

Abstract
Mutants lacking the ψ (HolD) subunit of the Escherichia coli DNA Polymerase III holoenzyme (Pol III HE) have poor viability, but a residual growth allows the isolation of spontaneous suppressor mutations that restore ΔholD mutant viability. Here we describe the isolation and characterization of two suppressor mutations in the trkA and trkE genes, involved in the main E. coli potassium import system. Viability of ΔholD trk mutants is abolished on media with low or high K+ concentrations, where alternative K+ import systems are activated, and is restored on low K+ concentrations by the inactivation of the alternative Kdp system. These findings show that the ΔholD mutant is rescued by a decrease in K+ import. The effect of trk inactivation is additive with the previously identified ΔholD suppressor mutation lexAind that blocks the SOS response indicating an SOS-independent mechanism of suppression. Accordingly, although lagging-strand synthesis is still perturbed in holD trkA mutants, the trkA mutation allows HolD-less Pol III HE to resist increased levels of the SOS-induced bypass polymerase DinB. trk inactivation is also partially additive with an ssb gene duplication, proposed to stabilize HolD-less Pol III HE by a modification of the single-stranded DNA binding protein (SSB) binding mode. We propose that lowering the intracellular K+ concentration stabilizes HolD-less Pol III HE on DNA by increasing electrostatic interactions between Pol III HE subunits, or between Pol III and DNA, directly or through a modification of the SSB binding mode; these three modes of action are not exclusive and could be additive. To our knowledge, the holD mutant provides the first example of an essential protein-DNA interaction that strongly depends on K+ import in vivo.
Author Summary
Replication polymerases are responsible for genome duplication; they are ubiquitous and show high levels of functional and structural conservation across all species. The HolC-HolD (χψ) complex is a component of the replicative polymerase in the model bacteria Escherichia coli, and is crucial for normal growth. We isolated suppressor mutations that restore the viability of the holD mutant and we found that inactivating the Trk system, responsible for the main pathway of potassium import, renders the entire χψ complex dispensable for growth. Activation of alternative pathways of potassium import abolishes the suppression. The viability of the holD trk mutant is due in large part to a better capacity of the χψ-less polymerase to compete with other polymerases. Potassium glutamate is the major intracellular ionic osmolyte in E. coli, and we propose that mutations that affect potassium concentration in vivo stabilize the χψ-less polymerase by increasing electrostatic interactions between the different polymerase subunits and between polymerase and DNA. Stabilized by the lower intracellular potassium concentration, the χψ-less polymerase becomes functional in trk mutants, to a level that permits cell growth with this defective polymerase. Our results imply that K+ import can play an important role in the stability of protein complexes on DNA.
Introduction
Chromosome replication is performed in Escherichia coli by a replicase called the DNA Polymerase III holoenzyme (Pol III HE) and composed of 9 different polypeptides [1]. DNA synthesis is realized by the core polymerase, composed of a polymerase subunit (α, encoded by dnaE), associated with a proof-reading activity (ε, encoded by dnaQ) and a stabilizing subunit (HolE). Each Pol III HE contains two core polymerases, one for the continuously synthesized leading-strand, one for the lagging-strand synthesized as 1–2 kilobase (kb) Okazaki fragments. The presence of an additional spare one to form a trimeric polymerase was proposed and is still debated [2–4]. The lagging-strand template is transiently single-stranded during Okazaki fragment synthesis and covered by single-stranded DNA binding proteins (SSB). The stability of each core polymerase on DNA is ensured by its interaction with a polymerase clamp, in E. coli this is a β-dimer (encoded by dnaN) which encircles the DNA and is structurally homologous to PCNA in eukaryotes [5]. The β-clamp is loaded onto DNA by a clamp loader complex, functionally homologous to the RFC complex in eukaryotes, which, in addition to loading the β-clamp, ensures the cohesion of the replisome by interacting with the three core polymerases and with the DNA helicase. In E. coli the clamp loader complex is composed of three τ subunits (encoded by dnaX), the δ and δ’ subunits (encoded by holA and holB, respectively), and a heterodimeric complex of χ and ψ proteins, encoded by holC and holD respectively [1, 2]. The ψχ complex forms a bridge between the τ3δδ’pentamer and SSB. Actually, ψ interacts with τ and χ, which itself interacts with SSB [6–9]. In addition, the ψ-τ interaction favors the assembly of the τ3δδ’pentamer at limiting δδ’concentrations, stabilizes an ATP-activated DNA-high affinity conformation of the clamp loader, and thus facilitates the clamp loading reaction in vitro [10–13].
Most Pol III HE subunits are essential for growth, with the notable exception of HolE and DnaQ [14]. Inactivating holC is not lethal but impairs growth, particularly at a high temperature; growth of holC mutants can be significantly improved if the induction of the SOS response, a set of repair genes induced by DNA damage or replication impairment, is prevented [15, 16]. Inactivating holD is strongly deleterious in all growth conditions; however, a residual growth on minimal medium at 30°C facilitates the selection of suppressor mutations. We previously reported that ΔholD mutant growth is improved by mutations that inactivate the SOS response and more specifically by the inactivation of dinB and polB genes, encoding the SOS-induced bypass polymerases Pol IV and Pol II, respectively [17]. We proposed that increased (SOS-induced) levels of these polymerases compete with HolD-less Pol III HE and destabilize its interaction with DNA. The viability of holD and holC mutants is also restored by a duplication of the ssb gene, which doubles the intra-cellular level of SSB proteins [16]. We proposed that increasing intracellular concentration of SSB favors the SSB-DNA binding mode where each SSB tetramer binds 35 nucleotides, which stabilizes Pol III bound to DNA and bypasses the need for HolD. In the present study, we describe the isolation of holD suppressor mutations that affect K+ import.
K+ is the major intracellular cation in E. coli, present at concentrations that vary from 100–150 to 500–600 mM, which is higher than that in the extracellular medium [18–20]. In the growth medium used here, about 45–50% of K+ ions are expected to be “free” and balance charges of small diffusible anions, including glutamate, and about 50–55% are thought to be “bound” and serve to balance charge on macromolecule anions, proteins and nucleic acids [21]. K+ glutamate is the intracellular ionic compound that ensures turgor and osmolarity. K+ is imported in E. coli by several independent systems [22, 23], [24] (Table 1). The major one is composed of TrkA, TrkH or TrkG, and TrkE [22, 25]. These four genes are unlinked and represent two separate pathways, TrkAEG and TrkAEH. A second system depends on one protein, TrkD later called Kup [22, 26]. Trk proteins and Kup have a low affinity for K+ (Km 1.5 mM), and Trk is the main active pathway when K+ concentration in the growth medium is 5 mM or more; Kup activity could only be studied in mutants lacking the other K+ import systems [27, 28]. At low K+ concentrations the kdpFABC operon is induced by a two component system, KdpD and KdpE; Kdp is a high affinity K+ import system (Km 2 μM), active below 5 mM K+ [29, 30], [31]. Finally, at high K+ concentrations the triple trk kup kdp mutant imports K+ with a system called TrkF, which is a combination of several minor nonspecific (called “illicit”) transport pathways [32].
Table 1. K+ uptake systems in E. coli.
| Name | Genetics | Km (mM) / Vmax (μmol.g-1 min-1) | Other properties |
|---|---|---|---|
| Kdp | 6 genes, 2 operons kdpFABC encodes the structural proteins kdpDE encodes the regulatory proteins | 0.002 / 100–150 | kdp FABC operon is regulated by the KdpD, sensor kinase and the KdpE response regulator. It is expressed when the growth rate of cells begins to become limited by K+ availability |
| Trk | Four unlinked genes, trkA, trkE (also called sapD), trkG and trkH | 1.5 / 300–500 | Constitutively expressed; Corresponds to two pathways: TrkAEG and TrkAEH. |
| Kup | kup (also called trkD) | 0.5 / 0–50 | Transports Cs as well as K+ |
| TrkF | Multiple “aberrant” K+ transport activities | Allows the kdp trk kup triple mutant to grow at high K+ concentrations (above 100 mM) |
In this work, we show that trkA and trkE mutations restore the ΔholD mutant growth. The viability of ΔholD Δtrk mutants requires the Kdp and the TrkF K+ import pathways to be of low or negligible activity. We propose that HolD-less Pol III HE can replicate E. coli chromosomes when K+ intracellular concentration is affected, owing to its stabilization on DNA by improved electrostatic interactions.
Results
Isolation of ΔholD suppressor mutations
As inactivation of the holD gene prevents E. coli growth, ΔholD mutants were constructed and propagated in the presence of pAM-holD, a plasmid that carries the holD wild-type gene and replicates only in the presence of IPTG. Suppressors of the ΔholD growth defects can be obtained by growing ΔholD [pAM-holD] cells in the absence of IPTG and selecting for plasmid-less fast growing clones [16, 17]. Four such ΔholD fast growing colonies were isolated on minimum medium (M9) at 37°C in an MG1655 background (JJC6376 to JJC6379) (unless otherwise indicated, all minimal media used in this work contain 0.4% glucose and 0.2% casamino acids). The SOS response is induced in the ΔholD mutant and a sfiA mutant was used to prevent cell division blockage by the SOS-induced SfiA protein. Suppressor mutations were also isolated in a ΔholD sfiA::Mu strain, and two fast growing colonies obtained on M9 at 30°C were studied (JJC6389 and JJC6390). Interestingly, whole genome sequencing identified the presence of a mutation affecting the trk pathway of K+ import in two of these six clones. These mutations were confirmed by re-sequencing the genes of interest. JJC6377 carries a 84 base pair (bp) in-frame deletion in the trkA gene, from nucleotides 513 to 597, with 9 bp microhomology at the junction (called hereafter ΔtrkA84, S1 Fig). It also carries a large duplication, from about 3 648 000 to around 4 167 000. It has to be noted that a holD mutation was shown to increase the frequency of recombination events between repeated sequences, which may account for the recurrent presence of deletions and duplications in suppressed holD mutants [16, 33]., JJC6389 carries a trkE point mutation at position 767 changing glutamine 255 to proline. This was the only sequence modification identified in this strain. The suppressor mutations identified in the four other sequenced genomes will be described in future publications.
The ΔtrkA84 deletion, a ΔtrkA or ΔtrkE full gene deletion is sufficient for suppression of the ΔholD growth defects
The two ΔholD clones affected for K+ import formed colonies on LB at 30°C, 37°C, and 42°C overnight. On M9, the strain carrying ΔtrkA84 formed colonies at all temperatures, while the strain mutated in trkE formed colonies at 30°C and 37°C only (Fig 1, the two original suppressed clones are called ΔholD trkA sup and ΔholD trkE sup). However, the spontaneous suppressor strain carrying ΔtrkA84 also carries a large duplication in addition to the ΔtrkA84 mutation, which could improve its viability, whereas the trkEQ255P mutation was the sole genome modification identified in JJC6389. Co-transduction of ΔtrkA84 with an adjacent marker (zhd-3082::Tn10) in a ΔholD [pAM-holD] or in a ΔholD sfiA [pAM-holD] background yielded clones that, after plasmid loss, were able to grow on LB at all temperatures and on M9 at 30°C and 37°C, and were only slightly more viable than the trkEQ255P suppressed strain at 37°C (Fig 1 ΔholD ΔtrkA84, ΔholD ΔtrkA84 sfiA). A ΔtrkA::Cm mutant lacking the entire trkA sequence was constructed by gene replacement and behaved as the ΔtrkA84 deletion (Fig 1 ΔholD trkA::Cm), suggesting that the ΔtrkA84 deletion is a null allele. The use of a null allele of trkE showed that the inactivation of trkE or trkA restores holD mutant growth to the same extent, as expected since these two genes belong to the same pathway. The trkEQ255P mutation was slightly less efficient than the ΔtrkE deletion in restoring holD mutant growth at 37°C, suggesting a residual activity of the mutant protein. We conclude that inactivation of the trk pathway of K+ import restores growth of the ΔholD mutant on LB at all temperatures and on M9 at 30°C and 37°C.
Fig 1. trkA and trkE mutations restore ΔholD viability.

In a first step pAM-holD containing cultures were grown for 8 hours in M9 in the absence of IPTG at 30°C, appropriate dilutions were plated on M9 and plates were incubated for 3 days at 30°C. Isolated colonies, cured of pAM-holD, were suspended in 1 ml M9 salt medium and 5 μl drops of serial dilutions were plated on M9 or LB. LB plates were incubated overnight at 42°C or 37°C, or two days at 30°C. M9 plates were incubated for two days at the indicated temperatures. Isolated colonies obtained by streaking holD trkA sup, JJC6377 and holD trkE sup, JJC6389 on M9 at 30°C were tested in parallel. The strains used to generate plasmid-less colonies are: holD, JJC6869; holD ΔtrkA84, JJC6669; holD ΔtrkA::cm, JJC6682; holD ΔtrkA84 sfiA, JJC6969; holD trkA+, JJC6670; holD ΔtrkE::kan JJC7173.
Rescue of ΔholD growth by trk inactivation is abolished at low K+ concentrations in a kdp-dependent way
To check that the ΔtrkA84, ΔtrkA::Cm, ΔtrkE::kan and trkEQ255P mutations allow ΔholD mutant growth by affecting K+ import, we tested the effect of different K+ concentrations in the external medium on the viability of the different ΔholD trk mutants. In K+-limiting conditions the kdpFABC operon is induced by the regulatory proteins KdpD and KdpE [30]. As shown in Figs 2 and S2, ΔholD ΔtrkA84, ΔholD ΔtrkE and ΔholD trkEQ255P did not form colonies on minimal medium containing 0.2 mM or 1 mM K+ (called MK0.2 and MK1, respectively), although, as expected, they could grow on the same medium containing 22 mM K+ (MK22), the K+ concentration in M9. Lethality of ΔholD trk mutants on plates containing low K+ concentrations may result from the activation of the kdp operon, which promotes active K+ uptake below 5 mM in the external medium [30]. Inactivation of kdpA restored the growth of ΔholD ΔtrkA84 and ΔholD ΔtrkE mutants on MK0.2 and MK1 (ΔholD ΔtrkA84 Δkdp, ΔholD ΔtrkE Δkdp, Figs 2 and S2), indicating that the lethality of holD trk mutants at low K+ concentrations results from the activity of Kdp. However, inactivation of kdp in a Trk+ ΔholD did not restore viability at low K+ concentrations, with the exception of a partial growth at 30°C on MK0.2 (ΔholD Δkdp Figs 2 and S2). Because the Trk system is constitutively expressed and has a high Vmax it is still partly active even on MK0.2 (the growth rate of the kdp mutant is only 20% lower in 0.2 mM than in 5 mM K+ [18, 34]). On MK0.2, the remaining activity of Trk in the holD kdp mutant is responsible for the growth defect, but this activity is limited, allowing a weak but significant residual growth at 30°C. Finally, it has to be noted that the K+ concentration in our LB was measured and found to be 10.9 ± 0.74 mM, but as shown below other components than K+ also play a role in the growth of ΔholD Δtrk mutants on LB.
Fig 2. Viability of ΔholD trkA and ΔholD trkE mutants is abolished by a high K+ concentration, and in a Kdp-dependent way at a low K+ concentration.

In a first step, isolated colonies were obtained by plating appropriate dilutions of wild-type, ΔtrkA, holD trkE sup or ΔtrkA Δkdp overnight cultures on M9, or appropriate dilutions of pAM-holD containing cultures grown for 8 hours in the absence of IPTG at 30°C, and incubating plates for 3 days at 30°C. These isolated colonies were suspended in 1 ml MK0 salt medium. Serial dilutions were made and 5 μl drops of each dilution were plated on MK plates containing different K+ concentrations as indicated (MK1 1 mM, MK22 22 mM, MK115 115 mM; see S2 Fig for growth on MK0.2, M9, MMA, and LB). Pictures of wild-type and ΔtrkA strains at 37°C were taken after 24 h incubation; all other plates were incubated for two days at the indicated temperatures. Wild-type, JJC1392; ΔtrkA::cm, JJC6800; holD trkE sup, JJC6389 (the original suppressed clone); ΔtrkA84 Δkdp::cm, JJC7030. The strains used to generate plasmid-less colonies are: holD, JJC6869; holD ΔtrkA::cm, JJC6898; holD ΔtrkA84, JJC6969; holD Δkdp::cm JJC6750; holD ΔtrkA84 Δkdp::cm, JJC6819; holD ΔtrkE::kan, JJC7173; holD ΔtrkE::kan Δkdp::cm, JJC7223.
The Trk system is the main K+ import system in E. coli. Therefore, inactivating trkA might decrease K+ intracellular concentration in growing cells. As shown in Table 2, intracellular K+ concentration was significantly decreased in trkA and holD trkA mutants compared to wild-type cells. This result suggests that a 12–17% decrease in intracellular K+ is sufficient to promote HolD-less Pol III stabilization on DNA.
Table 2. trkA gene inactivation decreases E. coli intracellular K+ concentration.
| Strain | Genotype | nmoles K+/mg DW | Ratio to wt | N |
|---|---|---|---|---|
| JJC1392 | wild-type | 502 ± 12 | 1 | 8 |
| JJC6800 | trkA | 443 ± 12 | 0.88 | 7 |
| JJC6898S | holD trkA | 418 ± 10 | 0.83 | 7 |
Cells grown to OD 0.4 in M9 were washed in hypertonic, potassium-free glucose medium and dried. K+ concentrations were measured in dried pellets with a flame spectrophotometer (see Material and Methods for details). Results are expressed in nmoles K+ per milligram of dry weight (DW). Averages of N independent experiments ± Standard Error of the Mean (SEM) are shown. Results were compared by a Tukey-Test (multiple comparisons) in conjunction with an ANOVA: trkA and holD trkA mutants were both significantly different from wild-type (P = 0.004 and P = 0.00014, respectively) and were not significantly different from each other (P = 0.307). (“JJC6898S” stands for “segregated” cultures).
In conclusion, when ΔholD trkA (or holD trkE) cells are grown on 22 mM K+ (M9 or MK22), K+ uptake is impaired and intracellular K+ concentration decreases to a level that rescues the ΔholD mutant. At low K+ concentrations (0.2 mM and 1 mM) the activation of the kdp operon increases K+ uptake to a level that prevents the growth of ΔholD ΔtrkA and ΔholD ΔtrkE mutants. By preventing Kdp-mediated K+ import, kdp gene inactivation restores growth of the ΔholD ΔtrkA Δkdp and ΔholD ΔtrkE Δkdp mutants. We conclude that viability of the ΔholD mutant can be restored by decreasing K+ import.
Growth of the ΔholD trk mutants is prevented by a high K+ concentration and unaffected by kup inactivation
Mutants lacking all three K+ import systems (trk, kdp, kup) require a high K+ concentration for growth and rely on multiple minor K+ import activities called TrkF [32]. To test whether TrkF activity prevents growth of the holD trkA and holD trkE mutants on medium containing a high level of K+, colony formation of the ΔtrkA, ΔholD ΔtrkA, ΔholD ΔtrkE and ΔholD trkEQ255P mutants was compared on synthetic medium containing 22 mM (M9, MK22), 115 mM (MK115) or 150 mM (MMA) K+. As shown in Figs 2 and S2, a high concentration of K+ prevented growth of the ΔholD trk mutants; therefore activating K+ import by a high concentration of K+ in the growth medium is lethal to the ΔholD trk mutants. It was not possible to inactivate TrkF, which is not a defined locus but a combination of several minor pathways [32].
Growth of the ΔholD ΔtrkA mutant was compared in different liquid media (Fig 3). For these experiments, the ΔholD ΔtrkA [pAM-holD] mutant was grown overnight at 30°C to saturation in LB, M9, MK1 or MK115 medium without IPTG, diluted to OD 0.002 in the same medium, and grown at 37°C; growth was monitored by plating appropriate dilutions on M9. As shown in Fig 3A, growth was rapid in LB, slower in M9, and stopped after two or three generations in MK1 and MK115, in agreement with the lack of colony formation at these K+ concentrations. Generation times were calculated from the slope of the best fit straight line during exponential growth (Fig 3B, generation times could not be calculated for the ΔholD ΔtrkA mutant grown in MK1 or MK115 owing to the rapid growth arrest). The growth rate of the single ΔtrkA mutant was similar in M9, MK1 and MK115, and not significantly different from wild-type (30 min), while the generation time of ΔholD ΔtrkA cells in M9 was nearly 50% longer (43 min). Surprisingly, the generation time of ΔholD ΔtrkA cells was similar to that of wild-type and ΔtrkA single mutant in LB (22–23 min), in agreement with overnight colony formation on LB, confirming that the rescue of the holD mutant by trkA is very efficient in LB.
Fig 3. Viability of the holD trkA mutant in liquid medium.

Overnight cultures of wild-type (JJC1392), trkA (JJC6800) and holD trkA [pAM-holD] (JJC6898) strains were grown at 30°C in LB, M9, MK1, MK115 medium, diluted, and further grown at 37° in the same medium for 7 hours, as described in the Materials and Methods. (A) Representative growth curves of holD trkA (JJC6898) in LB (full line, diamond), M9 (full line, square), MK1 (dashed line, circles), MK115 (dashed line, triangles). (B) The doubling time expressed in minutes was calculated from the exponential part of the growth curve, averages of three independent determinations are shown with standard deviations. No generation time could be calculated for the holD trkA mutant grown in MK115 and in MK1, as growth at 37°C stopped after 2 to 3 generations (NA = not applicable).
The kup gene, originally called trkD, is constitutively expressed and active in the same growth conditions as the Trk system, but Kup has a low level of activity [22, 26]. A Δkup mutant was used to test whether the Kup pathway plays a role in the viability of ΔholD and ΔholD ΔtrkA mutants at different K+ concentrations. Inactivating kup did not rescue the ΔholD mutant on M9 (S3A Fig), in agreement with the idea that Trk is the main K+ import pathway under these conditions. It did not affect the growth of the ΔholD ΔtrkA mutant at any K+ concentration tested (S3A Fig), including in 10.9 mM K+, the K+ concentration in our LB. These results are in agreement with the idea that Trk is the major K+ import system on M9, and that the Kdp K+ import system is activated at low K+ concentrations. They also suggest that at high K+ concentrations the poor viability of ΔholD trkA cells is caused by TrkF activity, since Kup and TrkF are the only active pathways at high K+ concentrations in a trk mutant, and the phenotype of the holD trk mutant is not affected by Kup inactivation.
All LB components participate to the holD trkA mutants growth on LB at 42°C
Surprisingly, at 42°C ΔholD trkA mutants formed colonies overnight (ON) on LB but not on minimal medium (Fig 1). This result was unexpected since the number of replication forks per cell is increased in rich medium compared to minimal medium, which is expected to disfavor a mutant that lacks a Pol III HE subunit. Furthermore, LB contains 10.9 mM K+, a concentration that does not allow growth of the ΔholD trkA mutant at 42°C in synthetic medium (S3A Fig). Consistent with the idea that a low intracellular K+ concentration restores viability by stabilizing HolD-less Pol III HE-DNA complexes, the lethality on M9 at 42°C could result from a destabilization of replication complexes by temperature. To understand why this destabilization is not observed in LB, we tested the three LB components individually (tryptone, yeast extract and NaCl). As shown on Fig 4, adding yeast extract to M9 casamino acids medium allowed growth of the ΔholD ΔtrkA and ΔholD trkEQ255P mutants at 42°C. Tryptone is the product of casein hydrolysis by trypsin, while casamino acids are the product of acidic hydrolysis of casein. Consequently, tryptone contains mainly peptides while casamino acids contain mainly free amino acids. Replacing casamino acids by tryptone in M9 improved growth of holD trk mutants, although colonies were heterogeneous in size, possibly because growth remains slow, favoring the appearance of new suppressor mutations. Therefore, although yeast extract is more efficient than tryptone, each of these two components can improve growth at 42°C in M9. Note that the presence of casamino acids increases growth rate, but does not affect the plating efficiency of holD trkA cells at any temperature (S3B Fig).
Fig 4. All LB components participate to the viability of holD trkA mutants at 42°C.

Isolated colonies obtained on M9 as described in the legend of Fig 2 were suspended in MK0 salts, and 5 μl drops of serial dilutions were plated on the indicated medium. LB and M9 casamino acids yeast extract plates were incubated overnight. M9 casamino acids and M9 tryptone plates were incubated for two days at 42°C. Wild-type, JJC1392; holD trkE sup, JJC6389. The strains used to generate plasmid-less colonies are: holD, JJC6869; holD ΔtrkA::cm, JJC6898; holD ΔtrkA84, JJC6969.
The salts in M9 are 92.5 mM Na+ and 22 mM K+, while LB is 171 mM NaCl (10 g/l) and 10.9 mM K+. The ΔholD ΔtrkA mutants formed slow growing colonies and ΔholD trkEQ255P did not grow at 42°C on low salt LB (0.5 g/l NaCl, 8.5 mM) (Fig 4). As on M9 at 37°C, the ΔholD trkEQ255P mutant was more impaired than the ΔholD ΔtrkA mutant, presumably because of a residual activity of the mutated TrkE protein. Nevertheless, the high Na+ concentration in LB improves growth of both ΔholD ΔtrkA and ΔholD trkEQ255P mutants at 42°C. In conclusion, all three components of LB, and particularly yeast extract in the presence of high Na+, participate to the viability of the ΔholD trk mutants at 42°C.
The ΔtrkA mutation suppresses the growth defect of holC and holC holD mutants at 30°C and 37°C
ψ (HolD) plays a dual role in the clamp loader complex: its interaction with τ (DnaX) stabilizes the complex, and its interaction with χ (HolC) connects clamp loading and Okazaki fragment synthesis through the χ-SSB interaction. Accordingly, a ΔholC mutation, which lacks only the clamp loader-SSB interaction, is less deleterious than the ΔholD mutation, particularly at 30°C [16] (Figs 1 and 5). The ΔtrkA mutation improved the ΔholC mutant growth at 30°C, 37°C, and 42°C on LB, and at 30°C and 37° on M9. Results were variable on M9 at 42°C, with either no growth of the ΔholC ΔtrkA mutant or, as in the example shown in Fig 5, appearance of new suppressor mutations. ΔholC ΔholD ΔtrkA mutant viability was similar to that of the ΔholD ΔtrkA mutant, showing that suppression of the ΔholD growth defects by trkA inactivation does not require χ (Fig 5).
Fig 5. trkA inactivation restores ΔholC and ΔholC ΔholD viability.

Isolated colonies obtained on M9 as described in the legend of Fig 2 were suspended in MK0 salts and 5 μl drops of serial dilutions were plated on the indicated medium (see S4 Fig for growth on MK1, MK22 and MK115). LB plates were incubated overnight at 37°C and 42°C, and for two days at 30°C. M9 casamino acids plates were incubated for two days at all temperatures. The strains used to generate plasmid-less colonies are: holC, JJC6748; holC ΔtrkA84, JJC6827; holC holD, JJC6774; holC holD ΔtrkA84, JJC6828.
The effects of K+ concentration on the suppression of the holC mutant growth defects by trk inactivation were tested (S4 Fig). At 30°C, presumably owing to a significant growth of the ΔholC mutant, the ΔtrkA mutation improved growth at all K+ concentrations, forming smaller colonies only on MK1. Therefore K+ import by Kdp or TrkF did not prevent ΔholC ΔtrkA mutant growth. However, at 37°C where growth of the ΔholC mutant is severely impaired, rescue by the ΔtrkA mutation was efficient only on 22 mM K+, while on low and high K+ concentrations colonies were highly heterogeneous, indicating that improvement of residual growth mainly favored the appearance of new suppressor mutations. As expected, rescue of ΔholC ΔholD mutant was weak or null at low or high K+ concentrations compared to rescue at 22 mM (S4 Fig). Therefore, a defect in K+ import allows chromosome replication in the absence of either ψ, or χ or both.
trkA does not restore holD mutant growth by affecting SOS induction and does not restore a wild-type level of primer elongation
We previously showed that growth of the ΔholD mutant is improved upon inactivation of SOS by a lexAind or a recF mutation [17]. These suppressor mutations were more efficient in the AB1157 context than in the context used here, MG1655, where ΔholD lexAind and ΔholD recF viability was mainly improved at 37°C ([16] (Fig 6). We tested whether the trk mutations act through a decrease of SOS induction. Measures of SOS induction using a sfiA::lacZ fusion showed that the SOS response was induced in the ΔholD ΔtrkA mutant as in the ΔholD mutant (Table 3). SOS induction was RecF-dependent indicating that it results from the accumulation of single-stranded DNA gaps in the ΔholD ΔtrkA mutant as in the ΔholD mutant. Furthermore, preventing SOS induction and trk inactivation showed an additive effect on the viability of the holD mutant, as ΔholD ΔtrkA lexAind and ΔholD ΔtrkA recF mutants were viable on M9 at 42°C (Fig 6). Rescue of the ΔholD lexAind and ΔholD recF mutants by ΔtrkA was only efficient on 22 mM K+ and was not observed at low or high K+ concentrations, indicating that it requires alternative K+ import systems to be of low or negligible activity (S5 Fig). We conclude from these experiments that ΔtrkA did not restore the viability of the ΔholD mutant by preventing RecF-dependent SOS induction, and that the ΔholD ΔtrkA mutant still accumulates single-stranded DNA gaps during replication.
Fig 6. trkA inactivation does not rescue the ΔholD mutant by affecting SOS induction or SSB binding.

Isolated colonies obtained on M9 as described in the legend of Fig 2 were suspended in MK0 salts and 5 μl drops of serial dilutions were plated on M9 and LB (see S5 Fig for growth on MK1, MK22, MK115). LB plates were incubated overnight at 37°C and 42°C, and for two days at 30°C. M9 casamino acids plates were incubated for two days at all temperatures. The strains used here to generate plasmid-less colonies are: holD ΔtrkA::cm, JJC6898; holD lexAind, JJC6420; holD lexAind ΔtrkA::cm, JJC7008; holD recF, JJC7058; holD ΔtrkA::cm recF, JJC7063; holD argE::ssb, JJC6394; holD argE::ssb ΔtrkA::cm, JJC7000.
Table 3. The holD trkA mutant constitutively expresses the SOS response.
| strain | relevant genotype | Β-gal Miller Units | N |
|---|---|---|---|
| JJC6478 | Wild-type | 35 ± 1.7 | 3 |
| JJC6897 | ΔtrkA | 33.5 ± 2 | 4 |
| JJC6545S | ΔholD | 168 ± 28 | 6 |
| JJC6969S | ΔholD ΔtrkA | 228 ± 25 | 7 |
| JJC7058S | ΔholD recF | 81 ± 7 | 3 |
| JJC7067 | ΔholD ΔtrkA recF | 81 ± 9 | 3 |
Strains harboring a lacZ deletion and a sfiA::lacZ fusion were used to measure SOS constitutive expression in different mutants as described in Materials and Methods. JJC7067 was constructed by introduction of the sfiA::MudAplacZ fusion in a ΔholD ΔtrkA recF clone previously cured of pAM-holD. For JJC6545, JJC6969 and JJC7058, overnight cultures were grown in the absence of IPTG at 30°C in M9, diluted 50-fold in M9 at 30°C and grown to OD 0.2 to 0.6 for β-galactosidase tests (“S” stands for “segregated” cultures). Averages ± standard deviations are shown. N indicates the number of independent experiments.
If these gaps result from a delay in the use of RNA primers for the synthesis of Okazaki fragments, the strain might be sensitive to an excess of RNase H, which could destroy the RNA primers prior to their elongation. The results in Table 4 shows that the presence of a 20 copy plasmid that expresses RNase H prevented the formation of ΔholD ΔtrkA colonies, although it did not affect the growth of HolD+ cells, suggesting that single-stranded DNA gaps are, at least in part, caused by a defect in RNA primer elongation.
Table 4. The holD trkA mutant is sensitive to RNaseH overexpression.
| Strain | relevant genotype | % of plasmid-less colonies |
|---|---|---|
| JJC6669 | ΔholD ΔtrkA | 85 ± 7.7 |
| JJC6736 | ΔholD ΔtrkA [pACYC184] | 91.4 ± 3 |
| JJC6737 | ΔholD ΔtrkA [pEM001] | 0.5 |
| JJC7267 | HolD+ ΔtrkA [pACYC184] | 91 ± 7 |
| JJC7278 | HolD+ΔtrkA [pEM001] | 97.6 ± 1 |
The indicated strains were propagated in M9 at 30°C for 8 hours for curing pAM-holD. The ratios of SpcS colonies were calculated from the plating efficiency on M9 and M9 IPTG Spc. Averages and standard deviations of three independent experiments are shown. Colony forming efficiency on M9 IPTG Spc was not affected by the presence of pACYC184 or pEM001, and the number of colonies cured of pAM-holD on M9 was similar in the presence or absence of the vector pACYC184. A total of only five colonies cured of pAM-holD were obtained in three independent experiments in the presence of pEM001 in the holD trkA mutant (JJC6737), these colonies were highly heterogeneous upon streaking, indicating that pEM001 is highly deleterious for holD trkA cells.
We previously showed that two SOS-induced proteins play an important role in the ΔholD mutant growth defects, the bypass polymerases DinB (Pol IV) and Pol II, and we proposed that these SOS-induced proteins are deleterious in the holD mutant because they compete efficiently with HolD-less Pol III for β-clamps binding at replication forks [17]. Accordingly, the over-production of DinB with a deletion of the last 5 amino acids (DinBΔC5), which fails to bind DnaN [35], was not lethal in a ΔholD ssb-duplicated strain [16]. The amount of DinB is expected to be similar in holD trkA, where DinB is 5- to 8-fold over-expressed owing to SOS-induction, and in holD trkA lexAind [pGB-DinB], where it is not SOS-induced but expressed from a 8–10 copy pSC101 vector [16, 36]. pGB2-DinB could be introduced by transformation in the ΔholD ΔtrkA lexAind mutant, in which SOS is inactivated, (Fig 7), however it slightly slowed down growth, as cells harboring pGB2-DinB formed smaller colonies than those harboring the vector pGB2 or the control plasmid pGB-DinBΔC5. In the ΔholD ΔtrkA mutant, where the SOS response is induced, pGB2 and pGB-DinBΔC5 could be introduced while pGB-DinB could not (Fig 7). We conclude that the ΔtrkA mutation restores growth by preventing SOS-induced DinB proteins from destabilizing the HolD-less Pol III HE, but ΔholD ΔtrkA [pGB-DinB] cells remain sensitive to the large excess of DinB resulting from the combination of plasmid-borne expression and SOS induction.
Fig 7. The holD trkA mutant is resistant to an increased level of DinB, but less than wild-type.
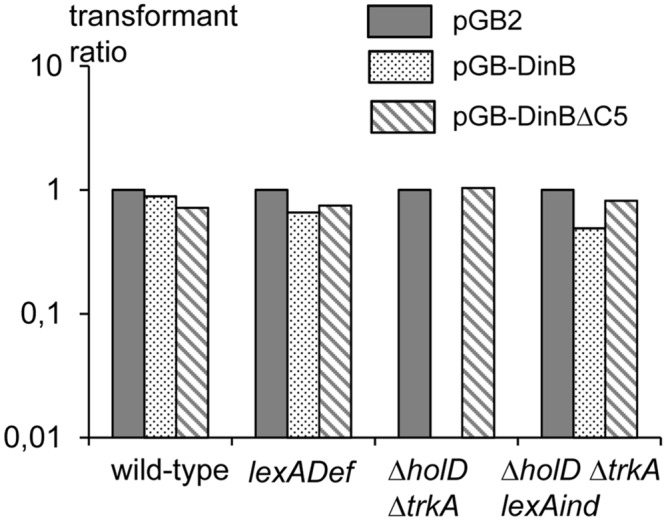
Wild-type, (JJC6534), lexADef (JJC6904), holD ΔtrkA::cm (JJC6927) and holD ΔtrkA::cm lexAind (JJC7004) strains were transformed with plasmids pGB2 (plain bars), pGB-DinB (dotted bars) and pGB-DinBΔC5 (hatched bars). Transformants were selected on M9 spc plates. For each experiment, the number of pGB-DinB and pGB-DinBΔC5 transformants was normalized to the number of pGB2 transformants and the averages of three independent experiments are shown. Colonies obtained with pGB-DinB in the holD trkA lexAind mutant were smaller than pGB2 and pGB-DinBΔC5 colonies in the same strain, and than pGB-DinB colonies in wild-type and LexADef strains.
trkA and the ssb gene duplication are partially additive
The ΔholD mutant is rescued by a duplication of the ssb gene, and we proposed that this duplication allows a modification of the SSB-binding mode, from 65 nucleotides to 35 nucleotides bound by an SSB tetramer, which stabilizes HolD-less Pol III on DNA [16]. Low salts favor the (SSB)35 DNA binding mode in vitro ([37] and references therein), and to test whether the decrease in intracellular concentration of K+ in trk mutants suppressed the ΔholD mutant defects through a modification of the SSB binding mode, the ssb duplication was introduced in the ΔholD ΔtrkA mutant. The ssb gene duplication showed an additive effect with ΔtrkA mutation, allowing growth of the ΔholD ΔtrkA argE::ssb triple mutant on M9 at 42°C (Fig 6). Rescue of the ΔholD argE::ssb mutants by ΔtrkA was only efficient on 22 mM K+ and was not observed at low or high K+ concentrations, as expected for the role of intracellular K+ concentration in this phenomenon (S5 Fig). The additive effects of ssb gene duplication and trkA inactivation on ΔholD viability at 42°C suggests that ΔtrkA does not act only by modifying the SSB binding mode. In fact, variations of temperature from 25°C to 37°C only modestly affect the SSB binding mode in vitro [38], 42°C was not tested), which disfavors the hypothesis that in vivo the ssb gene duplication promotes the (SSB)35 binding mode at 37°C and not at 42°C. If indeed in strains carrying the ssb duplication SSB binds DNA in the 35 base-pair mode at 42°C as at 37°C, the lethality of the holD argE::ssb mutant at 42°C results from a lack of stabilization of HolD-less Pol III by (SSB)35 at this temperature, and, in turn, the viability of the holD argE::ssb trkA triple mutant at 42°C suggests an effect of the trkA mutation on Pol III stability. However, we cannot exclude that trk mutations could promote a shift to the (SSB)35 at 30°C and 37°C on their own, and at 42°C in cells that carry the ssb gene duplication. Because these two modes of action are not exclusive and may have cumulative effects, the trkA mutation could stabilize Pol III both by enhancing directly its binding to DNA and by promoting the replication-favorable (SSB)35 binding mode, as discussed below.
Discussion
In this study, we show that ΔholD and ΔholC mutants are viable when K+ import is affected. trk inactivation does not improve ΔholD viability by affecting SOS induction. We propose that decreasing K+ import improves electrostatic interactions between subunits in the defective Pol III HE and/or between Pol III and DNA, and may in addition stabilize Pol III on DNA indirectly by favoring the (SSB)35 binding mode, the combination of all these effects allowing chromosome replication by a defective Pol III HE and restoring viability.
HolD plays a crucial role in strengthening DNA Pol III HE binding on DNA
The finding that decreasing the intracellular K+ concentration rescues the ΔholD mutant is in line with the idea that ψ (HolD) plays an important role in vivo in the stabilization of Pol III HE on DNA during replication. In vitro, ψ increases the affinity of τ for δδ’ in the core loading clamp pentameric complex, stabilizes the ATP-activated clamp loader complex conformational state, and increases the affinity of the clamp loader complex for the primer-template junction, thus favoring clamp loading [10–12]. Therefore ψ improves both protein-protein and protein-DNA interactions. The association of Pol III HE with DNA is intrinsically salt-dependent and strongly depends on the anion used for the reaction, with glutamate, the most physiological anion, protecting against high salt destabilizing effects [39]. Our results show that an intrinsically unstable Pol III HE can be stabilized by decreasing intracellular K+ concentration. However, our results argue that clamp loading during lagging-strand synthesis is still defective in the ΔholD ΔtrkA mutant, since the SOS response remains highly induced, and RNA primers remain sensitive to an excess of RNaseH. Furthermore, the inactivation of trk improves the viability of the ΔholC mutant, although in vitro clamp-loader activity is mainly stimulated by ψ alone and the presence of χ only weakly affects the reaction [11]. Therefore, we propose that lowering K+ import restores the viability of the ΔholD mutant mainly by improving leading-strand replication, by limiting replication arrest or Pol III HE disassembly after arrest, or by facilitating replication restart.
A duplication of the ssb gene also rescues the holD mutant, and we hypothesized that this rescue results from the promotion of the (SSB)35 binding mode by a two-fold increase in intracellular SSB protein concentration. Because in vitro a shift from the (SSB)65 to the (SSB)35 binding mode can be promoted by increasing SSB concentration or decreasing salt concentration [37], it is conceivable that the 12–17% decrease in intracellular K+ concentration observed in trk mutants promotes the formation of (SSB)35, which, in turn, stabilizes the HolD-less Pol III on DNA. In conclusion, the trkA mutation could allow chromosome replication either by a direct effect on Pol III, if affecting potassium import increases electrostatic interactions between Pol III subunits and/or between Pol III and DNA, or indirectly, if it favors a shift to the SSB binding mode to (SSB)35. These two models are not exclusive and the trk mutation could affect Pol III stability on DNA by cumulative effects of a direct and an SSB-mediated stabilization. Furthermore, the observation that the difference in K+ intracellular concentration between wild-type and trkA mutants is only 12–17% supports the idea that viability of holD trkA mutant might result from a combination of potassium concentration effects.
Inhibition of SOS induction by a lexAind or a recF mutation restores full viability to ΔholD ΔtrkA cells, including at high temperatures. This observation suggests that the remaining growth defects of ΔholD ΔtrkA cells are caused by SOS-induced polymerases and that the combination of a more stable Pol III and less abundant Pol II and DinB competitors is sufficient to restore full viability. Altogether, we conclude that electrostatic interactions are crucial for replisome stability in vivo and can be improved beyond the wild-type level by decreasing K+ import.
Effects of K+ on protein-nucleic acid interactions
K+ glutamate is the natural solute in E. coli, and is the major intracellular ionic osmolyte [20]. K+ intracellular concentration is regulated after hyper- or hypo-osmotic shocks by changes in the amount of water and by the action of several K+ import and efflux systems. In spite of a strong effect of salt concentration on protein-DNA interactions in vitro, K+ intracellular concentration can largely increase in vivo without affecting lac operator-repressor or phage λ RNA polymerase-promoter interactions [20]. Following a hyper-osmotic shock only some specific promoters are affected by the increased intracellular concentration of K+ glutamate: genes involved in osmotic protection are induced and ribosomal transcription is decreased [40]; effects of K+ transporters on the virulence of pathogenic bacteria were also reported [41, 42]. To account for the lack of effect of hyper-osmotic conditions on operator-repressor binding and promoter activity, it was proposed that increased intracellular K+ concentrations trigger a decrease in free cytoplasmic water, which enhances molecular crowding and thereby compensates for the destabilizing effect of the original K+ concentration increase [43–45]. Here the use of a mutant where the replication complex is intrinsically unstable on DNA allows us to show that protein-DNA interactions and possibly protein-protein interactions can be increased by lowering K+ import below the physiological wild-type level. We would like to propose the following hypotheses to account for the strong effect on viability in spite of a relatively weak K+ concentration decrease (i) either the replication machinery is highly sensitive to weak variations of intracellular K+ concentrations, for example because the effects on several replication proteins such as Pol III and SSB are cumulative as discussed above, (ii) or the 80 mM decrease in total K+ concentration that we observe in the holD trk mutants compared to wild-type affects only K+ ions free for exchange, leading to a 30% reduction of the potassium ions that affect DNA-protein interactions effectively, (iii) or trk and kdp mutations exert secondary effects on ions other than K+ that also control protein-DNA interactions. Strikingly, stabilization of Pol III HE on DNA, reflected by the ΔholD ΔtrkA mutant viability, is particularly efficient in LB. The effects of LB are likely the result of a combination of multiple factors, including the presence of molecules such as glycine betaine and glutamate that stabilize protein-DNA complexes [39, 46]. However, only ΔholD mutants that lack the trk import system formed colonies on LB at 42°C, and not ΔholD mutants that lack SOS induction or carry an ssb gene duplication. Therefore, whatever the compounds that favor growth in LB, they are active in the holD mutant when combined with a limited K+ import.
Replication fork arrest is a recognized source of genome rearrangements in all organisms, and any replication defect can have severe consequences [47–50]. The identification of factors that improve replication fork stability in perturbed conditions is therefore crucial. Furthermore, theoretically mutations that affect protein-protein and protein-nucleic acid interactions in processes other than replication could also be suppressed by limiting K+ import. Our work underlines the influence of chemical intracellular composition on essential processes.
Materials and Methods
Strains, plasmids and media
Strains, plasmids and oligonucleotides used in this work are described in S1 Table. Genes were inactivated by recombineering as described in [51] using DY330 [52]. Mutations were transferred by P1 transduction. Antibiotics were used at the following concentrations: kanamycin (Kan) 50 μg/ml, chloramphenicol (Cm) 20 μg/ml, tetracycline (Tet) 15 μg/ml, ampicillin (Ap) 100 μg/ml, spectinomycin (Spc) 60 μg/ml. All minimum media used in this work contain 0.4% glucose, 0.2% casamino acids and 1 mg/L thiamine, except M9 tryptone medium which contains 0.4% tryptone instead of casamino acids. LB broth (Miller) is from Sigma, yeast extract, tryptone and casamino acids are from Difco. M9 is Na2HPO4 42 mM, KH2PO4 22 mM, NaCl 8.5 mM, NH4Cl 18.7 mM, MgSO4 1 mM, CaCl2 0.1 mM [53]; MMA is K2HPO4 60.3 mM, KH2PO4 33 mM, (NH4)2SO4 7.6 mM, Na Citrate 1.7 mM, MgSO4 1 mM [53]; MK115 is K2HPO4 46 mM, KH2PO4 23 mM, (NH4)2SO4 8 mM, Na Citrate 1 mM, MgSO4 0.4 mM, FeSO4 6 μM [29]; MK0 is Na2HPO4 46 mM, NaH2PO4 23 mM, (NH4)2SO4 8 mM, Mg SO4 0.4 mM, FeSO4 6 μM. Different amounts of MMA 2X or MK115 2X were added to MK0 for MK0.2, MK1 and MK22, to adjust to 0.2, 1 and 22 mM K+ respectively [29]. Strains containing pAM-holD were routinely grown in M9 containing 500 mM IPTG and 60 μg/ml spectinomycin at 37°C. pAM-holD (or pAM-holCD) were cured prior to each experiment by growing cells in the absence of IPTG, and plasmid-less colonies were isolated on M9 [16, 17]. We determined that less than 10% cells in the culture contain pAM-holD and less than 1% had acquired a suppressor mutation. Because of the high frequency of appearance of suppressor mutations, all new holD derivatives were constructed in the presence of pAM-holD. All mutations introduced by P1 transduction were verified by PCR, and all mutations constructed by recombineering were verified by PCR and sequencing. lexAInd and recF mutations were checked by measuring UV sensitivity.
Viability measurement
For spot assays, plasmid-less colonies formed in three days on M9 at 30°C were suspended in M9 or MK0 salts. Serial 10-fold dilutions were performed and 5 μl of dilutions 10−1 to 10−5 were spotted on different media. Pictures of LB plates incubated at 37°C and 42°C were taken after 24 h incubation, for all holD mutants, pictures of LB plates incubated at 30°C and of minimum medium plates were taken after 2 days; for HolD+ strains for pictures of minimal medium plates incubated at 37°C were taken after 24 h incubation. All strains were tested at least three times independently. For growth curves, cultures of wild-type (JJC1392), trkA (JJC6800) and holD trkA [pAM-holD] (JJC6898) strains were grown overnight at 30°C in LB, M9, MK1, MK115 medium. Cells were diluted to O.D. 0.002 in the same medium and further grown at 37°C for 7 hours. This protocol was chosen because it allows overnight cultures and the subsequent growth curves to be performed in the same medium, without medium shift, and a direct comparison with the same protocol of viable (holD trkA in M9 and LB) and lethal (holD trkA in MK1 and MK115) growth conditions. The number of colony forming units per ml of culture (cfu/ml) was determined by plating appropriate dilutions on M9 and incubating plates at 30°C. The average percentage of plasmid-less cells, determined by plating appropriate dilutions on M9 with spectinomycin and IPTG, was independent of the medium, in average 74% after overnight propagation and 96% at the end of the growth curve. To verify that the holD trkA cultures did not acquire additional suppressor mutations during growth in M9, appropriate dilutions were also plated at 42°C, to check that cells were thermosensitive as expected.
Genome sequencing
Chromosomal DNA was extracted using Sigma GenElute bacterial genomic DNA kit. 5 μg of DNA were used to generate a genomic library according to Illumina's protocol. The libraries and the sequencing were performed by the High-throughput Sequencing facility of the I2BC (http://www.i2bc.paris-saclay.fr/spip.php?article399&lang=en, CNRS, Gif-sur-Yvette, France). Genomic DNA libraries were made with the ‘Nextera DNA library preparation kit’ (Illumina) following the manufacturer’s recommendations. Library quality was assessed on an Agilent Bioanalyzer 2100, using an Agilent High Sensitivity DNA Kit (Agilent technologies). Libraries were pooled in equimolar proportions. Paired-end 2x250 bp reads were generated on an Illumina MiSeq instrument, using a MiSeq Reagent kit V2 (500 cycles) (Illumina), with an expected depth of 217X. Reads from mutant genome were aligned on the Escherichia coli K12 MG1655 genome using Illumina's package CASAVA 1.8.2. The point mutation and the small indels were detected also using Illumina's package CASAVA 1.8.2 and the large indels with profil visualisation and Blast (Basic Local Alignment Search Tool).
Measurements of intracellular potassium concentration
Cells grown in M9 at 37°C until OD650 = 0.4 were cooled in ice, harvested by centrifugation, and washed three times in cold hyper-tonic medium: 1.mM Tris-Cl (pH 8), 1 mM MgSO4, and 0.4 M glucose [31]. Pellets were dried overnight at 56°C. Dry pellets were weighted, digested in 2 ml of HNO3 >68% (20 min at 80°C and 1h at 120°C), diluted 50 fold in H2O, and K+ was measured by flame spectrophotometry using a Varian AA240FS spectrophotometer and a range of 0.1 to 5 mg/L K+ standard solutions.
β-galactosidase assays
β-galactosidase assays for measures of SOS induction were performed as described previously [16, 53]. Since isolated JJC6545 and JJC7058 colonies could not be cultivated owing to the growth advantage of suppressor mutations, pAM-holD containing clones were grown overnight in M9 lacking IPTG and diluted 50 fold in M9 for the experiment. Cultures were tested for the loss of pAM-holD and for containing less than 1% suppressor mutations.
Supporting Information
Top and bottom lines show the wild-type trkA gene in the region of the deletion. Middle line is the sequence of trkAΔ84. The 9 pb microhomology is in bold, the upstream sequence is in blue and the downstream sequence in purple. Numbers refer to the position in trkA, when the A in the ATG is numbered 1.
(PDF)
Serial dilutions used in Fig 2 were plated on MM containing different concentrations of K+ and incubated at the indicated temperature.
(PDF)
Serial 10-fold dilutions were made and 5μl drops of each dilution were spotted on minimal medium M9, LB, or MK plates containing the indicated potassium concentration. A. Δkup mutation does not affect the growth of ΔholD or ΔholD ΔtrkA mutants. Plates were incubated for two days at the indicated temperature. holD, JJC6869; holD kup, JJC7001; holD trkA, JJC6898; holD trkA kup, JJC7002. B. The presence of casaminoacids in minimal medium does not affect viability In this work all minimal medium plates contain 0.2% casaminoacids. The presence of casaminoacids increased growth rates but did not affect viability. Strains are as in Fig 4: wild-type, JJC1392; holD trkE sup, JJC6389; strains used to get plasmid-less colonies: holD, JJC6869; holD trkA, JJC6898; holD trkAΔ84, JJC6969. Plates were incubated at 42°C or 37°C for two days or at 30°C for three days. Note that in this particular experiment the holD mutant colony that was used contained a higher than usual sub-population of suppressors allowing growth at 30°C (compare with S3A Fig). Such jackpots of suppressors were observed in less than 10% of plasmid-less holD colonies.
(PDF)
Serial dilutions used in Fig 5 were plated on MK medium containing different concentrations of K+ and incubated at the indicated temperature.
(PDF)
Serial dilutions used in Fig 6B were plated on MK medium containing different concentrations of K+ and incubated at the indicated temperature.
(PDF)
(PDF)
Acknowledgments
We are very grateful to Dr. Sébastien Thomine for his invaluable help in measuring intracellular potassium concentrations. We thank Pr Wolfgang Epstein (University of Chicago) for his advice and his helpful reading of the manuscript, Dr. Timothy Lohman (Washington University, Saint Louis) for communication of results prior to publication, and the four anonymous referees for very useful suggestions. We are also grateful to Dr. Christopher Herbert (I2BC, Gif, France) for helpful reading of the manuscript, and to Dr. J. Gowrishankar (CDFD, Hyderabad, India) for helpful discussion.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the Agence Nationale de la Recherche (FR) ANR 11 BSV5 006 01. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.McHenry CS. DNA replicases from a bacterial perspective. Annu Rev Biochem. 2011;80:403–36. 10.1146/annurev-biochem-061208-091655 [DOI] [PubMed] [Google Scholar]
- 2.Reyes-Lamothe R, Sherratt DJ, Leake MC. Stoichiometry and architecture of active DNA replication machinery in Escherichia coli. Science. 2010;328(5977):498–501. 10.1126/science.1185757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Georgescu RE, Kurth I, O'Donnell ME. Single-molecule studies reveal the function of a third polymerase in the replisome. Nat Struct Mol Biol. 2012;19(1):113–6. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dohrmann PR, Correa R, Frisch RL, Rosenberg SM, McHenry CS. The DNA polymerase III holoenzyme contains gamma and is not a trimeric polymerase. Nucleic Acids Res. 2016;44(3):1285–97. 10.1093/nar/gkv1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Indiani C, O'Donnell M. The replication clamp-loading machine at work in the three domains of life. Nat Rev Mol Cell Biol. 2006;7(10):751–61. . [DOI] [PubMed] [Google Scholar]
- 6.Glover BP, McHenry CS. The chi psi subunits of DNA polymerase III holoenzyme bind to single-stranded DNA-binding protein (SSB) and facilitate replication of an SSB-coated template. J Biol Chem. 1998;273(36):23476–84. [DOI] [PubMed] [Google Scholar]
- 7.Kelman Z, Yuzhakov A, Andjelkovic J, ODonnell M. Devoted to the lagging strand—the chi subunit of DNA polymerase III holoenzyme contacts SSB to promote processive elongation and sliding clamp assembly. EMBO J. 1998;17(8):2436–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gulbis JM, Kazmirski SL, Finkelstein J, Kelman Z, O'Donnell M, Kuriyan J. Crystal structure of the chi:psi sub-assembly of the Escherichia coli DNA polymerase clamp-loader complex. Eur J Biochem. 2004;271(2):439–49. . [DOI] [PubMed] [Google Scholar]
- 9.Marceau AH, Bahng S, Massoni SC, George NP, Sandler SJ, Marians KJ, et al. Structure of the SSB-DNA polymerase III interface and its role in DNA replication. Embo J. 2011;30(20):4236–47. 10.1038/emboj.2011.305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olson MW, Dallmann HG, McHenry CS. DnaX complex of Escherichia coli DNA polymerase III holoenzyme. The chi psi complex functions by increasing the affinity of tau and gamma for delta.delta' to a physiologically relevant range. J Biol Chem. 1995;270(49):29570–7. . [PubMed] [Google Scholar]
- 11.Anderson SG, Williams CR, O'Donnell M, Bloom LB. A function for the psi subunit in loading the Escherichia coli DNA polymerase sliding clamp. J Biol Chem. 2007;282(10):7035–45. . [DOI] [PubMed] [Google Scholar]
- 12.Simonetta KR, Kazmirski SL, Goedken ER, Cantor AJ, Kelch BA, McNally R, et al. The mechanism of ATP-dependent primer-template recognition by a clamp loader complex. Cell. 2009;137(4):659–71. 10.1016/j.cell.2009.03.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao DX, McHenry CS. tau binds and organizes Escherichia coli replication proteins through distinct domains—Domain III, shared by gamma and tau binds delta delta ' and x psi. J Biol Chem. 2001;276(6):4447–53. [DOI] [PubMed] [Google Scholar]
- 14.Slater SC, Lifsics MR, Odonnell M, Maurer R. HolE, the Gene Coding for the theta Subunit of DNA Polymerase III of Escherichia Coli—Characterization of HolE Mutant and Comparison with a DnaQ (epsilon-Subunit) Mutant. J Bacteriol. 1994;176(3):815–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saveson CJ, Lovett ST. Enhanced deletion formation by aberrant DNA replication in Escherichia coli. Genetics. 1997;146(2):457–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duigou S, Silvain M, Viguera E, Michel B. ssb gene duplication restores the viability of DeltaholC and DeltaholD Escherichia coli mutants. PLoS Genet. 2014;10(10):e1004719 10.1371/journal.pgen.1004719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Viguera E, Petranovic M, Zahradka D, Germain K, Ehrlich DS, Michel B. Lethality of bypass polymerases in Escherichia coli cells with a defective clamp loader complex of DNA polymerase III. Mol Microbiol. 2003;50(1):193–204. . [DOI] [PubMed] [Google Scholar]
- 18.Rhoads DB, Waters FB, Epstein W. Cation transport in Escherichia coli. VIII. Potassium transport mutants. J Gen Physiol. 1976;67(3):325–41. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cayley S, Lewis BA, Guttman HJ, Record MT Jr. Characterization of the cytoplasm of Escherichia coli K-12 as a function of external osmolarity. Implications for protein-DNA interactions in vivo. J Mol Biol. 1991;222(2):281–300. . [DOI] [PubMed] [Google Scholar]
- 20.Richey B, Cayley DS, Mossing MC, Kolka C, Anderson CF, Farrar TC, et al. Variability of the intracellular ionic environment of Escherichia coli. Differences between in vitro and in vivo effects of ion concentrations on protein-DNA interactions and gene expression. J Biol Chem. 1987;262(15):7157–64. . [PubMed] [Google Scholar]
- 21.McLaggan D, Naprstek J, Buurman ET, Epstein W. Interdependence of K+ and glutamate accumulation during osmotic adaptation of Escherichia coli. J Biol Chem. 1994;269(3):1911–7. . [PubMed] [Google Scholar]
- 22.Dosch DC, Helmer GL, Sutton SH, Salvacion FF, Epstein W. Genetic analysis of potassium transport loci in Escherichia coli: evidence for three constitutive systems mediating uptake potassium. J Bacteriol. 1991;173(2):687–96. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Epstein W, Buurman E, McLaggan D, Naprstek J. Multiple mechanisms, roles and controls of K+ transport in Escherichia coli. Biochem Soc Trans. 1993;21(4):1006–10. . [DOI] [PubMed] [Google Scholar]
- 24.Silver S. Transport of inorganic cations p 1091–1102 In Neidhardt F C, et al. (ed), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed American Society for Microbiology, Washington, DC: 1996. [Google Scholar]
- 25.Harms C, Domoto Y, Celik C, Rahe E, Stumpe S, Schmid R, et al. Identification of the ABC protein SapD as the subunit that confers ATP dependence to the K+-uptake systems Trk(H) and Trk(G) from Escherichia coli K-12. Microbiology. 2001;147(Pt 11):2991–3003. . [DOI] [PubMed] [Google Scholar]
- 26.Bossemeyer D, Schlosser A, Bakker EP. Specific cesium transport via the Escherichia coli Kup (TrkD) K+ uptake system. J Bacteriol. 1989;171(4):2219–21. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schleyer M, Bakker EP. Nucleotide sequence and 3'-end deletion studies indicate that the K(+)-uptake protein kup from Escherichia coli is composed of a hydrophobic core linked to a large and partially essential hydrophilic C terminus. J Bacteriol. 1993;175(21):6925–31. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato Y, Nanatani K, Hamamoto S, Shimizu M, Takahashi M, Tabuchi-Kobayashi M, et al. Defining membrane spanning domains and crucial membrane-localized acidic amino acid residues for K(+) transport of a Kup/HAK/KT-type Escherichia coli potassium transporter. J Biochem. 2014;155(5):315–23. 10.1093/jb/mvu007 [DOI] [PubMed] [Google Scholar]
- 29.Epstein W, Kim BS. Potassium transport loci in Escherichia coli K-12. J Bacteriol. 1971;108(2):639–44. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laermann V, Cudic E, Kipschull K, Zimmann P, Altendorf K. The sensor kinase KdpD of Escherichia coli senses external K+. Mol Microbiol. 2013;88(6):1194–204. 10.1111/mmi.12251 [DOI] [PubMed] [Google Scholar]
- 31.Epstein W. The KdpD Sensor Kinase of Escherichia coli Responds to Several Distinct Signals To Turn on Expression of the Kdp Transport System. J Bacteriol. 2016;198(2):212–20. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buurman ET, McLaggan D, Naprstek J, Epstein W. Multiple paths for nonphysiological transport of K+ in Escherichia coli. J Bacteriol. 2004;186(13):4238–45. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flores MJ, Bierne H, Ehrlich SD, Michel B. Impairment of lagging strand synthesis triggers the formation of a RuvABC substrate at replication forks. Embo J. 2001;20(3):619–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Epstein W, Davies M. Potassium-dependant mutants of Escherichia coli K-12. J Bacteriol. 1970;101(3):836–43. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lenne-Samuel N, Wagner J, Etienne H, Fuchs RP. The processivity factor beta controls DNA polymerase IV traffic during spontaneous mutagenesis and translesion synthesis in vivo. EMBO Rep. 2002;3(1):45–9. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heltzel JM, Maul RW, Wolff DW, Sutton MD. Escherichia coli DNA polymerase IV (Pol IV), but not Pol II, dynamically switches with a stalled Pol III* replicase. J Bacteriol. 2012;194(14):3589–600. 10.1128/JB.00520-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roy R, Kozlov AG, Lohman TM, Ha T. Dynamic structural rearrangements between DNA binding modes of E. coli SSB protein. J Mol Biol. 2007;369(5):1244–57. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bujalowski W, Lohman TM. Escherichia coli single-strand binding protein forms multiple, distinct complexes with single-stranded DNA. Biochemistry. 1986;25(24):7799–802. . [DOI] [PubMed] [Google Scholar]
- 39.Griep MA, McHenry CS. Glutamate overcomes the salt inhibition of DNA polymerase III holoenzyme. J Biol Chem. 1989;264(19):11294–301. . [PubMed] [Google Scholar]
- 40.Gralla JD, Vargas DR. Potassium glutamate as a transcriptional inhibitor during bacterial osmoregulation. Embo J. 2006;25(7):1515–21. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Y, Ho KK, Su J, Gong H, Chang AC, Lu S. Potassium transport of Salmonella is important for type III secretion and pathogenesis. Microbiology. 2013;159(Pt 8):1705–19. 10.1099/mic.0.068700-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valente RS, Xavier KB. The Trk Potassium Transporter Is Required for RsmB-Mediated Activation of Virulence in the Phytopathogen Pectobacterium wasabiae. J Bacteriol. 2015;198(2):248–55. 10.1128/JB.00569-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Record MT Jr., Courtenay ES, Cayley DS, Guttman HJ. Responses of E. coli to osmotic stress: large changes in amounts of cytoplasmic solutes and water. Trends Biochem Sci. 1998;23(4):143–8. . [DOI] [PubMed] [Google Scholar]
- 44.Record MT Jr., Courtenay ES, Cayley S, Guttman HJ. Biophysical compensation mechanisms buffering E. coli protein-nucleic acid interactions against changing environments. Trends Biochem Sci. 1998;23(5):190–4. . [DOI] [PubMed] [Google Scholar]
- 45.Wood JM. Bacterial osmoregulation: a paradigm for the study of cellular homeostasis. Annu Rev Microbiol. 2011;65:215–38. 10.1146/annurev-micro-090110-102815 [DOI] [PubMed] [Google Scholar]
- 46.Leirmo S, Harrison C, Cayley DS, Burgess RR, Record MT Jr. Replacement of potassium chloride by potassium glutamate dramatically enhances protein-DNA interactions in vitro. Biochemistry. 1987;26(8):2095–101. . [DOI] [PubMed] [Google Scholar]
- 47.Michel B, Boubakri H, Baharoglu Z, Lemasson M, Lestini R. Recombination proteins and rescue of arrested replication forks. DNA Repair (Amst). 2007;6(7):967–80. . [DOI] [PubMed] [Google Scholar]
- 48.Lambert S, Froget B, Carr AM. Arrested replication fork processing: interplay between checkpoints and recombination. DNA Repair (Amst). 2007;6(7):1042–61. . [DOI] [PubMed] [Google Scholar]
- 49.Tourriere H, Pasero P. Maintenance of fork integrity at damaged DNA and natural pause sites. DNA Repair (Amst). 2007;6(7):900–13. . [DOI] [PubMed] [Google Scholar]
- 50.Jacobs JZ, Rosado-Lugo JD, Cranz-Mileva S, Ciccaglione KM, Tournier V, Zaratiegui M. Arrested replication forks guide retrotransposon integration. Science. 2015;349(6255):1549–53. 10.1126/science.aaa3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97(12):6640–5. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A. 2000;97(11):5978–83. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miller JH. A short course in bacterial genetic: Cold Spring Harbor, New York: Cold Spring Harbor Press; 1992. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Top and bottom lines show the wild-type trkA gene in the region of the deletion. Middle line is the sequence of trkAΔ84. The 9 pb microhomology is in bold, the upstream sequence is in blue and the downstream sequence in purple. Numbers refer to the position in trkA, when the A in the ATG is numbered 1.
(PDF)
Serial dilutions used in Fig 2 were plated on MM containing different concentrations of K+ and incubated at the indicated temperature.
(PDF)
Serial 10-fold dilutions were made and 5μl drops of each dilution were spotted on minimal medium M9, LB, or MK plates containing the indicated potassium concentration. A. Δkup mutation does not affect the growth of ΔholD or ΔholD ΔtrkA mutants. Plates were incubated for two days at the indicated temperature. holD, JJC6869; holD kup, JJC7001; holD trkA, JJC6898; holD trkA kup, JJC7002. B. The presence of casaminoacids in minimal medium does not affect viability In this work all minimal medium plates contain 0.2% casaminoacids. The presence of casaminoacids increased growth rates but did not affect viability. Strains are as in Fig 4: wild-type, JJC1392; holD trkE sup, JJC6389; strains used to get plasmid-less colonies: holD, JJC6869; holD trkA, JJC6898; holD trkAΔ84, JJC6969. Plates were incubated at 42°C or 37°C for two days or at 30°C for three days. Note that in this particular experiment the holD mutant colony that was used contained a higher than usual sub-population of suppressors allowing growth at 30°C (compare with S3A Fig). Such jackpots of suppressors were observed in less than 10% of plasmid-less holD colonies.
(PDF)
Serial dilutions used in Fig 5 were plated on MK medium containing different concentrations of K+ and incubated at the indicated temperature.
(PDF)
Serial dilutions used in Fig 6B were plated on MK medium containing different concentrations of K+ and incubated at the indicated temperature.
(PDF)
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.