

Abstract
BACKGROUND
Higher-than-normal levels of circulating triglycerides are a risk factor for ischemic cardiovascular disease. Activation of lipoprotein lipase, an enzyme that is inhibited by angiopoietin-like 4 (ANGPTL4), has been shown to reduce levels of circulating triglycerides.
METHODS
We sequenced the exons of ANGPTL4 in samples obtain from 42,930 participants of predominantly European ancestry in the DiscovEHR human genetics study. We performed tests of association between lipid levels and the missense E40K variant (which has been associated with reduced plasma triglyceride levels) and other inactivating mutations. We then tested for associations between coronary artery disease and the E40K variant and other inactivating mutations in 10,552 participants with coronary artery disease and 29,223 controls. We also tested the effect of a human monoclonal antibody against ANGPTL4 on lipid levels in mice and monkeys.
RESULTS
We identified 1661 heterozygotes and 17 homozygotes for the E40K variant and 75 participants who had 13 other monoallelic inactivating mutations in ANGPTL4. The levels of triglycerides were 13% lower and the levels of high-density lipoprotein (HDL) cholesterol were 7% higher among carriers of the E40K variant than among noncarriers. Carriers of the E40K variant were also significantly less likely than noncarriers to have coronary artery disease (odds ratio, 0.81; 95% confidence interval, 0.70 to 0.92; P = 0.002). K40 homozygotes had markedly lower levels of triglycerides and higher levels of HDL cholesterol than did heterozygotes. Carriers of other inactivating mutations also had lower triglyceride levels and higher HDL cholesterol levels and were less likely to have coronary artery disease than were noncarriers. Monoclonal antibody inhibition of Angptl4 in mice and monkeys reduced triglyceride levels.
CONCLUSIONS
Carriers of E40K and other inactivating mutations in ANGPTL4 had lower levels of triglycerides and a lower risk of coronary artery disease than did noncarriers. The inhibition of Angptl4 in mice and monkeys also resulted in corresponding reductions in these values. (Funded by Regeneron Pharmaceuticals.)
The level of serum triglycerides is, in part, heritable, and elevated levels are associated with a risk of ischemic cardiovascular disease.1–3 Mendelian randomization studies of genetically determined triglyceride levels have suggested that this association is causal.4 Two lines of genetic evidence have further established a causal role for serum triglycerides in the risk of cardiovascular disease. First, inactivating mutations in the gene encoding apolipoprotein C3 (APOC3), a component of remnant particles, were reported to be associated with decreased serum triglyceride levels, a decreased burden of subclinical atherosclerosis, and a reduced risk of ischemic cardiovascular disease, which suggests that therapeutic modulation of APOC3 may reduce the risk of cardiovascular disease.5–7 Second, rare nonsynonymous variants in apolipoprotein A-V have been associated with increased serum triglyceride levels and a doubling of the risk of early-onset myocardial infarction.8
Lipoprotein lipase (LPL) is the primary enzyme that hydrolyzes lipoprotein triglycerides, releasing free fatty acids for utilization by and clearance from tissues.9 Truncating mutations that increase LPL activity decrease serum triglyceride levels and the risk of cardiovascular disease, whereas mutations that decrease LPL function have been shown to increase serum triglyceride levels.10,11 Among the endogenous modulators of LPL are angiopoietin-like 3 (ANGPTL3) and angiopoietin-like 4 (ANGPTL4), two related secreted proteins that inhibit LPL in vitro and in vivo and modulate the uptake of free fatty acids in fasting and fed states.12–15 Angptl4-deficient mice have better lipid metabolism and smaller atherosclerotic lesions than mice without this deficiency.16 Naturally occurring human mutations in ANGPTL4 that inactivate its protein product may provide a bridge between these observations in animal models and the possibility of a similar effect in humans and illuminate the potential clinical effects of therapeutic antagonism of ANGPTL4.
The amino acid–altering (missense) E40K variant in ANGPTL4 has been associated with decreased levels of triglycerides and increased levels of high-density lipoprotein (HDL) cholesterol.17 There have been conflicting reports regarding the association of this variant with the risk of coronary artery disease, with one report showing an increased risk18 and another showing a decreased risk.19 In this study, we initially examined associations between the presence of the E40K variant and lipid levels and coronary artery disease. We sought additional allelic evidence for effects of genetic inactivation of ANGPTL4 by examining associations of novel rare, inactivating mutations in ANGPTL4 with lipid levels and coronary artery disease. Finally, we explored whether therapeutic antibody inhibition of Angptl4 had similar effects on lipid metabolism in murine and nonhuman primate models.
METHODS
STUDY OVERSIGHT
The human genetics studies were conducted as part of the DiscovEHR project of the Regeneron Genetics Center and the Geisinger Health System. The study was approved by the institutional review board at the Geisinger Health System. The Regeneron Genetics Center funded the collection of study samples, the generation of sequence data, and the analysis of clinical and sequence data. The first three and last three authors vouch for the accuracy and completeness of the data and all analyses. All participants provided written informed consent.
PARTICIPANTS, CLINICAL MEASUREMENTS, AND DISEASE DEFINITIONS
The population for the DiscovEHR human genetics study that we analyzed in this study included the first 42,930 enrollees of predominantly European ancestry from the MyCode Community Health Initiative in the Geisinger Health System. From 2007 through 2015, we recruited participants from outpatient primary care and specialty clinics and from the cardiac catheterization laboratory, as well as patients who were referred for bariatric or abdominal vascular surgery. We extracted clinical laboratory measurements, disease diagnosis codes from the International Classification of Diseases, Ninth Revision (ICD-9), information on medication history, and procedural codes from the electronic health records, which covered a median of 15 years of clinical care.
ICD-9–based diagnosis codes were collapsed into clinical disease categories. ICD-9–based diagnoses required one or more of the following: a problem-list entry of the diagnosis code, an inpatient hospitalization-discharge diagnosis code, or an encounter diagnosis code entered for two separate outpatient visits on separate calendar days. We developed additional phenotype definitions of coronary artery disease, hypertension, and type 2 diabetes to capture cases with greater specificity (Table 1). Controls without coronary artery disease were defined as participants who had no case criteria or single-encounter or problem-list diagnosis code indicating coronary artery disease.
Table 1.
Clinical Characteristics of the 42,930 Study Participants.*
| Characteristic | Value |
|---|---|
| Median age (IQR) — yr | 58 (48–68) |
| Female sex — no. (%) | 25,326 (59.0) |
| Median body-mass index (IQR)† | 31 (27–36) |
| Current smoker — no. (%) | 6,494 (15.1) |
| Medications — no. (%) | |
| Lipid-lowering | 16,156 (37.6) |
| Antihypertensive | 24,812 (57.8) |
| Hypoglycemic | 8,163 (19.0) |
| Medical history — no. (%) | |
| Coronary artery disease‡ | 10,552 (24.6) |
| Type 2 diabetes§ | 9,946 (23.2) |
| Hypertension¶ | 23,700 (55.2) |
IQR denotes interquartile range.
The body-mass index is the weight in kilograms divided by the square of the height in meters.
Participants were considered to have coronary artery disease if they had a history of coronary revascularization in the electronic health records or a history of the acute coronary syndrome, ischemic heart disease, or exertional angina with angiographic evidence of obstructive coronary atherosclerosis (>50% stenosis in at least one major epicardial vessel according to the catheterization report).
Participants were considered to have diabetes if they had a history of type 2 diabetes in the electronic health records, were taking an antidiabetic medication, or had a fasting glucose level of more than 126 mg per deciliter (7.0 mmol per liter) or a glycated hemoglobin level of more than 6.5%.
Participants were considered to have hypertension if they had a history of hypertension in the electronic health records, were taking an antihypertensive medication, or had a systolic blood pressure of more than 140 mm Hg or a diastolic blood pressure of more than 90 mm Hg.
We calculated median values for serially measured laboratory and anthropometric traits, including levels of total cholesterol, low-density lipoprotein (LDL) cholesterol, HDL cholesterol, and triglycerides, along with body-mass index, for all participants with two or more measurements in the electronic health records after the removal of values that were probably spurious (i.e., >3 SD from the median value among participants). Levels of total cholesterol and LDL cholesterol were adjusted for the use of lipid-altering medication by dividing by 0.8 and 0.7, respectively, to estimate pretreatment lipid values on the basis of the average reduction in total cholesterol and LDL cholesterol with the average statin dose.20 Levels of HDL cholesterol and triglycerides were not adjusted for the use of lipid-altering medication.
SEQENCING OF ANGPTL4
We extracted sequence data for ANGPTL4 from exome sequences generated and processed at the Regeneron Genetics Center using protocols described in the Methods section in the Supplementary Appendix, available with the full text of this article at NEJM.org. Sequence reads were aligned to the human reference build GRCh37.p13. Single-nucleotide variants (SNVs) and insertion– deletion (indel) sequence variants were identified with the use of the Genome Analysis Toolkit21,22 and annotated with the use of SnpEff.23 Inactivating mutations were defined as any of the following: SNVs leading to a premature stop codon, loss of a start codon, or loss of a stop codon; SNVs or indels disrupting canonical splice acceptor or donor dinucleotides; and indels with a shift in the open reading frame leading to the formation of a premature stop codon. Carriers of the E40K variant, which has been shown to block the inhibition of LPL by ANGPTL4,24 were also identified. All positions are reported on the basis of the reference sequence for Ensembl transcript ENST00000301455 (RefSeq messenger RNA sequence, NM_139314). We used Sanger sequencing to confirm the presence of certain inactivating mutations, especially indel variants (see the Methods section in the Supplementary Appendix).
ANTIBODY CHARACTERIZATION
The ANGPTL4-neutralizing, fully human monoclonal antibody (REGN1001) that was used in this study was derived with the use of Regeneron’s Velocimmune technology.25 Binding of REGN1001 human immunoglobulin G1 to human and monkey ANGPTL4 was measured by means of Biacore.26 Reversal by REGN1001 of ANGPTL4-induced inhibition of LPL activity was determined with the use of the Confluolip Continuous Fluorometric Lipase Test (Progen).26
INHIBITION OF ANGPTL4 IN MICE
We tested the effect of REGN1001 in male mice with a mixed genetic background (C578Bl/6NTac for 62.5% and 129S6/SvEvTac for 37.5%) that were homozygous for human ANGPTL4 and for ApoE deficiency. Mice were fed ad libitum with a high-fat Western diet (Research Diets, D12492; 60% fat by calories). Mice received subcutaneous injections of REGN1001 or a control antibody of the same isotype as REGN1001. Blood was collected for lipid measurements after a 4-hour fast.26 All procedures in animals were conducted in compliance with protocols approved by the institutional animal care and use committee of Regeneron Pharmaceuticals.
MONOCLONAL ANTIBODY INHIBITION OF ANGPTL4 IN OBESE MONKEYS
Five obese rhesus macaques (body weight, >5 kg; age, >10 years) that were being maintained on a high-fat diet (TAD Primate Diet, Lab Diet formulation 5L0P) received a single intravenous injection of REGN1001 on day 1 at a dose of 10 mg per kilogram of body weight. Serum samples were collected through day 35 and assessed for serum lipids. In a separate study, REGN1001 was administered in a single intravenous dose of 5 mg per kilogram once a week for 13 weeks to male and female cynomolgus monkeys (10 per group) that were placed on a high-fat diet (Western Primate Diet, 5S2T) before the initiation of the study. A separate control group received the same dose of vehicle control. Blood samples for lipid measurements were collected at baseline and at 4, 8, and 14 weeks of the study. Six animals from each group underwent necropsy 1 week after the last injection, whereas the remaining four animals were euthanized after a 15-week recovery period. Necropsies were performed to assess organ-specific effects of therapeutic ANGPTL4 inhibition.
STATISTICAL ANALYSIS
We used mixed linear models of association to test for associations between genotype and levels of total cholesterol, LDL cholesterol, log10 HDL cholesterol, and log10 triglycerides under an additive model (alleles coded 0, 1, and 2 for E40 homozygotes, E40/K40 heterozygotes, and K40 homozygotes, respectively). We included age, age squared, sex, and log10-transformed body-mass index as fixed-effects covariates and a genetic-relationship matrix, which captures population structure from ancestry and relatedness, as a random-effects covariate. We then tested for associations between the E40K variant (with coding as described) and the risk of coronary artery disease using the same set of covariates except for log10-transformed body-mass index. Odds ratios for coronary artery disease were estimated with the use of Firth’s penalized likelihood method of logistic regression27 after adjustment for age, age squared, sex, and the first four principal components of ancestry. We estimated Wald 95% confidence intervals for odds ratios using standard error estimates calculated from P values from the mixed linear models of association.
We next used the same statistical-testing framework to identify associations between novel inactivating mutations in ANGPTL4 found in our study, aggregated over the gene,28 and the above-mentioned traits. Alleles were coded 0 for non-carriers and 1 for carriers of inactivating mutations. As expected on the basis of the low allele frequencies, we did not observe any homozygotes for other inactivating mutations at any variant site. Exomewide quantile–quantile plots for single-marker and gene-based burden tests are provided in Figures S1 through S3 in the Supplementary Appendix.
An alpha level of 0.05 was considered to indicate statistical significance. All statistical analyses were performed with the use of GTCA software, version 1.2.4,29 and R software, version 3.2.1 (R Project for Statistical Computing). For the mouse and monkey studies, data are expressed as means and standard errors. Mean values were compared with the use of unpaired t-tests or two-way analysis of variance, as implemented in Graphpad Prism software, version 6.0 (Graphpad Software).
RESULTS
INACTIVATING MUTATIONS IN ANGPTL4
The clinical characteristics of MyCode participants in the DiscovEHR study are described in Table 1. Among 42,930 DiscovEHR study participants, we identified 1661 (3.9%) who were heterozygous for the E40K mutation, which had been reported to have decreased the ability of ANGPTL4 to inhibit LPL activity.24 There were 17 participants (0.04%) who were homozygous for E40K (overall allele frequency, 2.0%), which indicates that E40K is viable as a homozygous variant and which provided a unique opportunity to study clinical characteristics extracted from the electronic health records of homozygotes. We also identified 13 other distinct mutations that were predicted to inactivate ANGPTL4, including 6 premature stop mutations, 5 indel mutations with a shift in the open reading frame, 1 splice donor mutation, and 1 splice acceptor mutation (Table S3 in the Supplementary Appendix). A total of 75 participants were heterozygous for these inactivating mutations, corresponding to a total carrier frequency of 1 in 572 participants. We did not observe homozygotes or compound heterozygotes for these genetic variants in the DiscovEHR population. The most frequently observed inactivating mutation was a single-nucleotide deletion that was predicted to result in a frameshift in the codon encoding glycine at position 313 (G313fs), which was observed in 46 participants (carrier frequency, 1 in 933).
Association with Lipid Levels
Fasting lipid measurements were available for 33,090 participants; 27,781 of these participants had two or more serially gathered measurements, with a median of six measurements per participant. Medians for each trait are shown according to the presence of E40K and other inactivating mutations in Table 2 and according to the inactivating allele in Fig. 1. Results of linear regression analysis are summarized in Table S4 in the Supplementary Appendix. The levels of triglycerides per allele were 13% lower among carriers of the E40K variant than among E40 homozygotes (P = 2.0×10−23), and the levels of HDL cholesterol per allele were 7% higher among carriers of the E40K variant than among E40 homozygotes (P = 1.6×10−17) (Table 2). Similar to E40K heterozygotes, the 75 heterozygous carriers of inactivating mutations in ANGPTL4 had significantly lower triglyceride levels (by 13%) than noncarriers (P = 0.02) and higher HDL cholesterol levels (by 9%) than noncarriers (P = 0.009) (Table 2). There were no significant differences in levels of LDL cholesterol or total cholesterol between carriers of E40K or other inactivating mutations and noncarriers.
Table 2.
Association between ANGPTL4 E40K or Other Inactivating Mutations and Lipid Levels.*
| Lipid | Noncarriers (N = 41,177) | E40K Heterozygotes (N = 1661) | E40K Homozygotes (N = 17) | P Value† | Heterozygotes with Other Inactivating Mutation (N = 75) | P Value‡ |
|---|---|---|---|---|---|---|
| median (IQR) | median (IQR) | |||||
|
| ||||||
| Triglycerides — mg/dl | 132 (95–182) | 115 (85–157) | 81 (61–122) | 2.0×10−23 | 115 (78–162) | 0.02 |
|
| ||||||
| HDL cholesterol — mg/dl | 48 (40–59) | 52 (43–63) | 67 (54–72) | 1.6×10−17 | 54 (44–62) | 0.009 |
|
| ||||||
| LDL cholesterol — mg/dl | 114 (94–135) | 116 (96–138) | 107 (89–132) | 0.20 | 119 (101–136) | 0.60 |
|
| ||||||
| Total cholesterol — mg/dl | 195 (172–218) | 196 (173–219) | 182 (168–209) | 0.90 | 193 (179–208) | 0.80 |
All P values were calculated by means of a mixed-linear-model association method after adjustment for age, age squared, sex, log10-transformed body-mass index, and genetic relatedness. Values for triglycerides and high-density lipoprotein (HDL) cholesterol were log10 transformed before the regression analysis. Levels of low-density lipoprotein (LDL) and total cholesterol were adjusted for the use of lipid-lowering medications by dividing by 0.7 and 0.8, respectively. To convert the values for triglycerides to millimoles per liter, multiply by 0.01129. To convert values for cholesterol to millimoles per liter, multiply by 0.02586.
The P value is for E40K heterozygotes and homozygotes combined as compared with E40 homozygotes. (Data for E40 homozygotes are not shown in the table.)
The P value is for carriers of inactivating mutations as compared with noncarriers.
Figure 1. Fasting Metabolic Phenotypes in Carriers of Inactivating Mutations in ANGPTL4.
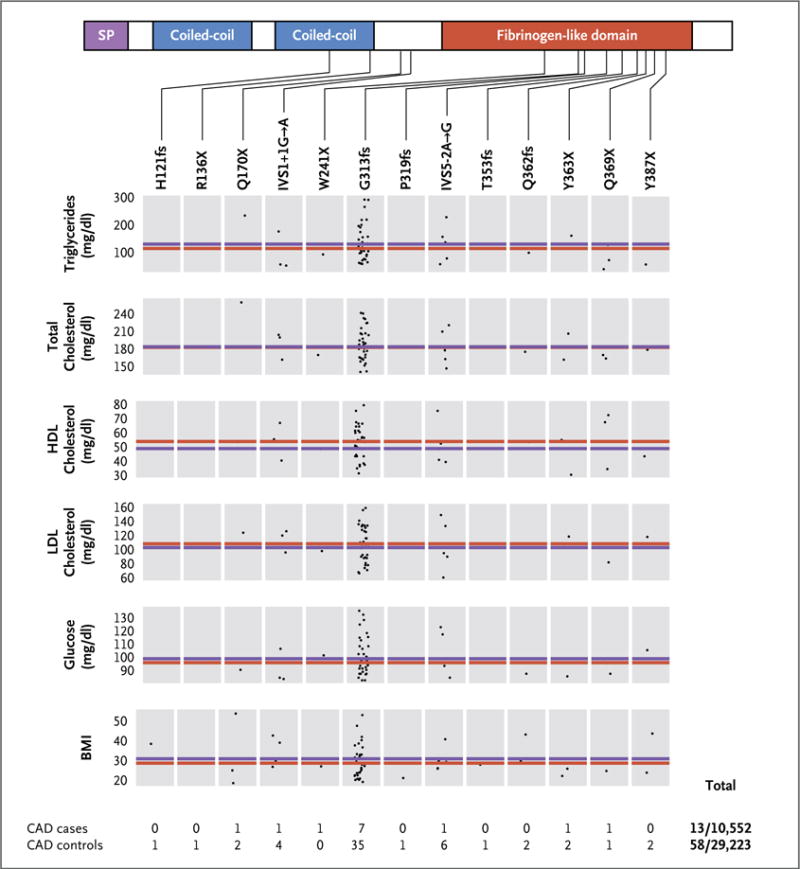
Shown are genotype-specific measures for each trait among study participants with inactivating mutations in ANGPTL4, according to the presence of an inactivating allele in the single-peptide (SP), coiled-coil, and fibrinogen-like domains. The red line indicates the median value for all carriers of inactivating mutations. Each data point represents the value for a single carrier of the mutation specified above each box. The blue line indicates the median value for all participants who did not have an inactivating mutation in ANGPTL4 on sequencing. Overall, 13 of 10,552 participants with coronary artery disease and 58 of 29,223 controls carried an inactivating mutation. The body-mass index (BMI) is the weight in kilograms divided by the square of the height in meters. To convert the values for triglycerides to millimoles per liter, multiply by 0.01129. To convert values for cholesterol to millimoles per liter, multiply by 0.02586. To convert the values for glucose to millimoles per liter, multiply by 0.05551. CAD denotes coronary artery disease, HDL high-density lipoprotein, and LDL low-density lipoprotein.
Association with Coronary Artery Disease
We then tested for the presence of an association (or inverse association) between coronary artery disease and ANGPTL4 mutations. The E40K variant was significantly less common among participants with angiographically defined coronary artery disease (allele frequency, 1.7%; 357 heterozygotes and 2 homozygotes among 10,552 cases) than among controls (allele frequency, 2.1%; 1196 heterozygotes and 15 homozygotes among 29,223 controls), corresponding to a 19% lower risk of coronary artery disease per allele than the risk among E40 homozygotes (odds ratio, 0.81; 95% confidence interval [CI], 0.70 to 0.92; P = 0.002) after adjustment for age and sex (Table 3). K40 homozygotes had a numerically lower risk of coronary artery disease than E40 homozygotes (odds ratio, 0.32; 95% CI, 0.06 to 1.09; P = 0.07).
Table 3.
Association between ANGPTL4 E40K or Other Inactivating Mutations and Coronary Artery Disease.*
| Variants | Allele Frequency | Odds Ratio (95% CI) | P Value | |
|---|---|---|---|---|
| CAD Cases | CAD Controls | |||
|
| ||||
| E40K mutation in 1661 heterozygotes and 17 homozygotes | 1.71 | 2.10 | 0.81 (0.70–0.92) | 0.002 |
|
| ||||
| Heterozygous inactivating mutations in 75 participants | 0.06 | 0.10 | 0.56 (0.32–1.00) | 0.05 |
Odds ratios were calculated by means of Firth’s penalized likelihood method of logistic regression after adjustment for age, age squared, sex, and four principal components of ancestry. An odds ratio of less than 1.00 indicates protection against coronary artery disease. P values were calculated by means of a mixed-linear-model association method after adjustment for age, age squared, sex, and genetic relatedness. CAD denotes coronary artery disease, and CI confidence interval.
The novel inactivating mutations in ANGPTL4 that we identified were also less common among participants with coronary artery disease than among controls. Overall, 13 of 10,552 participants with coronary artery disease (cumulative allele frequency, 0.06%) and 58 of 29,223 controls (cumulative allele frequency, 0.1%) carried an inactivating mutation. After adjustment for age, sex, and ancestry, the presence of an inactivating mutation resulted in 44% lower odds of coronary artery disease than the odds with no inactivating mutation (odds ratio, 0.56; 95% CI, 0.32 to 1.00; P = 0.05) (Table 3). Further adjustment for status with respect to type 2 diabetes, smoking, and hypertension did not appreciably alter associations between coronary artery disease and E40K or other inactivating mutations in ANGPTL4 (Table S5 in the Supplementary Appendix).
MODULATING ANGPTL4 IN MICE AND MONKEYS
We found that the binding affinity of REGN1001 to ANGPTL4 was similar in humans and monkeys (binding dissociation equilibrium constant [Kd] = 0.1 to 0.4 nM) (Table S6 in the Supplementary Appendix). REGN1001 effectively blocked the inhibition of LPL by ANGPTL4 with a 50% inhibitory concentration of 0.4 nM (Table S7 in the Supplementary Appendix). Because REGN1001 does not bind to mouse Angptl4, we tested REGN1001 in male mice that were homozygous for human ANGPTL4 and for ApoE deficiency and that had a genetic predisposition to hypertriglyceridemia. REGN1001 was associated with a rapid and sustained reduction in plasma triglyceride levels for the 17-day duration of the study (Fig. 2A). In a finding that was consistent with the results of previous studies,30–32 we observed accumulation of lipid-filled Touton cells and lesions in the mesenteric lymph nodes in 21 mice receiving a high-fat diet that were treated with multiple doses of REGN1001.
Figure 2. Effect of REGN1001 on Serum Triglyceride Levels in Mice and Monkeys.
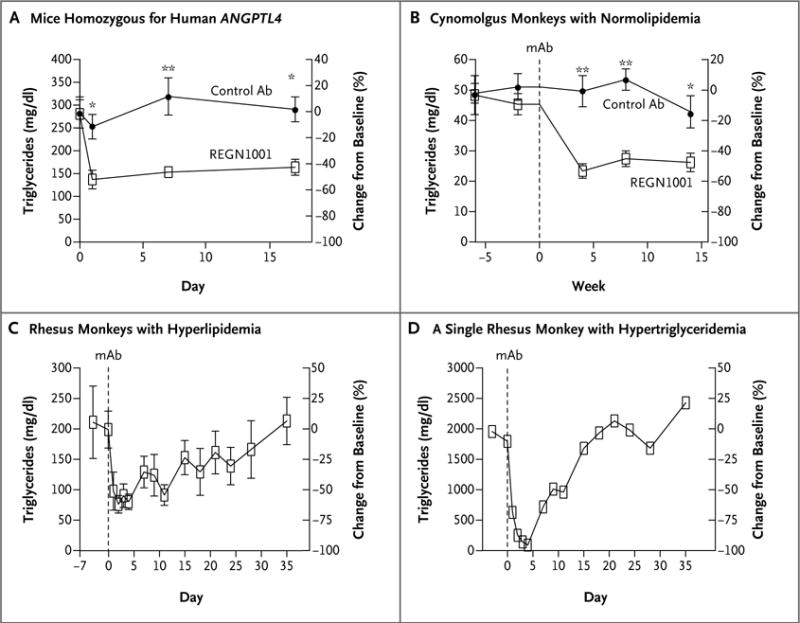
Panel A shows serum triglyceride levels in fasting male mice (five in each group) that were homozygous for human ANGPTL4 and for ApoE deficiency before and after a single subcutaneous injection of monoclonal antibody (mAb) REGN1001 or control antibody at a dose of 10 mg per kilogram of body weight. Panel B shows plasma triglyceride levels in male cynomolgus monkeys (10 in each group) that received weekly intravenous injections of REGN1001 or control antibody at a dose of 5 mg per kilogram for 13 weeks. The triglyceride levels were evaluated before and 4, 8, and 14 weeks after study initiation. Panel C shows changes from baseline in plasma triglyceride levels in 4 of 5 obese rhesus monkeys with hyperlipidemia that received a single intravenous dose of 10 mg of REGN1001 per kilogram. One of the 5 monkeys with a very high triglyceride level at baseline had a dramatic reduction in plasma levels (Panel D). In Panels A, B, and C, values are means, and I bars indicate standard errors. A single asterisk indicates P<0.05, and double asterisks indicate P<0.001.
In the study involving cynomolgus monkeys that were fed a high-fat diet, those that received REGN1001 had lower levels of serum triglycerides than the control monkeys (Fig. 2B). As we had found in mice, we observed accumulation of lipids in the mesenteric lymph nodes in three of the six euthanized female monkeys at week 14 and in one of the four female monkeys that underwent a 15-week recovery period. We did not observe any such lymphadenopathy in the male monkeys.
In the study involving five obese rhesus monkeys with dyslipidemia, four of those that received REGN1001 had a relative decrease from baseline of 60% in circulating triglyceride levels, which reached a nadir within 2 days after administration of the drug (Fig. 2C). The relative reduction was even more pronounced in a single monkey that had a baseline plasma triglyceride level of more than 1500 mg per deciliter (16.9 mmol per liter) (Fig. 2D). In this monkey, treatment with REGN1001 reduced triglyceride levels by more than 95%, to a mean (±SE) of 76±14 mg per deciliter (0.86±0.16 mmol per liter), which is close to the normal range of triglyceride levels in rhesus monkeys.33 The reduction in plasma triglycerides in the other four monkeys was maintained for 28 days before the levels returned to baseline levels (Fig. 2C).
OTHER CLINICAL FINDINGS IN CARRIERS
Our findings in preclinical treatment models prompted us to evaluate humans carrying inactivating mutations in ANGPTL4 to determine whether there was evidence of clinical manifestations mimicking the intestinal and mesenteric lymphadenopathy observed in mice and nonhuman primates treated with an ANGPTL4 antibody. We reviewed deidentified data from electronic health records for participants carrying E40K and other inactivating mutations in ANGPTL4 for information on abdominal or other lymphatic disease (Table S8 in the Supplementary Appendix). (A full description of the clinical characteristics, vital status, and problem list and diagnosis-code entries for K40 homozygotes is provided in Table S9 in the Supplementary Appendix.) We did not observe any significant difference among participants in the rates of diagnosis codes for abdominal lymphatic disorders, which were reported in 1 of 17 E40K homozygotes (5.9%), 5 of 75 heterozygotes with inactivating mutations (6.7%), 154 of 1661 E40K heterozygotes (9.3%), and 3831 of 41,777 noncarriers (9.2%). Similarly, we observed no significant difference in rates of diagnosis-code entries for ascites, peritonitis, malabsorption, abdominal discomfort, or diarrhea.
DISCUSSION
By sequencing the exons of ANGPTL4 in samples obtained from 42,930 participants in the DiscovEHR study population, we identified more than 1600 participants who were heterozygous and 17 who were homozygous for the E40K variant, which has been shown to block the inhibition of LPL by ANGPTL4.24 In addition, we discovered 13 mutations that were predicted to inactivate one allele of ANGPTL4. E40K and other inactivating mutations in ANGPTL4 were associated with decreased levels of serum triglycerides and increased levels of HDL cholesterol. Homozygotes had numerically lower levels of triglycerides and higher levels of HDL cholesterol than did heterozygotes, which suggests a gene-dosage effect of the variant on these traits.
Previous studies have shown that the E40K variant is associated with alterations in levels of triglycerides and HDL cholesterol,17–19 but there have been contradictory claims about the effect of inactivating mutations in ANGPTL4 on the risk of coronary disease. In this study, we found that the presence of the E40K variant was associated with a 19% lower risk of coronary artery disease than the risk among E40 homozygotes. Furthermore, we found that other rare novel mutations that were predicted to inactivate one ANGPTL4 allele were also associated with a reduced risk of cardiovascular disease. In an exomewide search for coding variants associated with coronary artery disease now reported in the Journal, Stitziel and colleagues34 found similar reductions in the risk of coronary artery disease among patients with E40K and other inactivating mutations in ANGPTL4. These results provide independent allelic evidence that the genetic loss of ANGPTL4 function confers favorable lipid profiles and protection from coronary artery disease. We found that monoclonal antibody inhibition of ANGPTL4 in mice and nonhuman primates lowered triglyceride levels — a finding consistent with results in Angptl4-deficient mice15,32 — which provides additional evidence that modulation of ANGPTL4 affects triglyceride metabolism.
Several mechanisms may link the loss of ANGPTL4 function to protection from atherosclerotic coronary artery disease. Lifelong, genetically determined lowering of triglyceride levels may reduce the burden of atherogenic triglyceride-rich lipoproteins. However, the magnitude of the reduction in the risk of coronary artery disease as a result of E40K and other inactivating mutations may be greater than would be expected from triglyceride effects alone, given previous observations of a reduction of approximately 0.7 to 0.8% in relative risk for every genetically associated reduction of 1 mg per deciliter (0.01 mmol per liter) in triglyceride levels.4,6 We cannot rule out an effect of increased HDL cholesterol levels on the risk of coronary artery disease, though emerging evidence suggests that certain mechanisms of alteration in HDL cholesterol may not affect the risk of coronary artery disease.35 Non-HDL types of cholesterol, which are epidemiologically associated with coronary artery disease,36 may also mediate the apparent association between the presence of E40K or other inactivating mutations in ANGPTL4 and coronary artery disease that was observed in our study.
Our data support ANGPTL4 as a therapeutic target for inhibition through an antibody or other mechanism for reducing the risk of cardiovascular disease in humans. On the other hand, the data from our studies in mice receiving a high-fat diet or in obese monkeys also raise serious concern about whether ANGPTL4 inhibition will result in abdominal lymphadenopathy secondary to granulomatous lipid accumulation, as has been reported previously.32 It is not clear whether therapeutic antagonism of ANGPTL4 will have similar effects in humans. Carriers of E40K (including homozygotes) or other inactivating mutations in ANGPTL4 did not have increased rates of abdominal lymphadenopathy or other abdominal disorders, as determined from electronic health records. The potential for recontact and consent for additional clinical phenotyping of E40K homozygotes and carriers of other inactivating mutations facilitated by the DiscovEHR collaboration affords a unique opportunity to investigate this concern. Even if the potential benefits of ANGPTL4 blockade turn out to be limited by the risk of lymphadenopathy in humans, similar therapeutic benefits may still be achieved by blocking ANGPTL4-related proteins, such as ANGPTL8 and ANGPTL3, neither of which appears to be associated with lymphadenopathy in animals. Whether similar reductions in the risk of cardiovascular disease will be observed for triglyceride-lowering mutations affecting other angiopoietin-like proteins such as ANGPTL837 and ANGPTL338–40 remains to be shown.
Our study has several limitations. First, we focused on the lipid levels and risk of coronary artery disease in participants with the known reduced-function–associated E40K variant and in carriers of splice-disrupting or frameshift mutations or premature stop, start, or loss-of-stop codons. The likelihood that these mutations will have a significant effect on the function of the ANGPTL4 protein is high. However, we cannot exclude the possibility that other coding variants or regulatory noncoding variants may yield additional insight into the phenotypic effects of ANGPTL4 antagonism. Second, it is not known whether our observations will be generalizable to persons of non-European ancestry. Third, the associations with lipid levels and coronary artery disease were observed in heterozygotes. Whether such observations accurately reflect the phenotypic effects of a more robust antibody blockade is not clear.
In conclusion, we observed that inactivating mutations in ANGPTL4 were associated with favorable lipid profiles and that the risk of coronary artery disease was lower among persons with those inactivating mutations than in those without them, a finding that mirrors the effects of monoclonal antibody inhibition in mice and nonhuman primate models. Whether inhibition of ANGPTL4 will safely reduce the risk of cardiovascular disease in humans remains to be determined.
Acknowledgments
Supported by Regeneron Pharmaceuticals.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Nordestgaard BG, Varbo A. Triglycerides and cardiovascular disease. Lancet. 2014;384:626–35. doi: 10.1016/S0140-6736(14)61177-6. [DOI] [PubMed] [Google Scholar]
- 2.Sarwar N, Danesh J, Eiriksdottir G, et al. Triglycerides and the risk of coronary heart disease: 10,158 incident cases among 262,525 participants in 29 Western prospective studies. Circulation. 2007;115:450–8. doi: 10.1161/CIRCULATIONAHA.106.637793. [DOI] [PubMed] [Google Scholar]
- 3.Miller M, Stone NJ, Ballantyne C, et al. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123:2292–333. doi: 10.1161/CIR.0b013e3182160726. [DOI] [PubMed] [Google Scholar]
- 4.Do R, Willer CJ, Schmidt EM, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45:1345–52. doi: 10.1038/ng.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pollin TI, Damcott CM, Shen H, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science. 2008;322:1702–5. doi: 10.1126/science.1161524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.The TG and HDL Working Group of the Exome Sequencing Project. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371:22–31. doi: 10.1056/NEJMoa1307095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjærg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med. 2014;371:32–41. doi: 10.1056/NEJMoa1308027. [DOI] [PubMed] [Google Scholar]
- 8.Do R, Stitziel NO, Won HH, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518:102–6. doi: 10.1038/nature13917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H, Eckel RH. Lipoprotein lipase: from gene to obesity. Am J Physiol Endocrinol Metab. 2009;297(2):E271–88. doi: 10.1152/ajpendo.90920.2008. [DOI] [PubMed] [Google Scholar]
- 10.Jensen MK, Rimm EB, Rader D, et al. S447X variant of the lipoprotein lipase gene, lipids, and risk of coronary heart disease in 3 prospective cohort studies. Am Heart J. 2009;157:384–90. doi: 10.1016/j.ahj.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rip J, Nierman MC, Ross CJ, et al. Lipoprotein lipase S447X: a naturally occurring gain-of-function mutation. Arterioscler Thromb Vasc Biol. 2006;26:1236–45. doi: 10.1161/01.ATV.0000219283.10832.43. [DOI] [PubMed] [Google Scholar]
- 12.Yoshida K, Shimizugawa T, Ono M, Furukawa H. Angiopoietin-like protein 4 is a potent hyperlipidemia-inducing factor in mice and inhibitor of lipoprotein lipase. J Lipid Res. 2002;43:1770–2. doi: 10.1194/jlr.c200010-jlr200. [DOI] [PubMed] [Google Scholar]
- 13.Yau MH, Wang Y, Lam KS, Zhang J, Wu D, Xu A. A highly conserved motif within the NH2-terminal coiled-coil domain of angiopoietin-like protein 4 confers its inhibitory effects on lipoprotein lipase by disrupting the enzyme dimerization. J Biol Chem. 2009;284:11942–52. doi: 10.1074/jbc.M809802200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sukonina V, Lookene A, Olivecrona T, Olivecrona G. Angiopoietin-like protein 4 converts lipoprotein lipase to inactive monomers and modulates lipase activity in adipose tissue. Proc Natl Acad Sci U S A. 2006;103:17450–5. doi: 10.1073/pnas.0604026103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Köster A, Chao YB, Mosior M, et al. Transgenic angiopoietin-like (angptl)4 overexpression and targeted disruption of angptl4 and angptl3: regulation of triglyceride metabolism. Endocrinology. 2005;146:4943–50. doi: 10.1210/en.2005-0476. [DOI] [PubMed] [Google Scholar]
- 16.Adachi H, Fujiwara Y, Kondo T, et al. Angptl 4 deficiency improves lipid metabolism, suppresses foam cell formation and protects against atherosclerosis. Biochem Biophys Res Commun. 2009;379:806–11. doi: 10.1016/j.bbrc.2008.12.018. [DOI] [PubMed] [Google Scholar]
- 17.Romeo S, Pennacchio LA, Fu Y, et al. Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat Genet. 2007;39:513–6. doi: 10.1038/ng1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Talmud PJ, Smart M, Presswood E, et al. ANGPTL4 E40K and T266M: effects on plasma triglyceride and HDL levels, postprandial responses, and CHD risk. Arterioscler Thromb Vasc Biol. 2008;28:2319–25. doi: 10.1161/ATVBAHA.108.176917. [DOI] [PubMed] [Google Scholar]
- 19.Folsom AR, Peacock JM, Demerath E, Boerwinkle E. Variation in ANGPTL4 and risk of coronary heart disease: the Atherosclerosis Risk in Communities Study. Metabolism. 2008;57:1591–6. doi: 10.1016/j.metabol.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–78. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- 21.Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;11:11.10.1–11.10.133. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cingolani P, Platts A, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yin W, Romeo S, Chang S, Grishin NV, Hobbs HH, Cohen JC. Genetic variation in ANGPTL4 provides insights into protein processing and function. J Biol Chem. 2009;284:13213–22. doi: 10.1074/jbc.M900553200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murphy AJ, Macdonald LE, Stevens S, et al. Mice with megabase humanization of their immunoglobulin genes generate antibodies as efficiently as normal mice. Proc Natl Acad Sci U S A. 2014;111:5153–8. doi: 10.1073/pnas.1324022111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gusarova V, Alexa CA, Wang Y, et al. ANGPTL3 blockade with a human monoclonal antibody reduces plasma lipids in dyslipidemic mice and monkeys. J Lipid Res. 2015;56:1308–17. doi: 10.1194/jlr.M054890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heinze G, Schemper M. A solution to the problem of separation in logistic regression. Stat Med. 2002;21:2409–19. doi: 10.1002/sim.1047. [DOI] [PubMed] [Google Scholar]
- 28.Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet. 2008;83:311–21. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lichtenstein L, Mattijssen F, de Wit NJ, et al. Angptl4 protects against severe proinflammatory effects of saturated fat by inhibiting fatty acid uptake into mesenteric lymph node macrophages. Cell Metab. 2010;12:580–92. doi: 10.1016/j.cmet.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mattijssen F, Alex S, Swarts HJ, Groen AK, van Schothorst EM, Kersten S. Angptl4 serves as an endogenous inhibitor of intestinal lipid digestion. Mol Metab. 2014;3:135–44. doi: 10.1016/j.molmet.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Desai U, Lee EC, Chung K, et al. Lipid-lowering effects of anti-angiopoietin-like 4 antibody recapitulate the lipid phenotype found in angiopoietin-like 4 knockout mice. Proc Natl Acad Sci U S A. 2007;104:11766–71. doi: 10.1073/pnas.0705041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yin W, Carballo-Jane E, McLaren DG, et al. Plasma lipid profiling across species for the identification of optimal animal models of human dyslipidemia. J Lipid Res. 2012;53:51–65. doi: 10.1194/jlr.M019927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators. Coding variation in ANGPTL4, LPL, and SVEP1 and the risk of coronary disease. N Engl J Med. 2016;374:1134–44. doi: 10.1056/NEJMoa1507652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Voight BF, Peloso GM, Orho-Melander M, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–80. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rana JS, Boekholdt SM, Kastelein JJ, Shah PK. The role of non-HDL cholesterol in risk stratification for coronary artery disease. Curr Atheroscler Rep. 2012;14:130–4. doi: 10.1007/s11883-011-0224-x. [DOI] [PubMed] [Google Scholar]
- 37.Peloso GM, Auer PL, Bis JC, et al. Association of low-frequency and rare coding-sequence variants with blood lipids and coronary heart disease in 56,000 whites and blacks. Am J Hum Genet. 2014;94:223–32. doi: 10.1016/j.ajhg.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Musunuru K, Pirruccello JP, Do R, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med. 2010;363:2220–7. doi: 10.1056/NEJMoa1002926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Minicocci I, Montali A, Robciuc MR, et al. Mutations in the ANGPTL3 gene and familial combined hypolipidemia: a clinical and biochemical characterization. J Clin Endocrinol Metab. 2012;97(7):E1266–75. doi: 10.1210/jc.2012-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Romeo S, Yin W, Kozlitina J, et al. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J Clin Invest. 2009;119:70–9. doi: 10.1172/JCI37118. [DOI] [PMC free article] [PubMed] [Google Scholar]