

Supplemental Digital Content is available in the text
Abstract
There is increasing evidence that circular RNAs (circRNAs) are involved in cancer development; however, their role in hepatocellular carcinoma (HCC) remains unclear. Here, we aimed to determine the circRNA expression profile in HCC, and investigate relevant mechanisms for cancer progression. The global circRNA expression profile between HCC (n = 3) and adjacent normal liver (n = 3) tissue was significantly different. Three circRNAs (hsa_circ_0000520, hsa_circ_0005075, and hsa_circ_0066444) showed significantly different expression levels in HCC tissues, which were further validated in 60 matched tissue samples using real-time qRT-PCR. Only hsa_circ_0005075 exhibited significant difference in expression (P <0.001) between HCC and normal tissues. Hsa_circ_0005075 expression correlated with HCC tumor size (P = 0.042), and showed good diagnostic potential (AUROC = 0.94). Finally, we constructed a network of hsa_circ_0005075-targeted miRNA–gene interactions, including miR-23b-5p, miR-93-3p, miR-581, miR-23a-5p, and their corresponding mRNAs. Gene oncology analysis revealed that hsa_circ_0005075 could participate in cell adhesion during HCC development. In summary, we identified hsa_circ_0005075 as a potential HCC biomarker; however, further studies are required to confirm the role of this circRNA, and others, in HCC development.
INTRODUCTION
Circular RNAs (circRNAs) are a unique class of RNA with a stable structure formed by special loop splicing. To date, thousands of highly expressed circRNAs have been detected in humans, often with tissue and developmental-phase-specific expression.1–3 CircRNAs tend to accumulate in the cytoplasm of some cells, and may even be present in larger number than associated linear mRNAs.4 Compared with microRNAs (miRNAs) and long noncoding RNAs (lncRNAs), circRNAs have a higher degree of stability and sequence conservation among mammalian cells.5 CircRNAs can bind to miRNAs, acting as “miRNA sponges,” and can regulate gene expression at the transcriptional or post-transcriptional level.6 For example, the circular RNA sponge for miR-7 (ciRS-7), which contains more than 70 conserved miRNA target sites, acts as a miR-7 sponge leading to strong suppression of miR-7 activity, and the overexpression of miR-7 targets.6 In addition, increasing evidence suggests that circRNAs are involved in disease, including cancer, and therefore, they may be promising as diagnostic or predictive biomarkers.7
Although circRNAs are becoming a new research hotspot in the field of RNA, little is known about their associations with various cancers. For example, hsa_circ_002059 has been reported as a potential diagnostic biomarker for gastric cancer,8 but how it regulates cancer progression at a mechanistic level is unclear. To investigate the proposed interactions between the circRNA-miRNA-mRNA regulatory axis and disease, further researches can be conducted focusing on this point. For example, Ghosal previously performed a gene ontology (GO) enrichment analysis combined with statistical analysis to detect the enrichment of genes related to specific biological processes. Among these mRNAs, he identified 43 cell cycle-related genes involved in the biological process of breast cancer, 194 genes in association with cervical cancer, and 68 genes associated with gastric cancer.9 However, the role of circRNAs in hepatocellular carcinoma (HCC) has not been well studied.
HCC is one of the leading causes of cancer-related death worldwide.10,11 With the improvement of diagnostic approaches and treatment strategies, HCC patients can be detected at early stage and accepted radical surgeries with favorable prognosis. However, there were still many patients with HCC who are missed by current diagnostic standards and therefor lost the best operation opportunity.12,13 To improve the therapeutic effect and prognosis of HCC patients, it is imperative to search for more efficient biomarkers to increase the early diagnostic rate of HCC. It has been reported that circRNA might serve as a novel potential biomarker for cancer diagnosis.8,14 Additionally, hsa_circ_0001649 has been suggested as a biomarker for use in diagnosing HCC.15 The involvement of circRNAs in HCC progression has recently become a hot topic among cancer researchers. Indeed, an integrated high-throughput data analysis showed that a circRNA resulted in miRNA overexpression (miR-181a-1_3p), which in turn inhibited an enzyme related to DNA damage (MGMT), and this may lead to HCC progression over time.16 Therefore, circRNAs may play a role in regulating HCC through regulation of miRNAs.17 Despite this potential link with circRNAs, the global circRNA expression profile in HCC has not been fully uncovered. In the present study, via circRNA microarray detection, we identified 61 differentially expressed circRNAs (including upregulated and downregulated genes) in HCC tissues compared with adjacent normal tissues. From these 61 differentially expressed circRNAs, 3 (hsa_circ_0000520, hsa_circ_0005075, and hsa_circ_0066444) were confirmed by real-time qRT-PCR. The miRNA binding sites of these confirmed circRNAs were revealed, and the related mRNAs were predicted. Our results indicate that hsa_circ_0005075 expression shows significant association with some clinicopathological factors of HCC patients, and may serve as a novel potential biomarker for HCC diagnosis.
METHODS
Clinical Population and Sample Collection
A total of 66 HCC tissues and paired adjacent nontumorous tissues were collected from HCC surgical specimens between March 2015 and August 2015 in the operating room of Shan Dong University affiliated with Shan Dong Provincial Hospital. During the operation, a professional pathologist excised these specimens. After removal from the body, the specimens were immediately preserved in RNA-fixer reagent (Bioteke, Beijing, China), and stored at −80°C until use. For excluding confounding factors affecting the steady of patients’ circRNA profiling, all recruited participants accepted no adjunctive treatment prior to the surgery, including chemotherapy, radiotherapy, or targeted therapy. The Medical Ethics Commission of Shan Dong Provincial Hospital approved our protocol and before the surgery, written informed consent was signed by the participant.
In total, 66 tissue samples were obtained from 33 patients (6 females and 27 males), including 33 histopathological confirmed HCC tissues and 33 normal liver tissues. For circRNA microarray analysis, 3 HCC samples and corresponding 3 normal liver tissues were randomly selected. And other 60 samples (30 HCC tissue + 30 normal tissue) were prepared for qRT-PCR verification experiments. The clinical characteristics of 3 patients whose samples were selected for microarray were displayed in Supplementary Table 1.
Total RNA Extraction
Total tissue RNA was extracted from the HCC tissues and paired adjacent normal tissues using TRIzol reagent (Invitrogen, Karlsruhe, Germany), according to the manufacturer's instructions. The purity and concentration of RNA samples were determined with the NanoDrop ND-1000 (Thermo Fisher Scientific, Wilmington, DE). RNA was prepared and stored at −80°C for the validation experiments.
Sample Preparation and Microarray Analysis
For enriching circRNAs, Rnase R (Epicentre, Madison, WI) was used to digest total RNAs extractive in order to remove linear RNAs. And then, the microarray analysis of circRNAs was conducted according to the manufacturer's protocol (Arraystar, Rockville, MD). The enriched circRNAs were amplified and transcribed into fluorescent cRNA by using the random priming method (Arraystar Super RNA Labeling Kit; Arraystar) to process the treated RNA extractive of 6 samples (3 HCC samples and 3 paired adjacent non-tumorous samples). And the enriched circRNAs were amplified and transcribed into fluorescent cRNA; the labeled cRNAs were hybridized onto the Arraystar Human circRNA Array (8×15K, Arraystar). And the cRNAs were incubated for 17 hours at 65°C in an Agilent Hybridization Oven. After washing the slides, the arrays were scanned by the Agilent Scanner G2505C (Agilent, Santa Clara, CA).
CircRNA Microarray Data Analysis
Data was extracted using Agilent Feature Extraction software. A series of data processing, including quantile normalization, were performed using the R software package. The differentially expressed circRNAs were selected according to fold change cutoff and P value with statistical significance (P value <0.05) or through the Volcano Plot filtering. The homemade computer program of Arraystar based on TargetScan18 and miRanda19 was applied to predict miRNA targets of circRNAs and the circRNA/miRNA interaction. To focus the targeted miRNA profile, the miRNA support vector regression (mirSVR) algorithm was used to score and rank the efficiency of the predicted miRNA targets.20 Accordingly, for each circRNA, we identified 5 miRNAs with the highest mirSVR score to establish a “Top-5” circRNA-miRNA network (1 circRNA connecting to 5 miRNAs).
Validation of Candidate circRNAs Using qRT-PCR
According to the value of the raw signal intensity tested in the microarray analysis, the selected circRNAs for further validation should meet the criteria that the group raw intensity of all tested samples was rather than 500. And because the circRNA may affect HCC development through the microRNAs, circRNAs whose predicted target miRNAs proved to be in related with cancer progression in previous research results were selected for further study. Finally, 3 circRNAs (hsa_circ_0000520, hsa_circ_0005075, and hsa_circ_0066444) showed close relationships with HCC progression, and were selected for further investigation. Validation of these circRNAs was performed using qRT-PCR (in triplicate) in the remaining 60-paired samples. Divergent primers, rather than the more commonly used convergent primers, were designed and optimized for these 3 circRNAs. The sequences of the 3 circRNAs were acquired from the database “circBase” (http://circbase.mdc-berlin.de). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), a housekeeping gene, was used as a control. Primers were synthesized by Sangon Biotech (Shanghai, China), with the following sequences: 5′-GGGAAACTGTGGCGTGAT-3′ (sense) and 5′- GAGTGGGTGTCGCTGTTGA -3′ (anti-sense) for GAPDH; 5′- CCTACCCCATCCCCTTATTC -3′ (sense) and 5′- ACCGTGCTGTAGACTGCTGAG -3′ (anti-sense) for hsa_circ_0005075. Divergent primer sequences for other circRNAs are displayed in Supplementary Table 2. The appearance of a single peak in the melting curve indicated the specificity of the PCR results. The data were analyzed using the ΔCt method, 2−ΔΔCT, to represent a relative expression level of circRNAs.
Annotation and Function Prediction for the hsa_circ_0005075
Validated candidate circRNAs were used as seeds to enrich a circRNA-miRNA-gene network according to the analysis of TargetScan (http://www.targetscan.org/) combined with miRanda (http://www.microrna.org/). Cytoscape (http://www.cytoscape.org/) was applied to build a circRNA–miRNA–mRNA interaction network of hsa_circ_0005075. The predicted gene functions in the networks were annotated using GO and KEGG pathway analysis.
Statistical Analysis
Quantile normalization and subsequent data processing were performed using the Kangcheng homemade R software package (Kangcheng Bio-tech, Shanghai, China). All other statistical data were analyzed and visualized by GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA). The significance of qRT-PCR validation between the HCC tissue group and the normal tissue group was tested by the Student t test, and a P value <0.05 was considered statistically significant. An unpaired Student t test was also used to determine correlations between expression levels of hsa_circ_0005075 and various clinicopathological parameters of HCC.
RESULTS
CircRNA Expression Profiles in HCC Tissues Relative to Adjacent Nontumorous Tissues
The circRNA expression patterns between HCC tissues and adjacent nontumorous tissues were found to be significantly different. After microarray scanning and normalization, 61 circRNAs were found to be differentially expressed in HCC tissues (fold change (FC) in expression >1.5; P <0.05). Among them, 26 circRNAs were upregulated and 35 were downregulated in tumor tissues. Unsupervised hierarchical clustering of circRNA expression patterns obviously classified the HCCs and adjacent normal tissues (Figure 1). The circRNAs that could significantly discriminate the tumor tissues of HCC from adjacent nontumorous tissues and other related details, including the predicted potential miRNA binding sites by bioinformatics, are summarized in Supplementary Table 3.
FIGURE 1.

Differentially expressed circRNAs between HCC tissues and adjacent nontumorous tissues. The result from unsupervised hierarchical clustering analysis shows distinguishable circRNA expression profiling among samples (H for HCC and N for normal adjacent nontumorous tissues). Each column represents the expression profile of a tissue sample, and each row corresponds to a circRNA. “Red” indicates higher expression level, and “green” indicates lower expression level. HCC = hepatocellular carcinoma.
Amplification of hsa_circ_0000520, hsa_circ_0005075, and hsa_circ_0066444
Considering the significant difference of raw signal intensity (>8.0) and the fold change of expression (FC > 2.0), 3 typically differential expression circRNAs (hsa_circ_0000520, hsa_circ_0005075, and hsa_circ_0066444) were selected for validation experiments. As this was the first time that divergent primers for these 3 circRNAs were designed, it was necessary to test the specificity of the amplified circRNA product. Our melting curve analysis showed a single peak for each circRNA (Supplementary Figure 1), proving that no primer dimers or nonspecific amplified product existed. These tests confirmed that hsa_circ_0000520, hsa_circ_0005075, and hsa_circ_0066444 existed in HCC tissues and could be amplified by qRT-PCR.
Upregulation of hsa_circ_expression in HCC Tissues
We explored the expression levels of hsa_circ_0000520, hsa_circ_0005075, and hsa_circ_0066444 in 60 samples of HCC tissues and their paired adjacent liver tissues by qRT-PCR, using GAPDH as the internal standard. The intratissue expression of these 3 circRNAs was examined by detecting their levels in both cancer and adjacent nontumorous tissues (Figure 2A). At the intratissue level, only hsa_circ_0005075 expression differed between the HCC tissues and the corresponding nontumorous tissues with statistical significance (P <0.001). As shown in Figure 2B, the expression levels of hsa_circ_0005075 in each cancer and adjacent nontumorous tissue pair were determined. We found that the hsa_circ_0005075 expression levels in HCC tissues were significantly higher than those in corresponding nontumorous tissues (Figure 2C).
FIGURE 2.
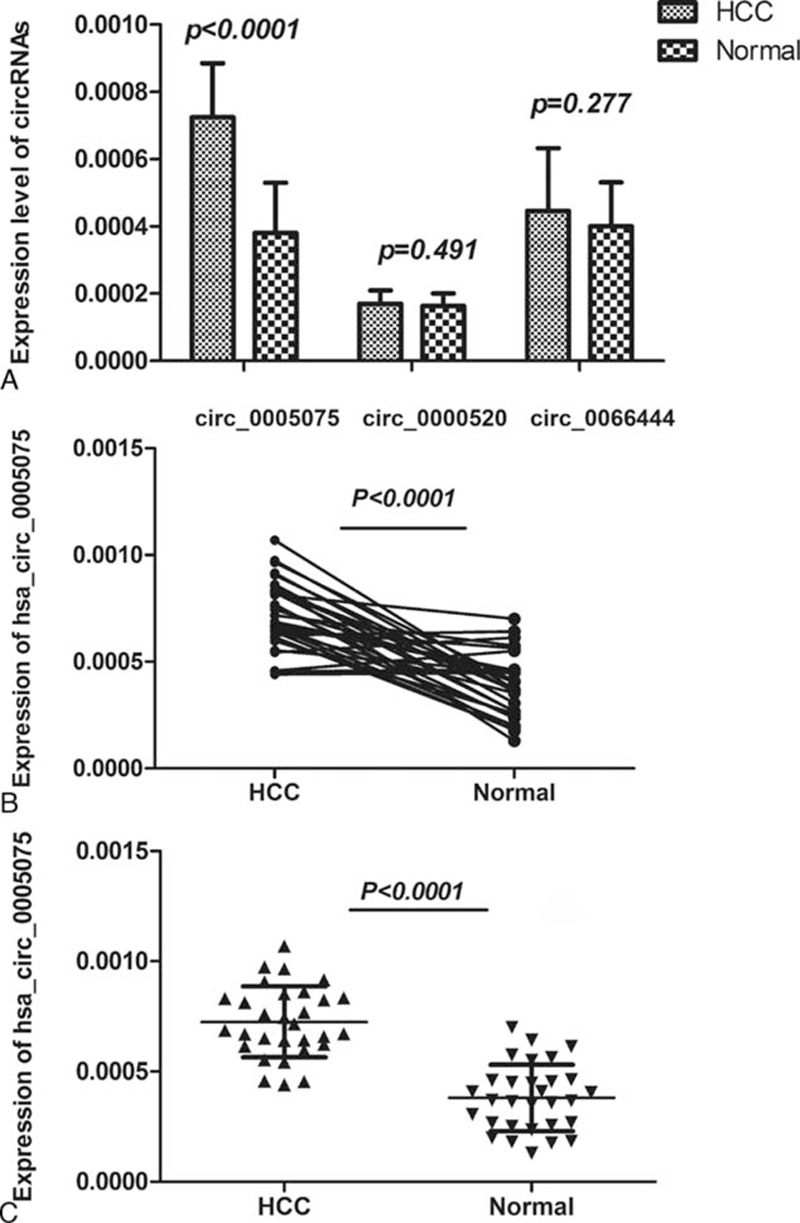
The expression levels of candidate circRNAs for validation in HCC tissue and adjacent liver tissue samples. A, Hsa_circ_0005075, hsa_circ_0000520, and hsa_circ_0066444 expression levels were examined in 60 paired tissue samples by qRT-PCR; only hsa_circ_0005075 was significantly differentially expressed between the 2 groups (P <0.001). B, The expression levels of hsa_circ_0005075 in each patient with comparison between cancer (HCC) and adjacent normal tissues (n = 30). C, The expression levels of hsa_circ_0005075 in the HCC group are significantly higher than those in corresponding nontumorous tissues (P <0.001).
Hsa_circ_0005075 as a Biomarker for HCC
In this study, we showed that hsa_circ_0005075 expression was significantly upregulated in HCC tissues. Therefore, we examined the potential diagnostic value of this circRNA by performing a correlation analysis between several clinicopathological parameters of HCC patients and hsa_circ_0005075 expression. We found that the level of hsa_circ_0005075 expression was associated with HCC tumor size (P = 0.042, Table 1). However, no association between hsa_circ_0005075 expression levels and other clinicopathological characteristics (including differentiation, tumor node metastasis stage, blood alpha fetal protein, and so) was found. In the future, further investigations with an increased sample size should be performed to confirm this relationship.
TABLE 1.
Correlation Between hsa_circ_0005075 Expression and Clinical Parameters in HCC

We then used the receiver-operating characteristic (ROC) curve to investigate the diagnostic effect of hsa_circ_0005075 in distinguishing HCC tissues from adjacent nontumorous tissues. When the expression level of hsa_circ_0005075 was analyzed for this purpose, the area under the ROC curve (AUROC) was 0.94 (Figure 3), indicating that hsa_circ_0005075 has a good potential as a biomarker. Moreover, hsa_circ_0005075 showed good sensitivity and specificity of 83.3% and 90.0%, respectively.
FIGURE 3.
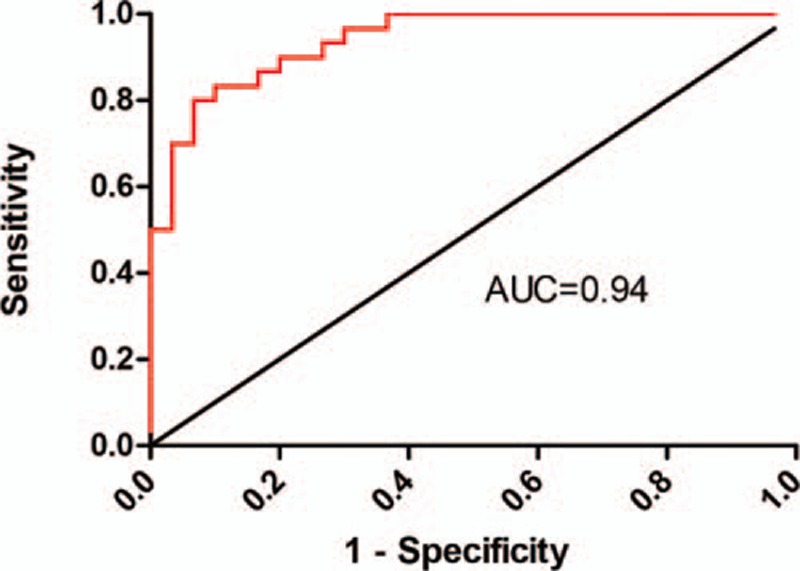
Hsa_circ_0005075 could serve as a biomarker for HCC. A larger area under the curve by ROC analysis for hsa_circ_0005075 in HCC indicates a greater potential as a biomarker (AUROC = 0.94). The cutoff value was 0.000586, and the sensitivity and specificity were 83.3% and 90.0%, respectively.
Prediction and Annotation of hsa_circ_0005075 Targeted miRNA-mRNA Network
Next, we identified and ranked the target miRNAs for hsa_circ_0005075 based on mirSVR scores. We identified the 5 highest-ranking candidate miRNA binding targets (“Top 5”) for further analysis. Specific details of the molecular interactions between hsa_circ_0005075 and its miRNA targets are depicted in Supplementary Figure 2. In accordance with the initial analysis data of the microarray, the genomic locus of hsa_circ_0005075 is on chromosome 1, and the predicted gene sequence of its best transcript is uc001bee.3. Through specific base pairing, we displayed the molecular interaction between hsa_circ_0005075 and its predicted Top-5 miRNA targets detected by mirSVR. We assumed that hsa_circ_0005075 act as a miRNA sponge to regulate its circRNA-miRNA-mRNA network, and that the interactions could be predicted by TargetScan and miRanda. Based on these analysis tools, a total of 4 miRNAs and 121 mRNAs were predicted to have an interaction with hsa_circ_0005075 in this study. Moreover, 4 miRNAs were conserved between the TargetScan and miRanda predictions, including hsa-miR-23b-5p, hsa-miR-93-3p, hsa-miR-581, and hsa-miR-23a-5p.
Cytoscape analysis of the circRNA-miRNA-mRNA interaction network of hsa_circ_0005075 indicated that miR-23b-5p exhibited the largest interaction network, followed by miR-93-3p, miR-581, and miR-23a-5p (Figure 4). To gain further insights into the functions of hsa_circ_0005075, the GO and KEGG pathway analysis was utilized based on the predicted results from TargetScan and miRanda. Hsa_circ_0005075 showed a strong relationship with the biological process of cell adhesion (Figure 5).
FIGURE 4.
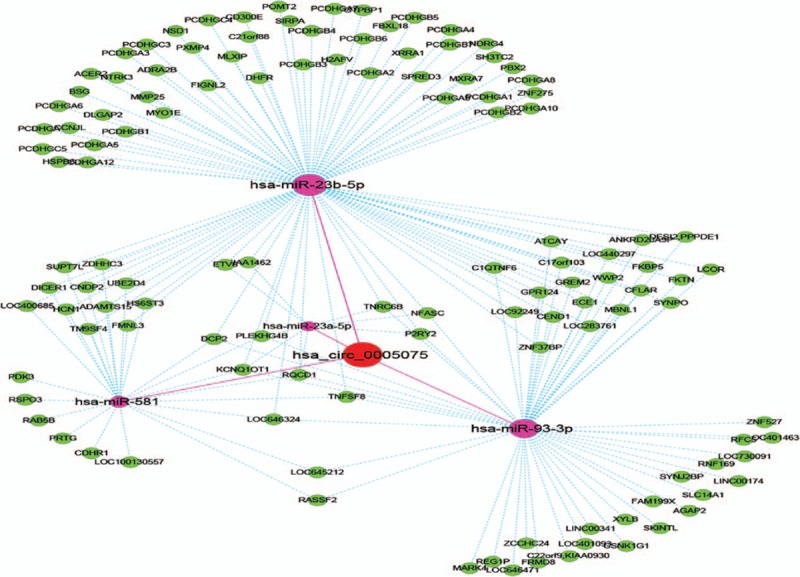
The predicted hsa_circ_0005075 targeted circRNA-miRNA-mRNA/gene network based on sequence-pairing prediction. The results of miRNA-binding sites predicted by mirSVR, and targeted miRNAs and mRNAs predicted by TargetScan and miRanda, were considered. Four miRNAs were found with overlapping results. As shown in this figure, miR-23b-5p exhibited the largest interaction network followed by miR-93-3p, miR-581, and miR-23a-5p.
FIGURE 5.

Gene ontology (GO) analysis based on the hsa_circ_0005075-miRNA-mRNAs network. The top 10 significantly enriched biological processes and their scores (negative logarithm of P value) are listed as the x-axis and the y-axis, respectively. The horizontal axis represents the significant level of GOs.
DISCUSSION
The rapid advances in high-throughput RNA sequencing for noncoding RNAs have sparked new interest in circRNA research. CircRNAs can act as miRNA sponges, and have the potential to be ideal biomarkers in the diagnosis of disease. However, little was known about the role of circRNAs in HCC. In the present study, we utilized the high-throughput circRNA microarray to detect 3 circRNAs (hsa_circ_0000520, hsa_circ_0005075, and hsa_circ_0066444) that were differentially expressed in HCC tissues (n = 3) compared with adjacent nontumor tissues (n = 3). After validating the expression of these 3 circRNAs in additional tissue samples (n = 60), only hsa_circ_0005075 was confirmed to be significantly upregulated in HCC (P <0.001). Moreover, we found that hsa_circ_0005075 expression was associated with tumor size (P = 0.042). Interestingly, HCC tumors with a larger size showed higher hsa_circ_0005075 expression, indicating that hsa_circ_0005075 may play a role in promoting tumor growth. Moreover, using GO and pathway analysis, hsa_circ_0005075 was predicted to be associated strongly with the biological process of cell adhesion, which is involved in cancer cell proliferation and metastasis.
To date, few studies have focused on the relationship between circRNAs and cancer. CircRNAs may regulate gene expression by acting as miRNA sponges, thereby regulating linear RNA transcription and protein production.21,22 Compared with linear miRNA sponges, circRNAs have more miRNA binding sites and higher expression levels, and may be more effective in sequestering miRNAs.23,24 Therefore, the carcinogenic mechanisms of circRNAs may occur through their miRNA-mediated effects on gene expression. Indeed, the circRNA identified in this study (hsa_circ_0005075) was found to potentially interact (i.e., contain complementary base-pair sites) with 4 miRNAs, including hsa-miR-23b-5p, hsa-miR-93-3p, hsa-miR-581, and hsa-miR-23a-5p. As hsa_circ_0005075 may function as a potent miRNA sponge, next we considered the regulatory network in which these miRNAs participate.
MiRNA-23b-5p has been found to be downregulated in a microarray analysis of gastric cancer cells, and a potential interaction between miRNA-23b-5p and the Wnt/β-catenin signaling pathway was identified.25 In addition, miR-23b-5p was significantly downregulated in adenocarcinoma of the esophagus compared with the corresponding normal tissue.26 In this study, through our Cytoscape analysis, we found that miR-23b-5p has significant association with hsa_circ_0005075. A large number of mRNAs may take part in this interaction network, such as dihydrofolate reductase, myosin-1E, and fidgetin-like 2. Therefore, based on the previous studies on miR-23b-5p in cancer, we hypothesize that hsa_circ_0005075 inhibits the expression and function of miR-23b-5p, by acting as a miRNA sponge.
Similar to miR-23b-5p, miR-93-3p was shown to be downregulated during an expression profile analysis in radioresistant nasopharyngeal carcinoma cells.27 Moreover, miR-581 was reportedly downregulated in HCC based on a prior pathogenesis-related microRNAs expression profiling analysis.28 Therefore, the decreased expression and inhibited function of miR-93-3p and miR-581 in cancer further support our hypothesis that hsa_circ_0005075 functions as a miRNA sponge to regulate the more comprehensive circRNA-miRNA-mRNA network, and aid HCC development.
Many of the predicted binding sites of circRNAs on miRNAs have proven to be functional, and appear to be under less selective pressure compared with the corresponding miRNA binding sites in mRNAs.29 Based on our hypothesis that hsa_circ_0005075 functions as a miRNA sponge for its predicted miRNA binding partners described above, we functionally examined the target genes using GO and KEGG pathway analysis. We found that hsa_circ_0005075 has a high possibility of participating in the biological function process of cell adhesion in HCC development. Since cell adhesion is strongly associated with cancer cell proliferation, invasion, and metastasis, it is possible to speculate that hsa_circ_0005075 functions in the regulation of some components of the tumor cell membrane, leading to increased HCC malignancy. Accordingly, we propose the hypothesis that the mRNA-microRNA-circRNA axis may be the possible mechanism promoting the growth of tumor, but further studies are needed for this mechanism.
In summary, the present study identified hsa_circ_0005075 as being associated with HCC. While we speculate that a high level of hsa_circ_0005075 expression in HCC tissues is possibly correlated with tumor progression, the detailed molecular mechanisms by which this circRNA contributes to HCC proliferation, invasion, and metastasis requires further research. Despite this, our results indicate that hsa_circ_0005075 may be utilized as a novel biomarker for HCC with a high degree of accuracy, specificity, and sensitivity. In the future, studying the specific molecular mechanisms by which circRNAs function as miRNA sponges to regulate HCC occurrence and development will be a promising research field.
Supplementary Material
Footnotes
Abbreviations: AUROC = area under the ROC curve, CircRNAs = circular RNAs, ciRS-7 = circular RNA sponge for miR-7, GAPDH = glyceraldehyde 3-phosphate dehydrogenase, GO = gene ontology, HCC = hepatocellular carcinoma, miRNA = micro RNA, mirSVR = miRNA support vector regression, qRT-PCRs = real-time quantitative reverse transcription-polymerase chain reactions, ROC = receiver-operating characteristic.
This work was supported by the National Natural Science Foundation of China (Nos. 81473483, 81472685), the Natural Science Foundation of Shandong Province (BS2013YY048, ZR2014HM106), and the China Postdoctoral Science Foundation (2015M572056).
Supplemental Digital Content is available for this article.
The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Memczak] S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013; 495:333–338. [DOI] [PubMed] [Google Scholar]
- 2.Wang PL, Bao Y, Yee MC, et al. Circular RNA is expressed across the eukaryotic tree of life. PLoS One 2014; 9:e90859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salzman J, Gawad C, Wang PL, et al. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 2012; 7:e30733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeck WR, Sorrentino JA, Wang K, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013; 19:141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liang D, Wilusz JE. Short intronic repeat sequences facilitate circular RNA production. Genes Dev 2014; 28:2233–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hansen TB, Jensen TI, Clausen BH, et al. Natural RNA circles function as efficient microRNA sponges. Nature 2013; 495:384–388. [DOI] [PubMed] [Google Scholar]
- 7.Qu S, Yang X, Li X, et al. Circular RNA: a new star of noncoding RNAs. Cancer Lett 2015; 365:141–148. [DOI] [PubMed] [Google Scholar]
- 8.Li P, Chen S, Chen H, et al. Using circular RNA as a novel type of biomarker in the screening of gastric cancer. Clin Chim Acta 2015; 444:132–136. [DOI] [PubMed] [Google Scholar]
- 9.Ghosal S, Das S, Sen R, et al. Circ2Traits: a comprehensive database for circular RNA potentially associated with disease and traits. Front Genet 2013; 4:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhanasekaran R, Limaye A, Cabrera R. Hepatocellular carcinoma: current trends in worldwide epidemiology, risk factors, diagnosis, and therapeutics. Hepat Med 2012; 4:19–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. International journal of cancer. J Int Cancer 2010; 127:2893–2917. [DOI] [PubMed] [Google Scholar]
- 12.Kinoshita A, Koike K, Nishino H. Clinical features and prognosis of elderly patients with hepatocellular carcinoma not indicated for surgical resection. Geriatr Gerontol Int 2016; doi: 10.1111/ggi.12747. [DOI] [PubMed] [Google Scholar]
- 13.Bruix J, Reig M, Sherman M. Evidence-based diagnosis, staging and treatment of patients with hepatocellular carcinoma. Gastroenterology 2016; doi: 10.1053/j.gastro.2015.12.041. [DOI] [PubMed] [Google Scholar]
- 14.Li Y, Zheng Q, Bao C, et al. Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res 2015; 25:981–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qin M, Liu G, Huo X, et al. Hsa_circ_0001649: a circular RNA and potential novel biomarker for hepatocellular carcinoma. Cancer Biomark 2016; 16:161–169. [DOI] [PubMed] [Google Scholar]
- 16.Caiment F, Gaj S, Claessen S, et al. High-throughput data integration of RNA-miRNA-circRNA reveals novel insights into mechanisms of benzo[a]pyrene-induced carcinogenicity. Nucleic Acids Res 2015; 43:2525–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang N, Ekanem NR, Sakyi CA, et al. Hepatocellular carcinoma and microRNA: new perspectives on therapeutics and diagnostics. Adv Drug Deliv Rev 2015; 81:62–74. [DOI] [PubMed] [Google Scholar]
- 18.Enright AJ, John B, Gaul U, et al. MicroRNA targets in Drosophila. Genome Biol 2003; 5:R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pasquinelli AE. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nature reviews. Genetics 2012; 13:271–282. [DOI] [PubMed] [Google Scholar]
- 20.Betel D, Koppal A, Agius P, et al. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol 2010; 11:R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen I, Chen CY, Chuang TJ. Biogenesis, identification, and function of exonic circular RNAs. Wiley interdisciplinary reviews. RNA 2015; 6:563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li J, Yang J, Zhou P, et al. Circular RNAs in cancer: novel insights into origins, properties, functions and implications. Am J Cancer Res 2015; 5:472–480. [PMC free article] [PubMed] [Google Scholar]
- 23.Guo JU, Agarwal V, Guo H, et al. Expanded identification and characterization of mammalian circular RNAs. Genome BiolV 15 2014; 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilusz JE, Sharp PA. Molecular biology. A circuitous route to noncoding RNA. Science 2013; 340:440–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong L, Deng J, Sun ZM, et al. Interference with the beta-catenin gene in gastric cancer induces changes to the miRNA expression profile. Tumour Biol 2015; 36:6973–6983. [DOI] [PubMed] [Google Scholar]
- 26.Warnecke-Eberz U, Chon SH, Holscher AH, et al. Exosomal onco-miRs from serum of patients with adenocarcinoma of the esophagus: comparison of miRNA profiles of exosomes and matching tumor. Tumour Biol 2015; 36:4643–4653. [DOI] [PubMed] [Google Scholar]
- 27.Li G, Qiu Y, Su Z, et al. Genome-wide analyses of radioresistance-associated miRNA expression profile in nasopharyngeal carcinoma using next generation deep sequencing. PLoS One 2013; 8:e84486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katayama Y, Maeda M, Miyaguchi K, et al. Identification of pathogenesis-related microRNAs in hepatocellular carcinoma by expression profiling. Oncol Lett 2012; 4:817–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas LF, Saetrom P. Circular RNAs are depleted of polymorphisms at microRNA binding sites. Bioinformatics 2014; 30:2243–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.