs by a similar mechanism. Additionally, whether non-vesicular release of soluble MMPs or TIMPs occurs in neurons or glia also remains to be explored.
MMPs drive synaptic remodelling
Our emerging understanding of how MMPs contribute to normal synaptic and behavioural plasticity comes mostly from detailed studies of MMP9. The focus on MMP9 reflects the availability of a broad set of investigative reagents for this MMP and the fact that it has a well-recognized role in pathophysiological neural circuit remodelling19, 25, 28. These new insights into the role of MMP9 in normal synaptic plasticity have been borne largely from experiments examining loss- and gain-of-function effects of MMP9 on persistent forms of synaptic structural and functional modifications such as LTP (BOX 5).
Box 5. Hippocampal long-term potentiation.
Hippocampal long-term potentiation (LTP) is the archetypal cellular model of synaptic functional and structural plasticity that enables memory formation63, information processing149 and skill learning150. Tetanic or theta-burst stimulation protocols applied to the Schaffer collaterals (axons of CA3 pyramidal neurons) elicit NMDA receptor (NMDAR)-dependent LTP at CA3→CA1 glutamatergic synapses, which are located mostly on dendritic spines. At these synapses, LTP reflects functional changes in the magnitude of synaptic neurotransmission that are coordinated with structural changes in synaptic spine morphology, size and density151. Functionally, LTP has at least two mechanistically distinct phases, the occurrence of which depends on the strength of the induction protocol. Weak stimulation protocols elicit so-called early phase (E-LTP), which is transient and involves post-translational modifications of synaptic proteins but is protein synthesis-independent. By contrast, stronger, repetitive stimulation protocols elicit E-LTP followed by a late-phase (L-LTP), which is protein synthesis-dependent and persistent152. In L-LTP, spines undergo actin-dependent increases in spine-head volume, changes in density, and enlargement of synapses. These phenomena are all thought to be important structural correlates of stable changes in synaptic strength153.
LTP and the accompanying dendritic spine plasticity, particularly those evoked by theta-burst stimulation, which mimics endogenous patterns of neural activity during learning154, are initially labile and subject to reversal by a number of protocols before transitioning, via a consolidation-like process, to a stable form that is resistant to reversal protocols155.
LTP-inducing stimuli rapidly activate MMP9
Levels of both the pro-form and the active form of MMP9 are notably elevated in CA1 60 min after induction of late-phase LTP (L-LTP) at CA1 synapses by a strong tetanic stimulation protocol, and 15 min following the end of a chemical induction protocol that selectively elicits L-LTP 18, 21. The increase in MMP9 levels induced by these L-LTP induction protocols is transient, as MMP9 levels return to baseline values by 120 min post-induction. Weaker tetanic stimulation that induces only early phase LTP (E-LTP), or any one of several protocols that induce LTD at CA1 synapses, does not cause any detectable changes in the levels of the pro-form or active-form of MMP921. Collectively, these observations suggest that upregulation of MMP9 is related mechanistically to L-LTP. Consistent with this idea, NMDAR antagonists and protein synthesis inhibitors abrogate the L-LTP-associated increase in protein levels21.
The elevation in MMP9 levels that results from L-LTP-inducing stimuli has been documented in both rats and mice and is consistent across ex vivo (acute hippocampal slices) and in vivo preparations (urethane-anaesthetized animals). Moreover, the increase in MMP9 levels with L-LTP is readily evident in preparations from animals of different ages (from 3 postnatal weeks to adulthood). These observations suggest that MMP9 plays a fundamental role in synaptic plasticity across species and the lifespan. Additionally, in terms of the gelatinases (that is, MMP2 and MMP9), L-LTP-inducing stimuli affect MMP9 levels selectively, as LTP induction has no effect on the levels of the pro-form or active form of MMP221. Whether the levels of other MMPs are altered following the induction of LTP (or other forms of synaptic plasticity) is unknown.
Anatomical localization methods have shown that following induction of L-LTP, marked elevations in the numbers of MMP9 immunoreactive puncta and gelatinolytic puncta can be seen in the neuropil of CA1 — the location of the potentiated synapses (FIG. 1B–D)18, 21. The majority of the LTP-associated gelatinolytic puncta colocalize with MMP9 (FIG. 1E–G) and these puncta are found mostly at or surrounding synapses, or along short stretches of dendrites. When LTP is blocked by NMDAR antagonists administered just before the tetanic stimulation used to induce LTP, the increase in the number of gelatinolytic puncta is also blocked18 (FIG. 1B–D). This result indicates that the appearance of gelatinolytic puncta is not a non-specific effect of high-frequency stimulation, but rather is related specifically to the LTP that results from it. Whether protein synthesis is also required to generate such L-LTP-associated hotspots of proteolytic activity has not been determined.
When these data are considered together, a striking picture emerges which indicates that MMP9 is upregulated and perisynaptically activated on a timescale (~15 min) that is roughly an order of magnitude faster than the timescales of induction and duration of MMP activity (several hours to weeks) following injury or onset of disease (BOX 2). What does a proteolytically active MMP9 contribute to synaptic form and function?
Blocking MMP9 impairs L-LTP and spine remodelling
The contribution of MMP9 to synaptic plasticity has been investigated through acute loss-of-function (exogenous reagents that inhibit MMP9) and chronic loss-of-function (genetic deletion) experimental approaches. Field recordings in CA1 show that L-LTP-inducing stimuli applied to the Schaffer collaterals in the presence of pharmacological MMP inhibitors or MMP9-specific neutralizing antibodies induce LTP at CA1 synapses that is indistinguishable from that induced in controls (by L-LTP-inducing stimuli applied without inhibitors) for 30–45 min, but which then declines rapidly to baseline values18, 21, 29, 30. This suggests that MMP9 inhibitors have no effect on E-LTP, but block L-LTP selectively.
The contribution of MMP9 activity to the spine enlargement that accompanies persistent LTP has been examined by combining whole-cell recordings from CA1 pyramidal cells with simultaneous two-photon imaging of their dendritic spines29. In a theta-burst stimulation paradigm applied in the presence of MMP inhibitors, spine enlargement and LTP were both induced normally but both subsequently collapsed to baseline, which is consistent with the timecourse of the effects of the inhibitors observed in the field recording experiments discussed above. The impaired stability of both LTP and spine structural plasticity was evident only when inhibitors were applied for a period of up to ~30 min post-induction; when they were applied after this point, L-LTP and spine enlargement were resistant to decay and therefore MMP9-independent effects. These observations suggest that MMP9 contributes to a consolidation-like process during the first 30 min of LTP in which coordinated functional and structural plasticity becomes stable and immutable. Similar time-dependent effects of pharmacological MMP inhibitors on LTP persistence have been described for synapses in other hippocampal circuits31 and in other brain regions, including the amygdala5 and prefrontal cortex32.
Interestingly, the acute MMP inhibitors that interfere with LTP persistence have no effect on the magnitude or timecourse of paired-pulse facilitation (PPF), a form of presynaptic plasticity. These inhibitors also have no effect on E-LTP that is induced by weaker tetanic stimulation protocols, or on NMDAR- or metabotropic glutamate receptor (mGluR)-dependent forms of LTD. Thus, MMP9 seems to have a specific role in the persistence of L-LTP, which is consistent with the lack of upregulation or activation of MMP9 in response to LTD, PPF or E-LTP as discussed above. MMP inhibitors also have no effects on the baseline properties of synaptic neurotransmission, spine size or spine morphology, which suggests that MMPs become operative only under the demands of dynamic synaptic remodelling, and is consistent with the observation that much of the MMP9 present within the neuropil under basal conditions is in the inactive pro-form. This notion stands in contrast to a recent study showing that members of the ADAM (a disintegrin and metalloproteinase) family of metalloproteinases may have a constitutive role in regulating baseline levels of synaptic neurotransmission, at least in cultured neurons26.
The lack of any direct involvement of MMP9 in hippocampal baseline neurotransmission, PPF or LTD is also supported by studies of MMP9-deficient mice (Mmp9−/−), in which none of these processes is affected by the absence of this MMP21. However, results from other studies using broad-spectrum metalloproteinase inhibitors (FN439 and GM6001) suggest that other metalloproteinases (that is, other MMPs or ADAMs) may contribute to these forms of plasticity. In one study, hippocampal slices were exposed to FN439 and exhibited deficits in both PPF and LTD30. Speculatively, deficits in PPF might arise through FN439-mediated inhibition of MMP7, as exogenous application of active MMP7 impairs synaptic vesicle release and uptake dynamics33, possibly through intracellular cleavage of synaptic vesicle proteins34. Similarly, diminished presynaptic vesicle release has also been reported following long-term (overnight) exposure of hippocampal cultures to GM600126. Deficits in LTD could arise if broad-spectrum pharmacological blockers affected the activity of ADAM17 (also known as TACE), as this enzyme is required for mGluR-dependent LTD35.
Two studies have reported that non-specific MMP inhibitors impair LTP induction in CA130, 36, which is in contrast to the data discussed above that suggests that MMP9 does not contribute to LTP induction. As most MMP inhibitors lack selectivity, it is possible that such discrepant results reflect off-target effects of the inhibitors on synaptic physiology. For example, barring clear differences across studies in LTP induction protocols and other experimental conditions, other metalloproteinases (MMPs and/or ADAMs) that are affected by the broad-spectrum inhibitors may be contributing to the induction of LTP. It is notable, however, that acute slices prepared from Mmp9−/− mice also exhibit impaired LTP induction, as well as maintenance, both of which are restored to wild-type levels in the presence of bath-applied proteolytically active MMP921. One potential confounding factor in interpreting results from MMP9 null-mutant mice is the possibility of developmental compensation for the chronic loss of MMP9, which could include compensation by other MMPs or by other types of molecules. Indeed, recent studies suggest, for example, that ECM-dependent regulation of neuronal survival in developing hippocampus is modulated by MMP9 and is therefore altered in Mmp9−/− mice37. The observation that adding proteolytically active MMP9 to hippocampal slices from Mmp9−/− mice restores normal levels of LTP could reflect an MMP9-dependent enhancement of synaptic neurotransmission (discussed below).
MMP9 activation proactively drives synaptic remodelling
Although loss-of-function approaches indicate that MMP9 is necessary for the persistence of functional and structural synaptic plasticity, acute gain-of-function experiments conducted in slices or in vivo show that MMP9 is also sufficient for driving functional and structural synaptic remodelling in the absence of conventional LTP-inducing stimuli21, 29.
Brief, local application of proteolytically active MMP9 to unstimulated dendritic spines of CA1 pyramidal neurons induces an LTP-like increase in synaptic signal strength and concurrent spine enlargement, both of which reach a maximum in the 15–20 min period after the spines are first exposed to the enzyme. Such plasticity is not observed when spines are exposed to pro-MMP9. Moreover, spines and synapses are unresponsive to active MMP2. Both functional and structural plasticity induced by MMP9 depends on signalling through β1 subunit-containing integrins, and are associated with integrin-dependent phosphorylation (and hence inactivation) of the actin-depolymerizing factor cofilin within dendritic spines. Additionally, such MMP9-induced synaptic and spine plasticity occludes subsequent potentiation and spine enlargement that are induced by theta-burst or tetanic stimulation18, 29, which suggests that common mechanisms of synaptic potentiation and spine remodelling are shared by MMP9 and electrical stimulation protocols. Consistent with this suggestion, MMP9-induced potentiation and persistent spine enlargement are both sensitive to inhibitors of protein synthesis as well as reagents that interfere with postsynaptic actin polymerization and blockers of SNARE-dependent exocytocis in postsynaptic neurons29. Such sensitivity to these reagents is identical to those of electrically evoked LTP and spine plasticity38.
Together, these findings support a working model of the molecular cascade of MMP9 activity in driving synaptic remodelling: from MMP9 activation by LTP induction to its downstream target effects driving persistent potentiation. According to this model (Fig. 2), perisynaptic pro-MMP9 is rapidly activated by NMDAR-mediated signalling that occurs during LTP induction. Active MMP9 then signals, via proteolysis of a yet unidentified target (see section below for candidates), through β1-containing integrins on dendritic spines39 to couple actin-dependent structural changes in spine shape with actin-dependent functional changes in synaptic neurotransmission.
FIGURE 2. A model of MMP9 activity in driving synaptic structural and functional remodelling.

Under basal conditions, mostly pro-forms of matrix metalloproteinase 9 (MMP9) are localized perisynaptically in CA1. Upon LTP induction, postsynaptic NMDA-type glutamate receptor (NMDAR) activity triggers rapid and local nitric oxide (NO) release into the perisynaptic microenvironment, which in turn converts pro-MMP9 to proteolytically active MMP9. Once proteolytically active, MMP9 cleaves perisynaptic intracellular adhesion molecule 5 (ICAM5). The ICAM5 ectodomain then binds to and activates postsynaptic β1-containing integrins, which in turn triggers actin polymerization in part through phosphorylation of cofilin. Actin polymerization is the basis for an integrin-mediated consolidation process of spine structural enlargement as well as AMPA receptor (AMPAR) trafficking into the synaptic membrane. New pro-MMP9 synthesis and release by pre- and postsynaptic neurons and/or perisynaptic astrocytes replenishes the extracellular pool of pro-MMP9. In time, tissue inhibitors of metalloproteinases (TIMPs) are released to terminate MMP9 activity.
Integrins are crucial for the consolidation of LTP40, 41; they activate downstream signaling cascades that increase synaptic glutamate receptor-mediated currents42, 43, regulate surface expression and composition of AMPARs44, and promote spine actin polymerization45, 46. Persistence of LTP requires the insertion of AMPARs into the synaptic membrane38, 47, which in turn stabilizes LTP-associated spine enlargement38, 48. Consequently, MMP9-mediated potentiation of synaptic transmission probably arises ultimately from integrin-directed trafficking of AMPARs into the synaptic membrane, which in turn stabilizes the spine enlargement, thereby consolidating both forms of MMP9-driven plasticity (Fig. 2).
In contrast to the potentiating and spine-enlarging effects of local, transient MMP9 exposure, widespread, chronic application of active MMP9 produces seemingly opposing effects, at least on spine morphology. In dissociated hippocampal cell or organotypical-slice cultures, active MMP9 that is bath-applied for up to 90 min induces the production of long, thin dendritic spines with smaller spine-heads24, 49, which represents an immature spine morphology. Additionally, although bath-applied MMP9 has no apparent effects on synaptic physiology for the first 90 min of exposure, after this point mEPSCs (miniature excitatory postsynaptic currents) display longer deactivation time-constants. Other studies involving cultured neurons have revealed that active MMP9 increases the lateral mobility50 and desensitization kinetics51 of surface NMDARs, without having any effects on surface mobility of AMPARs. This observation is interesting in light of the fact that enzymatic degradation of the ECM that normally surrounds neurons and synapses increases the surface mobility of AMPARs, but not that of NMDARs52. These differences in glutamate receptor-subtype mobility following degradation of the ECM may indicate that the loss of the ECM is permissive for AMPAR mobility, while NMDAR mobility may be regulated selectively by an MMP9-mediated mechanism. Indeed, although the precise relationships between the altered kinetics of synaptic and NMDAR-mediated currents, the increased rates of NMDAR diffusion and the transition towards an immature spine morphology observed in cultures is presently uncertain, all these MMP9-linked effects in neuronal cultures are dependent on β1-containing integrins.
Such stark differences in MMP9-driven, β1-integrin-dependent effects on synaptic form and function between the different gain-of-function experimental approaches might be attributed to the nature and timecourse of MMP9 application. Global exposure of an entire network of cultured neurons to an active MMP could generate homeostatic adjustments in synaptic currents or spine morphology that differ substantially from local and delimited effects on a small subset of spines53, as all neurons and their synapses would be affected simultaneously during bath-exposure to the active MMP. Indeed, bath-application of MMP7 to dissociated hippocampal cultures also results in a transition from mature to immature-looking spines, global increases in the phosphorylation of the NMDAR subunits NR2A and NR2B54, and in the ectodomain cleavage of NR1 and NR2A55. An increasingly more widespread exposure to active MMPs, coupled with long exposure times, may underlie how MMPs transition from adaptive, acute mediators of local synaptic remodelling to maladaptive, deleterious effectors of aberrant synaptic and neuronal networks (Fig. 3). Indeed, transgenic rats that constitutively overexpress an autocatalytically active form of MMP9 not only possess hippocampal neurons with immature-looking spines, but also exhibit a lower threshold for epileptogenesis25, which suggests that global and/or chronic MMP activity contributes to a pathophysiological profile of cellular and synaptic architecture.
FIGURE 3. Transition of MMP activity to pathophysiological processes.

a | Both experimentally induced155 and behaviorally induced63 forms of synaptic plasticity affect a minority of synapses that are widely distributed. Thus, during long-term potentiation LTP, matrix metalloproteinases (MMPs) are activated locally at a minority of synapses distributed widely throughout the neuropil. Within ~15 min, active MMP9 coordinately drives dendritic spine enlargement (arrow) and potentiation as part of an integrin-dependent consolidation process. After 1–2 hours, MMP activity is terminated, most likely by increased tissue inhibitor of metalloproteinase (TIMP) activity.
b | By contrast, under pathophysiological conditions, ischaemic or inflammatory disruption of the blood–brain barrier (BBB) results initially from aberrant expression of a number of MMPs within the neurovascular unit. Such MMP-mediated activity leads to subsequent invasion into the brain parenchyma of inflammatory and other cell types, which in turn triggers further aberrant MMP expression and secretion by astrocytes, neurons and microglia. Under these conditions, MMPs are activated by each other and by free radicals such as nitric oxide. Such excessive and active MMPs then infiltrate networks of neurons and their synapses. Broad and sustained MMP exposure at all or most synapses of a network lead to conversion of dendritic spines to immature-like filopodia (inset), followed by synapse breakdown and, eventually, neuronal apoptosis.
It should be noted that the different states of neuronal development represented by culture models versus intact circuitry of adult hippocampus in vivo or in acute ex vivo slices could also contribute to different outcomes of MMP9-driven integrin activation. Direct activation of integrins on young cultured neurons produces spine elongation and decreased filamentous actin content within dendritic protrusions56, whereas integrin activation in mature intact circuitry drives actin polymerization in spine-heads and is associated with spine enlargement45. In addition, important differences between model systems in the extracellular milieu may also be relevant to different outcomes, as some of the MMP7-mediated effects on neurons in culture vary depending on the substrate upon which the neurons are grown54.
MMPs contribute to behavioural and experience-dependent plasticity
The mounting evidence indicating that MMPs are a necessary component of the molecular mechanisms that couple and consolidate synaptic functional and structural synaptic remodelling suggests that MMP activity would also influence cognitive tasks that depend on such synaptic plasticity. This is indeed the case, as several associative and non-associative learning and memory paradigms, conducted in conjunction with in vivo delivery of pharmacological blockers, or MMP antisense oligonucleotides, or MMP overexpression, have shown that MMPs, particularly MMP3 and MMP9, have important roles in cognitive performance20, 21, 57–61.
Inhibitory avoidance (IA) learning, which is a single-trial fear-conditioning type of learning and memory task62 that elicits an LTP-like increase in the strength of synaptic transmission in CA163, leads to markedly elevated levels of both pro-forms and active forms of MMP360 and MMP920. In the case of MMP9, the increase in levels of the active form is matched by increased gelatinolytic activity: both are markedly higher 6 hours after IA training and remain elevated for ~48 hours. Importantly, such upregulation is not evident following exposure to either the chamber (the context) or the aversive stimulus (the shock) alone, but only when these two are coupled in time. Functionally, blockade of MMP activity with intrahippocampal administration of pharmacological blockers or MMP9-specific neutralizing antisera starting ~3 hours after IA training, just before the onset of training-induced MMP activity, completely abrogates memory for the IA response when tested days later. These data, when allied with the emerging understanding of how MMPs contribute at the cellular and molecular levels to synapse and spine remodelling, suggest that learning induces a period of focal MMP-mediated proteolysis that is vital for driving long-lasting synaptic modifications of the kind that underlie memory consolidation.
Considerable synaptic remodelling also occurs during rapid-eye movement (REM) sleep64, and recent studies suggest that MMPs may be involved in this remodeling process. Sleep deprivation in rats is associated with diminished levels of Mmp9 mRNA in cortical structures65, and treatment of sleep-deprived rats with modafinil, a wake-promoting pharmacological reagent that is used to treat narcolepsy, increases Mmp9 mRNA levels and restores cognitive function of sleep-deprived animals66.
Finally, recent in vivo studies of visual67 and somatosensory (barrel) cortex68 suggest that MMPs facilitate functional and structural changes in connectivity that underlie experience-dependent alterations in representational maps.
Together, the data discussed above indicate that MMPs have an active role in remodelling synaptic structure and function during LTP, a role that is crucial for cognitive and sensory function. This raises important questions about how the onset and termination of MMP activity is coupled to LTP and the target molecules through which MMPs influence synaptic form and function.
Timecourse and targets of MMP activity
There are several distinct mechanisms that could activate, sustain and terminate perisynaptic MMP proteolysis following LTP induction. Although the precise onset of proteolysis relative to the onset of LTP induction remains to be definitively determined, in principle, the latent, perisynaptic pool of pro-MMPs could be activated quickly by molecules that are both capable of converting pro-MMPs to their active forms and rapidly released by LTP induction. One attractive candidate molecule is nitric oxide (NO), a gaseous, diffusible cellular messenger. NO can activate MMP969, 70, is released from postsynaptic neurons by theta-burst-stimulated LTP in an NMDAR-dependent manner, and contributes to L-LTP71. Additionally, the release of NO is presumably localized to potentiated synapses, where it could activate MMP proteolysis locally (Fig. 2). MMP9 can also be activated by the serine protease tissue plasminogen activator (tPA) in some cell types72. tPA is encoded by an immediate early gene that is rapidly upregulated by LTP induction73; tPA is then released from dendritic spines74. However, the gain-of-function effects of tPA on neuronal activity and morphology are markedly different to those induced by MMP975. When applied to hippocampal slices, active tPA converts a normally transient potentiation induced by a weak stimulus protocol into persistent LTP, but does not, by itself, induce synaptic potentiation. Moreover, in cultured neurons, tPA increases axon growth and the formation of presynaptic boutons. These different effects on synaptic plasticity and morphology in comparison with those described above for active MMP9 make it unlikely that tPA is immediately upstream of MMP9 in the context of LTP-triggered proteolytic remodelling. Instead, such differences suggest that tPA, MMPs and other LTP-activated extracellular proteases76, 77 operate along largely independent pathways, but act synergistically to remodel the synaptic microenvironment.
Once pro-MMPs are activated at the synapse following LTP induction, new MMP synthesis and secretion probably replenishes the pro-MMP pool, since the LTP-triggered increase in MMP9 levels is sensitive to protein synthesis inhibitors (discussed above). Studies suggested that Mmp9 mRNA is localized to dendrites and dendritic spines of granule cells in rat dentate gyrus. Moreover, 12—24 hours following kainate-induced epileptogenesis in rats, levels of Mmp9 mRNA increased within the molecular (dendritic) layer of the dentate gyrus, attributed to both dendritic and glial expression19, 78. Similarly, increases in Mmp9 mRNA levels have been detected in hippocampus 2 hours post-seizure onset following administration of the GABAA receptor antagonist pentylenetetrazole79. Together, these findings suggest that Mmp9 mRNA could be translocated into dendrites and locally translated at synaptic sites under activity-dependent conditions. However, in CA1, there is no evidence yet for increased levels of Mmp9 mRNA, either in somata or dendrites, at 75 min following LTP induction by strong tetanic stimuli delivered in vivo18. Thus, LTP-related transcriptional regulation probably occurs at later time points when it might serve to sustain MMP activity levels. More studies are required to characterize potential transcriptional regulation of MMPs in the context of LTP.
Behaviourally, the upregulation in MMP9 levels following IA training20 probably results from increases in translation and transcription. The Mmp9 promoter contains binding sites for the transcription factor nuclear factor-kappa B (NF-κB)80, and IA training increases the transcriptional activity of NF-κB during the period over which MMP9 levels rise81. Other studies79 suggest that Mmp9 mRNA synthesis in neurons is subject to epigenetic regulation of the transcriptional repressor YY1 (Yin Yang 1), while under conditions of epileptogenesis, transcriptional regulation of mRNAs encoding MMP9 or tissue inhibitor of metalloproteinases 1 (TIMP1) may be controlled by the transcription factor activator protein 1 (AP1)82, 83. At the translational level, recent studies implicate microRNA-dependent control of MMP9 levels84. An important goal is to determine how these various transcriptional and translational mechanisms contribute to MMP-driven modulation of synaptic or behavioural plasticity.
It also remains to be determined precisely how focal MMP-mediated proteolysis is terminated following synaptic remodelling. TIMPs are likely to be key regulators of this termination step (Fig. 2), as Timp1 mRNAs are rapidly upregulated following LTP induction or epileptogenesis in the hippocampus85, 86. Importantly, viral-mediated TIMP1 overexpression in rat prefrontal cortex (PFC) abolishes the persistence but not the induction of LTP at subiculum→PFC synapses32. Behaviourally, the results of several studies, using a variety of approaches to interfere with or augment TIMP activity, are consistent with the idea that TIMPs are critical modulators of MMP activity that is necessary for performance of a variety of behaviours, including olfactory learning87, 88, pre-pulse inhibition of the startle reflex89, spatial navigation in a water maze and habituation in an open field90.
Targets of MMP proteolysis underlying synaptic remodeling
Several molecules have been identified or suggested to be MMP targets through which MMP-mediated proteolysis contributes to synaptic remodelling.
The involvement of integrins as mediators of almost all aspects of MMP9-driven forms of synapse and spine plasticity discovered to date casts the spotlight on endogenous integrin ligands as MMP proteolytic targets that drive synaptic remodelling. In this context, recent studies show that intercellular adhesion molecule 5 (ICAM5; also known as telencephalin) may be an important MMP target. ICAM5 is a transmembrane protein that is a member of the immunoglobulin superfamily and binds to and activates integrins in the immune system91. In brain, ICAM5 localizes to dendrites and filopodia92, where it negatively regulates spine number and maturation during development93. Recent studies show that LTP-inducing tetanic stimuli of CA1 synapses promotes MMP-dependent shedding of an amino-terminal ectodomain fragment of ICAM5 that is detectable at 15 min after LTP induction36 (Fig. 2). Such cleavage can be mimicked by exposing cultured neurons to NMDA and can be blocked by the application of MMP inhibitors36, 94, which is consistent with the idea that NMDARs are upstream of MMP activation as discussed above. Similarly, active forms of MMP3, MMP7 and MMP9 can promote ICAM5 ectodomain shedding in cultured neurons36. Biochemical studies demonstrate that the cleaved ICAM5 fragment associates with β1-integrins, and when applied to cultured neurons, stimulates β1-integrin-dependent phosphorylation of cofilin95.
Members of the cadherin family of cell adhesion molecules are also targets of MMP-mediated perisynaptic proteolysis. MMP-dependent ectodomain cleavage of neuronal- (N-) and epithelial- (E-) cadherin has been demonstrated in several cell types, including cultured neurons96, and may reflect the activity of MMP2423. Nevertheless, it is not clear at present how MMP-mediated processing of synaptic cadherins would contribute to LTP and associated spine enlargement, since L-LTP-inducing stimuli promote N-cadherin synthesis and recruitment to synapses97, where it has a vital function in stabilizing both LTP and spine enlargement in CA1 neurons97–99. One possibility is that such MMP-mediated proteolysis may affect only a subset of cadherin molecules, perhaps ones that are highly localized to a particular synaptic or perisynaptic domain.
Another activity-dependent target of MMPs that has been identified in neuronal culture assays is β-dystroglycan100. This molecule is a transmembrane protein that, together with α-dystroglycan, couples the ECM to the actin cytoskeleton101. The onset of seizure activity in hippocampus increases the levels of a proteolytic fragment of β-dystroglycan, but this fragment is absent in Mmp9−/− mice100. These data suggest that such cleavage is MMP9-dependent, and occurs in an activity-dependent manner under pathophysiological conditions. Whether such MMP-directed proteolysis of β-dystroglycan contributes to synaptic remodelling in non-pathophysiological or other contexts has not been studied.
Several studies indicate that myelin-associated, axon growth-inhibitory molecules (that is, MAG (myelin-associated glycoprotein), NOGO66, and OMGP (oligodendrocyte myelin glycoprotein)) and one of their associated receptors, NGR1 (NOG66 receptor 1), are important in gating the expression of ocular dominance plasticity in the visual cortex102, and LTP and spine plasticity in the hippocampus103. Cell culture assays demonstrate that MAG can be cleaved by MMP2, MMP7 or MMP9, releasing bioactive fragments that inhibit axonal outgrowth from dorsal root ganglion neurons104. Similarly, NGR1 can be cleaved by MMP14, MMP15, MMP16 or MMP24, which diminishes growth-cone collapse in response to myelin-associated inhibitors105. Interestingly, the cleaved ectodomain fragment of NGR1 retains the ability to bind NOGO66, suggesting it can act as a dominant-negative entity under physiological conditions105. These data suggest that MMP-mediated proteolytic regulation of this inhibitory signalling system directly influences the molecular cascade through which myelin-derived inhibitors and their receptors regulate experience-dependent synaptic plasticity.
Members of the family of insulin-like growth factors and their related receptors and binding proteins have recently been implicated in the molecular mechanisms underlying memory consolidation106, and some of these are targets of MMP-mediated proteolysis. Levels of insulin-like growth factor 2 (IGF2) mRNA and protein are upregulated in rat hippocampus 20 hours following IA training107. Antisense-mediated knockdown of IGF2 or its interacting IGF2 receptor (IGF2R) markedly impairs IA memory retention, whereas administration of exogenous IGF2 promotes persistent hippocampal LTP. In other systems, the bioavailability of IGF2 is regulated by sequestration through binding to insulin-like growth factor binding protein 2 (IGFBP2)108. Studies show that the IGF2–IGFBP2 complex is proteolytically processed by MMP9109, thereby releasing IGF2 for full bioactivity. Notably, MMP9 upregulation by IA training coincides with the period over which IGF2 is operative in memory consolidation, which raises the possibility that MMP9 critically regulates bioavailability of IGF2 as part of an orchestrated sequence of molecular mechanisms that consolidate and enhance memory.
Finally, in addition to candidate MMP targets described above, there is a growing list of additional molecules with documented roles in synapse development, plasticity and function that are also known targets of MMPs110. These include: tenascins; synaptogenic proteins such as the thrombospondins, hevin and SPARC; neurotrophins; tumor necrosis factor-α (TNFα); and ephrins and their eph receptors. Whether MMP-mediated processing of these molecules contributes to synaptic remodelling during LTP or in any other context remains to be rigorously tested.
MMP proteolysis in brain disorders
It is increasingly recognized that several neurological and psychiatric brain disorders have deficient or maladaptive synaptic functional and structural plasticity at their core 111–115. The growing recognition that fast, focal MMP-mediated perisynaptic proteolysis is a crucial driver of ongoing, adaptive synaptic remodelling, with direct consequences on behavioural plasticity, has established a new conceptual framework for linking MMP-mediated proteolysis to pathophysiological, maladaptive synaptic remodelling. Some recent studies that examine the relationship between MMP activity and potentially maladaptive synaptic remodelling capture these emerging links, and are briefly highlighted here.
Repeated exposure to the illicit psychostimulant methamphetamine is associated, in rodent models, with persistent abnormalities in the density and morphology of dendrites and spines116, and alterations in LTP117 in a number of forebrain structures. Protein and mRNA levels of MMP2, MMP9 and TIMP2 are elevated in rat prefrontal cortex, nucleus accumbens and hippocampus following repeated methamphetamine exposure95, 118, whereas abrogating MMP expression or function blocks behavioural sensitization to methamphetamine119. Moreover, the reinstatement of cocaine-induced conditioned place preference (CPP) by cocaine-priming is associated with increased expression and activity of MMP9 in rat medial prefrontal cortex120 and blocking such MMP9 activity impairs acquisition of cocaine-induced CPP or subsequent reinstatement of CPP upon cocaine-priming121. Mechanistically, acute methamphetamine-treatment in vivo stimulates cleavage of ICAM5 in striatum and hippocampus by ~6 hours post-treatment; such methamphetamine-induced cleavage is blocked by MMP inhibitors when it is recapitulated in vitro95. Additionally, the expression and activity levels of β1- and β3-containing integrins — critical signalling mediators of MMP9 remodelling as well as of ICAM5 activity — are bidirectionally regulated by cocaine treatment, withdrawal and relapse in the nucleus accumbens122. Additionally, exposure to morphine drives aberrant MMP9 activity in neurons and glia in the spinal cord dorsal horn, whereas inhibiting MMP9 blocks morphine dependence70. As persistent drug craving resembles dysfunctional synaptic plasticity processes that are involved in memory consolidation111, these studies together suggest that drugs of abuse co-opt normal, ongoing and adaptive MMP-mediated synaptic plasticity by driving maladaptive MMP expression, circuit remodeling and addictive behaviour.
Neuropathic pain is also associated with aberrant synaptic circuit rewiring in the spinal cord113. Following ligation of the fifth lumbar spinal nerve — a standard rodent model for neuropathic pain — levels of MMP9 are rapidly but transiently elevated in injured dorsal root ganglion neurons, whereas upregulation of MMP2 occurs at later time points, which suggests a role for these different MMPs in different phases of neuropathic pain123. Consistent with this, suppression of MMP9 inhibits the initial phase of aberrant pain sensation, but suppression of MMP2 inhibits persistence. Intrathecal delivery of active MMP2 or MMP9 delivery causes behavioral changes that mimic behavioural manifestations of neuropathic pain. The MMP-driven effects may be mediated through proteolytic processing of the proinflammatory cytokine interleukin-1β123.
Fragile X syndrome (FXS) is the most common inherited form of intellectual disability and autism124, and is caused by a trinucleotide expansion that inactivates the fragile X mental retardation 1 gene (FMR1), leading to the loss of fragile X mental retardation protein (FMRP). FMRP is a postsynaptically localized RNA-binding protein that regulates local translation of a select subset of mRNAs at synapses125. One abnormal structural hallmark of humans and mice lacking FMRP is an overabundance of long, thin, immature-appearing dendritic spines with corresponding defects in synaptic and experience-dependent plasticity126. The spine structural abnormalities associated with FXS are very similar to those described above following global and prolonged MMP9 exposure in hippocampal cultures, which suggests that aberrant expression, localization and/or activity of MMP9 could contribute to spine and plasticity deficits in FXS. Indeed, recent studies show that levels of MMP9 as well as gelatinolytic activity are both elevated in hippocampal lysates from Fmr1−/− mice in comparison with those from wild-type mice24. Importantly, treatment of Fmr1−/− mice with minocycline, a tetracyline derivative that — among many of its targets — blocks MMP9 activity127, reduced levels of endogenous MMP9 activity, reversed the abnormal behaviours that these mice display and restored dendritic spines to normal morphologies24. These findings have recently been largely validated in a Drosophila melanogaster model of FXS128. Together, these data indicate that a strong correlation exists between aberrant MMP9 function and abnormal spine and circuit-level architecture in FXS, and potentially offers a realistic therapeutic approach to reversing dendritic spine, circuit and behavioural abnormalities that are driven by excessive MMP proteolysis.
Conclusions and perspective
MMPs can no longer be regarded solely as injury-related molecules in the mature nervous system. Multiple and compelling lines of evidence now show that MMPs are vital molecular drivers of synaptic structural and functional remodelling of the kind that supports ongoing cognitive functions.
One important area for future investigation is to determine precisely how MMP activity transitions from its normal, adaptive roles in synaptic circuit remodelling to its aberrant and deleterious roles that have important pathophysiological cellular and synaptic consequences. At its core, the transition in MMP-mediated cellular responses is probably dictated by the relative scale in amount, duration and spatial extent of MMP activity. In the case of disease or injury, this scale is potentially quite large (Fig. 3). Neurons, glia and a variety of invading inflammatory cell types all contribute to a massive elevation in MMP activity that spans days to weeks or longer, affects many cells and cell types over large areas, and permits broad access of MMPs to potential proteolytic targets that they may not normally encounter under non-pathophysiological conditions5, 129. It will therefore be crucial in the future to define precisely the regulatory mechanisms that govern focal release, localization, activation and termination of perisynaptic MMP activity, as these become overridden and dysregulated under conditions of injury or disease, leading to aberrant proteolytic processing of synaptic molecules such as NR1130, 131. A thorough characterization of the regulatory mechanisms that govern MMP activity at the synapse will also serve the design of therapeutic strategies aiming to harness the adaptive roles of MMPs as molecular drivers of synaptic circuit remodelling to promote restoration and repair of damaged brain circuits, while at the same time diminishing the deleterious effects of these molecules that could impede recovery of synaptic function following injury132.
Another area of future research will be to see whether other metalloproteinases have functional roles in synaptic structural and behavioural plasticity. Pursuing this question is potentially daunting because of the sheer number of MMPs and other metalloproteinases and the relative lack of reliable reagents to detect them. However, one potential strategy to identify families of metalloproteinases whose proteolytic activity is regulated by plasticity-inducing stimuli is to use MMP active site-directed chemical probes133 in a high-throughput proteomic screen for plasticity-regulated metalloproteinases, akin to how such probes have been used to identify and distinguish metalloproteinases that are active in certain cancers134.
Relatedly, another limitation — both experimentally and therapeutically — is the lack of MMP-specific inhibitors. It is already known that different MMPs have distinct cell-type expression patterns, react differently to synaptic stimulation, affect synaptic structural and functional plasticity differently, and exert different pathophysiological effects, and so the need for advancing the development of MMP type-specific inhibitors is self-evident. The formidable obstacle to developing MMP-specific small-molecule antagonists of the catalytic site is the high structural homology across MMPs and other metzincins in this region. However, recent advances in next-generation small-molecule inhibitors have been described that target exosites or other allosteric regions135, and these strategies may prove effective in overcoming the barriers to producing MMP-specific antagonists.
Online summary.
Matrix metalloproteinases (MMPs) are a large family of mostly secreted, extracellularly acting proteolytic enzymes. In brain, they have well described roles in slowly emerging, but long-lasting pathophysiological processes of cell loss and synaptic dysfunction associated with acute injury, ischaemia, neurodegeneration, and demyelination.
Remodelling of synapse structure and function also underlies normal cognitive processes, such as learning and memory. This Review focuses on recent studies that indicate that MMPs have important roles in driving such synapse plasticity under non-pathological conditions that is distinct from their roles in neuropathophysiology.
MMPs are secreted as inactive pro-enzymes (zymogens). Under basal conditions, a large pool of mostly pro-MMPs are situated perisynaptically, poised for activation by plasticity-inducing stimuli such as long-term potentiation (LTP).
Upon induction of LTP, but not other forms of short- or long-lasting plasticity, pro-MMPs are rapidly (within ~15 min) converted to proteolytically active MMPs through an NMDAR-dependent mechanism. Such proteolytically active MMPs then signal through β1-containing integrins to promote dendritic spine enlargement and synaptic potentiation concurrently.
Intercellular adhesion molecule 5 (ICAM5), which binds to and activates integrins, may be a direct target of perisynaptic MMP proteolysis during LTP. LTP-associated MMP proteolysis is probably then terminated by an increase in the activity of endogenous inhibitors called tissue inhibitors of metalloproteinases (TIMPs).
When MMP activity is blocked pharmacologically or genetically, LTP, spine enlargement and behavioral measures of cognitive function are all impaired.
Several psychiatric and neurological disorders, including drug addiction, neuropathic pain sydromes, and Fragile X mental retardation syndrome, are associated with abnormal or deficient synaptic plasticity. Recent studies indicate that aberrant MMP expression, localization and function may contribute to synaptic plasticity deficits associated with such disorders.
A key area for future research is to elucidate how MMP activity transitions from normal, adaptive roles in local synaptic remodelling to deleterious roles that have important pathophysiological cellular and synaptic consequences. This transition likely involves abnormal regulatory mechanisms leading to excessive, prolonged, and widespread MMP activity.
Acknowledgments
I thank Drs. Deanna L. Benson and Vincent Vialou for helpful comments on the manuscript, and Drs. Vanja Nagy, Ozlem Bozdagi, Paven Aujla, Xiao-bin Wang and Qiang Zhou for their scientific contributions to the personal work discussed in this review. I am ever grateful to Ted Jones and Dave Colman—two prolific savants no longer with us—for their guidance and friendship over many years. My research was supported by NIMH grant MH075783.
Glossary
- Metzincin
a clan of metalloproteases comprising the matrix metalloproteinases (MMPs), the ADAMs (a disintegrin and metalloproteinase) and ADAMs-TSs (ADAMs with thrombospondin repeats). It is so-named by an amalgam of two structural hallmarks of the active-site region: a conserved methionine-containing turn (Met-turn) downstream of—and positioned underneath to stabilize—a conserved zinc-binding motif (HExxHxxGxxH), in which the three histadine residues are zinc-binding ligands within the catalytic site.
- Dendritic spines
small actin-rich dendritic protrusions that harbour most of the excitatory glutamatergic synapses.
- Gelatinases
any proteolytic enzyme capable of cleaving gelatin (denatured collagen). In the MMP lexicon, the gelatinases are matrix metalloproteinase 2 (MMP2) (gelatinase A) and MMP9 (gelatinase B), as gelatin is a canonical substrate for these MMPs.
- Integrins
heterodimers composed of an α- and a β-subunit. In mammals, there are eighteen α- and eight β-subunits. They are canonical receptors of extracellular matrix and other proteins. In hippocampus, most integrin heterodimers contain the β-subunit.
- Cofilin
an actin binding protein that is enriched in dendritic spines. Cofilin is a member of the ADF (actin-depolymerizing factor)/cofilin family, and regulates the disassembly of filamentous actin. Cofilin is negatively regulated by phosphorylation at a single site (Ser3).
- SNARE
soluble NSF (N-ethylmaleimide-sensitive factor) attachment protein (SNAP) receptors. A family of proteins comprising vesicular (v)-snares and target (t)-snares that mediate intracellular membrane fusion.
- mEPSCs
miniature excitatory postsynaptic currents. These are the postsynaptic responses to a quantum of excitatory neurotransmitter (usually glutamate).
- Conditioned place preference
a behavioural task used to evaluate an animal’s preference for stimuli or an environment associated with postitive or negative reward.
- Exosites
a secondary substrate-binding site on an enzyme that is distinct from the catalytic active site. Exosites are often important for positioning substrates for full proteolytic cleavage.
Biography
George W. Huntley is Associate Professor of Neuroscience and a member of the Friedman Brain Institute at the Mount Sinai School of Medicine, New York, USA. His research focuses on development and plasticity of cortical connectivity, the role of adhesion proteins in synaptic function, and extracellular proteolytic remodelling of synaptic circuits.
Footnotes
Further Information
George W. Huntley’s homepage: http://neuroscience.mssm.edu/huntley
References
- 1.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. This is a comprehensive review of MMP biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Butler GS, Overall CM. Updated biological roles for matrix metalloproteinases and new “intracellular” substrates revealed by degradomics. Biochemistry. 2009;48:10830–45. doi: 10.1021/bi901656f. [DOI] [PubMed] [Google Scholar]
- 3.Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8:221–33. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McFarlane S. Metalloproteases: carving out a role in axon guidance. Neuron. 2003;37:559–62. doi: 10.1016/s0896-6273(03)00089-8. [DOI] [PubMed] [Google Scholar]
- 5.Rivera S, Khrestchatisky M, Kaczmarek L, Rosenberg GA, Jaworski DM. Metzincin proteases and their inhibitors: foes or friends in nervous system physiology? J Neurosci. 30:15337–57. doi: 10.1523/JNEUROSCI.3467-10.2010. This is an up-to-date review on MMPs, ADAMs and ADAMs-TSs in nervous system function and pathophysiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu M, Zuo Y. Experience-dependent structural plasticity in the cortex. Trends Neurosci. 2011;34:177–87. doi: 10.1016/j.tins.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Florence SL, Taub HB, Kaas JH. Large-scale sprouting of cortical connections after peripheral injury in adult macaque monkeys. Science. 1998;282:1117–21. doi: 10.1126/science.282.5391.1117. [DOI] [PubMed] [Google Scholar]
- 8.Nudo RJ, Wise BM, SiFuentes F, Milliken GW. Neural substrates for the effects of rehabilitative training on motor recovery after ischemic infarct. Science. 1996;272:1791–4. doi: 10.1126/science.272.5269.1791. [DOI] [PubMed] [Google Scholar]
- 9.Buonomano DV, Merzenich MM. Cortical plasticity: from synapses to maps. Annu Rev Neurosci. 1998;21:149–86. doi: 10.1146/annurev.neuro.21.1.149. [DOI] [PubMed] [Google Scholar]
- 10.Gundelfinger ED, Frischknecht R, Choquet D, Heine M. Converting juvenile into adult plasticity: a role for the brain’s extracellular matrix. Eur J Neurosci. 31:2156–65. doi: 10.1111/j.1460-9568.2010.07253.x. A comprehensive review of current information about how ECM regulates synaptic plasticity. [DOI] [PubMed] [Google Scholar]
- 11.Dityatev A, Schachner M. Extracellular matrix molecules and synaptic plasticity. Nat Rev Neurosci. 2003;4:456–68. doi: 10.1038/nrn1115. [DOI] [PubMed] [Google Scholar]
- 12.Vecil GG, et al. Interleukin-1 is a key regulator of matrix metalloproteinase-9 expression in human neurons in culture and following mouse brain trauma in vivo. J Neurosci Res. 2000;61:212–24. doi: 10.1002/1097-4547(20000715)61:2<212::AID-JNR12>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 13.Ulrich R, Gerhauser I, Seeliger F, Baumgartner W, Alldinger S. Matrix metalloproteinases and their inhibitors in the developing mouse brain and spinal cord: a reverse transcription quantitative polymerase chain reaction study. Dev Neurosci. 2005;27:408–18. doi: 10.1159/000088455. [DOI] [PubMed] [Google Scholar]
- 14.Forsyth PA, et al. Gelatinase-A (MMP-2), gelatinase-B (MMP-9) and membrane type matrix metalloproteinase-1 (MT1-MMP) are involved in different aspects of the pathophysiology of malignant gliomas. Br J Cancer. 1999;79:1828–35. doi: 10.1038/sj.bjc.6990291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ayoub AE, Cai TQ, Kaplan RA, Luo J. Developmental expression of matrix metalloproteinases 2 and 9 and their potential role in the histogenesis of the cerebellar cortex. J Comp Neurol. 2005;481:403–15. doi: 10.1002/cne.20375. [DOI] [PubMed] [Google Scholar]
- 16.Sekine-Aizawa Y, et al. Matrix metalloproteinase (MMP) system in brain: identification and characterization of brain-specific MMP highly expressed in cerebellum. Eur J Neurosci. 2001;13:935–48. doi: 10.1046/j.0953-816x.2001.01462.x. [DOI] [PubMed] [Google Scholar]
- 17.Jaworski DM. Developmental regulation of membrane type-5 matrix metalloproteinase (MT5-MMP) expression in the rat nervous system. Brain Res. 2000;860:174–7. doi: 10.1016/s0006-8993(00)02035-7. [DOI] [PubMed] [Google Scholar]
- 18.Bozdagi O, Nagy V, Kwei KT, Huntley GW. In vivo roles for matrix metalloproteinase-9 in mature hippocampal synaptic physiology and plasticity. J Neurophysiol. 98:334–344. doi: 10.1152/jn.00202.2007. This study demonstrates loss- and gain-of-function effects of MMP9 in adult rat hippocampal synaptic physiology in vivo, which validates the relevance of slice studies, as well as introduces a new modification of in vivo zymography. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szklarczyk A, Lapinska J, Rylski M, McKay RD, Kaczmarek L. Matrix metalloproteinase-9 undergoes expression and activation during dendritic remodeling in adult hippocampus. J Neurosci. 22:920–30. doi: 10.1523/JNEUROSCI.22-03-00920.2002. This study links neural activity with MMP upregulation and activity in the context of epileptogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagy V, Bozdagi O, Huntley GW. The extracellular protease matrix metalloproteinase-9 is activated by inhibitory avoidance learning and required for long-term memory. Learn Mem. 2007;14:655–64. doi: 10.1101/lm.678307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagy V, et al. Matrix metalloproteinase-9 is required for hippocampal late-phase long-term potentiation and memory. J Neurosci. 26:1923–34. doi: 10.1523/JNEUROSCI.4359-05.2006. This study makes several important advances. It demonstrates that LTP increases MMP9 activity and requires MMP activity; that active MMP9 drives synaptic potentiation; and that the potentiating effects of MMP9 are integrin-mediated. It also shows the behavioral deficits in fear memory formation in Mmp9 knockout mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meighan SE, et al. Effects of extracellular matrix-degrading proteases matrix metalloproteinases 3 and 9 on spatial learning and synaptic plasticity. J Neurochem. 2006;96:1227–41. doi: 10.1111/j.1471-4159.2005.03565.x. [DOI] [PubMed] [Google Scholar]
- 23.Monea S, Jordan BA, Srivastava S, DeSouza S, Ziff EB. Membrane localization of membrane type 5 matrix metalloproteinase by AMPA receptor binding protein and cleavage of cadherins. J Neurosci. 26:2300–12. doi: 10.1523/JNEUROSCI.3521-05.2006. This study provides molecular details of synaptic regulation and trafficking of a transmembrane MMP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bilousova TV, et al. Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J Med Genet. 2009;46:94–102. doi: 10.1136/jmg.2008.061796. [DOI] [PubMed] [Google Scholar]
- 25.Wilczynski GM, et al. Important role of matrix metalloproteinase 9 in epileptogenesis. J Cell Biol. 2008;180:1021–35. doi: 10.1083/jcb.200708213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Restituito S, et al. Synaptic autoregulation by metalloproteases and gamma-secretase. J Neurosci. 2011;31:12083–93. doi: 10.1523/JNEUROSCI.2513-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gawlak M, et al. High resolution in situ zymography reveals matrix metalloproteinase activity at glutamatergic synapses. Neuroscience. 158:167–76. doi: 10.1016/j.neuroscience.2008.05.045. This study introduces a new method for high-resolution in situ zymography in brain tissue that can be combined with immunolocalization of synaptic proteins. [DOI] [PubMed] [Google Scholar]
- 28.Lee SR, Tsuji K, Lo EH. Role of matrix metalloproteinases in delayed neuronal damage after transient global cerebral ischemia. J Neurosci. 2004;24:671–8. doi: 10.1523/JNEUROSCI.4243-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang XB, et al. Extracellular proteolysis by matrix metalloproteinase-9 drives dendritic spine enlargement and long-term potentiation coordinately. Proc Natl Acad Sci U S A. 105:19520–5. doi: 10.1073/pnas.0807248105. This study combines whole-cell recording with simultaneous 2-photon imaging to demonstrate that MMP9 acts through integrins to coordinate both synaptic potentiation and spine enlargement. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meighan PC, Meighan SE, Davis CJ, Wright JW, Harding JW. Effects of matrix metalloproteinase inhibition on short- and long-term plasticity of schaffer collateral/CA1 synapses. J Neurochem. 2007;102:2085–96. doi: 10.1111/j.1471-4159.2007.04682.x. [DOI] [PubMed] [Google Scholar]
- 31.Wojtowicz T, Mozrzymas JW. Late phase of long-term potentiation in the mossy fiber-CA3 hippocampal pathway is critically dependent on metalloproteinases activity. Hippocampus. 2010;20:917–21. doi: 10.1002/hipo.20787. [DOI] [PubMed] [Google Scholar]
- 32.Okulski P, et al. TIMP-1 abolishes MMP-9-dependent long-lasting long-term potentiation in the prefrontal cortex. Biol Psychiatry. 62:359–62. doi: 10.1016/j.biopsych.2006.09.012. This study shows that MMPs and TIMPs regulate LTP in prefrontal cortex, indicating that the involvement of MMPs in plasticity extends beyond hippocampal synapses. [DOI] [PubMed] [Google Scholar]
- 33.Szklarczyk A, et al. Matrix metalloproteinase-7 modulates synaptic vesicle recycling and induces atrophy of neuronal synapses. Neuroscience. 2007;149:87–98. doi: 10.1016/j.neuroscience.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 34.Szklarczyk A, Oyler G, McKay R, Gerfen C, Conant K. Cleavage of neuronal synaptosomal-associated protein of 25 kDa by exogenous matrix metalloproteinase-7. J Neurochem. 2007;102:1256–63. doi: 10.1111/j.1471-4159.2007.04625.x. [DOI] [PubMed] [Google Scholar]
- 35.Cho RW, et al. mGluR1/5-dependent long-term depression requires the regulated ectodomain cleavage of neuronal pentraxin NPR by TACE. Neuron. 2008;57:858–71. doi: 10.1016/j.neuron.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conant K, et al. Matrix metalloproteinase-dependent shedding of intercellular adhesion molecule-5 occurs with long-term potentiation. Neuroscience. 166:508–21. doi: 10.1016/j.neuroscience.2009.12.061. This study identifies an LTP-related, MMP proteolytic target. The MMP-cleaved ICAM5 fragment is the best candidate yet for mediating the MMP9-integrin-dependent effects on synaptic plasticity, although this remains to be tested. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murase S, McKay RD. Matrix Metalloproteinase9 Regulates Survival of Neurons in Newborn Hippocampus. J Biol Chem. 2012 doi: 10.1074/jbc.M111.297671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Y, Wang XB, Frerking M, Zhou Q. Spine expansion and stabilization associated with long-term potentiation. J Neurosci. 2008;28:5740–51. doi: 10.1523/JNEUROSCI.3998-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mortillo S, et al. Compensatory redistribution of neuroligins and N-cadherin following deletion of synaptic β1-integrin. J Comp Neurol. 2011 doi: 10.1002/cne.23027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan CS, et al. Beta1-integrins are required for hippocampal AMPA receptor-dependent synaptic transmission, synaptic plasticity, and working memory. J Neurosci. 2006;26:223–32. doi: 10.1523/JNEUROSCI.4110-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chun D, Gall CM, Bi X, Lynch G. Evidence that integrins contribute to multiple stages in the consolidation of long term potentiation in rat hippocampus. Neuroscience. 2001;105:815–29. doi: 10.1016/s0306-4522(01)00173-7. [DOI] [PubMed] [Google Scholar]
- 42.Lin B, Arai AC, Lynch G, Gall CM. Integrins regulate NMDA receptor-mediated synaptic currents. J Neurophysiol. 2003;89:2874–8. doi: 10.1152/jn.00783.2002. [DOI] [PubMed] [Google Scholar]
- 43.Kramar EA, Bernard JA, Gall CM, Lynch G. Integrins modulate fast excitatory transmission at hippocampal synapses. J Biol Chem. 2003;278:10722–30. doi: 10.1074/jbc.M210225200. [DOI] [PubMed] [Google Scholar]
- 44.Cingolani LA, et al. Activity-dependent regulation of synaptic AMPA receptor composition and abundance by beta3 integrins. Neuron. 2008;58:749–62. doi: 10.1016/j.neuron.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kramar EA, Lin B, Rex CS, Gall CM, Lynch G. Integrin-driven actin polymerization consolidates long-term potentiation. Proc Natl Acad Sci U S A. 103:5579–84. doi: 10.1073/pnas.0601354103. This is one of a series of papers from the Lynch and Gall laboratories that define the role of integrins in consolidation of hippocampal LTP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin B, et al. Theta stimulation polymerizes actin in dendritic spines of hippocampus. J Neurosci. 2005;25:2062–9. doi: 10.1523/JNEUROSCI.4283-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kopec CD, Li B, Wei W, Boehm J, Malinow R. Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci. 2006;26:2000–9. doi: 10.1523/JNEUROSCI.3918-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kopec CD, Real E, Kessels HW, Malinow R. GluR1 links structural and functional plasticity at excitatory synapses. J Neurosci. 2007;27:13706–18. doi: 10.1523/JNEUROSCI.3503-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Michaluk P, et al. Influence of matrix metalloproteinase MMP-9 on dendritic spine morphology. J Cell Sci. 2011;124:3369–80. doi: 10.1242/jcs.090852. [DOI] [PubMed] [Google Scholar]
- 50.Michaluk P, et al. Matrix metalloproteinase-9 controls NMDA receptor surface diffusion through integrin beta1 signaling. J Neurosci. 29:6007–12. doi: 10.1523/JNEUROSCI.5346-08.2009. This study shows that MMP9 signals through integrins to control surface mobility of NMDARs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gorkiewicz T, et al. Matrix metalloproteinase-9 reversibly affects the time course of NMDA-induced currents in cultured rat hippocampal neurons. Hippocampus. 2010;20:1105–8. doi: 10.1002/hipo.20736. [DOI] [PubMed] [Google Scholar]
- 52.Frischknecht R, et al. Brain extracellular matrix affects AMPA receptor lateral mobility and short-term synaptic plasticity. Nat Neurosci. 2009;12:897–904. doi: 10.1038/nn.2338. [DOI] [PubMed] [Google Scholar]
- 53.Pozo K, Goda Y. Unraveling mechanisms of homeostatic synaptic plasticity. Neuron. 2010;66:337–51. doi: 10.1016/j.neuron.2010.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bilousova TV, Rusakov DA, Ethell DW, Ethell IM. Matrix metalloproteinase-7 disrupts dendritic spines in hippocampal neurons through NMDA receptor activation. J Neurochem. 2006;97:44–56. doi: 10.1111/j.1471-4159.2006.03701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Szklarczyk A, et al. MMP-7 cleaves the NR1 NMDA receptor subunit and modifies NMDA receptor function. FASEB J. 2008;22:3757–67. doi: 10.1096/fj.07-101402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shi Y, Ethell IM. Integrins control dendritic spine plasticity in hippocampal neurons through NMDA receptor and Ca2+/calmodulin-dependent protein kinase II-mediated actin reorganization. J Neurosci. 2006;26:1813–22. doi: 10.1523/JNEUROSCI.4091-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fragkouli A, et al. Enhanced neuronal plasticity and elevated endogenous sAPPalpha levels in mice over-expressing MMP9. J Neurochem. 2011 doi: 10.1111/j.1471-4159.2011.07637.x. [DOI] [PubMed] [Google Scholar]
- 58.Wiediger RV, Wright JW. Influence of dorsal hippocampal lesions and MMP inhibitors on spontaneous recovery following a habituation/classical conditioning head-shake task. Neurobiol Learn Mem. 2009;92:504–11. doi: 10.1016/j.nlm.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 59.Wright JW, et al. Habituation-induced neural plasticity in the hippocampus and prefrontal cortex mediated by MMP-3. Behav Brain Res. 2009;203:27–34. doi: 10.1016/j.bbr.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 60.Olson ML, et al. Hippocampal MMP-3 elevation is associated with passive avoidance conditioning. Regul Pept. 2008;146:19–25. doi: 10.1016/j.regpep.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 61.Wright JW, Brown TE, Harding JW. Inhibition of hippocampal matrix metalloproteinase-3 and -9 disrupts spatial memory. Neural Plast. 2007;2007:73813. doi: 10.1155/2007/73813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Izquierdo I, et al. Sequential role of hippocampus and amygdala, entorhinal cortex and parietal cortex in formation and retrieval of memory for inhibitory avoidance in rats. Eur J Neurosci. 1997;9:786–93. doi: 10.1111/j.1460-9568.1997.tb01427.x. [DOI] [PubMed] [Google Scholar]
- 63.Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–7. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- 64.Yang G, Gan WB. Sleep contributes to dendritic spine formation and elimination in the developing mouse somatosensory cortex. Dev Neurobiol. 2011 doi: 10.1002/dneu.20996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Taishi P, et al. Conditions that affect sleep alter the expression of molecules associated with synaptic plasticity. Am J Physiol Regul Integr Comp Physiol. 2001;281:R839–45. doi: 10.1152/ajpregu.2001.281.3.R839. [DOI] [PubMed] [Google Scholar]
- 66.He B, Peng H, Zhao Y, Zhou H, Zhao Z. Modafinil treatment prevents REM sleep deprivation-induced brain function impairment by increasing MMP-9 expression. Brain Res. 2011;1426:38–42. doi: 10.1016/j.brainres.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 67.Spolidoro M, Putignano E, Munafo C, Maffei L, Pizzorusso T. Inhibition of matrix metalloproteinases prevents the potentiation of nondeprived-eye responses after monocular deprivation in juvenile rats. Cereb Cortex. 2011;22:725–34. doi: 10.1093/cercor/bhr158. [DOI] [PubMed] [Google Scholar]
- 68.Kaliszewska A, Bijata M, Kaczmarek L, Kossut M. Experience-Dependent Plasticity of the Barrel Cortex in Mice Observed with 2-DG Brain Mapping and c-Fos: Effects of MMP-9 KO. Cereb Cortex. 2011 doi: 10.1093/cercor/bhr303. ePub ahead of print. [DOI] [PubMed] [Google Scholar]
- 69.Gu Z, et al. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–90. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- 70.Liu WT, et al. Spinal matrix metalloproteinase-9 contributes to physical dependence on morphine in mice. J Neurosci. 2010;30:7613–23. doi: 10.1523/JNEUROSCI.1358-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schuman EM, Madison DV. A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science. 1991;254:1503–1506. doi: 10.1126/science.1720572. [DOI] [PubMed] [Google Scholar]
- 72.Wang X, et al. Lipoprotein receptor-mediated induction of matrix metalloproteinase by tissue plasminogen activator. Nat Med. 2003;9:1313–7. doi: 10.1038/nm926. [DOI] [PubMed] [Google Scholar]
- 73.Qian Z, Gilbert ME, Colicos MA, Kandel ER, Kuhl D. Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature. 1993;361:453–7. doi: 10.1038/361453a0. [DOI] [PubMed] [Google Scholar]
- 74.Lochner JE, et al. Activity-dependent release of tissue plasminogen activator from the dendritic spines of hippocampal neurons revealed by live-cell imaging. J Neurobiol. 2006;66:564–77. doi: 10.1002/neu.20250. [DOI] [PubMed] [Google Scholar]
- 75.Baranes D, et al. Tissue plasminogen activator contributes to the late phase of LTP and to synaptic growth in the hippocampal mossy fiber pathway. Neuron. 1998;21:813–25. doi: 10.1016/s0896-6273(00)80597-8. [DOI] [PubMed] [Google Scholar]
- 76.Komai S, et al. Neuropsin regulates an early phase of schaffer-collateral long-term potentiation in the murine hippocampus. Eur J Neurosci. 12:1479–86. doi: 10.1046/j.1460-9568.2000.00035.x. One of a series of studies from the Shiosaka laboratory on the role, regulation and targets of the serine protease neuropsin in [Au:in?] LTP. [DOI] [PubMed] [Google Scholar]
- 77.Matsumoto-Miyai K, et al. Coincident pre- and postsynaptic activation induces dendritic filopodia via neurotrypsin-dependent agrin cleavage. Cell. 2009;136:1161–71. doi: 10.1016/j.cell.2009.02.034. [DOI] [PubMed] [Google Scholar]
- 78.Konopacki FA, et al. Synaptic localization of seizure-induced matrix metalloproteinase-9 mRNA. Neuroscience. 2007;150:31–9. doi: 10.1016/j.neuroscience.2007.08.026. [DOI] [PubMed] [Google Scholar]
- 79.Rylski M, et al. Yin Yang 1 is a critical repressor of matrix metalloproteinase-9 expression in brain neurons. J Biol Chem. 2008;283:35140–53. doi: 10.1074/jbc.M804540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sato H, Seiki M. Regulatory mechanism of 92 kDa type IV collagenase gene expression which is associated with invasiveness of tumor cells. Oncogene. 1993;8:395–405. [PubMed] [Google Scholar]
- 81.Freudenthal R, et al. NF-kappaB transcription factor is required for inhibitory avoidance long-term memory in mice. Eur J Neurosci. 2005;21:2845–52. doi: 10.1111/j.1460-9568.2005.04126.x. [DOI] [PubMed] [Google Scholar]
- 82.Rylski M, et al. JunB is a repressor of MMP-9 transcription in depolarized rat brain neurons. Mol Cell Neurosci. 2009;40:98–110. doi: 10.1016/j.mcn.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 83.Jaworski J, et al. Neuronal excitation-driven and AP-1-dependent activation of tissue inhibitor of metalloproteinases-1 gene expression in rodent hippocampus. J Biol Chem. 1999;274:28106–12. doi: 10.1074/jbc.274.40.28106. [DOI] [PubMed] [Google Scholar]
- 84.Konopka W, et al. MicroRNA loss enhances learning and memory in mice. J Neurosci. 2010;30:14835–42. doi: 10.1523/JNEUROSCI.3030-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nedivi E, Hevroni D, Naot D, Israeli D, Citri Y. Numerous candidate plasticity-related genes revealed by differential cDNA cloning. Nature. 363:718–22. doi: 10.1038/363718a0. One of the earliest indications that the MMP–TIMP system is an activity-regulated one in neurons. [DOI] [PubMed] [Google Scholar]
- 86.Rivera S, et al. Tissue inhibitor of metalloproteinases-1 (TIMP-1) is differentially induced in neurons and astrocytes after seizures: evidence for developmental, immediate early gene, and lesion response. J Neurosci. 1997;17:4223–35. doi: 10.1523/JNEUROSCI.17-11-04223.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jourquin J, et al. Tissue inhibitor of metalloproteinases-1 (TIMP-1) modulates neuronal death, axonal plasticity, and learning and memory. Eur J Neurosci. 2005;22:2569–78. doi: 10.1111/j.1460-9568.2005.04426.x. [DOI] [PubMed] [Google Scholar]
- 88.Chaillan FA, et al. Involvement of tissue inhibition of metalloproteinases-1 in learning and memory in mice. Behav Brain Res. 2006;173:191–8. doi: 10.1016/j.bbr.2006.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jaworski DM, Boone J, Caterina J, Soloway P, Falls WA. Prepulse inhibition and fear-potentiated startle are altered in tissue inhibitor of metalloproteinase-2 (TIMP-2) knockout mice. Brain Res. 2005;1051:81–9. doi: 10.1016/j.brainres.2005.05.057. [DOI] [PubMed] [Google Scholar]
- 90.Baba Y, et al. Timp-3 deficiency impairs cognitive function in mice. Lab Invest. 2009;89:1340–7. doi: 10.1038/labinvest.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gahmberg CG, Tian L, Ning L, Nyman-Huttunen H. ICAM-5--a novel two-facetted adhesion molecule in the mammalian brain. Immunol Lett. 2008;117:131–5. doi: 10.1016/j.imlet.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 92.Benson DL, Yoshihara Y, Mori K. Polarized distribution and cell-type specific localization of telencephalin, an intercellular adhesion molecule. J Neurosci Res. 1998;52:43–53. doi: 10.1002/(SICI)1097-4547(19980401)52:1<43::AID-JNR5>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 93.Matsuno H, et al. Telencephalin slows spine maturation. J Neurosci. 2006;26:1776–86. doi: 10.1523/JNEUROSCI.2651-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tian L, et al. Activation of NMDA receptors promotes dendritic spine development through MMP-mediated ICAM-5 cleavage. J Cell Biol. 2007;178:687–700. doi: 10.1083/jcb.200612097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Conant K, et al. Methamphetamine-associated cleavage of the synaptic adhesion molecule intercellular adhesion molecule-5. J Neurochem. 2011;118:521–32. doi: 10.1111/j.1471-4159.2010.07153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Marambaud P, et al. A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. Embo J. 2002;21:1948–56. doi: 10.1093/emboj/21.8.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bozdagi O, Shan W, Tanaka H, Benson DL, Huntley GW. Increasing numbers of synaptic puncta during late-phase LTP: N-cadherin is synthesized, recruited to synaptic sites, and required for potentiation. Neuron. 2000;28:245–59. doi: 10.1016/s0896-6273(00)00100-8. [DOI] [PubMed] [Google Scholar]
- 98.Bozdagi O, et al. Persistence of coordinated long-term potentiation and dendritic spine enlargement at mature hippocampal CA1 synapses requires N-cadherin. J Neurosci. 2010;30:9984–9. doi: 10.1523/JNEUROSCI.1223-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tang L, Hung CP, Schuman EM. A role for the cadherin family of cell adhesion molecules in hippocampal long-term potentiation. Neuron. 1998;20:1165–75. doi: 10.1016/s0896-6273(00)80497-3. [DOI] [PubMed] [Google Scholar]
- 100.Michaluk P, et al. Beta-dystroglycan as a target for MMP-9, in response to enhanced neuronal activity. J Biol Chem. 2007;282:16036–41. doi: 10.1074/jbc.M700641200. [DOI] [PubMed] [Google Scholar]
- 101.Higginson JR, Winder SJ. Dystroglycan: a multifunctional adaptor protein. Biochem Soc Trans. 2005;33:1254–5. doi: 10.1042/BST0331254. [DOI] [PubMed] [Google Scholar]
- 102.McGee AW, Yang Y, Fischer QS, Daw NW, Strittmatter SM. Experience-driven plasticity of visual cortex limited by myelin and Nogo receptor. Science. 2005;309:2222–6. doi: 10.1126/science.1114362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lee H, et al. Synaptic function for the Nogo-66 receptor NgR1: regulation of dendritic spine morphology and activity-dependent synaptic strength. J Neurosci. 2008;28:2753–65. doi: 10.1523/JNEUROSCI.5586-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Milward E, et al. Cleavage of myelin associated glycoprotein by matrix metalloproteinases. J Neuroimmunol. 2008;193:140–8. doi: 10.1016/j.jneuroim.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ferraro GB, Morrison CJ, Overall CM, Strittmatter SM, Fournier AE. Membrane-type matrix metalloproteinase-3 regulates neuronal responsiveness to myelin through Nogo-66 receptor 1 cleavage. J Biol Chem. 2011;286:31418–24. doi: 10.1074/jbc.M111.249169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Alberini CM, Chen DY. Memory enhancement: consolidation, reconsolidation and insulin-like growth factor 2. Trends Neurosci. 2012 doi: 10.1016/j.tins.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen DY, et al. A critical role for IGF-II in memory consolidation and enhancement. Nature. 2011;469:491–7. doi: 10.1038/nature09667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rajaram S, Baylink DJ, Mohan S. Insulin-like growth factor-binding proteins in serum and other biological fluids: regulation and functions. Endocr Rev. 1997;18:801–31. doi: 10.1210/edrv.18.6.0321. [DOI] [PubMed] [Google Scholar]
- 109.Rorive S, et al. Matrix metalloproteinase-9 interplays with the IGFBP2-IGFII complex to promote cell growth and motility in astrocytomas. Glia. 2008;56:1679–90. doi: 10.1002/glia.20719. [DOI] [PubMed] [Google Scholar]
- 110.Ethell IM, Ethell DW. Matrix metalloproteinases in brain development and remodeling: synaptic functions and targets. J Neurosci Res. 2007;85:2813–23. doi: 10.1002/jnr.21273. [DOI] [PubMed] [Google Scholar]
- 111.Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–58. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- 112.Russo SJ, et al. The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci. 2011;33:267–76. doi: 10.1016/j.tins.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. 2009;32:1–32. doi: 10.1146/annurev.neuro.051508.135531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Christoffel DJ, Golden SA, Russo SJ. Structural and synaptic plasticity in stress-related disorders. Rev Neurosci. 2011;22:535–49. doi: 10.1515/RNS.2011.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Arnsten AF. Prefrontal cortical network connections: key site of vulnerability in stress and schizophrenia. Int J Dev Neurosci. 2011;29:215–23. doi: 10.1016/j.ijdevneu.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jedynak JP, Uslaner JM, Esteban JA, Robinson TE. Methamphetamine-induced structural plasticity in the dorsal striatum. Eur J Neurosci. 2007;25:847–53. doi: 10.1111/j.1460-9568.2007.05316.x. [DOI] [PubMed] [Google Scholar]
- 117.Swant J, Chirwa S, Stanwood G, Khoshbouei H. Methamphetamine reduces LTP and increases baseline synaptic transmission in the CA1 region of mouse hippocampus. PLoS One. 2011;5:e11382. doi: 10.1371/journal.pone.0011382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mizoguchi H, Yamada K, Nabeshima T. Neuropsychotoxicity of abused drugs: involvement of matrix metalloproteinase-2 and -9 and tissue inhibitor of matrix metalloproteinase-2 in methamphetamine-induced behavioral sensitization and reward in rodents. J Pharmacol Sci. 2008;106:9–14. doi: 10.1254/jphs.fm0070139. [DOI] [PubMed] [Google Scholar]
- 119.Mizoguchi H, et al. Reduction of methamphetamine-induced sensitization and reward in matrix metalloproteinase-2 and -9-deficient mice. J Neurochem. 2007;100:1579–88. doi: 10.1111/j.1471-4159.2006.04288.x. [DOI] [PubMed] [Google Scholar]
- 120.Brown TE, Forquer MR, Harding JW, Wright JW, Sorg BA. Increase in matrix metalloproteinase-9 levels in the rat medial prefrontal cortex after cocaine reinstatement of conditioned place preference. Synapse. 2008;62:886–9. doi: 10.1002/syn.20562. [DOI] [PubMed] [Google Scholar]
- 121.Brown TE, et al. Role of matrix metalloproteinases in the acquisition and reconsolidation of cocaine-induced conditioned place preference. Learn Mem. 14:214–23. doi: 10.1101/lm.476207. This and the preceding study make the important link between maladaptive plasticity of drug addiction and aberrant MMP regulation and activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wiggins A, Smith RJ, Shen HW, Kalivas PW. Integrins modulate relapse to cocaine-seeking. J Neurosci. 2011;31:16177–84. doi: 10.1523/JNEUROSCI.3816-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kawasaki Y, et al. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med. 2008;14:331–6. doi: 10.1038/nm1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Garber KB, Visootsak J, Warren ST. Fragile X syndrome. Eur J Hum Genet. 2008;16:666–72. doi: 10.1038/ejhg.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60:201–14. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Portera-Cailliau C. Which comes first in fragile X syndrome, dendritic spine dysgenesis or defects in circuit plasticity? Neuroscientist. 2011;18:28–44. doi: 10.1177/1073858410395322. [DOI] [PubMed] [Google Scholar]
- 127.Brundula V, Rewcastle NB, Metz LM, Bernard CC, Yong VW. Targeting leukocyte MMPs and transmigration: minocycline as a potential therapy for multiple sclerosis. Brain. 2002;125:1297–308. doi: 10.1093/brain/awf133. [DOI] [PubMed] [Google Scholar]
- 128.Siller SS, Broadie K. Neural circuit architecture defects in a Drosophila model of Fragile X syndrome are alleviated by minocycline treatment and genetic removal of matrix metalloproteinase. Dis Model Mech. 2011;4:673–85. doi: 10.1242/dmm.008045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yong VW. Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci. 2005;6:931–44. doi: 10.1038/nrn1807. [DOI] [PubMed] [Google Scholar]
- 130.Kim HJ, Fillmore HL, Reeves TM, Phillips LL. Elevation of hippocampal MMP-3 expression and activity during trauma-induced synaptogenesis. Exp Neurol. 2005;192:60–72. doi: 10.1016/j.expneurol.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 131.Pauly T, et al. Activity-dependent shedding of the NMDA receptor glycine binding site by matrix metalloproteinase 3: a PUTATIVE mechanism of postsynaptic plasticity. PLoS One. 2008;3:e2681. doi: 10.1371/journal.pone.0002681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cybulska-Klosowicz A, et al. Matrix metalloproteinase inhibition counteracts impairment of cortical experience-dependent plasticity after photothrombotic stroke. Eur J Neurosci. 2011;33:2238–46. doi: 10.1111/j.1460-9568.2011.07713.x. [DOI] [PubMed] [Google Scholar]
- 133.Sieber SA, Niessen S, Hoover HS, Cravatt BF. Proteomic profiling of metalloprotease activities with cocktails of active-site probes. Nat Chem Biol. 2006;2:274–81. doi: 10.1038/nchembio781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jessani N, Liu Y, Humphrey M, Cravatt BF. Enzyme activity profiles of the secreted and membrane proteome that depict cancer cell invasiveness. Proc Natl Acad Sci U S A. 2002;99:10335–40. doi: 10.1073/pnas.162187599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sela-Passwell N, Rosenblum G, Shoham T, Sagi I. Structural and functional bases for allosteric control of MMP activities: can it pave the path for selective inhibition? Biochim Biophys Acta. 2010;1803:29–38. doi: 10.1016/j.bbamcr.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 136.Hadler-Olsen E, Fadnes B, Sylte I, Uhlin-Hansen L, Winberg JO. Regulation of matrix metalloproteinase activity in health and disease. FEBS J. 2011;278:28–45. doi: 10.1111/j.1742-4658.2010.07920.x. [DOI] [PubMed] [Google Scholar]
- 137.Gomis-Ruth FX. Catalytic domain architecture of metzincin metalloproteases. J Biol Chem. 2009;284:15353–7. doi: 10.1074/jbc.R800069200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Basbaum CB, Werb Z. Focalized proteolysis: spatial and temporal regulation of extracellular matrix degradation at the cell surface. Curr Opin Cell Biol. 1996;8:731–8. doi: 10.1016/s0955-0674(96)80116-5. [DOI] [PubMed] [Google Scholar]
- 139.Mantuano E, et al. The hemopexin domain of matrix metalloproteinase-9 activates cell signaling and promotes migration of schwann cells by binding to low-density lipoprotein receptor-related protein. J Neurosci. 2008;28:11571–82. doi: 10.1523/JNEUROSCI.3053-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Van Wart HE, Birkedal-Hansen H. The cysteine switch: a principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc Natl Acad Sci U S A. 1990;87:5578–82. doi: 10.1073/pnas.87.14.5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sbai O, et al. Differential vesicular distribution and trafficking of MMP-2, MMP-9, and their inhibitors in astrocytes. Glia. 58:344–66. doi: 10.1002/glia.20927. This and Ref 147 provide detailed information on cellular trafficking and vesicular release of MMPs and TIMPs in neurons and glia. [DOI] [PubMed] [Google Scholar]
- 142.Cuadrado E, et al. Matrix metalloproteinase-13 is activated and is found in the nucleus of neural cells after cerebral ischemia. J Cereb Blood Flow Metab. 2009;29:398–410. doi: 10.1038/jcbfm.2008.130. [DOI] [PubMed] [Google Scholar]
- 143.Eguchi T, et al. Novel transcription-factor-like function of human matrix metalloproteinase 3 regulating the CTGF/CCN2 gene. Mol Cell Biol. 2008;28:2391–413. doi: 10.1128/MCB.01288-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yang Y, et al. Increased intranuclear matrix metalloproteinase activity in neurons interferes with oxidative DNA repair in focal cerebral ischemia. J Neurochem. 2010;112:134–49. doi: 10.1111/j.1471-4159.2009.06433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhang H, Adwanikar H, Werb Z, Noble-Haeusslein LJ. Matrix metalloproteinases and neurotrauma: evolving roles in injury and reparative processes. Neuroscientist. 2010;16:156–70. doi: 10.1177/1073858409355830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Henley JM, Barker EA, Glebov OO. Routes, destinations and delays: recent advances in AMPA receptor trafficking. Trends Neurosci. 2011;34:258–68. doi: 10.1016/j.tins.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sbai O, et al. Vesicular trafficking and secretion of matrix metalloproteinases-2, -9 and tissue inhibitor of metalloproteinases-1 in neuronal cells. Mol Cell Neurosci. 39:549–68. doi: 10.1016/j.mcn.2008.08.004. This and Ref 141 provide detailed information on cellular trafficking and vesicular release of MMPs and TIMPs in neurons and glia. [DOI] [PubMed] [Google Scholar]
- 148.Kean MJ, et al. VAMP3, syntaxin-13 and SNAP23 are involved in secretion of matrix metalloproteinases, degradation of the extracellular matrix and cell invasion. J Cell Sci. 2009;122:4089–98. doi: 10.1242/jcs.052761. [DOI] [PubMed] [Google Scholar]
- 149.Neves G, Cooke SF, Bliss TV. Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat Rev Neurosci. 2008;9:65–75. doi: 10.1038/nrn2303. [DOI] [PubMed] [Google Scholar]
- 150.Rioult-Pedotti MS, Friedman D, Donoghue JP. Learning-induced LTP in neocortex. Science. 2000;290:533–6. doi: 10.1126/science.290.5491.533. [DOI] [PubMed] [Google Scholar]
- 151.Bourne JN, Harris KM. Coordination of size and number of excitatory and inhibitory synapses results in a balanced structural plasticity along mature hippocampal CA1 dendrites during LTP. Hippocampus. 2011;21:354–73. doi: 10.1002/hipo.20768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Frey U, Krug M, Reymann KG, Matthies H. Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res. 1988;452:57–65. doi: 10.1016/0006-8993(88)90008-x. [DOI] [PubMed] [Google Scholar]
- 153.Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–89. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]
- 154.Buzsaki G. Theta oscillations in the hippocampus. Neuron. 2002;33:325–40. doi: 10.1016/s0896-6273(02)00586-x. [DOI] [PubMed] [Google Scholar]
- 155.Lynch G, Rex CS, Gall CM. LTP consolidation: substrates, explanatory power, and functional significance. Neuropharmacology. 2007;52:12–23. doi: 10.1016/j.neuropharm.2006.07.027. [DOI] [PubMed] [Google Scholar]